

Abstract
Connexin 43 (Cx43) is a protein expressed in a variety of mammalian tissues. However, the lack of specific blockers and the absence of known genetic mutants have hampered the investigation of the function of this protein. Cx43-null mice die shortly after birth, thus preventing functional studies in vivo. Here, we report the generation and characterization of a vascular endothelial cell-specific deletion of the Cx43 gene (VEC Cx43 KO) in mice by using the loxP/Cre system. Using homologous recombination, a mouse line was created carrying loxP sites flanking exon 2 of the Cx43 gene (“floxed” mice). To produce cell specific deletion of the Cx43 gene, these mice were crossed with animals from a line carrying the Tie 2-Cre transgene. The homozygous VEC Cx43 KO mice survived to maturity. However, they were hypotensive and bradycardic when compared with heterozygous VEC Cx43 KO mice, or to the floxed Cx43 gene mice. The hypotension was associated with marked elevation of plasma nitric oxide (NO) levels as well as elevated plasma angiotensin (Ang) I and II. We hypothesize that endothelial cell Cx43 plays a key role in the formation and/or action of NO, and that the elevation of Ang II is a secondary event. The specific cellular basis for the hypotension remains to be established, but our findings support the idea that endothelial Cx43 gap junctions are involved in maintaining normal vascular function; moreover, these animals provide the opportunity to determine more clearly the role of endothelial Cx43 in vascular development and homeostasis.
Gap junctions have been found in almost all mammalian tissues, with the exception of circulating red blood cells and adult skeletal muscle. In many organs, such as the heart, they play key roles in coordinating cellular function, by providing a pathway for passage of electrical and/or second messenger signals from one cell to another (1). Four connexins—Cx37, -40, -43, and -45—are expressed in the cardiovascular system (2). Cardiac muscle cells express Cx43 and -40, vascular smooth muscle (VSM) cells express Cx40, -43, and -45, and endothelial cells express all but Cx45 (3). The common presence of connexins in the vasculature has led to the speculation that gap junctional communication may play a key role in other aspects of cardiovascular function. Correlative studies have shown that changes in Cx43 expression are associated with hypertension (4, 5), postischemic damage (6), and the development of atherosclerosis (7). However, causal relationships linking the pathologies with connexin function have yet to be established. The lack of specific blockers or well defined animal models has precluded a clear demonstration of the specific role that these molecules may play in vascular function.
A knockout strategy would be an attractive approach to study gap junctional function in the vasculature, but knockout of Cx43 is lethal in early postnatal life (8). Moreover, knockout of Cx40 results in only slight alterations in vasomotor conduction and a modest elevation of blood pressure (9), and Cx37 knockout animals have yet to manifest any vascular phenotype (10). These facts led us to attempt to overcome the disadvantages of a global gene deletion and associated developmental defects or premature death, by producing cell-specific deletions by using the Cre/loxP recombination system (11). The structures of the 14 different connexin genes identified to date are quite similar, consisting of two exons separated by a long intron. All of the protein coding information resides in the second exon (12, 13). Based on this information, we created a mouse line in which exon 2 of Cx43 was flanked by loxP sites. Cell-specific gene knockout was then achieved by breeding these animals with a line of mice carrying the Tie 2 promoter-enhancer driving the Cre gene. The Tie 2 promoter has been shown to be highly specific for endothelium (14). This approach yielded an animal with the gene deletion restricted to those cells in which Cre recombinase was expressed. Here, we report the generation and characterization of the resultant vascular endothelial cell-specific Cx43 knockout (VEC Cx43 KO) mice.
Materials and Methods
Production of Targeted Embryonic Stem Cells and the Floxed Cx43 Gene Mice.
The Cre recombinase encoded by the phage P1 is a 38-kDa protein that efficiently catalyzes deletion of a segment of DNA bounded by two 34-bp nucleotide sequences termed loxP sites (15). Cre recombinase deletes the DNA segment bounded by the loxP sites and recombines the two ends of the DNA strand (16, 17). To produce selective deletion of the 2nd exon of Cx43, we used the targeting construct shown in Fig. 1. The construct contains a DNA sequence homologous for the 2nd exon of Cx43, three loxP sites, a neomycin/thymidine kinase cassette (neo/TK), and the gene for diphtheria toxin (DTA). The neo/TK and DTA were used as selection markers. The construct was 14 kb in length, and the 5′ and 3′ homologous sequences for targeting were 2 and 1.2 kb, respectively. Embryonic stem cells (ES cells, AB2.2-prime, Lexicon Genetics, Woodlands, TX) were transfected with the linearized targeting construct, and subjected to G418 selection (400 μg/ml, GIBCO/BRL) for 10 days to identify cells that had internalized the construct. Cells that retained the DTA (i.e., had not undergone homologous recombination at the 3′ end) did not survive. The remaining clones were screened for homologous recombination by using two sets of PCR primers, YL30/YL34 and YL35/YL16, to detect 5′ and 3′ recombination events, respectively (see Fig. 1). The PCR-positive clones were then confirmed by Southern blot by using a 340-bp HaeIII-AccI fragment of the Cx43 intron sequence as a probe (Probe A, Fig. 1). To eliminate potential deleterious effects of the selection marker genes, we excised the neo/TK fragments from those ES cell clones with confirmed homologous recombination. Excision was accomplished by transfection with supercoiled Cre-encoding plasmid, and then the cells were subjected to gancyclovir selection for 7 days (1 × 106 M). The treatment produced resistant clones of two types, as shown in the Lower section of Fig. 1. We identified three of the desired type II deletion ES cell clones by PCR screens, and these were injected into C57BL/6 blastocysts; two yielded chimeric mice. The chimeric mice were backcrossed to C57BL/6 mice to produce heterozygous and homozygous floxed Cx43 gene mice.
Figure 1.

Targeting construct and screening strategies for the generation of floxed Cx43 mice. Schematic maps for the wild-type Cx43 gene-targeting construct, the homologous recombinant allele, and type I and type II deletions produced by transient Cre expression in stem cells after homologous recombination are shown. The large arrows represent the selection marker genes: neo, TK, and DTA. The thick solid black line in each map represents the native intron sequence of Cx43 gene, and the red dashed line represents the Cx43 gene intron sequence, which is absent in the targeting construct. The positions of PCR primers and the StuI restriction enzyme cut sites used for each screening step are displayed at the approximate locations. The red triangles represent the loxP sites; the thick pink line (Probe A) represents the labeled DNA fragment used for Southern blot analysis.
Immunohistochemistry.
Mice were anesthetized and perfused with 2% paraformaldehyde through the left ventricle; the hearts and aortas were removed and subjected to 2 h of post-fixation. One set of cardiac samples was frozen, embedded in OCT (Sakura Finetek, Torrance, CA) and sectioned (12-μm thickness). A second set of hearts and the aortas were embedded in paraffin, sectioned (6 μm), and deparaffinized by standard procedures. Sections were placed on charge-coated slides, washed three times in PBS, placed in blocking/permeabilization solution (2% BSA/4% normal goat serum/0.02% Triton X-100 in PBS) for 1 h and then incubated with rabbit anti-Cx43 (1:500 dilution in PBS; Chemicon) overnight at 4°C. The slides were then washed three times with PBS and incubated with Cy 5-labeled goat anti-rabbit secondary antibody (Molecular Probes) for 1 h at room temperature. The immunoreactivity was examined by using the Olympus (New Hyde Park, NY) Fluoview confocal microscope.
Southern Blot—Confirmation of Gene Deletion.
Endothelium comprises only a small fraction of the vessel wall, and, to establish the extent of deletion, we used endothelial cells that were isolated from mouse lungs of floxed Cx43 and VEC Cx43 KO mice (18). Endothelial cells were cultured on Matrigel (BD Biosciences, Franklin Lakes, NJ) coated plates and incubated in DMEM-F12 medium (GIBCO/BRL) supplemented with 15% FBS, 100 units/ml of penicillin, 100 μg/ml of streptomycin, 90 units/ml of heparin, 10 μg/ml of mouse epidermal growth factor (EGF), and 150 μg/ml of endomitogen (Biological Technologies, Stoughton, MA). Endothelial cells were confirmed by positive staining with platelet-endothelial cell adhesion molecule (PECAM) antibody (Santa Cruz Biotechnology). Genomic DNA extracted from the primary cultured lung endothelial cells was used for Southern blot analysis by using Probe A (see Fig. 1).
Western Blot—Assay of Protein Production.
Tissue samples were homogenized in lysis buffer. Approximately 20 μg of protein for each were separated by 12% SDS/PAGE, transferred to a nitrocellulose filter, and incubated with rabbit anti-Cx43 (1:300 dilution). Samples were treated with peroxidase-conjugated secondary antibody (1:1000 dilution), and immunoreactivity was detected by using the ECL chemiluminescent detection kit according to the manufacturer's specifications (Amersham Pharmacia). The intensity of the resulting bands was quantified by densitometry.
Measurement of Blood Pressure and Heart Rate.
As a first screen for a cardiovascular phenotype, mean systolic blood pressure (MSBP) and heart rate were obtained from tail cuff measurements on 11- to 16-wk-old mice by using the Visitech System (Physiological Research Instruments, Apex, NC). Mice were grouped based on genotypes (floxed, heterozygous, and homozygous VEC Cx43 KO); each group contained 10 to 12 mice. The measurements were conducted in a dark, warm, and quiet environment. Two to four records were collected for each mouse on each day, and these procedures were repeated three to four times on different days. At least six measurements were collected for each mouse. Data were pooled to obtain a single mean value for each mouse.
Nitric-Oxide Synthase (NOS) Activity and Plasma NO (Nitrite/Nitrate) Concentration.
Prior work from this lab has shown that gap junctional communication in the arterioles can modify NO synthesis (19). We therefore estimated NO production by measuring plasma nitrite and nitrate concentration. Mice (11 to 20 wk old) were anesthetized by pentobarbital sodium (80 mg/kg), and blood samples were withdrawn by cardiac puncture by using EDTA to prevent coagulation. One hundred microliters of plasma was deproteinized by adding 2 vol. of cold ethanol and centrifuged at 13,000 rpm for 30 min. The supernatant was transferred to a new 1.5-ml tube. After reduction of NOx to NO by VCl3 according to the manufacture's protocol, a 50-μl aliquot was used for the measurement of the nitrite and nitrate concentration with the NO analyzer (NO A280, Sievers, Boulder, CO).
Measurement of Plasma Ang I and Ang II Levels.
The VSM and endothelium of the afferent arterioles are extensively connected by gap junctions, and this is a site of very dense gap junctional plaque formation (20). We therefore assessed plasma Ang I and II concentration. Samples were transferred into ice-cold 1.5-ml tubes, and spun at 12,000 rpm for 10 min at 4°C. Twelve microliters of ice-cold plasma containing 25 mM EDTA was used for RIA of Ang I (estimate of plasma renin activity). Two hundred fifty microliters of plasma containing 25 mM EDTA, 0.44 mM o-phenanthroline, 1 mM p-hydroxy-mercuribenzoic acid, and 0.12 mM pepstatin A was used for measurement of Ang II concentration by using a RIA kit (SPI Bio, Paris). Duplicate measurements were conducted for each sample, and the average values were used for statistical analysis.
Statistics.
All values are expressed as mean ± SEM. One-way ANOVA was used for statistical analysis of group comparisons. P < 0.05 was considered as significant.
Results
Generation and Characterization of Floxed Cx43 Gene Mice.
The neo-resistant ES cell clones were tested for 5′ and 3′ homologous recombination by using the PCR pairs YL30/YL34 and YL35/YL16, respectively. As shown in Fig. 1, the sense primer YL30 is upstream of the Cx43 exon 2 but located outside the targeting construct, and the antisense primer YL34 is located downstream of the first loxP site. This PCR pair produces a 2.2-kb band from the native allele and a 2.3-kb band as a result of the 5′-homologous recombination. No PCR product would be generated from the targeting construct because of the lack of the sense primer. Thus, the clones in which 5′-homologous recombination occurred would show double-bands of 2.2 kb and 2.3 kb (Fig. 2A). Similarly, at the 3′-end, the sense primer YL35 is located in the neo gene, and the antisense primer YL16 is located downstream of the Cx43 exon 2 but not present in the targeting construct. Therefore, clones bearing a homologous recombination at the 3′-end produce a 1.4-kb band (Fig. 2B). Candidate clones identified by PCR analysis were confirmed by Southern blot analysis by using Probe A. After digestion by StuI, the homologous recombination clones were predicted to give a 7.8-kb band in addition to a 14-kb band from the native allele (Fig. 2C). A total of 456 neo-resistant ES cell clones were obtained and characterized, and 6 clones were confirmed to have undergone expected homologous recombination, representing a targeting frequency of 1 in 76 neo-resistant clones.
Figure 2.

PCR and Southern blot analysis used in the identification of the homologous recombination event. (A) ES cell clones showing homologous recombination in the 5′-region show double-bands. (B) Samples with a 1.4-kb band represent ES cell clones with homologous recombination at the 3′-region. (C) Southern blot analysis with Probe A (see Fig. 1). Both the 14-kb (solid arrow) and 7.8-kb (open arrow) bands were present in clones in which homologous recombination occurred at the 5′-end whereas wild-type ES cells or clones that obtained the targeting construct through random integration have only the 14-kb band. (D and E) PCR analyses focused on the loxP sites. Samples with double-bands represent EC cell clones possessing two lox P sites. (F) Samples with or without the 686-bp band represent type I and II deletion, respectively. Primer locations are shown in Fig. 1 and their sequences are as follows: YL16, GAAGCCATGGCAAGAGAATGTAGAGGC; YL18, CTTTGACTCTGATTACAGAGCTTAA; YL30, CATCCTTCAATGTGTATGTACAGG; YL34, GTCTCACTGTTACTTAACAGCTTGA; YL35, CTACCCGGTAGAATTGACCTGCAG; YL45, CCCACCTTTGTGTCTTCCATATATT; YL46, TATCATTCAAGCATCAATGATTATA; YL49, GCTACTTCTTGCTTTGACTCTGATTA; and YL50, GCTCACTTGATAGTCCACTCTAAGC.
Three PCR primer sets (for locations, see Fig. 1) were used to confirm the type II deletion (Fig. 2 D, E, and F). A total of 300 gancyclovir-resistant clones were characterized, and 34 clones showed type II deletion, indicating approximately a 1:10 ratio of type II–type I recombination, a frequency of type II deletion similar to that reported previously (21).
Expression of Cx43 Gene in Floxed Cx43 Gene Mice Is Unaltered.
To achieve cell specific and/or conditional gene knockout, it is critical that the insertions of loxP sites not interfere with expression of the floxed gene and normal protein production. Six months of inbreeding of heterozygous mice for the floxed Cx43 gene yielded 103 pups—27 wild-type (23.5%), 65 heterozygous (56.5%), and 23 homozygous (20%)—which is close to the Mendelian ratio. Survival, Mendelian breeding patterns, and an apparently normal phenotype of mice homozygous for the floxed Cx43 gene are all indications of normal gene expression in the presence of loxP sites. We also used immunohistochemistry and Western blot analysis to assess the levels of Cx43 gene expression in heart tissue from floxed gene mice and wild-type mice. Results showed that the expression level and cellular distribution of the Cx43 gene in hearts of heterozygous or homozygous floxed Cx43 gene mice were unchanged, as compared with those of wild-type mice (Fig. 3).
Figure 3.

(A) Immunohistochemical examination showing the cellular distribution of Cx43 protein in mouse hearts. (A1) Negative control stained with secondary antibody only. A2, -3, and –4 represent, respectively, wild-type, heterozygous, and homozygous mice for the floxed Cx43 gene. The Cx43 immunostaining (red) is mainly located at the intercalated discs and no apparent difference was observed among mice from these three genotypes. (B) Quantitative measurement of Cx43 gene expression by Western blot analysis. Approximately 20 μg of protein from each sample was used. Lanes 1, 2, and 3 are, respectively, wild-type, heterozygous, and homozygous mice for the floxed Cx43 gene. The Cx43 expression in the floxed Cx43 gene mice was unchanged as compared with the wild-type control mice.
Generation of VEC Cx43 KO Mice.
To accomplish the endothelial specific Cx43 gene deletion, mice carrying a transgene in which the Tie 2 promoter-enhancer was used regulate Cre expression selectively in endothelial cells (see below for discussion of specificity). To identify the correct genotypes at each mating step, three PCR primer sets—Cre-PCR (Cre SP: AGGTGTAGAGAAGGCACTTAGC and Cre ASP: CTAATCGCCATCTTCCAGCAGG), YL18/YL34, and YL45/YL46—were used to detect the Cre gene and the loxP sites.
Deletion of the Cx43 Gene Is Efficient and Restricted to the Vasculature.
To assess the specificity of the deletion of the Cx43 gene, genomic DNA samples were isolated from white blood cells, tail, aorta, brain gray matter, and brain vessels of mice homozygous for the VEC Cx43 KO. Equal amounts of DNA (100 ng) from each sample were used for PCR analysis by using primer set YL49/YL50, which was designed to amplify a 686-bp fragment from the deleted Cx43 allele (see Fig. 1). As shown in Fig. 4A, the intensity of the 686-bp deletion band was proportional to the richness of the endothelium in the tissue and was highest in DNA from brain vessels, second in aorta, and not detectable in samples from white cells and brain gray matter.
Figure 4.

Specificity and efficiency of the deletion of Cx43 gene in the endothelium. (A) PCR analysis of Cx43 deletion in a variety of tissues of homozygous VEC Cx43 KO mice. DNA preparations from white blood cells (WBC), brain gray matter (BGM), tail, aorta (AA), and brain vessels (BV) were analyzed. (B) Southern blot analyses. Genomic DNA samples were isolated from BGM, AA, and cultured lung endothelial cells (LEC) and assayed with Probe A. Open and closed arrows represent the positions of floxed (8-kb) and deleted (5-kb) Cx43 alleles, respectively. The first three lanes represent samples from floxed Cx43 gene mice; the next four lanes represent samples isolated from VEC Cx43 KO mice. AA− represents aortas with endothelial cells removed by scraping. (C) Cx43 immunostaining in floxed and VEC Cx43 KO mice. Sections from aortas were stained with Cx43 antibody (red) and visualized under confocal microscopy. The location of internal elastic lumen (IEL) is indicated by the arrow.
Southern blot was used to assess the efficiency of Cx43 deletion in cultured lung endothelial cells. After EcoRI digestion, Probe A hybridized with an 8-kb and a 5-kb band from the floxed and deleted Cx43 allele, respectively. As show in Fig. 4B, in VEC Cx43 KO mice, ≈80% of the Cx43 gene was deleted in the lung endothelial cells and the deletion of Cx43 gene was detectable in aorta, whereas no deletion of Cx43 gene was observed in DNA preparations from brain gray matter. After removal of the endothelium, no deletion band could be detected in DNA samples of aortas from VEC Cx43 KO mice (Fig. 4B, AA−). The deletion band was never detected in samples from the floxed Cx43 gene mice.
Further confirmation of the efficacy of the deletion was confirmed by immunohistochemistry of aortic sections, which showed virtual elimination of Cx43 immunostaining in endothelium of VEC Cx43 KO mice as compared with that of floxed Cx43 gene mice. A remarkable finding in these experiments however, was that the Cx43 immunostaining was also significantly reduced in the VSM of the VEC Cx43 KO mice (Fig. 4C).
Hypotension and Bradycardia in Homozygous VEC Cx43 KO Mice.
Obviously, the endothelium plays a critical role in the regulation of cardiovascular function. A possible involvement of gap junctional communication in maintaining the vessel tone and blood pressure is suggested by recent work on the Cx40-null mice (9). Moreover, in previous work, this laboratory has shown that endothelial cell NO synthesis in the arterioles is at least in part determined by interchange of calcium between VSM and endothelium (19). Therefore, MSBP and heart rate were obtained from tail cuff measurements (Fig. 5), and statistical analysis showed that both were significantly lower in homozygous VEC Cx43 KO mice than in floxed Cx43 gene mice or heterozygous VEC Cx43 KO mice.
Figure 5.

Mean systolic blood pressure (MSBP) and heart rate of floxed Cx43 gene mice (Floxed), heterozygous (Hetero) mice, and homozygous (Homo) mice for VEC Cx43 KO. Graphs represent MSBP (Left) or heart rate (Right); error bars represent standard error (SE). The number of mice in each group appears in parentheses. *, Difference between the two groups is significant (P < 0.05).
We were concerned that the hypotension in the VEC Cx43 KO mice might have been related to strain differences because the stem cells were derived from 129Sv strain and the blastocysts were obtained from the C57BL/6 strain. Therefore, we also measured the blood pressure and heart rate in these two strains as well as the Tie 2-Cre transgenic animal (BL6/SJL strain). Blood pressures were 106 ± 4, 102 ± 3, and 108 ± 4 for the 129Sv, C57BL/6, and Tie 2-Cre transgenic mice, respectively, and none of the pressures or heart rates were significantly different from the data from the homozygous floxed allele animals.
Changes of Plasma NO, Ang I, and Ang II Concentration in VEC Cx43 KO Mice.
To investigate the causes of the reduced blood pressure in the VEC Cx43
KO animals, we measured plasma levels of NO and of Ang I and II
concentrations, two central regulators of blood pressure. NO has a
short half-life (T1/2 is a few seconds to a
minute), and it is rapidly oxidized to the stable, inactive end
products, nitrite and nitrate (NO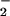 and
NO
and
NO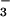 ). We estimated the NO concentration by measuring
the sum of the plasma concentrations of nitrite and nitrate. Fig.
6 shows that the plasma NO concentrations
were markedly increased in both heterozygous and homozygous VEC Cx43 KO
mice, as compared with the floxed Cx43 mice. Unexpectedly, both plasma
Ang I and Ang II concentration were also markedly increased after
endothelial Cx43 gene deletion (Fig.
7).
). We estimated the NO concentration by measuring
the sum of the plasma concentrations of nitrite and nitrate. Fig.
6 shows that the plasma NO concentrations
were markedly increased in both heterozygous and homozygous VEC Cx43 KO
mice, as compared with the floxed Cx43 mice. Unexpectedly, both plasma
Ang I and Ang II concentration were also markedly increased after
endothelial Cx43 gene deletion (Fig.
7).
Figure 6.

Plasma NO concentration in floxed Cx43 gene mice (Floxed), heterozygous (Hetero) mice, and homozygous (Homo) mice for VEC Cx43 KO. The number of mice in each group appears in parentheses. *, Difference between the two groups is significant (P < 0.05).
Figure 7.

Plasma Ang I (Left) and Ang II (Right) concentration of floxed Cx43 gene mice (Floxed), heterozygous (Hetero) mice, and homozygous (Homo) mice for VEC Cx43 KO. The number of mice in each group appears in parentheses. *, Difference between the two groups is significant (P < 0.05).
Discussion
Alterations in Blood Pressure in the VEC Cx43 KO Mice.
Correlation between Cx43 and blood pressure has previously been noted, in the case of hypertension (22) and elasticity of the vascular wall (23). However, the finding of hypotension in the VEC Cx43 KO mice was unexpected, especially in view of the fact that Cx40 knockout mice demonstrate hypertension (9). As shown above, the change in blood pressure may be quite complex, with alterations in both circulating dilators and constrictors. Our working hypothesis is that the initial event is an elevation in NO synthesis and a compensatory elevation in Ang II. The cellular basis for the initial event remains to be established, however. Also, it is important to note that, at this time, an unknown developmental change cannot be distinguished from an ongoing failure in gap junctional communication as a cause for the hypotension.
Elevated Plasma NO Concentration May Be a Primary Effect of Deletion of Cx43 Gene in Endothelium.
The plasma concentration of NO derivatives increased almost 50% in the VEC Cx43 KO animals as compared with the floxed Cx43 gene control mice (Fig. 6). Because plasma NO is mainly generated by endothelial NOS (eNOS), it is most likely that the plasma NO concentration is the result of a change in eNOS activity rather than inducible or neuronal NOS (24, 25).
Recent evidence from our laboratory shows the potential for a strong association between myoendothelial junctions and the formation of NO in the endothelium (19). We have shown that gap junctional communication provides a structural basis for coordinated vasoconstriction or vasodilation, and for extensive cross-talk between cells of vascular wall. Moreover, molecular signaling between endothelium and VSM cells can be achieved through myoendothelial junctions, and we now recognize that VSM cells and endothelium share cytoplasmic Ca2+ pools via gap junctions (19). Thus, NO synthesis is, at least under some circumstances, dependent on gap junctional exchange between VSM and endothelium (19). Therefore, we hypothesize that the hypotension observed in homozygous VEC Cx43 KO mice may have resulted in an elevation of endothelial cell calcium as a result of deletion of Cx43 in either the myoendothelial junctions or the homocellular endothelial cell junctions.
Elevation of Plasma Ang I and II.
Our results showed that the plasma Ang I and II levels in homozygous VEC Cx43 KO mice were doubled as compared with the floxed Cx43 gene control mice (Fig. 7). It seems most likely that the elevated Ang II may be a compensatory response to an initial blood pressure drop induced by NO. We note, however, that Cx43 might be more directly involved in the control of renin secretion from the juxtaglomerular apparatus (JGA) because Cx43 proteins are extremely abundant in kidney (26, 27), where gap junctional communication is extensive, including connections between VSM and endothelium (20). Thus, the deletion of Cx43 in the endothelium might inhibit the communication between endothelium and JGA and result in an altered secretion of renin.
Observations on Bradycardia in VEC Cx43 KO Mice.
Obviously, the bradycardia was unexpected and the mechanism is unknown. This is an important observation, however, because bradycardia causes a decrease in cardiac output and consequently could contribute to the hypotensive phenotype. It is possible that the bradycardia is secondary to the complex, multifactorial cardiovascular adaptations that are obviously occurring. An alternative explanation, however, is suggested by the fact that the Tie 2 promoter is also active in the endocardium. This acitivity of Tie 2 promoter is clearly demonstrable by observing the tissues of Tie 2-enhanced green fluorescent protein (EGFP) transgenic mice (28), and is not unexpected because the endocardium has the same embryonic origin as vascular endothelium. One can speculate that the endocardium might participate in the electrical conduction of the heart, and that the bradycardia is in some way associated with the deletion of Cx43 gene in the endocardium.
Specificity in Cre Recombinase Deletions.
A serious concern with any Cre recombinase-based knockout approach is the selectivity of the deletion, especially because the production of Cre recombinase, even during a very brief period at any stage of life, has the potential to permanently delete a floxed gene (29). At present, it is very difficult to examine the distribution of the Cre protein directly, because no satisfactory Cre antibody has been produced that functions adequately in mouse tissues. One must therefore resort to indirect strategies to determine Cre expression. Motoike et al. have reported that the expression of the indicator protein EGFP, under control of the Tie 2 promoter, is restricted to endothelium over a wide range of developmental stages (28). We have also exhaustively examined the distribution of GFP under the control of the Tie 2 promoter in the adult mice (unpublished results). We found it only in the endothelium and in the endocardium.
In addition, we have evaluated the specificity of Cre expression by using a so-called “indicator mouse” carrying a transgene for LacZ, driven by a promoter whose activity is blocked by a loxP-flanked stop codon (CAG-CAT-Z; ref. 30). In the presence of Cre recombinase, the CAT element is deleted, which allows expression of LacZ. A cross of the Tie 2-Cre mouse with the CAG-CAT-Z mouse has been reported to result in selective LacZ staining in the endothelium (31), and we have confirmed this finding by using the mouse line that produced the endothelial cell specific deletion. Thus, we feel that the likelihood is high that the effects of the genetic manipulations that we have accomplished are due to selective deletion of Cx43 in the endothelium.
In addition to high selectivity, the Cre-based approach is quite effective. Based on the Southern blot shown in Fig. 4B, we estimate the efficacy of the deletion to be 80–90%. The wild-type DNA that remains might reflect several sources. It may be that Cre was not adequately expressed in a subset of the population. Alternatively, Cre might delete only a single copy of the floxed gene because of low levels of expression or to unidentified cellular differences. Finally, it might be due to a contaminant cell type other than endothelial cells.
Examination of Fig. 4C shows that immunostaining in VSM is reduced, as well as that in the endothelium. This was a consistent observation in several animals examined. This reduction is not due to Cre reaching the smooth muscle, however, because no Cx43 deletion band was observed in VEC Cx43 KO aortas after the endothelial cells were removed (Fig. 4B, lane AA−). Thus, the reduction in Cx43 in the smooth muscle is the result of altered transcription or translation, but not the absence of the gene. Recently, Nelles et al. reported an effect that may explain the alteration of Cx43 in VSM with deletion in the endothelium. They found that deletion of Cx32 in the liver caused a reduction of Cx26, and they suggested that the connexins might be coordinately regulated (32). We therefore hypothesize that a similar regulatory process might occur between the VSM and the endothelium, such that the pools of Cx43 in the two cells are coordinated, most likely via the myoendothelial junctions.
Potential Impact of the Floxed Cx43 Gene Mice.
Cx43 gap junctional communication is widely distributed in many different cell types and may be involved in many biological events, such as coordination of the contraction of cardiac and VSM cells (33), control of cell growth (6, 34), carcinogenesis, transmission of neuronal signals at electronic synapses, and coordination of developmental events between pancreatic epithelia cells and between cells during embryogenesis (35, 36). The floxed Cx43 gene mouse can be used with any transgenic animal possessing a cell-specific promoter driving the production of Cre recombinase. The availability of these floxed Cx43 gene mice, combined with a large number of tissue-specific Cre mice will provide a unique opportunity to study the function of Cx43 gap junction in diverse cell lineages.
In summary, our initial observations show that the VEC Cx43 KO mice produce a complex phenotype that has profound implications for our understanding of cardiovascular function. The simplest interpretation of the data is that deletion of Cx43 gap junctions in the endothelium causes a primary rise in NO that tends to lower the blood pressure and that the Ang II levels are elevated as a secondary event. We do not yet know how NO synthesis is connected to endothelial cell gap junctional function. Other possibilities, such as alteration in sympathetic/parasympathetic nerve activities, changes in peripheral resistance, and impairment of the baroreflex system, might also explain the hypotensive and bradycardic phenotype in VEC Cx43 KO mice. As the cellular and molecular basis for these changes is investigated, the importance of Cx43 to cardiovascular function should be clarified.
Acknowledgments
We thank Ms. Valerie Stewart (Transgenic/Knockout Facility, Lerner Institute at Cleveland Clinic Foundation) for the injection of ES cells, Dr. Janet Rossant (Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, Canada) for the Cx43 genomic DNA fragment, Dr. Jun-ichi Miyazaki (Osaka University Medical School, Osaka, Japan) for the CAG-CAT-Z indicator mouse, Dr. Yaz Kisanuki and Dr. Masashi Yanagisawa (University of Texas Southwestern Medical Center) for the Tie 2-Cre transgenic mice, Dr. Thomas N. Sato (University of Texas Southwestern Medical Center) for the Tie 2-EGFP transgenic mice, the laboratory of Dr. Robert Carey for Ang I and Ang II measurement, the University of Virginia Research Histology Core for embedding tissues, and Dr. Cindy McKinney, Nick Douris, and Jennifer Bryant for technical assistance. This work was supported by National Institutes of Health Grants HL12792 and HL23531 (B.R.D.) and Academic Enhancement program on Gene Transfer and Gene Therapy and the Cardiovascular Research Center at the University of Virginia.
Abbreviations
- VSM
vascular smooth muscle
- neo
neomycin
- TK
thymidine kinase
- DTA
diphtheria toxin
- ES cell
embryonic stem cell
- MSBP
mean systolic blood pressure
- NOS
nitric-oxide synthase
- ANG
angiotensin
- VEC Cx43 KO
vascular endothelial cell-specific Cx43 knockout
References
- 1.Hotz-Wagenblatt A, Shalloway D. Crit Rev Oncog. 1993;4:541–588. [PubMed] [Google Scholar]
- 2.Simon A M, Goodenough D A. Trends Cell Biol. 1998;8:477–483. doi: 10.1016/s0962-8924(98)01372-5. [DOI] [PubMed] [Google Scholar]
- 3.Kruger O, Plum A, Kim J S, Winterhager E, Maxeiner S, Hallas G, Kirchhoff S, Traub O, Lamers W H, Willecke K. Development (Cambridge, UK) 2000;127:4179–4193. doi: 10.1242/dev.127.19.4179. [DOI] [PubMed] [Google Scholar]
- 4.Bastide B, Neyses L, Ganten D, Paul M, Willecke K, Traub O. Circ Res. 1993;73:1138–1149. doi: 10.1161/01.res.73.6.1138. [DOI] [PubMed] [Google Scholar]
- 5.Yamasaki H, Naus C C G. Carcinogenesis. 1996;17:1199–1213. doi: 10.1093/carcin/17.6.1199. [DOI] [PubMed] [Google Scholar]
- 6.Jara P I, Boric M P, Saez J C. Proc Natl Acad Sci USA. 1995;92:7011–7015. doi: 10.1073/pnas.92.15.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polacek D, Bech F, Mckinsey J F, Davies P F. J Vasc Res. 1997;34:19–30. doi: 10.1159/000159198. [DOI] [PubMed] [Google Scholar]
- 8.Reaume A G, De Sousa P A, Kulkarni S, Langille B L, Zhu D, Davies T C, Juneja S C, Kidder G M, Rossant J. Science. 1995;267:1831–1844. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 9.de Wit C, Roos F, Bolz S S, Kirchhoff S, Kruger O, Willecke K, Pohl U. Circ Res. 2000;86:649–655. doi: 10.1161/01.res.86.6.649. [DOI] [PubMed] [Google Scholar]
- 10.Simon A M, Goodenough D A, Li E, Paul D L. Nature (London) 1997;385:525–529. doi: 10.1038/385525a0. [DOI] [PubMed] [Google Scholar]
- 11.Sauer B, Henderson N. Proc Natl Acad Sci USA. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finbow M E, Pitts J D. J Cell Sci. 1993;106:463–471. doi: 10.1242/jcs.106.2.463. [DOI] [PubMed] [Google Scholar]
- 13.Sullivan R, Ruangvoravat C, Joo D, Morgan J, Wang B L, Wang X K, Lo C W. Gene. 1993;130:191–199. doi: 10.1016/0378-1119(93)90419-4. [DOI] [PubMed] [Google Scholar]
- 14.Schlaeger T M, Bartunkova S, Lawitts J A, Teichmann G, Risau W, Deutsch U, Sato T N. Proc Natl Acad Sci USA. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauer B, Henderson N. Gene. 1988;70:331–341. doi: 10.1016/0378-1119(88)90205-3. [DOI] [PubMed] [Google Scholar]
- 16.Abremski K, Hoess R, Sternberg N. Cell. 1983;32:1301–1311. doi: 10.1016/0092-8674(83)90311-2. [DOI] [PubMed] [Google Scholar]
- 17.Abremski K, Hoess R. J Biol Chem. 1984;259:1509–1514. [PubMed] [Google Scholar]
- 18.Gerritsen M E, Shen C P, McHugh M C, Atkinson W J, Kiely J M, Milstone D S, Luscinskas F W, Gimbrone M A. Microcirculation. 1995;2:151–163. doi: 10.3109/10739689509146763. [DOI] [PubMed] [Google Scholar]
- 19.Dora K A, Doyle M P, Duling B R. Proc Natl Acad Sci USA. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taugner R, Buhrle C P, Nobiling R. Cell Tissue Res. 1984;237:459–472. doi: 10.1007/BF00228430. [DOI] [PubMed] [Google Scholar]
- 21.Marth J D. J Clin Invest. 1996;97:1999–2002. doi: 10.1172/JCI118634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haefliger J A, Castillo E, Waeber G, Aubert J F, Nicod P, Waeber B, Meda P. Adv Exp Med Biol. 1997;432:71–82. doi: 10.1007/978-1-4615-5385-4_8. [DOI] [PubMed] [Google Scholar]
- 23.Haefliger J A, Meda P, Formenton A, Wiesel P, Zanchi A, Brunner H R, Nicod P, Hayoz D. Arterioscler Thromb Vasc Biol. 1999;19:1615–1622. doi: 10.1161/01.atv.19.7.1615. [DOI] [PubMed] [Google Scholar]
- 24.Geller D A, Billiar T R. Cancer Metastasis Rev. 1998;17:7–23. doi: 10.1023/a:1005940202801. [DOI] [PubMed] [Google Scholar]
- 25.Laubach V E, Shesely E G, Smithies O, Sherman P A. Proc Natl Acad Sci USA. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo R, Liu L, Barajas L. Am J Physiol. 1998;44:R439–R447. doi: 10.1152/ajpregu.1998.275.2.R439. [DOI] [PubMed] [Google Scholar]
- 27.Barajas L, Liu L, Tucker M. Kidney Int. 1994;46:621–626. doi: 10.1038/ki.1994.314. [DOI] [PubMed] [Google Scholar]
- 28.Motoike T, Loughna S, Perens E, Roman B L, Liao W, Chau T C, Richardson C D, Kawate T, Kuno J, Weinstein B M, et al. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 29.Regan C P, Manabe I, Owens G K. Circ Res. 2000;87:363–369. doi: 10.1161/01.res.87.5.363. [DOI] [PubMed] [Google Scholar]
- 30.Sakai K, Miyazaki J. Biochem Biophys Res Commun. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 31.Kisanuki Y Y, Hammer R E, Miyazaki J, Williams S C, Richardson J A, Yanagisawa M. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 32.Nelles E, Butzler C, Jung D, Temme A, Gabriel H D, Dahl U, Traub O, Stumpel F, Jungermann K, Zielasek J, et al. Proc Natl Acad Sci USA. 1996;93:9565–9570. doi: 10.1073/pnas.93.18.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruzzone R, White T W, Paul D L. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhu D, Kidder G M, Caveney S, Naus C C. Proc Natl Acad Sci USA. 1992;89:10218–10221. doi: 10.1073/pnas.89.21.10218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bennett M V, Barrio L C, Bargiello T A, Spray D C, Hertzberg E, Saez J C. Neuron. 1991;6:305–320. doi: 10.1016/0896-6273(91)90241-q. [DOI] [PubMed] [Google Scholar]
- 36.Bennett M V. J Neurocytol. 1997;26:349–366. doi: 10.1023/a:1018560803261. [DOI] [PubMed] [Google Scholar]