

SUMMARY
Select lectins have powerful anti-viral properties that effectively neutralize HIV-1 by targeting the dense glycan shield on the virus. Here, we reveal the mechanism by which one of the most potent lectins, BanLec, achieves its inhibition. We identify that BanLec recognizes a subset of high-mannose glycans via bidentate interactions spanning the two binding sites present on each BanLec monomer that were previously considered separate carbohydrate recognition domains. We show that both sites are required for high-affinity glycan binding and virus neutralization. Unexpectedly we find that BanLec adopts a tetrameric stoichiometry in solution whereby the glycan-binding sites are positioned to optimally target glycosylated viral spikes. The tetrameric architecture, together with bidentate binding to individual glycans, leads to layers of multivalency that drive viral neutralization through enhanced avidity effects. These structural insights will prove useful in engineering successful lectin therapeutics targeting the dense glycan shield of HIV.
Graphical abstract
INTRODUCTION
An estimated 37 million people worldwide are HIV positive, with the majority living in sub-Saharan Africa where infection rates disproportionately affect women due to gender inequality and social norms restricting intervention practices (World Health Organization, 2011). Eradicating HIV will therefore require parallel interventions to limit viral transmission, including physical barrier methods, anti-retrovirals, and continuing vaccine development, as well as social and behavioral education (Ananworanich and Fauci, 2015; Burton et al., 2012; François and Balzarini, 2012). A key role in this battle will likely be played by HIV microbiocides, agents that act to reduce the infectivity of the virus during sexual contact (Brichacek et al., 2013). Lectins have recently emerged as attractive microbiocide candidates which could be applied topically or even produced in situ by engineered commensal microbes (Lagenaur et al., 2015), and work by limiting transmission at mucosal surfaces (François and Balzarini, 2012; Lagenaur et al., 2015; Liu et al., 2006). Several lectins have potent anti-HIV properties in vitro and are therefore desirable starting points for development (Boyd et al., 1997; François and Balzarini, 2012; Mori et al., 2005; Swanson et al., 2010; Xiong et al., 2006).
Env, the viral spike of HIV, is a trimer of heterodimers of extensively glycosylated gp120 and gp41 subunits responsible for host-cell recognition and fusion (Julien et al., 2013). Glycosylation of Env acts to form a “glycan shield” that impedes antibody neutralization (Dacheux et al., 2004). However, infected individuals can develop broadly neutralizing antibodies (bnAbs) against the virus, some of which specifically target the glycan surface (Crispin and Doores, 2015; Ward and Wilson, 2015). Discrimination of the bnAbs against self-recognition is achieved by the large proportion of high-mannose glycans within the glycan shield (Behrens et al., 2016; Bonomelli et al., 2011; Doores et al., 2010; Pritchard et al., 2015a). This arises because, while the number of N-glycosylation sites varies between viral strains (Korber et al., 2001), their density is consistently high relative to human glycoproteins and results in limited cellular processing (Bonomelli et al., 2011). As a result, high-mannose type structures, which are a conserved feature of the virus, may be exploited in the design of vaccines, therapeutics, and prophylactics (Burton et al., 2012).
Lectins are carbohydrate-binding proteins and several members of the family have been shown to bind the HIV virus via its surface glycoproteins, blocking glycan-mediated interactions with the CD4 receptor and/or CCR5/CXCR4 co-receptors on host cells. One of the most potent anti-viral lectins, BanLec, has been shown to inhibit HIV fusion to HeLa cells with half-maximal inhibitory concentration (IC50) values in the low-nanomolar range (Swanson et al., 2010). While N-glycan specificity has been well characterized for many anti-viral lectins (Bewley and Otero-Quintero, 2001; Botos et al., 2002; Koharudin and Gronenborn, 2011; Moulaei et al., 2010; Shenoy et al., 2002), there is no equivalent understanding of the intermolecular interactions between BanLec and HIV glycans. However, from X-ray crystallography structures it has been suggested that BanLec is a dimeric protein with each monomer containing two carbohydrate-binding sites (Meagher et al., 2005; Sharma and Vijayan, 2011; Singh et al., 2004, 2005). Crystallographic and molecular dynamics (MD) studies have reported BanLec-binding specificity to disaccharides with α-1,3-mannosyl linkages in both binding sites (Sharma and Vijayan, 2011), and BanLec-HIV recognition is expected via mannose epitopes on high-mannose N-glycans. A binding model was proposed recently whereby each binding site acts independently to engage viral carbohydrates (Swanson et al., 2015), but was not tested with high-mannose N-glycans. Protein-carbohydrate interactions are typically weak, with milli- or micromolar affinity (Weis and Drickamer, 1996), but can be strengthened through multivalent interactions and glycan clustering (e.g., the Env glycan shield).
In this study we investigate BanLec-glycan recognition through a combination of structural and functional studies, and provide an explanation for the extraordinary specificity and anti-viral activity. We propose the first tetrameric structure of BanLec and show that carbohydrate-binding sites present on each monomer are not independent, but function jointly to engage a single, specific high-mannose glycan that bridges both sites simultaneously. These key mechanistic insights will aid successful development of lectin therapeutics with superior HIV neutralization through the modulation of multivalent interactions to N-linked glycans.
RESULTS
BanLec Inhibits HIV by Targeting Specific High-Mannose gp 120 Glycans
We first sought to identify the gp120 N-glycans targeted by BanLec. To do this we designed a depletion assay where fluorescently labeled N-glycans released from HIV gp120 (JRCSF strain) were incubated with increasing concentrations of BanLec. The unbound glycans were identified by hydrophilic interaction chromatography combined with high-performance liquid chromatography (HPLC). The glycans that possess the highest affinity for BanLec were then identified by their preferential depletion.
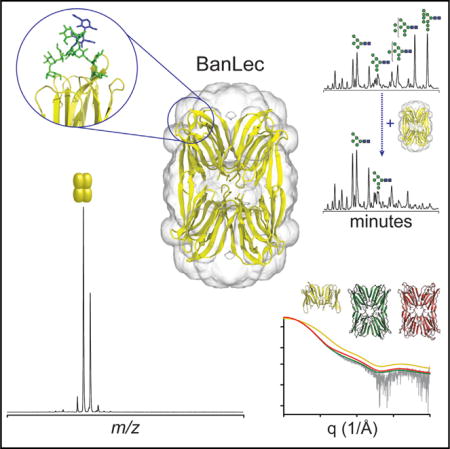
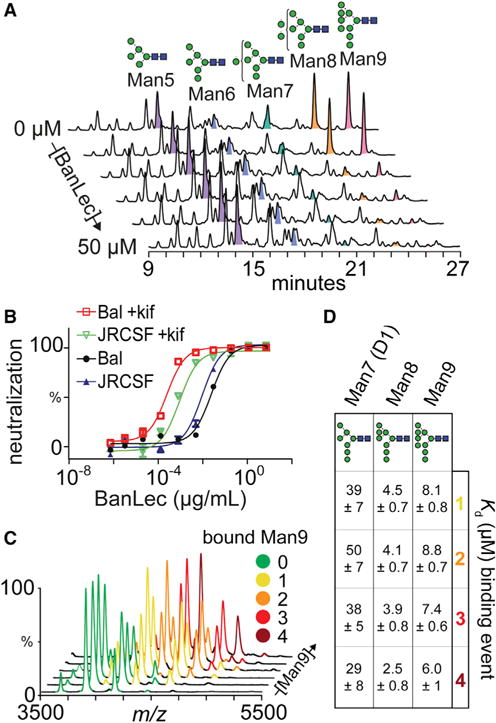
We found that the predominant N-glycans present were oligomannose structures (MannGlcNAc2, n = 5−9, referred to hereafter as Man5–9), with Man8 and Man9 the most abundant, in agreement with previous data (Bonomelli et al., 2011). Upon the addition of BanLec, the abundance of Man8 and Man9 structures, and to a lesser extent Man7, decreased considerably (Figure 1A). Surprisingly, we found that Man5 and Man6 were not depleted despite the presence of terminal mannose epitopes which have previously been reported to bind to BanLec (Mo et al., 2001; Sharma and Vijayan, 2011). Therefore, BanLec binds HIV primarily via the Man8 and Man9 glycans on Env.
Figure 1. N-Glycan-Binding Assays to BanLec and Inhibition Assays.
(A) HPLC depletion assay of released gp120 N-glycans incubated with 0, 1, 5, 10, 20, and 50 μM BanLec.
(B) Inhibition of HIV pseudovirus strains JRCSF and BaL expressed with and without kifunensine (kif).
(C) Native mass spectra of BanLec with different concentrations of Man9 (0, 2.5, 7.5, 15, 30, and 50 μM).
(D) Kds determined from MS experiments binding Man7, Man8, and Man9 to BanLec, and calculated from peak intensities.
We then tested the affinity of BanLec to Man8/9 glycans in vivo by employing an HIV neutralization assay. We used two pseudovirus strains (HIVJRCSF and HIVBaL) produced in the absence or presence of kifunensine (kif), a potent inhibitor of α-mannosidase. Treatment with kif generates virions decorated predominantly with Man8 and Man9 glycans (Doores and Burton, 2010). We found that neutralization potency was considerably increased against kif-treated virions, leading to IC50 values an order of magnitude lower for HIVJRCSF (25 pM) and HIVBaL (35 pM) compared with wild-type HIVJRCSF (0.5 nM) and HIVBaL (1.1 nM) virions (Figure 1B). This reveals that the potency of BanLec is significantly enhanced when the HIV envelope proteins are augmented with longer-chain high-mannose structures, specifically Man8 and Man9. The difference in IC50 between HIVJRCSF and HIVBaL in the presence of kif is likely attributed to the increased number of glycosylation sites on HIVBaL strains. Together, the depletion assay and neutralization data demonstrate that BanLec neutralizes HIV via binding primarily to Man9 glycans on Env.
To characterize binding to longer-chain high-mannose glycans, we used native mass spectrometry (MS) to measure the stoichiometry and affinity of binding of Man7–9 glycans to BanLec. Although BanLec has been previously reported to be a dimeric protein (Meagher et al., 2005; Sharma and Vijayan, 2011; Singh et al., 2005; Swanson et al., 2015), we found it to exist as a tetramer in the mass spectrum (Figure 2A) (discussed further below). Upon incubation with Man9 glycans, we observed BanLec-glycan complexes with between one and four Man9 bound (Figure 1C). Importantly, we observed no complexes with higher binding stoichiometries (even in the presence of 25 equivalents of glycan), suggesting that BanLec-glycan binding stoichiometry is 1:1.
Figure 2. Mass Measurements of BanLec.
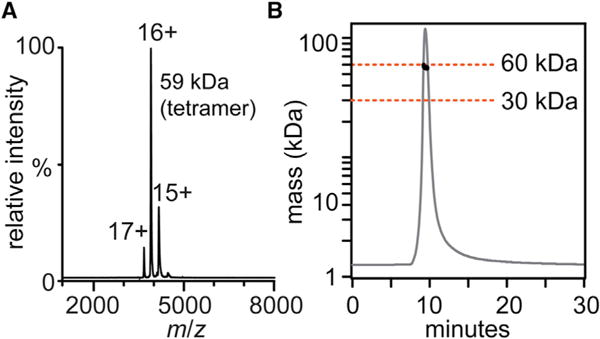
(A) Native mass spectrum of BanLec gives a mass of 59,394 Da.
(B) SEC-MALS of BanLec with the MALS trace (black circles) indicating a molecular mass of ∼60 kDa.
We then calculated dissociation constants (KD) for Man9 binding to BanLec by measuring the relative intensity of bound species at different glycan concentrations (Figures S1A–S1C). We found that the KDs for all four binding events of Man9 to BanLec are similar, with an average value of 7.6 ± 1.0 μM (Figure 1D). Performing the same binding experiments with Man8 and Man7 showed that these glycans had the same binding stoichiometry with average KDs of 3.8 ± 0.8 μM and 39 ± 7.5 μM for Man8 and Man7, respectively. These binding affinities, which are an order of magnitude lower for Man7 than for Man8 and Man9, are consistent with the results of the depletion assay. Notably, for all three glycans, each successive binding event has a similar KD, implying there is no cooperativity in binding.
Structural Model of the BanLec Tetramer
Native MS revealed BanLec is a tetrameric protein (with no other stoichiometries observed), which was unexpected as previous reports suggested it is a dimeric protein (Meagher et al., 2005; Sharma and Vijayan, 2011; Singh et al., 2005; Swanson et al., 2015). To validate our observation, we examined BanLec by size-exclusion chromatography/multi-angle light scattering (SEC-MALS). In agreement with the native MS experiment, BanLec eluted in a single peak with a molecular mass of approximately 60 kDa (Figure 2B). We conclude therefore that BanLec, in line with other members of the jacalin family of lectins (Pratap et al., 2002; Sankaranarayanan et al., 1996), assembles as a tetramer in solution.
To ascertain whether tetramers observed by native MS share an architecture similar to that of artocarpin/jacalin (jacalin family lectins) or an asymmetric crystal form, we measured BanLec by small-angle X-ray scattering (SAXS). Comparison of the experimental SAXS curve and radius of gyration with theoretical scattering and gyration values clearly indicates that BanLec is consistently tetrameric, with an architecture similar to that of artocarpin and jacalin (Pratap et al., 2002; Sankaranarayanan et al., 1996) (Figure 3).
Figure 3. Small-Angle X-ray Scattering of BanLec.
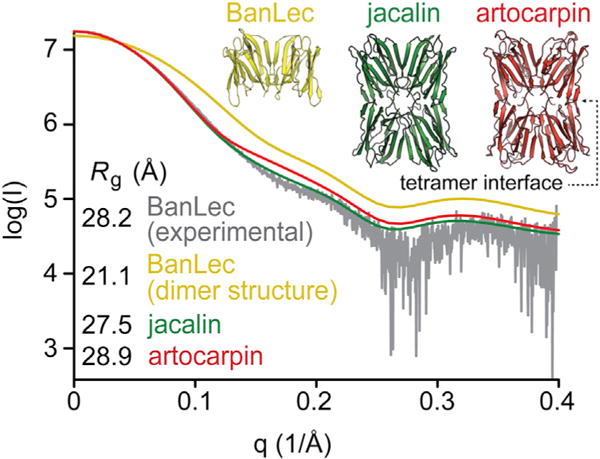
Experimental BanLec (gray) and theoretical SAXS curves of jacalin tetramer (green), artocarpin tetramer (red), and BanLec dimer (yellow). Radius of gyration values (Rg) of experimental BanLec and theoretical values calculated from BanLec (dimer), jacalin, and artocarpin crystal structures are shown.
The assembly of BanLec in previous X-ray crystal structures is a dimer and/or an asymmetric tetramer different to that observed for artocarpin and jacalin (Meagher et al., 2005; Sharma and Vijayan, 2011; Singh et al., 2004; Swanson et al., 2015). Using our preparation of BanLec, which we knew to be tetrameric in solution, we solved a crystal structure of BanLec in a new crystallization condition. This crystal form was equivalent to existing structures (Figure 4A and Table 1) with the arrangement of the dimer similar to that in other BanLec structures (root-mean-square deviation [RMSD] values of 0.475 Å [PDB: 2BMY], 0.464 Å [PDB: 3MIT], 0.464 Å, and 0.505 Å [PDB: 4PIF]), and likewise the presence of asymmetric tetramers in the crystal packing (Figure S2) (Meagher et al., 2005; Singh et al., 2005; Swanson et al., 2015).
Figure 4. BanLec Tetramer Model Generated from SAXS and Native MS.
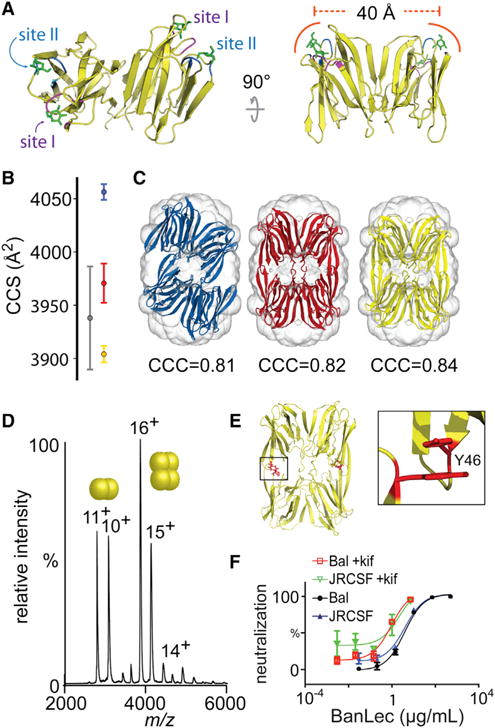
(A) Crystal structure of BanLec with mannose molecules (green, sticks) superimposed from PDB: 1X1V. Each of the two carbohydrate-binding domains present per monomer are labeled (site I, purple; site II, blue).
(B) Experimental CCS (gray), and theoretical CCS of artocarpin (red), symmetric BanLec model (yellow), and asymmetric tetramer (blue). Error bars are from three repeats averaging over three charge states.
(C) SAXS density envelope with asymmetric BanLec tetramer (blue), symmetric model obtained aligning two BanLec dimers onto artocarpin tetramer (red), and two independent BanLec dimers (symmetric tetramer, yellow) fitted into the density. CCC, cross-correlation coefficient.
(D) Intact mass spectrum of Y46K BanLec. Two balls indicate dimer charge series, four balls is the tetramer charge series.
(E) Symmetric tetramer model with tyrosine (Y46) residues located at the tetrameric interface.
(F) Neutralization assay of BanLec Y46K against HIV pseudovirus strains JRSCF and BaL expressed with and without kifunensine (+kif).
Table 1.
Crystal Parameters, Data Collection, and Refinement Statistics of BanLec
| Data Collection | |
|---|---|
| Space group | P21212 |
| Cell dimensions | |
| a, b, c (Å) | 63.33, 95.98, 47.53 |
| a = β = γ (°) | 90 |
| Resolution (Å) | 19.32−1.70 (1.74−1.70)a |
| Rmeas | 0.137 (1.283) |
| Mean I/σI | 15.77 (2.07) |
| Completeness (%) | 98.8 (96.5) |
| Redundancy | 12.5 (12.6) |
| Refinement | |
| Resolution (Å) | 19.32−1.70 (1.74−1.70) |
| No. of reflections | 30,641 |
| Rwork/Rfree | 0.1760/0.2062 |
| No. of atoms | |
| Protein | 2,091 |
| Ligand/ion | 42 |
| Water | 120 |
| Average B factors | |
| Protein | 19.5 |
| Ligand/ion | 21.7 |
| Water | 26.5 |
| RMSDs | |
| Bond lengths (Å) | 0.019 |
| Bond angles (°) | 1.919 |
Numbers in parentheses refer to the relevant outer-resolution shell.
Furthermore, a single tetramer form was distinguished by ion mobility MS (IM-MS) and correlated to the SAXS density envelope (Figures 4B and 4C). Two symmetric BanLec tetramer models were generated by aligning two copies of the dimeric form with artocarpin and by independently docking two BanLec dimers in the SAXS envelope, which had been generated from an ab initio structural model from the scattering data (Svergun et al., 2001). The symmetric tetramer created from independently docked dimers resulted in a higher cross-correlation coefficient than in any other model and represented the best fit to the SAXS density envelope. Using IM-MS, the rotationally averaged collision cross-section (CCS) was measured, which represents the size and shape of the protein. The CCS of BanLec was similar to theoretical CCSs for both symmetric BanLec models and significantly different from that of the asymmetric BanLec tetramer. The combined SAXS and IM-MS data therefore support a symmetric model of BanLec tetramers, in which the architecture is the same as in other members of the jacalin family of lectins (Pratap et al., 2002; Sankaranarayanan et al., 1996) rather than the asymmetric crystal form.
We then explored whether the tetrameric architecture of BanLec is important for HIV inhibition. A variant of BanLec (Y46K) has been reported to reduce tetrameric formation observed in the crystal packing (Swanson et al., 2015) and, consistent with this, in our model this residue is located at the tetrameric interface (Figure 4E). By native MS this variant is approximately 50% dimer/tetramer, and has noticeably diminished ability to neutralize pseudovirus strains compared with wild-type BanLec (Figures 4D and 4F). The IC50 values for this variant were 2.9 nM, 4.0 nM, 0.6 nM, and 0.6 nM for HIVJRCSF, HIVBaL, HIVJRCSF+kif, and HIVBaL+kif, respectively. These values are 5-, 3-, >24-, and >77-fold greater than for wild-type BanLec. Therefore, the tetrameric architecture of BanLec is essential for the high potency observed against HIV.
BanLec Binds High-Mannose Glycans via Bidentate Interactions
Notably, in the symmetric model presented here, all carbohydrate-binding sites (two per monomer) (Meagher et al., 2005) are on the exterior of the tetramer. The sites on each monomer are separated by 40 Å (Figure 4A) and are well positioned for interacting with multiple N-glycans simultaneously on different HIV trimers. This arrangement may help to explain the propensity of viral particles to become crosslinked and aggregated (Lusvarghi et al., 2016). Consistent with this mode of action, when we incubated BanLec and the soluble Env trimer mimic BG505 SOSIP.664, we observed aggregation by negative-stain electron microscopy (Figure S3) (Sanders et al., 2013, 2015).
The apparent number of glycan-binding sites (two per monomer) is intriguing, because the presence of eight independent-binding sites is in disagreement with four being the maximum number of high-mannose glycans we were able to observe binding by native MS. This raises the possibility that a single glycan can bind simultaneously to the pair of sites on each monomer.
To investigate whether a single glycan-binding interaction could involve both sites on each monomer, we performed MD simulations (Tessier et al., 2013) to sample sterically allowed configurations of glycan binding. Initially, we performed simulations of a small trisaccharide (Manα1-2Manα1-3Manα) bound to each binding site present on the BanLec monomer, and found that each site had a similar binding strength according to per-residue MM-GBSA (Molecular Mechanics/Generalized Born Surface Area) calculations (Table S1). We then performed simulations of Man9 binding to BanLec to determine whether two arms of the glycan structure could bind simultaneously to both glycan-binding sites on each monomer.
We uncovered seven unique bidentate interaction modes (Figure 5). Six of these modes involve the terminal Manα1–2 residue (here termed residue g) on the D1 arm of Man9, suggesting that binding is regulated by the ability of larger high-mannose glycans (i.e., Man7–9) to bridge both binding sites simultaneously. Using the same per-residue MM-GBSA MD analysis we calculated the Man9-binding energies for each mode (Table S2). Amino acids contributing to hydrogen bonding to mannose (site I: D133 and site II: D38) as observed from crystallography studies were also identified in this analysis (Meagher et al., 2005). However, other residues (G129, K130, and F131) located between each carbohydrate-binding site also appear to equally contribute to Man9 binding. Interestingly, we consistently observed binding-energy contributions from F131. This residue is positioned between the glycan-binding sites, suggesting that it may interact with a significant portion of the bound Man9.
Figure 5. Seven Predicted Bidentate Binding Modes of Man9 to BanLec.
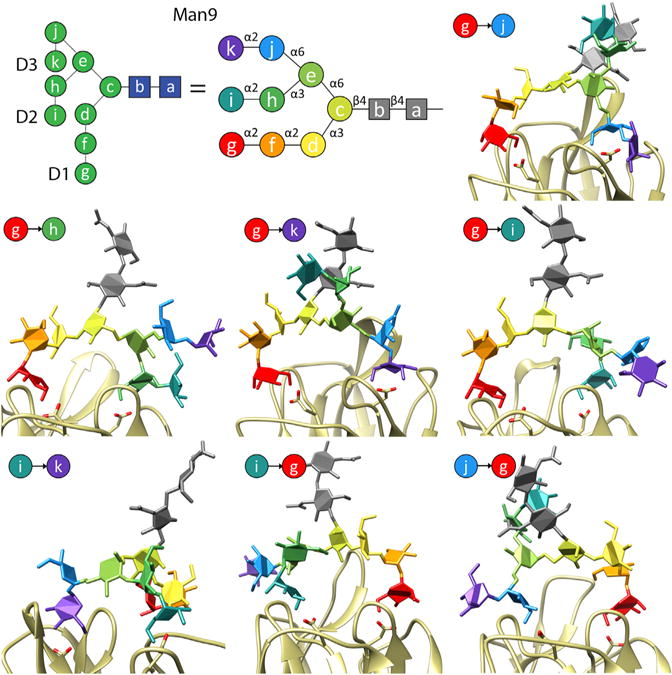
A cartoon representation of Man9 is shown in with D1, D2, and D3 arms of the structure indicated. The individual residues of Man9 are labelled with the letters a to k, and colored residues are used to clarify monosaccharide binding to BanLec. The particular Man9 residues bound in each binding site of the BanLec monomer are indicated in each image. An arrow points from the residue bound in site II (D38) to the residue bound in site I (D133).
To validate a bidentate model of binding, we created mutations (D38G and D133G) in each binding site of two residues that hydrogen bond to a mannose monosaccharide (Meagher et al., 2005) and that also bind to Man9 in our simulations (Figure 6A). We then recorded mass spectra of both variants, in which the proteins were tetrameric, and structurally unperturbed as suggested by identical charge-state distributions to the wild-type protein. We then mixed each variant with two equivalents of Man9 and compared the mass spectra with those obtained for the wild-type protein in the same conditions (Figure 6B). Whereas wild-type BanLec bound up to three Man9 glycans per tetramer, for both variants no significant glycan binding was detected. Consistent with an important role in glycan binding, the D38G and D133G variants possess no neutralization activity toward HIV pseudovirions (Figure 6C). D133G has previously been shown to abolish HIV neutralization (Swanson et al., 2015) but D38G has not been previously tested. We also characterized the behavior of a variant of F131, a residue located in the “saddle” between D38 and D133, which also under these conditions did not bind Man9 and for which HIV neutralization was abolished.
Figure 6. HIV Neutralization and Glycan Binding to BanLec Variants.
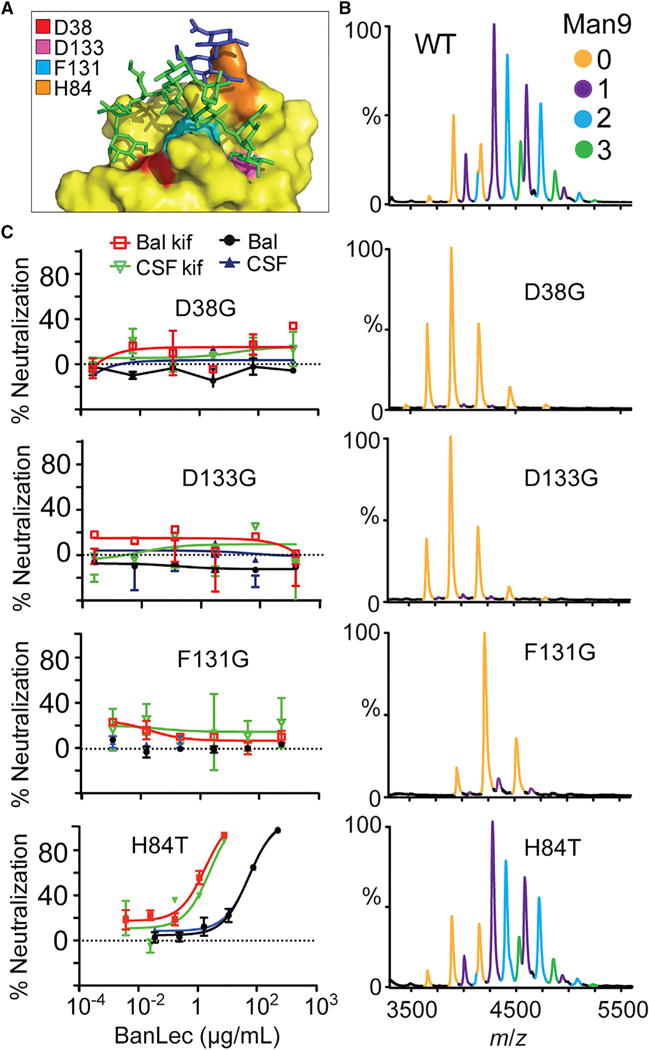
(A) MD-derived positions of Man9 binding to a single BanLec subunit show different glycan branches interacting simultaneously to form bidentate binding. BanLec is shown in surface representation (yellow) with residues contributing to glycan binding colored: D38, red; F131, cyan; D133, pink; H84, orange.
(B) Native mass spectra of wild-type (WT), D38G, D133G, F131G, and H84T variants with two molar equivalents Man9 substrate. Peaks corresponding to BanLec-Man9 complexes are independently colored.
(C) HIV neutralization assay of BanLec variants against JRCSF and BaL strains expressed with and without kif.
Interestingly, when we tested the binding of Man9 with that of another variant, H84T, which has been reported to have reduced multivalent interactions (Swanson et al., 2015), the binding was similar to that of wild-type BanLec. A maximum of four Man9 glycans could be observed bound, with an average KD of 9.5 ± 1.1 μM (Figure S1D). This is in contrast to the report by Swanson et al. (2015) of weakened affinity to dimannoside carbohydrates. The H84T mutation was proposed to disrupt “the wall that helps create the two independent sugar-binding sites” (Swanson et al., 2015), thus reducing multivalent interactions to N-glycans. Surprisingly, we observed diminished HIV neutralization with H84T variants with IC50 of 46.7 nM (HIVJRCSF), 46.8 nM (HIVBaL) 1.7 nM (HIVJRCSF+kifunensine), and 1.1 nM (HIVBaL+kifunensine). These results also contradict the reports of Swanson et al. in which H84T has neutralization ability equal to that of wild-type BanLec.
In summary, these results demonstrate that BanLec inhibits HIV through binding of Man7–9 glycans on gp120 and, while each high-mannose glycan interacts with both binding sites in a monomer, the two sites are not redundant, but rather are both necessary for competent binding and HIV neutralization.
DISCUSSION
How lectins achieve their extremely high potency and breadth of neutralization to HIV, as well as other enveloped viruses including Ebola (Barrientos et al., 2003) and influenza (O’Keefe et al., 2003), has been an important question in the field. N-linked glycan recognition has been reported for other anti-HIV lectins (Botos et al., 2002; Moulaei et al., 2010; Sato et al., 2011), but no equivalent observation had been made for BanLec. For potent neutralization, a lectin must exhibit high affinity and tight binding to the target viral spike to account for the nanomolar IC50 values reported. However, the extant biophysical understanding of BanLec (Weis and Drickamer, 1996) describes protein-monosaccharide interactions six or more magnitudes greater than this, which would be seemingly too weak to account for the anti-viral activity. To address this question, we used a variety of biophysical approaches to unravel the precise nature of the interactions between BanLec and the HIV virus.
Through a combination of HPLC glycan depletion and HIV neutralization assays, we discovered that BanLec binds only to Man7–9 structures, which typically represent >50% of the glycan shield (Pritchard et al., 2015b). These results refine the broader specificity established in previous lectin assays (Kanagawa et al., 2014; Singh et al., 2005; Swanson et al., 2015). By native MS we demonstrated that BanLec is a tetramer in solution, and by exploiting the ability of the technique to monitor individual binding stoichiometries (Hilton et al., 2013), found that each monomer is capable of binding only a single high-mannose N-linked glycan. We further quantified Man7–9 KDs to be in the low-micromolar range, approximately three orders of magnitude lower than for small mannose sugars (Mo et al., 2001). By performing MD simulations on Man9 binding we discovered modes by which different branches of large multi-antennary glycans can interact with a pair of sites on a BanLec monomer, which we could validate with native MS binding assays. This bidentate binding would account for the much lower KD of the large antennary glycans compared with that for small oligosaccharides, due to avidity effects of a single N-glycan. The avidity enhancement ratio (β) of 103 is in line with other bivalent interactions described in the literature (Mammen et al., 1998).
However, although it to some extent explains the strong interaction between BanLec and HIV, this intra-subunit mechanism does not fully account for viral inhibition at nanomolar concentrations. An explanation for this discrepancy comes from our discovery that BanLec is a tetramer in solution, rather than a dimer as previously thought. Our (symmetric) structural model of BanLec resembles other tetrameric members of the jacalin family of lectins and not the asymmetric BanLec tetramer frequently observed in crystal packing of the protein. The symmetric model shows that the glycan-binding sites are located at the four corners of the tetramer, allowing the lectin to bind Env glycans with minimal steric hindrance. As a result, BanLec should be considered as a tetravalent ligand for the HIV virion, with each binding site facilitating a strong bidentate interaction with a single high-mannose glycan.
We also found that both glycan-binding sites on each monomer are not partitioned as has been proposed (Swanson et al., 2015), but are equally essential for single glycan recognition. Notably the potency of the H84T variant, which was reported to maintain anti-viral properties (Swanson et al., 2015), was notably reduced in our hands. Disruption of tetramer assembly via the Y46K mutation reduces neutralization potency and highlights the importance of a tetrameric stoichiometry for anti-viral activity. These layers of multivalency appear to amplify each other, leading to the enhanced binding of single mannose moieties by a β factor of 106. The neutralization of HIV is therefore able to be achieved at nanomolar concentrations of BanLec.
The ability of BanLec to bind to high-mannose glycans presented across the surface of the viral spike underpins both the breadth of viral neutralization and its ability to recognize glycans of multiple bnAb epitopes. Furthermore, the large separation between binding sites on BanLec likely allows for the recognition of gp120 glycan clusters (Pritchard et al., 2015a), and raises the possibility that the lectin bridges virion spikes. Evidence for this comes from our observation of extensive aggregation when soluble mimics of the viral spike are incubated with BanLec. This broad and versatile nature of interaction explains the general potency across HIV strains through specific recognition of conserved high-mannose Env glycans, only elements of which are exploited by individual bnAbs. In addition, BanLec tetramers may promote interactions between Env proteins, which then could obstruct initial receptor recognition or the conformational rearrangements required for fusion of the HIV virion with the host membrane.
Recent efforts have established lectins as anti-HIV microbicides of great potential utility. However, the limited information on the precise nature of the interactions between BanLec and gp120 N-glycans has hampered protein-engineering strategies. The detailed understanding we have developed here, regarding both the native glycan targets and their binding mechanisms, will greatly inform future developments of successful lectin-based anti-viral therapies.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| goat anti-human Fab secondary antibody | Thermo Scientific Pierce | Cat # PA5-33290 |
| Bacterial and Virus Strains | ||
| E. coli BL21 Gold(DE3) cells | Agilent | Cat# 230132 |
| HEK 293T cells | ATCC | ATCC 11268 |
| TZM-bl cells | NIH ARRRP | NIH ARRRP 8129 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BanLec | This paper | N/A |
| gp120 (JR-CSF & Bal) | This paper | N/A |
| TEV protease | This paper | N/A |
| Ampicillin | Sigma-Aldrich | Cat# A0166 |
| Kanamycin | Sigma-Aldrich | Cat# K1377 |
| anthranliamide | Sigma-Aldrich | Cat# A89804 |
| ammonium acetate | Sigma-Aldrich | Cat# A2706 |
| IPTG | Sigma-Aldrich | Cat# I5502 |
| kifunensine | Sigma-Aldrich | Cat# K1140 |
| Protease inhibitor cocktail | Roche | Cat# 05056489001 |
| luciferase substrate | Promega | Cat# E1500 |
| PNGase F | New England BioLabs | Cat# P0705S |
| Critical Commercial Assays | ||
| Phusion High-Fidelity PCR kit | New England BioLabs | Cat# E0553 |
| Qiagen Gel Extraction kit | Qiagen | Cat# 28704 |
| In-Fusion HD Cloning Kit | Clontech | Cat# 639648 |
| Deposited Data | ||
| BanLec dimer | This paper | PDB: 5EXG |
| Oligonucleotides | ||
| Primers for BanLec D38G Forward: GTGACATCAACACCGCCAACCACATCGCCAC Reverse: GTGGCGATGTGGTTGGCGGTGTTGATGTCAC |
This paper | N/A |
| Primers for BanLec D133G Forward: GATAGACACCAATGGCACCCAGGAATTTACCACCA Reverse: TGGTGGTAAATTCCTGGGTGCCATTGGTGTCTATC |
This paper | N/A |
| Primers for BanLec H84T Forward: TGCTAACTACACCGGTGCGGTCGTGC Reverse: ACTTCACCTGCCATGCCC |
This paper | N/A |
| Primers for BanLec Y46K Forward: Forward: CACCTTCACGAAATACGGCAAAAC Reverse: ACATCAACACCGTCAACC |
This paper | N/A |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Recombinant DNA | ||
| BanLec template DNA | Clonetech (based on Peumans et al., 2000) | N/A |
| Plasmid: pET28b | Merck-Millipore | Cat# 69864 |
| Software and Algorithms | ||
| UniDec | Marty et al., 2015 | unidec.chem.ox.ac.uk. |
| Empower 3.0 | Waters | N/A |
| Masslynx v4.1 | Waters | N/A |
| Astra 6.1 software | Wyatt Technologies | N/A |
| ATSAS | Svergun et al., 2001 | https://www.embl-hamburg.de/biosaxs/software.html |
| Situs | Wriggers and Chacon, 2001 | http://situs.biomachina.org/fguide.html |
| Coot | Emsley et al., 2010 | http://www.ccp4.ac.uk/ |
| Phaser | McCoy et al., 2007 | http://www.phaser.com/ |
| Impact | Marklund et al., 2015 | http://impact.chem.ox.ac.uk/ |
| GLYCAM-Web | Kirschner et al., 2008 | www.glycam.org |
| Amber14SB | Case et al., 2014 | http://ambermd.org/ |
| Other | ||
| HisTrap HP column | GE Healthcare | Cat# 17-5248-02 |
| Superose 6 Increase 10/300 column | GE Healthcare | Cat# 29-0915-96 |
| Vivaspin 5000 MWCO filter | GE Healthcare | Cat# 28932223 |
| SDS-PAGE gel | Novex NuPAGE | Cat# NW04120 |
| Microtitre ELISA plates | Corning | Cat# 3369 |
| LudgerSep N2 amide HPLC column | Ludger Ltd. | Cat# LS-N2-4.6x150 |
| Bio SEC-3 SEC column | Agilent | Cat# 5190-2504 |
| Bio-spin 6 centrifuge columns | Bio-Rad | Cat# 7326221 |
METHOD DETAILS
Protein Expression and Purification
BanLec template DNA based on the reported sequence from Peumans et al. (Peumans et al., 2000) was purchased from Genscript (Piscataway, NJ). BanLec encoding DNA was amplified using a Phusion High-Fidelity PCR kit (New England Biolabs) and restriction sites for BamHI and XhoI were introduced and the PCR product was purified using a Qiagen Gel Extraction kit and cloned into a linearized pET28 vector (cut with BamHI and XhoI) containing a TEV protease-cleavable N-terminal polyhistidine tag using In-Fusion HD Cloning Kit (Clontech). Construction of green flourescent protein (GFP)-tagged BanLec was performed similarly but with NdeI and NheI specific inserts introduced to the PCR product. The PCR product was cloned into a linearized pET15 vector containing a TEV-GFP-His tag on the C-terminus. BanLec was transformed and expressed in E. coli BL21 Gold(DE3) competent cells (Agilent) and cells grown in LB media containing 50 μg/mL ampicillin (pET15) or 25 μg/mL kanamycin (pET28) until an OD of 0.6 was reached. Expression of BanLec was induced by the addition of isopropyl-D-thio-galactopyranoside (IPTG) to 1 mM, and cells cultured for a further 4 hours at 37 °C. Cells were pelleted by centrifugation at 5,000 g for 10 min and lysed by use of a microfluidiser. Soluble protein was separated from cell debris by centrifugation at 20,000 g for 20 min. The His-tag protein was purified from the lysate by affinity purification with a HisTrap HP column (GE Healthcare). The His-tag was then cleaved by incubation with tobacco etch virus (TEV) overnight at room temperature. The cleaved protein was purified by reverse Ni-affinity and the purity of the flow-through containing BanLec was assessed by SDS-PAGE (Novex NuPAGE) prior to MS.
HIV Inhibition Assays
Pseudovirus was generated in HEK293T cells as described (Montefiori, 2005; Singh et al., 2005). Briefly, 293T cells were transfected with plasmids expressing the virus backbone PSG-3Δenv and the functional envelope clone at a ratio of 2:1 using PEI (1 mg/mL, 1:3 PEI:total DNA, Polysciences) according to the manufacturer’s instructions. Virus supernatants were harvested after 72 h. Glycosidase inhibitor, kifunensine, was added at the time of transfection at a final concentration of 25 μM (Doores and Burton, 2010). Neutralization activity of BanLec or HIV bnAb wild-type and mutants against pseudovirus in TZM-bl cells was determined as described previously (Montefiori, 2005; Singh et al., 2005). Briefly, TZM-bl cells were seeded in a 96-well flat bottom plate and infected with pseudovirus in the presence of BanLec or HIV bnAb (200 μL total volume). Viruses were preincubated with BanLec or HIV bnAb for 1 hat37°C. Luciferase reporter gene expression was quantified 72 h after infection upon lysis and addition of luciferase substrate (Promega).
bnAb Competition ELISA
Microtitre ELISA plates (Corning) were coated with recombinant gp120JR-CSF (5 μg/mL in PBS) overnight at 4 °C. Plates were washed five times with a solution of PBS containing 0.05% Tween 20 (v/v) and then blocked for 1 h at room temperature with 5% non-fat milk in PBS + 0.05% Tween (blocking buffer). Plates were emptied before addition of a titration of BanLec (starting concentration, 500 μg/mL in blocking buffer using a 1:3 dilution series) and incubated for 30–60 min. HIV bnAbs were then added at a constant concentration (Ab concentration represented IC50 and were diluted in blocking buffer) and incubated for a further 1.5 h. Plates were then washed (×5) and alkaline phosphatase (AP)-conjugated goat anti-human Fab secondary antibody (Thermo Scientific Pierce) was added at a 1:1000 dilution in blocking buffer and incubated for 1 h. Plates were washed (×5) and then AP substrate (50 μL / well) was added. The OD at 405 nm was measured after 20 min.
Glycan HPLC
N-glycans were released from 150 μg recombinant gp120 (JRCSF) by incubation with PNGase F(New England BioLabs) according to manufacturer’s protocol. Glycans were separated from the protein by spin-filtration through 5000 Da MWCO filter (Vivaspin, GE Healthcare) and speedvac dried. Released glycans were 2-AB labelled as described previously (Struwe and Rudd, 2012). Briefly, glycans were incubated with 20 μL 1 % formic acid for 45 min at room temperature, and dried before addition of 5 μL 2-AB labelling solution (DMSO:glacial acetic acid (7:3 v/v), 1M sodium cyanoborohydride, 0.5 M anthranilamide (2-AB)), followed by incubation for 3 h at 65 °C. Excess dye was removed by purification with PhyNexus normal phase columns, equilibrated with 95% acetonitrile and eluted with 20% acetonitrile. 2-AB labelled glycans were incubated with BanLec for 1 h at 37°C for the depletion assays. Unbound 2-AB labelled glycans were separated from BanLec-glycan complexes using Vivaspin 500, 5000 Da MWCO spin columns (GE Healthcare). Glycans were dried and resuspended in 30 μL 65% CAN prior to analysis using a LudgerSep N2 amide HPLC column (Ludger Ltd). Each HPLC injection consisted of N-glycans released from 10 μg gp120. Analysis was performed using a Waters Acquity system with Empower software.
Native Mass Spectrometry
Initial stoichiometry measurements and carbohydrate titration experiments were performed on a hybrid quadrupole time-of-flight (q-TOF) mass spectrometer modified for high mass transmission (Sobott et al., 2002). Conformational analysis was performed using first generation Waters Synapt IM-MS instrument, employing a linear drift field to allow direct calculation of collisional cross-sections. Prior to all MS analyses, aliquots (50 μL) of BanLec were desalted using Bio-spin 6 (Bio-Rad) centrifuge columns, equilibrated with 200 mM ammonium acetate. Following buffer exchange protein concentration was measured by absorbance at 280 nm using a UV/Vis spectrophotometer. Protein concentrations were then adjusted to between 5 and 10 μM for all MS experiments. Typically, 3 μL of protein was loaded into gold-coated nanospray capillaries prepared in-house and mounted into a static nanospray source block (Hernandez and Robinson, 2007). Electrospray was induced by applying a voltage of between 1.1−1.3 kV to the capillary. Backing pressure in the source was raised to a pressure of 6−7×10−3 mbar by ‘choking’ the line to the roughing pump, improving the transmission of high molecular weight ions. For unbound BanLec stoichiometry measurements, sample cone voltages of 60−80 V were used, which provided efficient desolvation and narrower peak widths but with no evidence of any induced dissociation. During titration experiments, peak width was compromised to ensure no gas-phase dissociation of oligomannose ligands and sample cone was maintained below 40 V (typically 20 V). To this end, collision energies within the instrument’s collision cells were maintained at the minimum values necessary for adequate transmission. This was deemed as 10 V in the collision cell of the q-TOF and 5 V in the trap collision cell on the Synapt. Transfer cell voltages, which act to provide the linear drift field across the IMS cell, were varied over several values to allow calculation of the post-IMS flight time.
For titration experiments, protein and oligomannose aliquots were prepared at twice the required concentration and then mixed at a 1:1 ratio to give the desired concentration point. Oligomannose stocks were prepared to 150 μM and diluted to the required concentrations in milliQ water. Protein concentrations were determined by measuring absorbance at 280 nm. Titration experiments were performed at a final ammonium acetate concentration of 100 mM. Effort was made to ensure that capillary dimensions were kept constant, from the same batch and that spray was initiated from a similar capillary-cone distance. Collision energy ramps were performed to demonstrate that instrument conditions used during titrations did not lead to any gas-phase dissociation. Mass spectra were deconvolved using UniDec into zero-charge mass distributions (Marty et al., 2015). Using the UniDec Data Collector module, peak [heights] were extracted from the mass distributions and fit to a binding model using a least square fitting approach. The software and links to source code are available at unidec.chem.ox.ac.uk.
SEC-MALS
Analysis of purified BanLec was performed using an Agilent 1260 HPLC with an Agilent Bio SEC-3 SEC column (4.6×150 mm, 3 μm particle, 150 Å pore size) coupled to a DAWN HELEOS II scattering detector (Wyatt Technologies) and Optilab T-rEX (Wyatt Technologies) differential refractive index detector. The MALS detector was equilibrated overnight to ensure minimal background light scattering. Roughly 50 μg of protein was loaded on-column (column compartment was set to 20 °C) at a flow rate of 0.8 mL/min in 150 mM sodium phosphate pH 7.5. Data acquisition and analysis were performed with Astra 6.1 software (Wyatt Technologies).
X-ray Crystallography
BanLec crystals were grown by hanging-drop vapour diffusion. A protein solution [10 mg/mL, in 100 mM NaCl, 20 mM Tris (pH 7.4)] was mixed 1:1 (v/v) with a reservoir solution containing 0.05 M NaBr, 0.1 M Bis Tris and 19% PEG-3350 (pH 7.5). The drop sizes were 1 μL, and the volume of the reservoir solution was 1 mL. The crystallisation trays were left at 20 °C. Crystals were harvested and transferred briefly into a cryoprotectant composed of 10% glycerol in the reservoir solution. The plate-like crystals typically began to form after 24 hours and were fully formed in 72 hours. Data for BanLec were collected at Diamond Synchrotron using the I04-1 beamline and were processed using XDS and XSCALE. The results are summarised in Table EV1, along with key structure refinement details. The protein crystallised in the orthorhombic space group P21212 and diffracted to 1.70 Å, with the following unit cell dimensions: a = 63 Å, b = 96 Å, c = 48 Å. Cell content analysis indicated high probability of two monomers of BanLec in the asymmetric unit. A monomer of BanLec, from PDB 2BMY (stripped of all non-protein atoms) was used as a search molecule for molecular replacement using Phaser. Refinement was conducted with Refmac, and electron density maps were analysed with Coot. The validation tools of Coot and Molprobity were used to check for and correct conformational issues.
SAXS
Data was collected at the B21 bending magnet instrument at Diamond Light Source (Harwell, UK). Samples were prepared in200mM ammonium acetate to a concentration of 8 mg/mL in a 96 well plate at 5 °C. Protein and corresponding buffer solutions were exposed to the beam using an Arinax (Grenoble, France) BioSAXS automated sample changer robot, consisting of temperature controlled storage and exposure units. The exposure unit contained a 1.6 mm diameter quartz capillary in which the samples were illuminated with the X-ray beam; the exposure unit temperature was set to 15 °C. The sample capillary was held in vacuum and subjected to a cleaning cycle between each measurement. A Pilatus 2M two-dimensional detector was used to collect 180 frame exposures of 1 s from each sample and the corresponding buffer. The detector was placed at 3.9 m from the sample, giving a useful q-range of 0.012 Å−1 < 0.4 Å−1, where q = 4 π sin(θ)/λ, where 2θ is the scattering angle and λ is the wavelength, which was set to 1 Å. Two dimensional data reduction consisted of normalisation for beam current and sample transmission, radial sector integration, background buffer subtraction and averaging. Each frame was inspected for the presence of radiation induced protein damage; if this was found to be the case, the frames were not reduced and processed. We produced a bead model from our BanLec SAXS data using gasbor (Svergun et al., 2001). The resulting model was subsequently transformed into a density map using Situs’ pdb2vol tool, and all our BanLec structural models docked in it using Situs’ colores tool (Wriggers and Chacón, 2001).
An asymmetrical BanLec tetramer was observed from the crystal packing of our BanLec protein preparation. We first produced a BanLec symmetrical tetramer via a structural alignment of two of our BanLec dimers onto artocarpin tetrameric structure (pdb: 1J4S). The resulting model was subsequently relaxed with 2000 conjugate gradient steps in implicit solvent using the NAMD (Phillips et al., 2005) molecular dynamics engine and the Amber14SB (Case et al., 2014) force field. We produced a second symmetrical BanLec tetramer by directly docking two BanLec dimers into our SAXS density using Situs’ collage tool (Birmanns et al., 2011). For all the atomistic models detailed above, the SAXS signal was simulated using crysol (Svergun et al., 1995), the radius of gyration predicted with Hydropro (Ortega et al., 2011), and the collisional cross-section calculated using Impact (Marklund et al., 2015).
Molecular Grafting
Trimannose Structure
A model for bound trimannose oligosaccharide (Manα1-2Manα1-3Manα) was built by grafting (Grant et al., 2014; Tessier et al., 2013) the required residues onto the co-crystal structure of Manα bound to BanLec (PDBID: 2BMZ).
Bidentate Structure
The 3D structure of Man9 was generated by grafting the required branches onto the bound Manα in PDBID: 2BMZ. For grafting, initial conformations for Man9 were generated using GLYCAM-Web (www.glycam.org). Structure modelling was used to determine whether Man9 was capable of adopting a low energy conformation that would allow two branches of the Man9 oligosaccharide to bind simultaneously in both binding sites of BanLec (bidentate binding). A bound Manα residue in one arm was retained in the crystallographic orientation, while the intervening glycosidic torsion angles were varied within low energy bounds (± 20°) (Nivedha et al., 2013). Any binding modes that placed a Manα from a second arm within 2 Å of the second binding site were retained for further refinement by energy minimization and molecular dynamics (MD) simulation. This procedure was applied to each of the eight Manα residues in Man9.
Energy Minimization and MD Simulation
Bidentate Co-complexes
The MD simulations were performed using the Amber14 software suite (Case et al., 2014). The Glycam06h (Kirschner et al., 2008) and Amber14SB (Case et al., 2014) force fields were employed for carbohydrate and protein respectively. Hydrogen atoms were added and protonation states were assigned via tleap using the default parameters for Glycam06h and Amber14SB. Energy minimization was performed for 20,000 steps (10,000 steepest decent, 10,000 conjugant gradient). Following minimization, the system was heated over 100 ps from 5 K to 300 K and then equilibrated at 300 K for 300ps. During the final 200 ps, distance restraints were introduced to guide the desired Manα residue into the second binding site (Table S3). The restraints were then turned off over a 1 ns period, and the simulation performed without any restraints for a further 10 ns. Solvation was treated with the generalized Born approximation (igb = 2) (Onufriev et al., 2004) throughout with a non-bonded cut-off of 999 Å. The cuda (Gotz et al., 2012; Salomon-Ferrer et al., 2013) module was employed for the MD simulations. Pressure and temperature were regulated with a Berendsen-type barostat (1 ps constant) and Langevin thermostat (2 ps−1 collision frequency) respectively. Scaling factors for 1–4 electrostatic and van der Waals interactions were set to unity for glycans (Woods and Chappelle, 2000), and 1.2 and 2.0 for proteins (Hornak et al., 2006). Covalent bonds to hydrogen were constrained with the SHAKE algorithm allowing a 2 fs time step (Ryckaert et al., 1977).
Trimannose Co-complexes
A similar protocol was followed for simulating the trimannose co-complexes with the following variations. After the initial minimization, the structure was enclosed in a box of explicit TIP3P (Jorgensen et al., 1983) water, with an 8 Å buffer. This system was subjected to a second minimization round with 5 kcal/mol/Å2 restraints on non-hydrogen solute atoms. A 500 ns MD simulation was performed without distance or Cartesian restraints. A non-bonded interaction cut-off of 8 Å was applied, beyond which long-range electrostatics were treated with the particle-mesh Ewald (PME) method (Darden et al., 1993).
Per-residue MM-GBSA
An MM-GBSA calculation (molecular mechanics with generalised Born and surface area solvation) with igb = 2 (Onufriev et al., 2004) was performed on 1,000 snapshots taken at regular intervals from both the trimannose and Man9 simulations. The energies were decomposed on a per-residue basis (idecomp = 1).
CryoEM
Analysis of Lectin-SOSIP Complexes Negative Stain Electron Microscopy (EM)
Env proteins were prepared for negative stain EM analysis as previously described (Pugach et al., 2015; Sanders et al., 2013). Briefly, a 3 μL aliquot containing 0.01–0.05 mg/mL of Env protein (as determined by UV A280 using the theoretical extinction coefficient based on peptide sequence alone) or Env protein in complex with lectin was applied for 5 s onto a carbon-coated 400 Cu mesh grid that had been glow discharged at 20 mA for 30 s, then negatively stained with nano-W (Nanoprobes, USA) for about 5 s, blotted with filter paper, and stained for another 15 s with nano-W. Nano-W was chosen over the more common uranyl salts due to the more neutral pH of the stain, which should prevent dissociation between pH-sensitive complexes (such as Env-ligand interactions that are strongly governed by glycans). Grids were screened to assess stain quality Data were collected on either an FEI Tecnai T12 electron microscope operating at 120 keV, with an electron dose of ∼25 e-/Å2 and a magnification of 52,000 × that resulted in a pixel size of 2.05 Å at the specimen plane, or an FEI Talos electron microscope operating at 200 keV, with an electron dose of ∼25 e-/Å2 and a magnification of 73,000 × that resulted in a pixel size of 1.98 Å at the specimen plane. Images were acquired with a Tietz TemCam-F416 CMOS camera (FEI Tecnai T12) or FEI Ceta 16M camera (FEI Talos) using a nominal defocus range of 1000–1500. In the cases when trimers were visible in the raw images, data were processed using methods adapted from those used previously (Pugach et al., 2015). Resulting 2D class averages were visually inspected for any Env-lectin complexes.
Analysis of Lectin-SOSIP Complexes by Blue Native Gel Electrophoresis (BN-PAGE) and Size-Exclusion Chromatography (SEC)
Lectin and BG505 SOSIP.664 were combined at varying molar ratios (using the molecular weights of BG505 SOSIP.664 trimer and lectin tetramer) of 0.75:1, 1.5:1, 3.75:1, and 7.5:1, incubated at room temperature for 15 min, and then run on a 4–16% BN-PAGE gel according to manufacturer’s recommendations. A separate Lectin and BG505 SOSIP.664 incubated sample (6:1 molar ratio) was subjected to centrifugation (14,000 g, 10 min, 4 °C) and loaded onto a Superose 6 Increase 10/300 column (GE Healthcare). The higher molecular weight peak, corresponding to the expected MW for SOSIP.664 trimer, was pooled, concentrated, and analyzed by EM.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact WBS (weston.struwe@bioch.ox.ac.uk).
Supplementary Material
In Brief.
We present a tetrameric model of the potent anti-HIV lectin BanLec and identify its molecular mode of binding to specific high-mannose N-linked glycans. Recognition is achieved through bidentate interactions involving different arms of a single N-glycan. Together the tetrameric architecture and bidentate binding are critical for HIV neutralization.
Highlights.
Tetrameric structural model of BanLec
BanLec binds distinct HIV high-mannose N-glycans
Carbohydrate-binding domains function jointly to bind a single N-glycan
Altering stoichiometry and bidentate glycan binding affects HIV neutralization
Acknowledgments
The authors are grateful to Michael Marty, Joseph Gault, and Andrew Baldwin for helpful discussions. J.T.S.H, T.M.A. and C.V.R. acknowledge funding by the Medical Research Council (98101) and Wellcome Trust Investigator Award (104633/Z/14/Z). J.L.P.B. holds a Royal Society University Research Fellowship, C.V.R. is funded by an ERC grant (“IMPRESS”, 26851) and a Royal Society Professorship. W.B.S. and J.L.P.B. acknowledge funding from the Biotechnology and Biological Sciences Research Council (BB/L017733/1). R.J.W. thanks the NIH (U01 CA207824 and P41 GM103390) for support. M.C. is supported by an International AIDS Vaccine Initiative Neutralizing Antibody Center CAVD grant (Glycan characterization and Outer Domain glycoform design) and a National Institute of Allergy and Infectious Diseases grant (CHAVI-ID grant 1UM1AI100663). A.B.W. is supported by the International AIDS Vaccine Initiative and the Bill and Melinda Gates Foundation CAVD (OPP1115782 and OPP1084519). K.J.D. is funded by a Medical Research Council Career Development Award (MR/K024426/1). We also acknowledge the Diamond Light Source for time on Beamline B21 under Proposal [SM9384-2].
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures, three tables, and one 3D molecular model and can be found with this article online at http://dx.doi.org/10.1016/j.str.2017.03.015.
AUTHOR CONTRIBUTIONS
J.T.S.H., J.L.P.B., C.V.R., and W.B.S. designed the project. J.T.S.H., S.A., and W.B.S. performed all native mass spectrometry experiments. S.A., T.M.A., and W.B.S. performed crystallography experiments. O.C.G., R.J.W., and M.T.D. performed molecular grafting and structural data analysis. S.A.K. and K.J.D. performed inhibition assays. S.A., L.K.P., and W.B.S. designed and performed HPLC incubation/depletion assays. G.O. and A.B.W. performed electron microscopy experiments. J.T.S.H., M.C., J.L.P.B., and W.B.S. wrote the paper with contributions from all authors.
References
- Ananworanich J, Fauci A. HIV cure research: a formidable challenge. J Virus Erad. 2015;1:1–3. [PMC free article] [PubMed] [Google Scholar]
- Barrientos LG, O’Keefe BR, Bray M, Sanchez A, Gronenborn AM, Boyd MR. Cyanovirin-N binds to the viral surface glycoprotein, GP1, 2 and inhibits infectivity of Ebola virus. Antiviral Res. 2003;58:47–56. doi: 10.1016/s0166-3542(02)00183-3. [DOI] [PubMed] [Google Scholar]
- Behrens AJ, Vasiljevic S, Pritchard LK, Harvey DJ, Andev RS, Krumm SA, Struwe WB, Cupo A, Kumar A, Zitzmann N, et al. Composition and antigenic effects of individual glycan sites of a trimeric HIV-1 envelope glycoprotein. Cell Rep. 2016;14:2695–2706. doi: 10.1016/j.celrep.2016.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewley CA, Otero-Quintero S. The potent anti-HIV protein cyanovirin-N contains two novel carbohydrate binding sites that selectively bind to Man(8) D1D3 and Man(9) with nanomolar affinity: implications for binding to the HIV envelope protein gp120. J Am Chem Soc. 2001;123:3892–3902. doi: 10.1021/ja004040e. [DOI] [PubMed] [Google Scholar]
- Birmanns S, Rusu M, Wriggers W. Using Sculptor and Situs for simultaneous assembly of atomic components into low-resolution shapes. J Struct Biol. 2011;173:428–435. doi: 10.1016/j.jsb.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomelli C, Doores KJ, Dunlop DC, Thaney V, Dwek RA, Burton DR, Crispin M, Scanlan CN. The glycan shield of HIV is predominantly oligomannose independently of production system or viral clade. PLoS One. 2011;6:e23521. doi: 10.1371/journal.pone.0023521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botos I, O’Keefe BR, Shenoy SR, Cartner LK, Ratner DM, Seeberger PH, Boyd MR, Wlodawer A. Structures of the complexes of a potent anti-HIV protein cyanovirin-N and high mannose oligosaccharides. J Biol Chem. 2002;277:34336–34342. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- Boyd MR, Gustafson KR, McMahon JB, Shoemaker RH, O’Keefe BR, Mori T, Gulakowski RJ, Wu L, Rivera MI, Laurencot CM, et al. Discovery of cyanovirin-N, a novel human immunodeficiency virus-inactivating protein that binds viral surface envelope glycoprotein gp120: potential applications to microbicide development. Antimicrob Agents Chemother. 1997;41:1521–1530. doi: 10.1128/aac.41.7.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brichacek B, Lagenaur LA, Lee PP, Venzon D, Hamer DH. In vivo evaluation of safety and toxicity of a Lactobacillus jensenii producing modified cyanovirin-N in a rhesus macaque vaginal challenge model. PLoS One. 2013;8:e78817. doi: 10.1371/journal.pone.0078817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton DR, Ahmed R, Barouch DH, Butera ST, Crotty S, Godzik A, Kaufmann DE, McElrath MJ, Nussenzweig MC, Pulendran B, et al. A blueprint for HIV vaccine discovery. Cell Host Microbe. 2012;12:396–407. doi: 10.1016/j.chom.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Gohlke H, et al. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- Crispin M, Doores KJ. Targeting host-derived glycans on enveloped viruses for antibody-based vaccine design. Curr Opin Virol. 2015;11:63–69. doi: 10.1016/j.coviro.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacheux L, Moreau A, Ataman-Onal Y, Biron F, Verrier B, Barin F. Evolutionary dynamics of the glycan shield of the human immunodeficiency virus envelope during natural infection and implications for exposure of the 2G12 epitope. J Virol. 2004;78:12625–12637. doi: 10.1128/JVI.78.22.12625-12637.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N• log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089. [Google Scholar]
- Doores KJ, Burton DR. Variable loop glycan dependency of the broad and potent HIV-1-neutralizing antibodies PG9 and PG16. J Virol. 2010;84:10510–10521. doi: 10.1128/JVI.00552-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doores KJ, Bonomelli C, Harvey DJ, Vasiljevic S, Dwek RA, Burton DR, Crispin M, Scanlan CN. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc Natl Acad Sci USA. 2010;107:13800–13805. doi: 10.1073/pnas.1006498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- François K, Balzarini J. Potential of carbohydrate-binding agents as therapeutics against enveloped viruses. Med Res Rev. 2012;32:349–387. doi: 10.1002/med.20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theor Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant OC, Smith HMK, Firsova D, Fadda E, Woods RJ. Presentation, presentation, presentation! Molecular level insight into linker effects on glycan array screening data. Glycobiology. 2014;24:17–25. doi: 10.1093/glycob/cwt083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez H, Robinson CV. Determining the stoichiometry and interactions of macromolecular assemblies from mass spectrometry. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]
- Hilton GR, Hochberg GK, Laganowsky A, McGinnigle SI, Baldwin AJ, Benesch JL. C-terminal interactions mediate the quaternary dynamics of alphaB-crystallin. Philos Trans R Soc Lond B Biol Sci. 2013;368:20110405. doi: 10.1098/rstb.2011.0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagawa M, Liu Y, Hanashima S, Ikeda A, Chai WG, Nakano Y, Kojima-Aikawa K, Feizi T, Yamaguchi Y. Structural basis for multiple sugar recognition of jacalin-related human ZG16p Lectin. J Biol Chem. 2014;289:16954–16965. doi: 10.1074/jbc.M113.539114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ. GLYCAM06: a generalizable biomolecular force field. Carbohydrates J Comput Chem. 2008;29:622–655. doi: 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koharudin LM, Gronenborn AM. Structural basis of the anti-HIV activity of the cyanobacterial Oscillatoria agardhii agglutinin. Structure. 2011;19:1170–1181. doi: 10.1016/j.str.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br Med Bull. 2001;58:19–42. doi: 10.1093/bmb/58.1.19. [DOI] [PubMed] [Google Scholar]
- Lagenaur LA, Swedek I, Lee PP, Parks TP. Robust vaginal colonization of macaques with a novel vaginally disintegrating tablet containing a live biotherapeutic product to prevent HIV infection in women. PLoS One. 2015;10:e0122730. doi: 10.1371/journal.pone.0122730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lagenaur LA, Simpson DA, Essenmacher KP, Frazier-Parker CL, Liu Y, Tsai D, Rao SS, Hamer DH, Parks TP, et al. Engineered vaginal lactobacillus strain for mucosal delivery of the human immunodeficiency virus inhibitor cyanovirin-N. Antimicrob. Agents Chemother. 2006;50:3250–3259. doi: 10.1128/AAC.00493-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusvarghi S, Lohith K, Morin-Leisk J, Ghirlando R, Hinshaw JE, Bewley CA. Binding site geometry and subdomain valency control effects of neutralizing lectins on HIV-1 viral particles. ACS Infect Dis. 2016;2:882–891. doi: 10.1021/acsinfecdis.6b00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Marklund EG, Degiacomi MT, Robinson CV, Baldwin AJ, Benesch JL. Collision cross sections for structural proteomics. Structure. 2015;23:791–799. doi: 10.1016/j.str.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, Robinson CV. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem. 2015;87:4370–4376. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher JL, Winter HC, Ezell P, Goldstein IJ, Stuckey JA. Crystal structure of banana lectin reveals a novel second sugar binding site. Glycobiology. 2005;15:1033–1042. doi: 10.1093/glycob/cwi088. [DOI] [PubMed] [Google Scholar]
- Mo H, Winter HC, Van Damme EJ, Peumans WJ, Misaki A, Goldstein IJ. Carbohydrate binding properties of banana (Musa acuminata) lectin I. Novel recognition of internal alpha1,3-linked glucosyl residues. Eur J Biochem. 2001;268:2609–2615. doi: 10.1046/j.1432-1327.2001.02148.x. [DOI] [PubMed] [Google Scholar]
- Montefiori DC. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr Protoc Immunol. 2005 doi: 10.1002/0471142735.im1211s64. Chapter 12. Unit 12 11. [DOI] [PubMed] [Google Scholar]
- Mori T, O’Keefe BR, Sowder RC, 2nd, Bringans S, Gardella R, Berg S, Cochran P, Turpin JA, Buckheit RW, Jr, McMahon JB, et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J Biol Chem. 2005;280:9345–9353. doi: 10.1074/jbc.M411122200. [DOI] [PubMed] [Google Scholar]
- Moulaei T, Shenoy SR, Giomarelli B, Thomas C, McMahon JB, Dauter Z, O’Keefe BR, Wlodawer A. Monomerization of viral entry inhibitor griffithsin elucidates the relationship between multivalent binding to carbohydrates and anti-HIV activity. Structure. 2010;18:1104–1115. doi: 10.1016/j.str.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivedha AK, Makeneni S, Foley BL, Tessier MB, Woods RJ. The importance of ligand conformational energies in carbohydrate docking: sorting the wheat from the chaff. J Comput Chem. 2013;35:526–539. doi: 10.1002/jcc.23517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe BR, Smee DF, Turpin JA, Saucedo CJ, Gustafson KR, Mori T, Blakeslee D, Buckheit R, Boyd MR. Potent anti-influenza activity of cyanovirin-N and interactions with viral hemagglutinin. Antimicrob Agents Chemother. 2003;47:2518–2525. doi: 10.1128/AAC.47.8.2518-2525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- Ortega A, Amoros D, Garcia de la Torre J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys J. 2011;101:892–898. doi: 10.1016/j.bpj.2011.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peumans WJ, Zhang WL, Barre A, Astoul CH, Balint-Kurti PJ, Rovira P, Rouge P, May GD, Van Leven F, Truffa-Bachi P, et al. Fruit-specific lectins from banana and plantain. Planta. 2000;211:546–554. doi: 10.1007/s004250000307. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratap JV, Jeyaprakash AA, Rani PG, Sekar K, Surolia A, Vijayan M. Crystal structures of artocarpin, a Moraceae lectin with mannose specificity, and its complex with methyl-alpha-D-mannose: implications to the generation of carbohydrate specificity. J Mol Biol. 2002;317:237–247. doi: 10.1006/jmbi.2001.5432. [DOI] [PubMed] [Google Scholar]
- Pritchard LK, Spencer DI, Royle L, Bonomelli C, Seabright GE, Behrens AJ, Kulp DW, Menis S, Krumm SA, Dunlop DC, et al. Glycan clustering stabilizes the mannose patch of HIV-1 and preserves vulnerability to broadly neutralizing antibodies. Nat Commun. 2015a;6:7479. doi: 10.1038/ncomms8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard LK, Vasiljevic S, Ozorowski G, Seabright GE, Cupo A, Ringe R, Kim HJ, Sanders RW, Doores KJ, Burton DR, et al. Structural constraints determine the glycosylation of HIV-1 envelope trimers. Cell Rep. 2015b;11:1604–1613. doi: 10.1016/j.celrep.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugach P, Ozorowski G, Cupo A, Ringe R, Yasmeen A, de Val N, Derking R, Kim HJ, Korzun J, Golabek M, et al. A native-like SOSIP.664 trimer based on an HIV-1 subtype B env gene. J Virol. 2015;89:3380–3395. doi: 10.1128/JVI.03473-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of Cartesian equations of motion of a system with constraints: molecular-dynamics of N-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theor Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Sanders RW, Derking R, Cupo A, Julien JP, Yasmeen A, de Val N, Kim HJ, Blattner C, de la Pena AT, Korzun J, et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP. 664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013;9:e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RW, van Gils MJ, Derking R, Sok D, Ketas TJ, Burger JA, Ozorowski G, Cupo A, Simonich C, Goo L, et al. HIV-1 VACCINES. HIV-1 neutralizing antibodies induced by native-like envelope trimers. Science. 2015;349:aac4223. doi: 10.1126/science.aac4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan R, Sekar K, Banerjee R, Sharma V, Surolia A, Vijayan M. A novel mode of carbohydrate recognition in jacalin, a Moraceae plant lectin with a beta-prism fold. Nat Struct Biol. 1996;3:596–603. doi: 10.1038/nsb0796-596. [DOI] [PubMed] [Google Scholar]
- Sato Y, Hirayama M, Morimoto K, Yamamoto N, Okuyama S, Hori K. High mannose-binding lectin with preference for the cluster of alpha1-2-mannose from the green alga Boodlea coacta is a potent entry inhibitor of HIV-1 and influenza viruses. J Biol Chem. 2011;286:19446–19458. doi: 10.1074/jbc.M110.216655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Vijayan M. Influence of glycosidic linkage on the natureof carbohydrate bindinginbeta-prism I fold lectins:an X-ray and molecular dynamics investigation on banana lectin-carbohydrate complexes. Glycobiology. 2011;21:23–33. doi: 10.1093/glycob/cwq128. [DOI] [PubMed] [Google Scholar]
- Shenoy SR, Barrientos LG, Ratner DM, O’Keefe BR, Seeberger PH, Gronenborn AM, Boyd MR. Multisite and multivalent binding between cyanovirin-N and branched oligomannosides: calorimetric and NMR characterization. Chem Biol. 2002;9:1109–1118. doi: 10.1016/s1074-5521(02)00237-5. [DOI] [PubMed] [Google Scholar]
- Singh DD, Saikrishnan K, Kumar P, Dauter Z, Sekar K, Surolia A, Vijayan M. Purification, crystallization and preliminary X-ray structure analysis of the banana lectin from Musa paradisiaca. Acta Crystallogr D Biol Crystallogr. 2004;60:2104–2106. doi: 10.1107/S0907444904024114. [DOI] [PubMed] [Google Scholar]
- Singh DD, Saikrishnan K, Kumar P, Surolia A, Sekar K, Vijayan M. Unusual sugar specificity of banana lectin from Musa paradisiaca and its probable evolutionary origin. Crystallographic and modelling studies. Glycobiology. 2005;15:1025–1032. doi: 10.1093/glycob/cwi087. [DOI] [PubMed] [Google Scholar]
- Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- Struwe WB, Rudd PM. Aminoquinolines as fluorescent labels for hydrophilic interaction liquid chromatography of oligosaccharides. Biol Chem. 2012;393:757–765. doi: 10.1515/hsz-2012-0169. [DOI] [PubMed] [Google Scholar]
- Svergun D, Barberato C, Koch M. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]
- Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MD, Winter HC, Goldstein IJ, Markovitz DM. A lectin isolated from bananas is a potent inhibitor of HIV replication. J Biol Chem. 2010;285:8646–8655. doi: 10.1074/jbc.M109.034926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MD, Boudreaux DM, Salmon L, Chugh J, Winter HC, Meagher JL, Andre S, Murphy PV, Oscarson S, Roy R, et al. Engineering a therapeutic lectin by uncoupling mitogenicity from antiviral activity. Cell. 2015;163:746–758. doi: 10.1016/j.cell.2015.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier MB, Grant OC, Heimburg-Molinaro J, Smith D, Jadey S, Gulick AM, Glushka J, Deutscher SL, Rittenhouse-Olson K, Woods RJ. Computational screening of the human TF-glycome provides a structural definition for the specificity of anti-tumor antibody JAA-F11. PLoS One. 2013;8:e54874. doi: 10.1371/journal.pone.0054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AB, Wilson IA. Insights into the trimeric HIV-1 envelope glycoprotein structure. Trends Biochem Sci. 2015;40:101–107. doi: 10.1016/j.tibs.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis WI, Drickamer K. Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem. 1996;65:441–473. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- Woods RJ, Chappelle R. Restrained electrostatic potential atomic partial charges for condensed-phase simulations of carbohydrates. Theochem. 2000;527:149–156. doi: 10.1016/S0166-1280(00)00487-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. Global Health Sector Strategy on HIV/AIDS 2011–2015. World Health Organization; 2011. [Google Scholar]
- Wriggers W, Chacón P. Using Situs for the registration of protein structures with low-resolution bead models from X-ray solution scattering. J Appl Crystallogr. 2001;34:773–776. [Google Scholar]
- Xiong C, O’Keefe BR, Botos I, Wlodawer A, McMahon JB. Overexpression and purification of scytovirin, a potent, novel anti-HIV protein from the cultured cyanobacterium Scytonema varium. Protein Expr Purif. 2006;46:233–239. doi: 10.1016/j.pep.2005.09.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.