

Abstract
Idiopathic autism spectrum disorders (ASDs) are neurodevelopmental disorders with unknown etiology. An estimated 1:68 children in the U.S. are diagnosed with ASDs, making these disorders a substantial public health issue. Recent advances in genome sequencing have identified numerous genetic variants across the ASD patient population. Many genetic variants identified occur in genes that encode glycosylated extracellular proteins (proteoglycans or glycoproteins) or enzymes involved in glycosylation (glycosyltransferases and sulfotransferases). It remains unknown whether “glycogene” variants cause changes in glycosylation and whether they contribute to the etiology and pathogenesis of ASDs. Insights into glycan susceptibility factors are provided by studies in the normal brain and congenital disorders of glycosylation, which are often accompanied by ASD-like behaviors. The purpose of this review is to present evidence that supports a contribution of extracellular glycans and glycoconjugates to the etiology and pathogenesis of idiopathic ASDs and other types of pervasive neurodevelopmental disorders.
Keywords: Autism, Autism spectrum disorders, Glycans, Glycosaminoglycans, Proteoglycans, Glycosyltransferase, Brain extracellular matrix, Glycosylation, Dystroglycanopathies, Polysialic acid
1. Introduction
Autism spectrum disorders (ASDs) are neurodevelopmental disorders characterized by a wide range of symptoms that include abnormal social interactions, limited interests, and stereotypic and repetitive behaviors (American Psychiatric Association, 2013). Hallmark symptoms typically arise in the second or third year of life, following a period of normal development or accompanying prolonged developmental delay (Newschaffer et al., 2007). Currently 1 out of 68 children are diagnosed with ASDs, with males having a four times greater risk than females. In the past decade the prevalence of ASDs has more than doubled, which emphasizes the need for improved early diagnosis and therapeutic intervention (Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators and Centers for Disease Control and Prevention, 2012).
Idiopathic ASDs arise from an unknown cause, where as syndromic ASD is secondary to a primary condition caused by a single gene mutation, for example Fragile X syndrome. ASD patients exhibit a wide range of behaviors, which is mirrored by equally impressive genetic heterogeneity. Recent findings support a significant genetic contribution to idiopathic ASD (Geschwind, 2011; Geschwind and State, 2015; Murdoch and State, 2013); however disease etiology and pathophysiology remain largely unclear. Efforts to associate genetic risk factors into common biochemical pathways and developmental processes have been made (Geschwind, 2008; Parikshak et al., 2013; Rubenstein and Merzenich, 2003; Subramanian et al., 2015). This approach has led to new theories on the etiology of ASD, which place alterations in developmental transcriptional regulation, brain growth, changes in the excitatory/inhibitory balance of the neural network, and abnormalities in neural plasticity at the crux of disease pathogenesis. It is also known that inflammation in the developing brain can lead to ASD-like behaviors (Kern et al., 2015). Thus genetic heterogeneity in the patient population may reflect a series of different genetic insults that converge on common neurodevelopmental processes that when perturbed have a similar impact on brain function.
The genetic heterogeneity of ASD introduces a significant challenge in understanding disease etiology. The complexity of the genetic architecture arises from numerous factors including (i) many chromosomal loci and common and rare genetic variants, which are either inherited or acquired de novo; (ii) genetic perturbations that range from single nucleotide substitutions to large chromosomal deletions/duplications; and (iii) genetic perturbations that range from single (monogenic) to multiple genes (polygenic). Despite these challenges the identification of genetic variants, including single nucleotide polymorphisms (SNPs) and copy number variations (CNVs), provide insight into the factors that may contribute to ASDs. Interestingly a number of these variants occur in genes (“glycogenes”) that encode glycosylated extracellular proteins (proteoglycans or glycoproteins) and lipids (glycosphingolipids) or enzymes involved in glycosylation (glycosyltransferases and sulfotransferases).
Glycans and their conjugates (glycoproteins, proteoglycans and glycolipids) are major constituents of the neural extracellular matrix (ECM). In this context, glycans and glycoconjugates participate in nearly every biological process in the developing brain. A potential link between ASDs and changes in glycosylation was initially noted in patients with congenital disorders of glycosylation (CDGs) (Freeze et al., 2015). These disorders result from rare homozygous recessive mutations causing the loss-of-function of a specific glycoconjugate or glycosyltransferase. Studies in mouse models of CDGs and behavioral phenotypes observed in CDG patients support the idea that glycogene variants either cause or contribute to the development of idiopathic ASDs. The purpose of this review is to present evidence that supports a contribution of extracellular glycans and glycoconjugates to the etiology and pathogenesis of ASDs.
1.1. Organization and assembly of glycans and glycoconjugates in the brain
A glycan is defined generically as any sugar or assembly of sugars, in free form or attached to another molecule. Although some glycans are found as free chains (e.g. hyaluronan), most are found covalently linked to proteins or lipids, i.e. as glycoconjugates. These include glycoproteins, proteoglycans, and glycosphingolipids (Fig. 1). The assembly of glycans occurs in the endoplasmic reticulum and Golgi apparatus of cells by a series of glycotransferases. These enzymes catalyze glycan assembly using activated sugar nucleotide donor substrates (e.g. UDP-galactose, GDP-fucose, CMP-sialic acid) that are transferred to acceptor substrates. Many glycans are further modified by processing enzymes that catalyze removal of specific sugar residues, or sulfation, acetylation and phosphorylation (Fig. 1). These modifications fine-tune glycan structure and function. Regulation of glycan biosynthesis occurs at a variety of different levels, including the availability of high-energy nucleotide donors, enzyme expression levels, and competition among enzymes for common glycan precursors. The impact of reducing the expression or function of a glycosyltransferase gene, either through CNVs or a SNP, depends on the relationship between enzyme function and gene dosage. The majority of enzymes associated with ASDs show gene dosage effects, suggesting that they may be rate limiting in the formation of particular glycans.
Fig. 1.
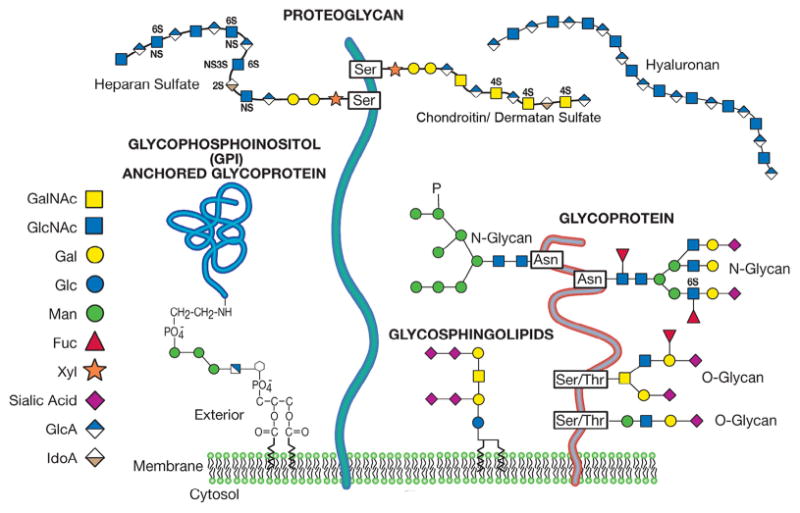
Common classes of animal glycans found in the extracellular environment of the brain. The glycans are depicted as parts of hypothetical glycoconjugates. The representative sugars are depicted by colored symbols as described in the legend. GalNAc, N-acetylgalactosamine; GlcNAc, N-acetylglucosamine; Gal, galactose; Glc, glucose; Man, mannose; Fuc, fucose; Xyl, xylose; GlcA, glucuronic acid; IdoA, iduronic acid. (Figure modified and reprinted with permission from Chapter 1 from Essentials of Glycobiology, 2nd edition).
Glycans and their glycoconjugates are abundant in the brain, in particular the ECM. All cell types including neurons, glia, and endothelial cells elaborate glycans and glycoconjugates. However, each cell type synthesizes a unique repertoire of glycan structures and glycoconjugates. For example, different antibody epitopes on different glycoforms of phosphacan label different types of cells in the developing cerebral cortex (Dwyer et al., 2015), supporting the idea that glycoform specialization may tailor protein function at the cellular level in the brain. Additional complexity arises from changes in the expression of different glycans and glycoconjugates in different brain regions and across developmental stages (Matthews et al., 2002; Morawski et al., 2012; Torii et al., 2014). The purpose of these differences is not fully understood.
The ECM of the brain can be divided into extracellular substructures comprising the pial basement membrane, interstitial neural extracellular matrix and cell surface glycocalyx (Fig. 2). The pial basement membrane covers the outermost surface of the brain and is comprised primarily of fibrillary proteins including laminin, fibronectin, collagen, and the secreted heparan sulfate proteoglycans agrin and perlecan (Fig. 2A). The predominant receptor for constituents of the pial basement membrane is the glycoprotein dystroglycan, which is expressed on the surface of radial glial cells and astrocytes comprising the limiting glial membrane. Interactions between dystroglycan and constituents of the pial basement membrane provide mechanical and structural integrity to the developing brain. The interstitial neural extracellular matrix fills the space between cells in the brain parenchyma and consists primarily of glycosaminoglycans and proteoglycans, hyaluronan and secreted chondroitin sulfate proteoglycans (Fig. 2B). The low abundance of fibrillary proteins such as laminin and collagen distinguishes the parenchymal neural ECM from matrices of other peripheral organs and tissue types. Nevertheless, the neural ECM regulates diffusion of growth factors, morphogens, and ions as in other organs. Constituents of the neural ECM are also ligands for many cell adhesion receptors, facilitating communication between the extracellular space and plasma membrane. The cell-surface glycocalyx is comprised of plasma membrane associated proteoglycans, glycoproteins and glycolipids (Fig. 2C). In the brain the cell-surface glycocalyx covers the surface of endothelial cells, astrocytes and neurons, including neuronal synapses. Glypicans (GPC, glycosylphosphatidylinositol-linked heparan sulfate proteoglycans) and syndecans (SDC, transmembrane heparan sulfate proteoglycans) predominate. Most cell adhesion molecules, cell surface receptors, and integral membrane proteins carry asparagine N-linked or serine/threonine O-linked glycans (e.g. PSA-NCAM, polysialylated neural cell adhesion molecule). Dystroglycan is also an abundant receptor/adhesion molecule in the glycocalyx of neurons and glia. Glycosphingolipids are abundant constituents of the cell surface glycocalyx. Constituents of the cell surface glycocalyx function as adhesion molecules, regulate local concentrations and the availability of growth factors and morphogens, and modulate receptor engagement and signaling.
Fig. 2.
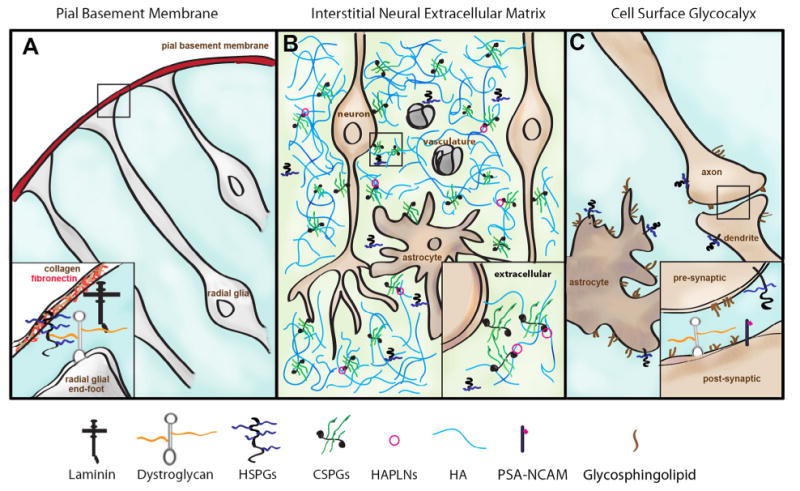
Extracellular substructures in the brain. The organization of free glycans and glycoconjugates in extracellular substructures in the brain is depicted. (A) Organization of the pial basement membrane is supported by interactions between glycosylated dystroglycan and extracellular matrix proteins. (B) The interstitial neural extracellular matrix fills the extracellular space between cells in the brain and is comprised predominately of secreted hyaluronan (HA), chondroitin sulfate proteoglycans (CSPGs), hyaluronan and link proteins (HAPLNs), and shed heparan sulfate proteoglycans (HSPGs). (C) The cell surface glycocalyx is comprised of glycoconjugates localized to the plasma membrane including glycosphingolipids, heparan sulfate proteoglycans (HSPGs), glycosylated dystroglycan, and glycoproteins carrying N- or O-linked glycans (e.g. polysialylated NCAM, PSA-NCAM). Constituents of the cell surface glycocalyx are also abundant in neuronal synapses (inset).
Glycans in the brain function as master regulators of nearly all neurodevelopmental processes including neurogenesis, neuronal migration, axon outgrowth and guidance, synaptogenesis, and neural plasticity. Previous studies have shown that deleting any glycan class has deleterious effects on brain development. Thus, it is not surprising that changes in glycan expression can underlie various diseases, including ASDs.
2. The dystrophin glycoprotein complex
Components of the dystrophin glycoprotein complex play essential roles in establishing gross brain architecture in the developing brain. Radial glial cells (neural stem cells of the developing cerebral cortex) residing in the ventricular zone of the developing brain extend processes outward to the marginal surface of the brain, physically anchoring the cells to the pial basement membrane (Fig. 2A). The glycoprotein, dystroglycan, expressed on the end feet of radial glial cells, binds to laminin present in the pial basement membrane. Anchoring provides mechanical strength to the radial glial scaffold as the brain expands during development. Campbell and colleagues determined that the “Large” glycan (also called matriglycan) modification on α-dystroglycan, synthesized by the Large glycosyltransferase complex, is required for binding of dystroglycan to its respective ECM constituents (Yoshida-Moriguchi and Campbell, 2015; Yoshida-Moriguchi et al., 2010). Newly generated neurons migrate along the radial glial scaffold and settle into their predetermined cortical layers, a process that gives rise to the lamination of the cerebral cortex.
Duplication and deletion CNVs at 22q12.3 encompassing the LARGE gene, encoding the Large glycosyltransferase, have been identified in cases of non-complex autism (van der Zwaag et al., 2009) (Table 1). Large is a dual-function glycosyltransferase, exhibiting the ability to synthesize repeating disaccharide units of xylose and glucuronic acid. To date, the only know protein modified by Large is dystroglycan. Recent work has shown that the length of the Large-glycan can be altered by changes in the expression of LARGE, which in turn affects the ligand-binding capacity of α-dystroglycan (Goddeeris et al., 2013). These results support the function of the Large-glycan as a tunable matrix scaffold and suggest that subtle changes in the expression of LARGE may have a substantial impact on Large-glycan biosynthesis, its ligand-binding properties, and biological function (Goddeeris et al., 2013).
Table 1.
Glycobiology related genes implicated in autism spectrum disorders.
| Gene/enzyme | Function | Localization of glycan/ glycoconjugates |
Association with ASDs |
References |
|---|---|---|---|---|
| LARGE (LARGE1) | Biosynthesis of the Large glycan (matriglycan) | Pial basement membrane; cell surface glycocalyx | Idiopathic ASD; syndromic ASD | Hehr et al. (2007), van der Zwaag et al. (2009) |
| LARGE xylosyltransferase and glucuronyltransferase 1 | ||||
| POMGNT1 | Biosynthesis of O-mannose linked glycans including the Large glycan (matriglycan) | Pial basement membrane; cell surface glycocalyx | Familial ASD; syndromic ASD | Hehr et al. (2007); Yu et al. (2013) |
| Protein O-linked mannose | ||||
| N-acetylglucosaminyltrasferase 1 | ||||
| B3GALNT2 | Biosynthesis of O- and N-linked glycans including the Large glycan (matriglycan) | Pial basement membrane; cell surface glycocalyx | Idiopathic ASD | van der Zwaag et al. (2009) |
| β1,3 N-acetylgalactosaminyltransferase 2 | ||||
| EXT1 | Biosynthesis of heparan sulfate chains | Interstitial neural extracellular matrix; cell surface glycocalyx | Idiopathic ASD; syndromic ASD | De Rubeis et al. (2014); Li et al. (2002) |
| Exostosin glycosyltransferase 1 | ||||
| B3GALT6 | Biosynthesis of heparan sulfate and chondroitin sulfate linkage region | Interstitial neural extracellular matrix; cell surface glycocalyx | Idiopathic ASD | van der Zwaag et al. (2009) |
| β1,3 galactosyltransferase 6 | ||||
| HS3ST5 | 3-O-sulfation of heparan sulfate chains | Interstitial neural extracellular matrix; cell surface glycocalyx | Idiopathic ASD | Connolly et al. (2013), Wang et al. (2009) |
| Heparan sulfate glucosaminyl-3-sulfotransferase 5 | ||||
| GPC5/GPC6 | Two GPI-anchored heparan sulfate proteoglycans | Interstitial neural extracellular matrix; cell surface glycocalyx | Idiopathic ASD | Pinto et al. (2010) |
| Glypican 5/6 | ||||
| SGSH | Lysosomal degradation of heparan sulfate | Lysosomal accumulation and possibly extracellular heparan sulfate | Syndromic ASD | Rumsey et al. (2014), Valstar et al. (2011) |
| Sulfamidase/N-sulfoglucosamine-N-sulfatase | ||||
| B3GALT1 | Biosynthesis of type I polylactosamine units found on N- and O-glycans | Cell surface glycocalyx | Idiopathic ASD | van der Zwaag et al. (2009) |
| β1,3 galactosyltransferase 1 | ||||
| GCNT2 | Branching of type II polylactosamine units found on N-glycans | Cell surface glycocalyx | Idiopathic ASD | van der Zwaag et al. (2009) |
| β1,6 N-acetylglucosaminyltransferase 2 | ||||
| SLC35A3 | Uridine diphosphate N-acetylglucosamine transporter | Cell surface glycocalyx | Familial ASD | Edvardson et al. (2013) |
| Solute carrier family 35 member A3 | ||||
| GAL3ST2 | Sulfation of terminal galactose residues on N- and O-glycans | Cell surface glycocalyx | Idiopathic ASD | van der Zwaag et al. (2009) |
| Galactose-3-O-sulfotransferase 2 | ||||
| ST8SIA2 | Biosynthesis of polysialic acid | Cell surface glycocalyx | Idiopathic ASD | Anney et al. (2010), Kamien et al. (2014) |
| ST8 N-acetyl-neuraminide α2,8 sialytransferase 2 | ||||
| B3GNT5 | Biosynthesis of lactosyltriosylceramide | Cell surface glycocalyx | Idiopathic ASD | van der Zwaag et al. (2009) |
| UDP-GlcNAcgalactose β1,3 | ||||
| N-acetylglucosaminyltransferase 5 | ||||
| GALNT9 | Biosynthesis of mucin-type O-linked glycans | Cell surface glycocalyx | Idiopathic ASD | van der Zwaag et al. (2009) |
| Polypeptide N-acetylgalactosaminyltransferase 9 | ||||
| GALNTL5 | Biosynthesis of mucin-type O-linked glycans | Unknown | Idiopathic ASD | van der Zwaag et al. (2009) |
| Polypeptide N-acetylgalactosaminyltransferase 5 |
Mutations in protein O-linked mannose N-acetylglucosaminyltransferase 1 (POMGNT1) have also been associated with inherited forms of ASDs (Yu et al., 2013). POMGnT1 catalyzes the elaboration of core M1 and M2 glycans on dystroglycan (Stalnaker et al., 2010) and other extracellular proteins such as phosphacan (Dwyer et al., 2012, 2015), CD-24 (Bleckmann et al., 2009) and Cadherins (Vester-Christensen et al., 2013). Interestingly loss of POMGnT1 activity affects the production of dystroglycan carrying the Large-glycan modification that is capable of functioning as an ECM receptor, suggesting a common glycosylation pathway. Homozygous recessive disorders caused by the loss-of-function mutations in LARGE or POMGNT1 give rise to dystroglycanopathies, a form of congenital muscular dystrophy with severe central nervous system abnormalities. Dystroglycanopathy patients and mouse models bearing mutations in Large or Pomgnt1 exhibit abnormalities in neuronal migration, cortical and cerebellar lamination defects, heterotopias, and hydrocephalus (Moore et al., 2002). Many dystroglycanopathy patients display ASD-like behavioral phenotypes (Hehr et al., 2007).
Aside from its role in neuronal migration, dystroglycan also has a synaptic function. Loss of dystroglycan or the Large glycan substantially impairs hippocampal long-term potentiation, a form of cellular learning and memory (Moore et al., 2002; Satz et al., 2010). Dystroglycan localizes to post-synaptic sites (Zaccaria et al., 2001) and is found on a subset of GABAergic inhibitory synapses in hippocampal neurons (Levi et al., 2002). The amount of glycosylated dystroglycan increases under conditions of chronically elevated neuronal activity, which enhances the scaling of inhibitory synaptic strength to maintain homeostatic plasticity (Pribiag et al., 2014). A role for dystroglycan in specification of inhibitory neural circuit subpopulations has also been proposed, as dystroglycan binds to presynaptic α-neurexins and competes with binding of α-neurexins to other post-synaptic adhesion molecules, including neurexophilin-1 and neuroligins (Reissner et al., 2014). Binding of dystroglycan to α-neurexins depends on its modification with the Large glycan (Reissner et al., 2014). These findings suggest that dystroglycan may contribute to maintaining the excitatory/inhibitory network balance. An imbalance in excitatory/inhibitory neural networks is believed to contribute to some types of ASDs. Furthermore, the placement of glycosylated dystroglycan in inhibitory synaptic specification pathways regulated by α-neurexins emphasizes a potential link to a complex ASD network, as neuroligins and neurexins are high-confidence ASD risk factors (Chih et al., 2004; Jamain et al., 2003; Tong et al., 2015). Together these data suggest convergence of glycosylated dystroglycan with a common synaptic pathway linked to ASDs.
Loss of function of the X-linked dystrophin gene is associated with syndromic autism and causes Duchenne muscular dystrophy (DMD). DMD patients have impaired cognition and are also diagnosed with ASDs more frequently than the normal population (Wu et al., 2005). The mdx mouse model of DMD also exhibits autistic-like behaviors (Miranda et al., 2015). A role for the dystrophin gene in idiopathic ASDs has not been firmly established; however many rare SNPs within the dystrophin gene have been identified (Koshimizu et al., 2013; Redin et al., 2014). A rare maternally inherited deletion of Xp21.2 encompassing the dystrophin gene was identified in a male ASD patient from a simplex family without accompanying muscular dystrophy (Pinto et al., 2014). In skeletal muscle, dystrophin interacts with dystroglycan to form the so-called dystrophin glycoprotein complex. A modified dystrophin glycoprotein complex is also present in the brain. While both dystroglycanopathy and DMD patients have congenital muscular dystrophy, DMD patients do not exhibit neuronal migration abnormalities. However dystrophin co-localizes with α and β-dystroglycan at inhibitory synapses in different brain regions (Levi et al., 2002; Waite et al., 2009). Reduced inhibitory synaptic function and clustering of the α1 subunit of GABAA receptor clusters has been described in mdx mice in hippocampal neurons and Purkinje cells of the cerebellum (Knuesel et al., 1999; Kueh et al., 2011). Recent evidence shows that deletion of dystrophin causes spatial reorganization of inhibitory synapses in the hippocampus, suggesting potential alterations at the level of inhibitory neural circuit function (Krasowska et al., 2014). These findings are consistent with the observation that GABAergic signaling is reduced in the brains of many ASD patients (Robertson et al., 2016), and suggest changes in inhibitory synaptic function that may contribute to ASD and abnormal cognitive behaviors in DMD patients. These studies of glycosylated dystroglycan and dystrophin emphasize the potential contribution of a synaptic dystrophin glycoprotein complex in the pathogenesis of ASDs. Studies to identify other synaptic proteins that interact with the dystroglycan/dystrophin complex could provide additional candidates to explain idiopathic ASDs and related behavioral phenotypes in congenital muscular dystrophy.
3. Heparan sulfate and heparan sulfate proteoglycans
Heparan sulfate (HS) regulates nearly every biological process in the developing embryonic and early postnatal brain. Conditional deletion of Ext1 using a Nestin-cre driver, which deletes HS in neural stem cells at the onset of neurogenesis, revealed that HS is required for cortical neurogenesis, patterning of the midbrain and cerebellum and axon guidance of major commissural tracts (Inatani et al., 2003). As a result conditional deletion of Ext1 in the neural stem cell population is lethal at birth (Inatani et al., 2003). Deficits in HS-modifying enzymes give rise to similar phenotypes. For example, inactivation of the sulfotransferases Ndst1 or Hs2st also impacts cortical neurogenesis (Grobe et al., 2005; McLaughlin et al., 2003). Similarly, inactivation of Ndst1, Hs2st or Hs6st in mice causes abnormalities in axon guidance at the developing optic chiasm and corpus callosum (Conway et al., 2011; Grobe et al., 2005; Pratt et al., 2006). HS also has a role in synapse formation and maintenance. Conditional deletion of Ext1 using a CAMKII-cre driver, which deletes HS in post-mitotic neurons, reduces excitatory synaptic function in pyramidal neurons of the basolateral amygdala (Irie et al., 2012).
HS chains are comprised of repeating disaccharide units of uronic acid (iduronic or glucuronic) and glucosamine. The HS chain is synthesized by the co-polymerase complex formed by Ext1 and Ext2 in the Golgi. A series of enzymes N-deacetylate, epimerize, and sulfate the HS chains at various positions, creating ligand-binding domains along the length of the chain. Many secreted growth factors and morphogens important for regulating neurogenesis, patterning of the brain, and axon guidance bind HS chains with high affinity, including members of the fibroblast growth factor, bone morphogenic protein, Wnt, hedgehog, and Slit families. In some cases ternary signaling complexes are created from secreted ligands, receptors and HS (e.g. Slit-Robo-HS (Hussain et al., 2006)). Chemokines and cytokines also bind HS. Alterations in HS chain structure or chain length alters ligandbinding properties and the activation of downstream signaling cascades (Bishop et al., 2007).
Systemic homozygous deletion of EXT1 results in complete loss of HS and early developmental arrest due to defective gastrulation (Lin et al., 2000). However, heterozygosity of EXT1 causes a 20–40% reduction in HS chain length and is tolerated, resulting in occasional osteochon-dromas on endochondral bones patients (Hereditary Multiple Exostoses, HME). Some HME patients have abnormal ASD-like social behaviors and are formally diagnosed with clinical autism (Li et al., 2002). In a remarkable study, Yamaguchi and colleagues showed a direct relationship between HS and autistic-like behavioral phenotypes in mice. Conditional deletion of Ext1 using a CAMKII-cre driver, which deletes HS in post-mitotic neurons, gave rise to social impairments, reduced anxiety, hyperactivity, and hypersensitivity to thermal stimuli (Irie et al., 2012). Rare CNVs and SNPs within EXT1 have been identified in patients with ASDs (De Rubeis et al., 2014; Kaminsky et al., 2011). The incomplete penetrance of ASD-like behaviors in HME patients suggests that neurological phenotypes caused by loss of EXT1 depend on the presence of other susceptibility traits with genetic, epigenetic or environmental origins.
B3GALT6, an enzyme involved in the biosynthesis of the HS linkage tetrasaccharide has also been associated with ASDs (van der Zwaag et al., 2009). A common intergenic variant of HS3ST5 has also been associated with ASDs as well (Connolly et al., 2013; Wang et al., 2009). HS3ST5 is one of seven 3-O-sulfotransferases expressed in the brain. To date, no mouse model of HS3ST5 has been generated. Recent work has shown that the function of 3-O-sulfation catalyzed by HS3ST2 depends on gene dosage, suggesting that 3-O-sulfation and 3-O-sulfate dependent activities may be generally regulated at the level of sulfotransferase expression (Thacker et al., 2016). Thacker and colleagues showed that neuropilin-1 binds to 3-O sulfated HS with high affinity and that genetic reduction of 3-O-sulfation desensitized neuropilin-1 to semaphorin3A induced growth cone collapse in dorsal root ganglion explants (Thacker et al., 2016). Interestingly, gene polymorphisms in neuropilin-2 have been associated with autism in Chinese Han population (Wu et al., 2007). Determining whether 3-O-sulfation also modulates neuropilin-2 function would be an important area of future investigation. Future studies to assess the function of 3-O-sulfation of HS in the brain will shed light on its potential role in ASDs.
Reductions in the immunoreactivity of HS antibodies in mouse models of ASD and human post mortem brain samples have also been described (Mercier et al., 2012; Meyza et al., 2012; Pearson et al., 2013). It is unclear whether these changes reflect a loss in HS content or alterations in HS fine structure that alters antibody affinity. Glycosaminoglycans in the urine of ASD patients have also been described (Endreffy et al., 2016). Release of HS also occurs in lysosomal storage disorders caused by mutations in lysosomal hydrolases that degrade glycosaminoglycans, suggesting abnormalities in lysosomal HS degradation may occur in ASD patients.
HS chains are covalently attached to a subset of extracellular proteins called HS proteoglycans (HSPGs), which are abundant in the cell surface glycocalyx. Unlike mutations of HS, the deletion of a single HSPG does not typically cause overt changes in gross brain structure. An exception to this generalization is Gpc1; Gpc1 knockout mice have reduced brain size due to abnormal neurogenesis (Jen et al., 2009). Recent work has revealed the contribution of HSPGs to synaptogenesis. A remarkable study by Allen and colleagues showed that astrocytes release the glycosylphosphatidylinositol-linked HSPGs Gpc4 and Gpc6, which enhance the insertion of AMPA receptors at the post-synaptic membrane of excitatory synapses (Allen et al., 2012). As predicted by this observation, Gpc4 knockout animals show reduced hippocampal excitatory synaptic strength (Allen et al., 2012). Presynaptic Gpc4 also functions as a cell-adhesion receptor for post-synaptic LRRTM4, thereby regulating the number of excitatory synaptic connections (de Wit et al., 2013). The HS chains of Gpc4 and Gpc6 play an essential role in synaptogenesis (Allen et al., 2012; Ko et al., 2015). Sdc2, a transmembrane HSPG, is required for maturation of dendritic spines in hippocampal neurons (Ethell and Yamaguchi, 1999). The biological functions of HSPGs in normal development suggest potential contributions of these molecules in pervasive developmental disorders.
Simpson–Golabi–Behmel is a rare overgrowth syndrome caused by the loss of function in the X-linked gene GPC3 and occasionally GPC4. Diagnosis of ASD and ADHD has been confirmed in one patient with Simpson–Golabi–Behmel (Halayem et al., 2016). Developmental delay has also been described in patients with autosomal-recessive omodysplasia, which is caused by homozygous loss of GPC6 (Campos-Xavier et al., 2009). Rare CNVs affecting GPC5/6, involving both deletions and duplications, have also been identified in several idiopathic ASD patients (Pinto et al., 2010). These findings suggest altered expression of GPCs may contribute to certain ASD subtypes.
Interestingly loss of function of lysosomal hydrolases that degrade HS is also associated with ASD-like behaviors. Patients with Sanfilippo Syndrome (Mucopolysaccharidosis III [MPS] A-D), a type of lysosomal storage disorder, are severely hyperactive and aggressive at disease onset. Many patients also display social behaviors consistent with ASDs (Rumsey et al., 2014; Valstar et al., 2011). Interestingly, these behaviors are not observed in other types of lysosomal storage disorders, suggesting a potential causative role of HS in their manifestation.
These data provide a circumstantial link between alterations in HS and ASD, but additional work is needed to establish a cause-and-effect relationship. It seems likely that the incomplete penetrance of ASD-like behaviors reflects in HME the contribution of other pathways and modulatory factors. Efforts to assemble a functional HS-interactome in the brain might lead to other genetic susceptibility factors that when compounded with deficiencies in HS cause synergistic/epistatic risk for ASDs.
4. N- and O-linked glycans
Several essential proteins involved in cell adhesion and migration, synaptic transmission, and signal transduction are decorated with asparagine N-linked or serine/threonine O-linked glycans. This form of glycosylation is common; as much as 85% of secreted and membrane proteins contain one or more N-linked and/or O-linked glycans, which are designated generically as glycoproteins. The glycan chains play many different roles, including protein folding and quality control, sites for ligand-recognition, protein oligomerization, protein stability and biological activity, and host–pathogen interactions. From the study of CDGs, it is known that N-linked glycosylation is important in brain structure and function. Many of the CDGs alter overall glycosylation, and have profound effects on cognitive function. In contrast, mutations in glycogenes that associate with ASDs affect downstream steps in N-glycan biosynthesis and presumably do not result in loss of chains, but rather in alterations in their structure. CNVs in B3GALT1, GCNT2, and GAL3ST2 have been identified (van der Zwaag et al., 2009) (Table 1). These CNVs also affect other genes, thus further work is needed to establish if ASD is directly related to loss-of-function of these glycosyltransferases.
B3GALT1 belongs to the β1,3 galactosyltransferase gene family that catalyzes the formation of Type I polylactosamine units (Galβ1,3GlcNAc) on N- and O-glycans. Early studies documented that B3GALT1 and B3GALT2 have similar kinetic properties and are both expressed in the brain. However, B3GALT1 expression is restricted to the brain; its association with ASD supports the idea that brain glycoproteins containing this structure are important (Amado et al., 1998).
GCNT2 encodes β1,6 N-Acetylglucosaminyltransferase, which initiates β1,6 branching of polylactosamine chains on type II polylactosamine containing N-glycans. Its expression is abundant in the olfactory neurons (Henion and Schwarting, 2014), but little is known about GCNT2 function in the brain. A reduction of β1,6 branching of N-glycans has been associated with familial ASD in which afflicted patients also presented with arthrogryposis and epilepsy. However, this was not linked to GCNT2, but rather to the uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) transporter, SLC35A3 (Edvardson et al., 2013), which is required for import of UDP-GlcNAc into the Golgi. These data support the idea that a reduction in β1,6 branched N-glycans as a risk factor for ASD.
GAL3ST2 encodes galactose-3-O-sulfotransferase, which is unrelated to the enzyme involved in 3-O-sulfation of HS. GAL3ST2 adds sulfate groups to terminal galactose residues on N- and O-glycans. The function of this modification is unknown.
Many proteins undergo N- and O-linked glycosylation, making it difficult to identify the specific glycoprotein targets that result in altered brain development or function. For additional information, see Scott and Panin (2014). Two examples are worth mentioning. Many voltage dependent ion channels are modified with N-glycans, which affect their expression and permeability. For example, rare missense SNP mutations have been identified in the voltage dependent calcium channel gene CACNA1H (T-type Cav3.2), which reduce Cav3.2 channel activity (D'Gama et al., 2015; Iossifov et al., 2014; Karaca et al., 2015; Splawski et al., 2006). N-glycans at N192 regulate surface expression of Cav3.2, while N-glycans at Asn1466 regulate activity by enhancing channel permeability or regulating pore opening (Ondacova et al., 2016; Weiss et al., 2013). Together these data provide circumstantial evidence to suggest that alteration in N-glycans on Cav3.2 might have an effect similar to SNP mutations on channel function and brain physiology.
A small set of N-linked glycoproteins contains polysialic acid (PSA, α2,8-linked) in the brain, including neural cell adhesion molecule (NCAM) and synaptic cell adhesion molecule SynCAM-1. PSA-NCAM is involved in neuronal migration, axon guidance and synaptic plasticity. Copy number loss and SNPs in the polysialic acid synthesizing enzyme, ST8SIAII, are associated with ASDs (Anney et al., 2010; Kamien et al., 2014). ST8SIAII is one of two enzymes involved in modifying NCAMs with PSA. In mice complete deletion of PSA (achieved by inactivation of ST8SIAII and ST8SIAIV) causes a gain of NCAM function and a variety of developmental phenotypes that can be restored by deleting NCAM (Eckhardt et al., 2000; Weinhold et al., 2005). These findings emphasize the importance of the PSA glycan in regulating glycoprotein function (Weinhold et al., 2005). More recent work has shown a direct connection between PSA and ASD-like behaviors, as mice deficient in ST8SIAII have reduced social motivation, increased aggression and hyperactivity (Calandreau et al., 2010).
5. Glycosphingolipids
Glycosphingolipids (GSLs) are the most abundant glycoconjugate in the brain, constituting ∼80% of brain glycans. Enriched in the outer leaflet of the plasma membrane, the GSLs mediate cell–cell interactions and modulate activities of proteins by way of clustering in so-called “lipid rafts.” In spite of their documented importance in my elination and nerve conduction, GSLs have not been associated with ASDs, with the exception of the enzyme B3GNT5 (van der Zwaag et al., 2009), which synthesizes lactosyltriosylceramide, the core of lactoseries derived glycosphingolipids. Mutations in ganglioside assembly (GM3 synthase and GM2/GD2 synthase) cause seizures, cognitive and motor decay, spastic paraplegia and intellectual disability. Thus, it seems likely that alterations in GSL biosynthesis might contribute to ASD etiology. GSL compositional studies in ASD patients are lacking.
6. Glycogenes as risk factors for ASDs
As should be clear from the above examples, additional work is needed to determine the contribution of glycosylation to the etiology and pathogenesis of ASDs. Table 1 outlines glycogenes that have been identified or associated with ASDs. Both gain of expression, induced by CNV duplication, as well as loss of expression due to SNPs and CNV loss could impact the glycan repertoire expressed by relevant cell types in the brain. Other mutations should be considered as well. For example, a SNP that introduces a single amino acid mutation in a glycan attachment site would interfere with the formation of specific protein glycoforms. Recent evidence suggests site-specific glycosylation is conferred in part by peptide sequence adjacent to a glycosylation site, as local protein surface influences enzyme accessibility to individual glycans during biosynthesis (Hang et al., 2015). Tools to study site-specific glycosylation (glycoproteomics) have matured over the last decade allowing analysis of site-specific glycan structures in brain glycoconjugates. Gain-of-glycosylation can also occur (Prada et al., 2012). It is also possible that alterations in glycosylation may arise indirectly from changes in cell metabolism or in the organization of cellular glycosylation machinery in the ER and Golgi.
The following multidisciplinary studies are needed to more firmly establish a link between glycosylation and ASD.
ASD-like behaviors should be studied in existing mutant mouse strains lacking specific enzymes and glycoconjugates. The incomplete deletion of glycosyltransferases and glycoconjugates genes in ASDs places emphasis on the importance of evaluating neurological phenotypes in heterozygous mice, which in general exhibit subtle glycan perturbations. About 1% of the human genome is dedicated to glycosylation, but only a small fraction of these genes have been studied in the context of neurological disorders and ASD.
The available association studies linking glycosylation to ASD are mostly correlative. Novel knock-in mouse models are needed bearing human genetic mutations associated with ASD. The availability of gene targeting methods, such as CRISPR/Cas9, makes this approach feasible.
Characterization of glycan structures in postmortem brain tissues from patients with ASD and other neuropsychiatric and neurodegenerative disorders might provide important insight into changes in the glycan landscape in these disorders. The long-term stability of glycans and ability to assess glycan structure in fixed specimens removes many technical limitations associated with evaluating post-mortem samples.
Studies should be undertaken to define high-confidence candidate ASD risk factors that create epistatic or synergistic risk when compounded with rare glycan-related risk factors. These studies will reveal important insights about comorbidity and further delineate key biochemical pathways that may confer heightened risk for ASD.
Diagnostic methods should be developed to examine plasma and urine glycans and related metabolites to identify potential biomarkers of ASD.
The identification of glycan susceptibility factors in ASDs will undoubtedly reveal new and exciting functions of brain glycans. Moreover, they may suggest glycan-based avenues for therapeutic intervention, such as gene therapy, glycoengineering, and development of drug-like agents for restoring glycan function.
Acknowledgments
This work was supported by grant NIH-T32 HL086344-08 (to C.A.D), and grants AR064202 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, HL107150 Program for Excellence in Glycosciences from the National Heart, Lung, and Blood Institute, and a grant from Cure Sanfilippo Foundation and the Lubkin Fund for MHE Research (to J.D.E). The authors would also like to thank Dr. Russell Matthews and Spencer Moore for their insightful comments.
Abbreviations
- ASDs
autism spectrum disorders
- CDGs
congenital disorders of glycosylation
- CNVs
copy number variations
- DMD
Duchenne muscular dystrophy
- ECM
extracellular matrix
- GPC
glypicans
- HS
heparan sulfate
- HSPGs
heparan sulfate proteoglycans
- MPSIII
mucopolysaccharidosis III A-D
- PSA-NCAM
polysialylated neural cell adhesion molecule
- SDC
syndecan
- SNPs
single nucleotide polymorphisms
Footnotes
C.A.D completed her PhD in neuroscience under the supervision of Dr. Russell Matthews. Her work led to the discovery that phosphacan is glycosylated with O-mannosyl glycans in a cell-type specific manner and abnormally glycosylated in dystroglycanopathy mouse models. Other work characterized the contribution of brevican to glioma initiating cell driven tumorigenesis. C.A.D joined the laboratory of Dr. Jeffrey Esko, where she identified neurodevelopmental changes in HS content and excitatory synaptic function in the somatosensory cortex of Sanfilippo Syndrome mouse models. Current work in collaboration with Dr. Alysson Muotri is focused on defining the contribution of HS to disease pathogenesis of syndromic ASDs.
J.D.E is a Distinguished Professor in the Department of Cellular and Molecular Medicine and Co-director of the Glycobiology Research and Training Center at the University of California, San Diego. J.D.E is renowned for his contribution in the elucidation of HS structure, assembly and function in a variety of biological systems, including the brain. J.D.E has authored over 250 scholarly papers and is a coeditor/coauthor of the Essentials of Glycobiology textbook.
References
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486(7403):410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amado M, Almeida R, Carneiro F, Levery SB, Holmes EH, Nomoto M, et al. A family of human beta3-galactosyltransferases. Characterization of four members of a UDP-galactose:beta-N-acetyl-glucosamine/beta-nacetyl-galactosamine beta-1,3-galactosyltransferase family. J Biol Chem. 1998;273(21):12770–12778. doi: 10.1074/jbc.273.21.12770. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. fifth. Arlington, VA: 2013. [Google Scholar]
- Anney R, Klei L, Pinto D, Regan R, Conroy J, Magalhaes TR, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet. 2010;19(20):4072–4082. doi: 10.1093/hmg/ddq307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators, Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders–Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. Morb Mortal Wkly Rep Surveill Summ. 2012;61(3):1–19. [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446(7139):1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Bleckmann C, Geyer H, Lieberoth A, Splittstoesser F, Liu Y, Feizi T, et al. O-glycosylation pattern of CD24 from mouse brain. Biol Chem. 2009;390(7):627–645. doi: 10.1515/BC.2009.044. [DOI] [PubMed] [Google Scholar]
- Calandreau L, Marquez C, Bisaz R, Fantin M, Sandi C. Differential impact of polysialyltransferase ST8SiaII and ST8SiaIV knockout on social interaction and aggression. Genes Brain Behav. 2010;9(8):958–967. doi: 10.1111/j.1601-183X.2010.00635.x. [DOI] [PubMed] [Google Scholar]
- Campos-Xavier AB, Martinet D, Bateman J, Belluoccio D, Rowley L, Tan TY, et al. Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia. Am J Hum Genet. 2009;84(6):760–770. doi: 10.1016/j.ajhg.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Afridi SK, Clark L, Scheiffele P. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet. 2004;13(14):1471–1477. doi: 10.1093/hmg/ddh158. [DOI] [PubMed] [Google Scholar]
- Connolly JJ, Glessner JT, Hakonarson H. A genome-wide association study of autism incorporating autism diagnostic interview-revised, autism diagnostic observation schedule, and social responsiveness scale. Child Dev. 2013;84(1):17–33. doi: 10.1111/j.1467-8624.2012.01838.x. [DOI] [PubMed] [Google Scholar]
- Conway CD, Howe KM, Nettleton NK, Price DJ, Mason JO, Pratt T. Heparan sulfate sugar modifications mediate the functions of slits and other factors needed for mouse forebrain commissure development. J Neurosci. 2011;31(6):1955–1970. doi: 10.1523/JNEUROSCI.2579-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, O'Sullivan ML, Savas JN, Condomitti G, Caccese MC, Vennekens KM, et al. Unbiased discovery of glypican as a receptor for LRRTM4 in regulating excitatory synapse development. Neuron. 2013;79(4):696–711. doi: 10.1016/j.neuron.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515(7526):209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Gama AM, Pochareddy S, Li M, Jamuar SS, Reiff RE, Lam AT, et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron. 2015;88(5):910–917. doi: 10.1016/j.neuron.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer CA, Baker E, Hu H, Matthews RT. RPTPzeta/phosphacan is abnormally glycosylated in a model of muscle-eye-brain disease lacking functional POMGnT1. Neuroscience. 2012;220:47–61. doi: 10.1016/j.neuroscience.2012.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer CA, Katoh T, Tiemeyer M, Matthews RT. Neurons and glia modify receptor protein-tyrosine phosphatase zeta (RPTPzeta)/phosphacan with cell-specific O-mannosyl glycans in the developing brain. J Biol Chem. 2015;290(16):10256–10273. doi: 10.1074/jbc.M114.614099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M, Bukalo O, Chazal G, Wang L, Goridis C, Schachner M, et al. Mice deficient in the polysialyltransferase ST8SiaIV/PST-1 allow discrimination of the roles of neural cell adhesion molecule protein and polysialic acid in neural development and synaptic plasticity. J Neurosci. 2000;20(14):5234–5244. doi: 10.1523/JNEUROSCI.20-14-05234.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Ashikov A, Jalas C, Sturiale L, Shaag A, Fedick A, et al. Mutations in SLC35A3 cause autism spectrum disorder, epilepsy and arthrogryposis. J Med Genet. 2013;50(11):733–739. doi: 10.1136/jmedgenet-2013-101753. [DOI] [PubMed] [Google Scholar]
- Endreffy I, Bjorklund G, Dicso F, Urbina MA, Endreffy E. Acid glycosaminoglycan (aGAG) excretion is increased in children with autism spectrum disorder, and it can be controlled by diet. Metab Brain Dis. 2016;31(2):273–278. doi: 10.1007/s11011-015-9745-2. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Yamaguchi Y. Cell surface heparan sulfate proteoglycan syndecan-2 induces the maturation of dendritic spines in rat hippocampal neurons. J Cell Biol. 1999;144(3):575–586. doi: 10.1083/jcb.144.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH, Eklund EA, Ng BG, Patterson MC. Neurological aspects of human glycosylation disorders. Annu Rev Neurosci. 2015;38:105–125. doi: 10.1146/annurev-neuro-071714-034019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135(3):391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH. Genetics of autism spectrum disorders. Trends Cogn Sci. 2011;15(9):409–416. doi: 10.1016/j.tics.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, State MW. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 2015;14(11):1109–1120. doi: 10.1016/S1474-4422(15)00044-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddeeris MM, Wu B, Venzke D, Yoshida-Moriguchi T, Saito F, Matsumura K, et al. LARGE glycans on dystroglycan function as a tunable matrix scaffold to prevent dystrophy. Nature. 2013;503(7474):136–140. doi: 10.1038/nature12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 2005;132(16):3777–3786. doi: 10.1242/dev.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halayem S, Hamza M, Maazoul F, Ben Turkia H, Touati M, Tebib N, et al. Distinctive findings in a boy with Simpson-Golabi-Behmel syndrome. Am J Med Genet A. 2016;170(4):1035–1039. doi: 10.1002/ajmg.a.37518. [DOI] [PubMed] [Google Scholar]
- Hang I, Lin CW, Grant OC, Fleurkens S, Villiger TK, Soos M, et al. Analysis of site-specific N-glycan remodeling in the endoplasmic reticulum and the Golgi. Glycobiology. 2015;25(12):1335–1349. doi: 10.1093/glycob/cwv058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehr U, Uyanik G, Gross C, Walter MC, Bohring A, Cohen M, et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle-eye-brain disease. Neurogenetics. 2007;8(4):279–288. doi: 10.1007/s10048-007-0096-y. [DOI] [PubMed] [Google Scholar]
- Henion TR, Schwarting GA. N-linked polylactosamine glycan synthesis is regulated by co-expression of beta3GnT2 and GCNT2. J Cell Physiol. 2014;229(4):471–478. doi: 10.1002/jcp.24467. [DOI] [PubMed] [Google Scholar]
- Hussain SA, Piper M, Fukuhara N, Strochlic L, Cho G, Howitt JA, et al. A molecular mechanism for the heparan sulfate dependence of slit-robo signaling. J Biol Chem. 2006;281(51):39693–39698. doi: 10.1074/jbc.M609384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302(5647):1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie F, Badie-Mahdavi H, Yamaguchi Y. Autism-like socio communicative deficits and stereotypies in mice lacking heparan sulfate. Proc Natl Acad Sci USA. 2012;109(13):5052–5056. doi: 10.1073/pnas.1117881109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jen YH, Musacchio M, Lander AD. Glypican-1 controls brain size through regulation of fibroblast growth factor signaling in early neurogenesis. Neural Develop. 2009;4:33. doi: 10.1186/1749-8104-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamien B, Harraway J, Lundie B, Smallhorne L, Gibbs V, Heath A, et al. Characterization of a 520 kb deletion on chromosome 15q26.1 including ST8SIA2 in a patient with behavioral disturbance, autism spectrum disorder, and epilepsy. Am J Med Genet A. 2014;164A(3):782–788. doi: 10.1002/ajmg.a.36345. [DOI] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13(9):777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, et al. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron. 2015;88(3):499–513. doi: 10.1016/j.neuron.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern JK, Geier DA, Sykes LK, Geier MR. Relevance of neuroinflammation and encephalitis in autism. Front Cell Neurosci. 2015;9:519. doi: 10.3389/fncel.2015.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM. Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice) Eur J Neurosci. 1999;11(12):4457–4462. doi: 10.1046/j.1460-9568.1999.00887.x. [DOI] [PubMed] [Google Scholar]
- Ko JS, Pramanik G, Um JW, Shim JS, Lee D, Kim KH, et al. PTPsigma functions as a presynaptic receptor for the glypican-4/LRRTM4 complex and is essential for excitatory synaptic transmission. Proc Natl Acad Sci USA. 2015;112(6):1874–1879. doi: 10.1073/pnas.1410138112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshimizu E, Miyatake S, Okamoto N, Nakashima M, Tsurusaki Y, Miyake N, et al. Performance comparison of bench-top next generation sequencers using microdroplet PCR-based enrichment for targeted sequencing in patients with autism spectrum disorder. PLoS ONE. 2013;8(9):e74167. doi: 10.1371/journal.pone.0074167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowska E, Zablocki K, Gorecki DC, Swinny JD. Aberrant location of inhibitory synaptic marker proteins in the hippocampus of dystrophin-deficient mice: implications for cognitive impairment in Duchenne muscular dystrophy. PLoS ONE. 2014;9(9):e108364. doi: 10.1371/journal.pone.0108364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh SL, Dempster J, Head SI, Morley JW. Reduced postsynaptic GABAA receptor number and enhanced gaboxadol induced change in holding currents in Purkinje cells of the dystrophin-deficient mdx mouse. Neurobiol Dis. 2011;43(3):558–564. doi: 10.1016/j.nbd.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Levi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci. 2002;22(11):4274–4285. doi: 10.1523/JNEUROSCI.22-11-04274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yamagata T, Mori M, Momoi MY. Association of autism in two patients with hereditary multiple exostoses caused by novel deletion mutations of EXT1. J Hum Genet. 2002;47(5):262–265. doi: 10.1007/s100380200036. [DOI] [PubMed] [Google Scholar]
- Lin X, Wei G, Shi Z, Dryer L, Esko JD, Wells DE, et al. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev Biol. 2000;224(2):299–311. doi: 10.1006/dbio.2000.9798. [DOI] [PubMed] [Google Scholar]
- Matthews RT, Kelly GM, Zerillo CA, Gray G, Tiemeyer M, Hockfield S. Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J Neurosci. 2002;22(17):7536–7547. doi: 10.1523/JNEUROSCI.22-17-07536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin D, Karlsson F, Tian N, Pratt T, Bullock SL, Wilson VA, et al. Specific modification of heparan sulphate is required for normal cerebral cortical development. Mech Dev. 2003;120(12):1481–1488. doi: 10.1016/j.mod.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Mercier F, Kwon YC, Douet V. Hippocampus/amygdala alterations, loss of heparan sulfates, fractones and ventricle wall reduction in adult BTBR T+ tf/J mice, animal model for autism. Neurosci Lett. 2012;506(2):208–213. doi: 10.1016/j.neulet.2011.11.007. [DOI] [PubMed] [Google Scholar]
- Meyza KZ, Blanchard DC, Pearson BL, Pobbe RL, Blanchard RJ. Fractone-associated N-sulfated heparan sulfate shows reduced quantity in BTBR T+tf/J mice: a strong model of autism. Behav Brain Res. 2012;228(2):247–253. doi: 10.1016/j.bbr.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda R, Nagapin F, Bozon B, Laroche S, Aubin T, Vaillend C. Altered social behavior and ultrasonic communication in the dystrophin-deficient mdx mouse model of Duchenne muscular dystrophy. Molecular autism. 2015;6:60. doi: 10.1186/s13229-015-0053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418(6896):422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Morawski M, Bruckner G, Arendt T, Matthews RT. Aggrecan: beyond cartilage and into the brain. Int J Biochem Cell Biol. 2012;44(5):690–693. doi: 10.1016/j.biocel.2012.01.010. [DOI] [PubMed] [Google Scholar]
- Murdoch JD, State MW. Recent developments in the genetics of autism spectrum disorders. Curr Opin Genet Dev. 2013;23(3):310–315. doi: 10.1016/j.gde.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Newschaffer CJ, Croen LA, Daniels J, Giarelli E, Grether JK, Levy SE, et al. The epidemiology of autism spectrum disorders. Annu Rev Public Health. 2007;28:235–258. doi: 10.1146/annurev.publhealth.28.021406.144007. [DOI] [PubMed] [Google Scholar]
- Ondacova K, Karmazinova M, Lazniewska J, Weiss N, Lacinova L. Modulation of Cav3.2 T-type calcium channel permeability by asparagine-linked glycosylation. Channels. 2016;10(3):175–184. doi: 10.1080/19336950.2016.1138189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155(5):1008–1021. doi: 10.1016/j.cell.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson BL, Corley MJ, Vasconcellos A, Blanchard DC, Blanchard RJ. Heparan sulfate deficiency in autistic postmortem brain tissue from the subventricular zone of the lateral ventricles. Behav Brain Res. 2013;243:138–145. doi: 10.1016/j.bbr.2012.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466(7304):368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada V, Passalacqua M, Bono M, Luzzi P, Scazzola S, Nobbio LA, et al. Gain of glycosylation: a new pathomechanism of myelin protein zero mutations. Ann Neurol. 2012;71(3):427–431. doi: 10.1002/ana.22695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt T, Conway CD, Tian NM, Price DJ, Mason JO. Heparan sulphation patterns generated by specific heparan sulfotransferase enzymes direct distinct aspects of retinal axon guidance at the optic chiasm. J Neurosci. 2006;26(26):6911–6923. doi: 10.1523/JNEUROSCI.0505-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H, Peng H, Shah WA, Stellwagen D, Carbonetto S. Dystroglycan mediates homeostatic synaptic plasticity at GABAergic synapses. Proc Natl Acad Sci USA. 2014;111(18):6810–6815. doi: 10.1073/pnas.1321774111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redin C, Gerard B, Lauer J, Herenger Y, Muller J, Quartier A, et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J Med Genet. 2014;51(11):724–736. doi: 10.1136/jmedgenet-2014-102554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner C, Stahn J, Breuer D, Klose M, Pohlentz G, Mormann M, et al. Dystroglycan binding to alpha-neurexin competes with neurexophilin-1 and neuroligin in the brain. J Biol Chem. 2014;289(40):27585–27603. doi: 10.1074/jbc.M114.595413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CE, Ratai EM, Kanwisher N. Reduced GABAergic action in the autistic brain. Curr Biol. 2016;26(1):80–85. doi: 10.1016/j.cub.2015.11.019. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2(5):255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumsey RK, Rudser K, Delaney K, Potegal M, Whitley CB, Shapiro E. Acquired autistic behaviors in children with mucopolysaccharidosis type IIIA. J Pediatr. 2014;164(5):1147–1151. doi: 10.1016/j.jpeds.2014.01.007. e1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satz JS, Ostendorf AP, Hou S, Turner A, Kusano H, Lee JC, et al. Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J Neurosci. 2010;30(43):14560–14572. doi: 10.1523/JNEUROSCI.3247-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott H, Panin VM. The role of protein N-glycosylation in neural transmission. Glycobiology. 2014;24(5):407–417. doi: 10.1093/glycob/cwu015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT. CACNA1H mutations in autism spectrum disorders. J Biol Chem. 2006;281(31):22085–22091. doi: 10.1074/jbc.M603316200. [DOI] [PubMed] [Google Scholar]
- Stalnaker SH, Hashmi S, Lim JM, Aoki K, Porterfield M, Gutierrez-Sanchez G, et al. Site mapping and characterization of O-glycan structures on alpha-dystroglycan isolated from rabbit skeletal muscle. J Biol Chem. 2010;285(32):24882–24891. doi: 10.1074/jbc.M110.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian M, Timmerman CK, Schwartz JL, Pham DL, Meffert MK. Characterizing autism spectrum disorders by key biochemical pathways. Front Neurosci. 2015;9:313. doi: 10.3389/fnins.2015.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker BE, Seamen E, Lawrence R, Parker MW, Xu Y, Liu J, et al. Expanding the 3-O-sulfate proteome-enhanced binding of neuropilin-1 to 3-O-sulfated heparan sulfate modulates its activity. ACS Chem Biol. 2016;11(4):971–980. doi: 10.1021/acschembio.5b00897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XJ, Hu Z, Liu Y, Anderson D, Kaplan JM. A network of autism linked genes stabilizes two pools of synaptic GABA(A) receptors. eLife. 2015;4:e09648. doi: 10.7554/eLife.09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii T, Yoshimura T, Narumi M, Hitoshi S, Takaki Y, Tsuji S, et al. Determination of major sialylated N-glycans and identification of branched sialylated N-glycans that dynamically change their content during development in the mouse cerebral cortex. Glycoconj J. 2014;31(9):671–683. doi: 10.1007/s10719-014-9566-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zwaag B, Franke L, Poot M, Hochstenbach R, Spierenburg HA, Vorstman JA, et al. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PLoS ONE. 2009;4(5):e5324. doi: 10.1371/journal.pone.0005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valstar MJ, Marchal JP, Grootenhuis M, Colland V, Wijburg FA. Cognitive development in patients with Mucopolysaccharidosis type III (Sanfilippo syndrome) Orphanet J Rare Dis. 2011;6:43. doi: 10.1186/1750-1172-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vester-Christensen MB, Halim A, Joshi HJ, Steentoft C, Bennett EP, Levery SB, et al. Mining the O-mannose glycoproteome reveals cadherins as major O-mannosylated glycoproteins. Proc Natl Acad Sci USA. 2013;110(52):21018–21023. doi: 10.1073/pnas.1313446110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite A, Tinsley CL, Locke M, Blake DJ. The neurobiology of the dystrophin-associated glycoprotein complex. Ann Med. 2009;41(5):344–359. doi: 10.1080/07853890802668522. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459(7246):528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhold B, Seidenfaden R, Rockle I, Muhlenhoff M, Schertzinger F, Conzelmann S, et al. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J Biol Chem. 2005;280(52):42971–42977. doi: 10.1074/jbc.M511097200. [DOI] [PubMed] [Google Scholar]
- Weiss N, Black SA, Bladen C, Chen L, Zamponi GW. Surface expression and function of Cav3.2 T-type calcium channels are controlled by asparagine-linked glycosylation. Pflugers Arch. 2013;465(8):1159–1170. doi: 10.1007/s00424-013-1259-3. [DOI] [PubMed] [Google Scholar]
- Wu JY, Kuban KC, Allred E, Shapiro F, Darras BT. Association of Duchenne muscular dystrophy with autism spectrum disorder. J Child Neurol. 2005;20(10):790–795. doi: 10.1177/08830738050200100201. [DOI] [PubMed] [Google Scholar]
- Wu S, Yue W, Jia M, Ruan Y, Lu T, Gong X, et al. Association of the neuropilin-2 (NRP2) gene polymorphisms with autism in Chinese Han population. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(4):492–495. doi: 10.1002/ajmg.b.30495. [DOI] [PubMed] [Google Scholar]
- Yoshida-Moriguchi T, Campbell KP. Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology. 2015;25(7):702–713. doi: 10.1093/glycob/cwv021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida-Moriguchi T, Yu L, Stalnaker SH, Davis S, Kunz S, Madson M, et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327(5961):88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TW, Chahrour MH, Coulter ME, Jiralerspong S, Okamura-Ikeda K, Ataman B, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77(2):259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccaria ML, Di Tommaso F, Brancaccio A, Paggi P, Petrucci TC. Dystroglycan distribution in adult mouse brain: a light and electron microscopy study. Neuroscience. 2001;104(2):311–324. doi: 10.1016/s0306-4522(01)00092-6. [DOI] [PubMed] [Google Scholar]