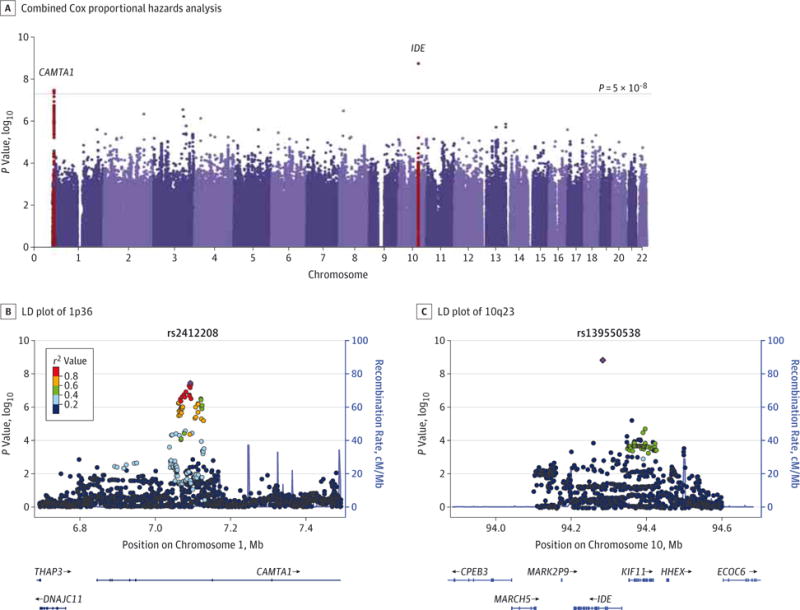
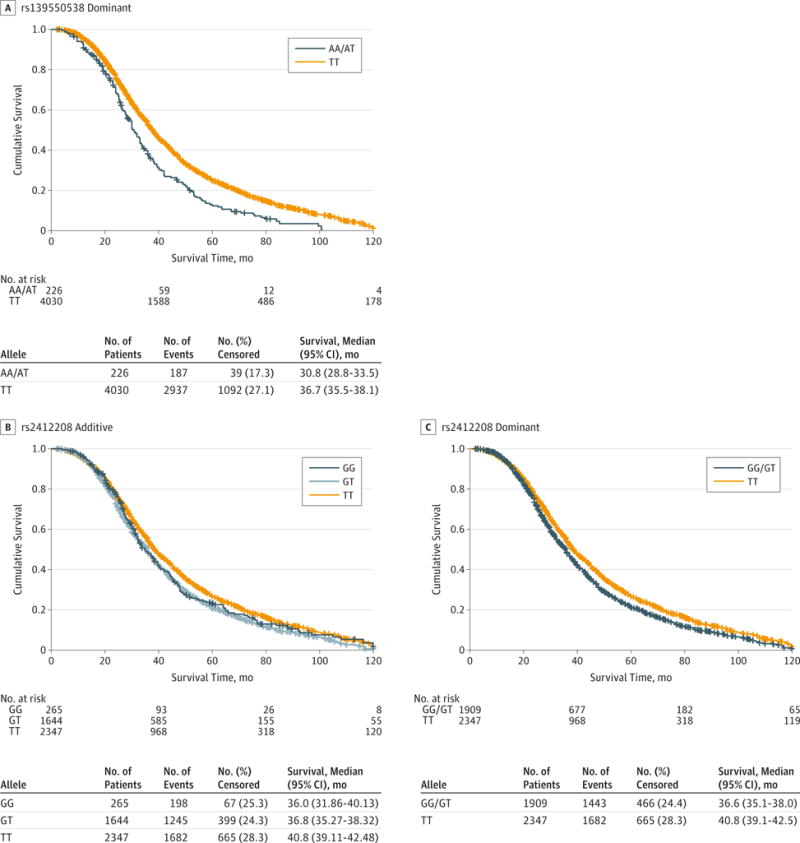
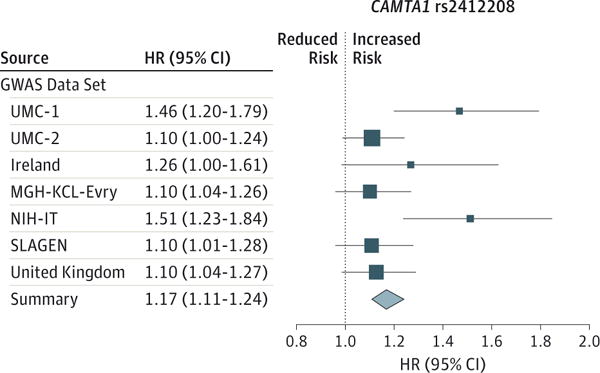
Isabella Fogh
Isabella Fogh, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Kuang Lin
Kuang Lin, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Cinzia Tiloca
Cinzia Tiloca, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
James Rooney
James Rooney, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Cinzia Gellera
Cinzia Gellera, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Frank P Diekstra
Frank P Diekstra, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Antonia Ratti
Antonia Ratti, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Aleksey Shatunov
Aleksey Shatunov, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Michael A van Es
Michael A van Es, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Petroula Proitsi
Petroula Proitsi, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Ashley Jones
Ashley Jones, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
William Sproviero
William Sproviero, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Adriano Chiò
Adriano Chiò, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Russell Lewis McLaughlin
Russell Lewis McLaughlin, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Gianni Sorarù
Gianni Sorarù, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Lucia Corrado
Lucia Corrado, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Daniel Stahl
Daniel Stahl, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Roberto Del Bo
Roberto Del Bo, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Cristina Cereda
Cristina Cereda, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Barbara Castellotti
Barbara Castellotti, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Jonathan D Glass
Jonathan D Glass, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Steven Newhouse
Steven Newhouse, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Richard Dobson
Richard Dobson, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Bradley N Smith
Bradley N Smith, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Simon Topp
Simon Topp, MSc
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Wouter van Rheenen
Wouter van Rheenen, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Vincent Meininger
Vincent Meininger, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Judith Melki
Judith Melki, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Karen E Morrison
Karen E Morrison, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Pamela J Shaw
Pamela J Shaw, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
P Nigel Leigh
P Nigel Leigh, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Peter M Andersen
Peter M Andersen, MD, DMSc
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Giacomo P Comi
Giacomo P Comi, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Nicola Ticozzi
Nicola Ticozzi, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Letizia Mazzini
Letizia Mazzini, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Sandra D’Alfonso
Sandra D’Alfonso, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Bryan J Traynor
Bryan J Traynor, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Philip Van Damme
Philip Van Damme, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Wim Robberecht
Wim Robberecht, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Robert H Brown
Robert H Brown, MD, DPhil
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
John E Landers
John E Landers, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Orla Hardiman
Orla Hardiman, MD, FRCPI
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Cathryn M Lewis
Cathryn M Lewis, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Leonard H van den Berg
Leonard H van den Berg, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Christopher E Shaw
Christopher E Shaw, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Jan H Veldink
Jan H Veldink, MD, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Vincenzo Silani
Vincenzo Silani, MD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
Ammar Al-Chalabi
Ammar Al-Chalabi, PhD, FRCP
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1,
John Powell
John Powell, PhD
1Department of Basic and Clinical Neuroscience, Maurice Wohl Clinical Neuroscience Institute, Institute of Psychiatry, Psychology, and Neuroscience (IoPPN), King’s College London, London, England (Fogh, Lin, Shatunov, Proitsi, Jones, Sproviero, Smith, Topp, C. E. Shaw, Al-Chalabi, Powell); Department of Neurology and Laboratory of Neuroscience, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Istituto Auxologico Italiano, Milano, Italy (Tiloca, Ratti, Ticozzi, Silani); Academic Unit of Neurology, Trinity College Dublin, Trinity Biomedical Sciences Institute, Dublin, Ireland (Rooney); Unit of Genetics of Neurodegenerative and Metabolic Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milano, Italy (Gellera, Castellotti); Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht, the Netherlands (Diekstra, van Es, van Rheenen, van den Berg, Veldink); Department of Pathophysiology and Tranplantation, Dino Ferrari Center, Università degli Studi di Milano, Milano, Italy (Ratti, Ticozzi, Silani); Rita Levi Montalcini Department of Neuroscience, ALS (Amyotrophic Lateral Sclerosis) Centre, University of Torino, Turin, Italy (Chiò); Azienda Ospedaliera Città della Salute e della Scienza, Torino, Italy (Chiò); Population Genetics Laboratory, Smurfit Institute of Genetics, Trinity College Dublin, Dublin, Ireland (McLaughlin, Hardiman); Department of Neurosciences, University of Padova, Padua, Italy (Sorarù); Department of Health Sciences, Interdisciplinary Research Center of Autoimmune Diseases, A. Avogadro University, Novara, Italy (Corrado, Mazzini, D’Alfonso); Department of Biostatistics, IoPPN, King’s College London, London, England (Stahl); Neurologic Unit, IRCCS Foundation Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy (Del Bo, Comi); Laboratory of Experimental Neurobiology, IRCCS C. Mondino National Institute of Neurology Foundation, Pavia, Italy (Cereda); Department of Neurology, Emory University, Atlanta, Georgia (Glass); National Institute for Health Research (NIHR) Biomedical Research Centre for Mental Health, IoPPN, King’s College London, London, England (Newhouse, Dobson); Department of Biostatistics, IoPPN, King’s College London, London, England (Newhouse); NIHR Biomedical Research Unit in Dementia, King’s College London, London, England (Dobson); Département des Maladies du Système Nerveux, Assistance Publique–Hôpitaux de Paris, Réseau SLA (Sclérose Latérale) Île de France, Hôpital Pitié-Salpêtrière, Paris, France (Meininger); Institut National de la Santé et de la Recherche Medicale Unité Mixte de Recherché–788 and University of Paris 11, Bicetre Hospital, Paris, France (Melki); School of Clinical and Experimental Medicine, College of Medicine and Dentistry, University of Birmingham, Birmingham, England (Morrison); Neurosciences Division, University Hospitals Birmingham National Health Service Foundation Trust, Birmingham, England (Morrison); Academic Neurology Unit, Department of Neuroscience, Faculty of Medicine, Dentistry and Health, University of Sheffield, Sheffield, England (P. J. Shaw); Section of Neurology, Division of Medicine, Brighton and Sussex Medical School, Trafford Centre for Biomedical Research, University of Sussex, East Sussex, England (Leigh); Institute of Clinical Molecular Biology, Kiel University, Kiel, Germany (Andersen); Department of Pharmacology and Clinical Neuroscience, Umeå University, Umeå, Sweden (Andersen); ALS Center Department of Neurology, Maggiore della Carità University Hospital, Novara, Italy (Mazzini); Neuromuscular Diseases Research Section, Laboratory of Neurogenetics, National Institute on Aging, National Institutes of Health, Bethesda, Maryland (Traynor); Department of Neurosciences, Experimental Neurology, Flanders Instititue for Biotechnology, Vesalius Research Center, Laboratory of Neurobiology, KU Leuven–University of Leuven, Leuven, Belgium (Van Damme, Robberecht); Department of Neurology, University Hospitals Leuven, Leuven, Belgium (Van Damme); Department of Neurology, University of Massachusetts Medical School, Worcester (Brown, Landers); IoPPN Genomics and Biomarker Core, Translational Genetics Group, Medical Research Council Social, Genetic and Developmental Psychiatry Centre, King’s College London, London, England (Lewis); Department of Medical and Molecular Genetics, King’s College London, London, England (Lewis)
1