

Abstract
The histone code hypothesis holds that covalent posttranslational modifications of histone tails are interpreted by the cell to yield a rich combinatorial transcriptional output. This hypothesis has been the subject of active debate in the literature. Here, we investigated the combinatorial complexity of the acetylation code at the four lysine residues of the histone H4 tail in budding yeast. We constructed yeast strains carrying all 15 possible combinations of mutations among lysines 5, 8, 12, and 16 to arginine in the histone H4 tail, mimicking positively charged, unacetylated lysine states, and characterized the resulting genome-wide changes in gene expression by using DNA microarrays. Only the lysine 16 mutation had specific transcriptional consequences independent of the mutational state of the other lysines (affecting ≈100 genes). In contrast, for lysines 5, 8, and 12, expression changes were due to nonspecific, cumulative effects seen as increased transcription correlating with an increase in the total number of mutations (affecting ≈1,200 genes). Thus, acetylation of histone H4 is interpreted by two mechanisms: a specific mechanism for lysine 16 and a nonspecific, cumulative mechanism for lysines 5, 8, and 12.
Keywords: chromatin, gene expression, histone code, nucleosomes
The “histone code” hypothesis was proposed as a generalization of the discovery that combinations of covalent histone modifications led to varied transcriptional outputs (1–4). One consequence of a combinatorial histone code is that DNA regulatory sequences provide only a partial explanation of gene regulation. Thus, deciphering the combinatorial complexity of histone modifications is crucial for a clearer understanding of transcriptional regulation. Here, we examined the consequences of single or multiple lysine-to-arginine substitutions in histone H4 on transcriptional output. We ask how many states are specified by all of the lysine residues in a given histone tail. In particular, we ask which genes fall into the following classes that are suggested by a consideration of plausible reading mechanisms: (i) the trivial class, where transcriptional output of a gene is independent of its acetylation state; (ii) a specific class in which genes are regulated if and only if specific residues are modified (which we refer to as “discriminators”); and (iii) a cumulative class in which gene regulation is monotonically dependent only on the total number of modification states (which we refer to as “counters”).
There are at least two mechanisms by which histone modifications can alter transcriptional states. The first mechanism involves recruitment of modification-specific histone tail-binding complexes. For example, the bromodomain, found in many chromatin-regulating proteins, specifically binds to acetylated lysines (5). Histone acetylation could thereby recruit a number of different sequence-specific acetyl-lysine-binding complexes. Genes regulated by this mechanism would fall into class ii, the discriminators. Alternately, because acetylation neutralizes the positive charge of the lysine residue, histone acetylation could be read through the decreased affinity of the modified histone tail for a negatively charged component in an adjacent nucleosome and either the resulting increased local accessibility (6, 7) or the subsequent destabilization of the chromatin fiber (8, 9). Originally, it was believed that the positively charged histone tail would interact with the negatively charged DNA in an adjacent nucleosome (10), but the discovery of an interaction between the H4 tail and an acidic patch on the H2A/H2B dimer of an adjacent nucleosome in the nucleosome crystal structure (11) demonstrated another possible mechanism for charge-dependent stabilization of compacted chromatin fibers. Charge-dependent compaction of chromatin structure and the resulting effects on transcription should yield a set of genes behaving as the class iii counters.
The N-terminal tail of histone H4 has four acetylated lysines: K5, K8, K12, and K16. Of these, only K16 is strongly associated with a specific regulatory function in yeast, where its acetylation state regulates the extent of silent heterochromatin. K16 is acetylated by Sas2 and deacetylated by Sir2, and the balance of these opposing activities determines the extent of spreading of silencing from the telomeres (12, 13). This spreading is in part due to Sir3's preferential association with H4 tails that have been deacetylated at K16 (14). In addition, the bromodomain factor Bdf1 preferentially associates with histone H4 deacetylated at K16 (15). The roles of K5, K8, and K12 are less clear. Newly synthesized histone H4 is diacetylated at K5 and K12 in multiple organisms (16), although in the absence of these residues, K8 acetylation performs a redundant function during chromatin deposition in yeast (17). In human cells, the bromodomain factor Bdf2 associates with acetylated K12 (18), whereas at the IFN-β locus hSwi/snf is recruited by acetylated K8 (19). Finally, it should be noted that yeast carrying mutant histone H4 tails without any lysines exhibits a G2/M cell cycle defect, and this defect can be corrected by the introduction of a single lysine, even one in a novel sequence context (20).
Recently, antibodies specific to individual acetylated histone lysines have been used in conjunction with microarrays to globally map histone acetylation patterns in yeast (15). Although these experiments identify the patterns of histone acetylation across the yeast genome, they cannot address questions concerning redundancy in the acetylation code. To this end, we have taken a complementary approach by mutagenizing the acetylated lysines in the histone H4 tail in all combinations. We used microarrays to characterize transcriptional changes in these mutants. This study reveals a general role for histone acetylation as a cumulative regulator of gene expression in discrete domains throughout the genome and supports a unique role for K16 in transcriptional control.
Materials and Methods
Yeast Strains and Plasmids. All Saccharomyces cerevisiae strains (Table 1, which is published as supporting information on the PNAS web site) used in this study are derivatives of S288C. Plasmid-containing strains were derived from the diploid strain BY4743. The diploid MDY450 was produced by using PCR targeting (21) to delete the entire coding regions of HHT1, HHF1, HHT2, and HHF2, using the plasmid pHHT2HHF2 (pRS316 containing the 1.8-kb SalI–SpeI fragment of HHT2–HHF2, a gift from S. Schreiber, Harvard University) to complement the lethal phenotype. Sporulation of MDY450 yielded the MATa haploid MDY451, the parental control strain for the studies described here. Mutant alleles of HHF2 were made by site-directed mutagenesis of pHHT2HHF2. Sequence-verified plasmids (pRS411 containing WT or mutated SalI–SpeI fragment from pHHT2HHF2) were transformed into the haploid strain MDY451, and Met+ transformants were cured of pHHT2HHF2 by isolating Ura– auxotrophs.
Microarray Hybridizations. Mutant and parental control strains were grown in 100 ml of yeast extract/peptone/glucose to a cell density of ≈0.75 × 107 cells per ml. Poly(A)+ RNA was prepared by using hot phenol extraction followed by binding to Oligotex resin (Qiagen, Valencia, CA), labeled by using aminoallyl labeling, and hybridized to yeast cDNA microarrays as previously described (protocols can be found at www.microarrays.org). Microarrays were scanned by using a Genepix 4000B scanner (Axon Instruments, Union City, CA), and images were gridded by using genepix 4.0. Normalization was carried out in the Rosetta Resolver database (mean log 2 ratio was normalized to zero for the entire array) (22). Each set of hybridizations was performed by using an independent RNA preparation. Hybridization of WT against independent WT cultures was also performed as a control and showed no significant changes in gene expression, confirming day-to-day reproducibility of culture and harvesting conditions (data not shown).
Analysis. To divide the set of experiments into those having a mutation at a specific residue r vs. those that do not have a mutation at r, we introduce the notation Fr and F–r, respectively. In addition, we introduce the notation Em{S} to denote the set of all experiments having exactly m K → R point mutations at the H4 tail positions given in the residue index set S. For example, if S = {5,8,12}, then E1{S} contains all of the experiments from the three single K5R, K8R, and K12R mutants, E2{S} contains all of the experiments from the three double K5,8R, K5,12R, and K8,12R mutants, and E3{S} contains all of the experiments from the one triple mutant K5,8,12R.
Discriminators. An ORF is defined to be a discriminator at residue r with confidence parameter α if the difference of expression in the sets of experiments Fr and F–r is statistically significant as determined by the nonparametric Wilcoxon rank sum test. We chose α = 0.01 (after Bonferroni correction) in Fig. 2B because it gave an acceptable number of total predicted discriminators with a relatively high expected accuracy (Fig. 4, which is published as supporting information on the PNAS web site).
Fig. 2.

K16R-specific gene expression. (A) Incremental gene expression values were plotted against each other for the residues noted. (B) K16R discriminators. ORFs having expression regulated by a specific residue mutation, independent of the mutation states of the other three residues, are shown for K16R, selected with α = 0.01 after Bonferroni correction (see Materials and Methods and Fig. 4).
Counters. An ORF is defined to be a (+) counter for a set of residues S with confidence parameter α if: (i) the mean of its log expression values from all single-point mutants is positive; i.e., mean(E1{S}) > 0, (ii) its mean expression levels increase monotonically with the number of mutations; i.e., mean(Em{S}) < mean(Em + 1{S}) for all m ≤ S – 1, and (iii) each expression increase in (ii) is statistically significant as determined by the nonparametric Wilcoxon rank sum test. A (–) counter is defined similarly, with the mean comparison inequalities reversed. To estimate the number of false positives for parameter α, we randomized the data set by permuting the experiments for each ORF and then determined the number of (±) counters as before.
For residue set S = {5,8,12}, we find for α = {0.01, 0.05, 0.1}: {532, 682, 776} (+) counters with false-positive number {1.4 ± 1.3, 9.6 ± 7.2, 25.3 ± 15.0} and {447, 585, 682} (–) counters with false-positive number {1.3 ± 1.6, 7.4 ± 7.5, 19.0 ± 12.7}. (We report the means and standard deviations for numbers of false positives from 100 randomizations.)
Count-Regulated Domains (CRDs). A region of a chromosome is defined to be a (±) CRD if it contains more (±) counters than expected by random chance. We searched for CRDs by using (i) a sliding window of 11 ORFs and (ii) a P-value threshold for statistical significance of enrichment for (±) counter vs. no (±) counter ORFs in the window. Enrichment was determined by using 5,000 randomized rearrangements of the counters on each chromosome.
Data Availability. Microarray data are available in Table 2, which is published as supporting information on the PNAS web site.
Results
Microarray Data Collection and Overview. To study the roles for specific lysines in transcriptional control, the N-terminal lysines of histone H4 that are acetylated in vivo (K5, K8, K12, and K16) were mutated to arginines, mimicking unacetylatable lysines as closely as possible. All 15 possible combinations of these four point mutants were created, and yeast strains were constructed carrying these mutant H4 genes in place of the WT histone H4 genes. Mutant yeast was grown to midlogarithmic phase, and RNA was extracted and hybridized to gene expression microarrays (yeast with WT histones was used as the reference). At least three replicate cultures were analyzed for each mutant, and the final data set was clustered hierarchically (23) in two dimensions (Fig. 1). We note here that in our strain background, the quadruple mutant (K5,8,12,16R) appears to be lethal (24), because every colony obtained carrying this mutation proved to be aneuploid (different colonies showed different chromosomal aberrations) as assayed by comparative genomic hybridization (data not shown). Data for several distinct aneuploid quadruple mutants are shown in Fig. 1, but the quadruple mutant was excluded from the analysis in the remainder of the report, preventing a detailed analysis of genes whose expression might depend solely on the presence of any single lysine (20).
Fig. 1.

Global view of gene expression in H4 tail mutants. Gene expression data for all replicates used in this study were filtered for genes changing >2-fold in at least five experiments (to avoid showing genes resulting solely from the K5,8,12,16R aneuploidies), and both experiments and genes were hierarchically clustered. (Upper) The height of the blue bar represents the number of K → R mutations in the columns below; dark blue represents mutants with K16R, and light blue represents mutants with K16 WT. (Lower) A dendrogram of the experiment cluster is shown. White boxes represent a WT K at the residue in question; black boxes represent K → R mutations. For example, the far right mutant is K8R. Arrow shows a cluster of genes regulated uniquely by K16R.
Mutation of the H4 N-terminal tail causes widespread transcriptional changes (Fig. 1), demonstrating the extensive role of histone modification in gene expression. Several additional features of these data are apparent. First, histone mutants clustered together largely on the basis of the number of tail mutations. The most notable exception to this rule was the clustering of the K16R single mutant together with double mutants containing K16R. This clustering was due to the strong, specific effects of the K16R mutation on gene expression (see arrow in Fig. 1) and will be discussed in more detail below. In the dendrogram (Fig. 1 Lower), the interspersing of replicates of the relevant single mutants amongst each other in the cluster demonstrates the lack of specific gene expression changes upon mutation of the other three lysines. Different single mutants are as correlated with each other as replicates from the same strain. The same is true of those double mutants that do not contain the K16R mutation.
Second, there were no genes whose expression level changed discordantly as more mutations were combined with an initial mutation (using a 2-fold change threshold). For example, there were no genes that were up-regulated in a double mutant and down-regulated in any of the triple mutants that included the two residues from the double among their mutations. This finding argues that there are no cases where two opposing activities are recruited by different tail residues to the same promoter.
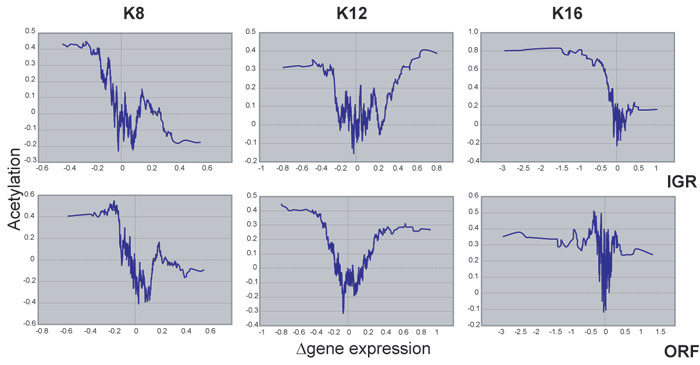
Our results were consistent with a number of previous observations. The genes whose expression changed in the K16R mutant were similar to the list of genes that change expression in yeast bearing mutations in components of the Sir (silent information regulator) complex, presumably because of K16R-dependent mislocalization of the Sir complex, which binds preferentially to histones deacetylated at K16 (Fig. 5, which is published as supporting information on the PNAS web site) (12, 13, 25, 26). Next, the set of genes whose expression changed in our histone mutants was enriched for telomere-proximal genes, consistent with the major role of histone modification in telomeric silencing (Fig. 6, which is published as supporting information on the PNAS web site) (27). In addition, the genes that change expression in these mutants, especially the genes that are up-regulated in the mutants, include a high percentage of genes with TATA boxes in their promoters, as suggested in a recent study showing that TATA-containing genes are frequently regulated by chromatin (28) (Fig. 7, which is published as supporting information on the PNAS web site).
Acetylation Status of Lysine-Regulated Genes. We compared our findings to recently published microarray data that globally mapped acetylation states in yeast by using chromatin immunoprecipitation (15). This comparison is important, given that the results reported here are subject to secondary effects of the mutations. For example, many components of the pheromone response pathway were down-regulated in the K16R mutant because of derepression of the silent mating-type locus rather than because of a direct role for K16 acetylation at the promoters of these genes (see below). Nonetheless, intergenic regions acetylated at K8, K12, or K16 (K5 was not examined in the previous study) were associated with genes strongly repressed by the corresponding K → R mutation (Fig. 8, which is published as supporting information on the PNAS web site), suggesting that many of the affected genes in our study are normally associated with acetylated histones. Interestingly, we also observed a correlation between genes strongly up-regulated (rather than repressed) in the K12R mutants and genes acetylated at K12. A similar but much less pronounced trend is seen with K8R mutants, consistent with previous reports of a direct role for histone deacetylation in gene activation (29). Taken together, these results suggest that a large number of the genes identified in this study are indeed acetylated at the relevant lysine and are therefore likely to be direct targets of acetylation.
Similar Effects on Gene Expression for K5, K8, and K12 Mutations. We further explored the role of each lysine in the transcriptional regulation of every gene assayed on the microarray. The incremental change in gene expression for a given K → R mutation was calculated by averaging the gene expression vector for the single point mutants together with the computed expression difference between strains differing only at the specific residue. For example, the incremental gene expression vector for K5 is given by the average of (K5R), (K5,8R – K8R), (K5,12R – K12R), (K5,16R – K16R), (K5,8,12R – K8,12R), (K5,8,16R – K8,16R), and (K5,12,16R – K12,16R). The incremental values for each ORF were plotted against each other for all possible lysine pairs in Fig. 2A. These plots demonstrate that there was little functional difference between K5, K8, and K12; the scatter plots of incremental gene expression for any pair of these residues fell very close to a straight line. Thus, although there were genes whose expression levels changed upon mutation of K5, for example, those genes also changed expression to a similar extent when K8 or K12 was mutated. In contrast, the plots for K16 against any of the other lysines demonstrate that mutation of K16 caused unique expression changes. As discussed below, the lack of specific incremental changes for lysines 5, 8, and 12 is consistent either with pure redundancy (i.e., there are no transcription changes in K5R or K8R, but when both lysines are absent in the K5,8R mutant, gene expression changes) or a mechanism where each K → R mutation contributes a partial change in transcription.
A Specific Transcriptional Outcome of the K16R Mutation. We explored the specificity of the transcriptional response to histone modification states by identifying genes whose expression differed significantly among the set of seven strains containing a mutation at a given lysine (excluding the quadruple mutant) and the remaining seven strains unmutated at the residue in question. We applied the Wilcoxon rank sum test to determine whether the expression patterns differed significantly between the two sets (see Materials and Methods and Fig. 4). Over a wide range of P values, there were many more genes that differed between K16R and K16 WT strains than would be expected by chance (Fig. 4). In contrast, the number of genes that differentiate K5R, K8R, or K12R strains from their respective WT strains was always less than the number expected by chance. For example, at P = 10–3, expected to return fewer than seven ORFs by chance, there were no genes whose expression was significantly different between K5 WT and K5R strains, or between K8 WT and K8R strains. Four genes differed in expression among K12 WT and K12R mutants. Conversely, there were 125 genes whose expression differed significantly in K16R mutants when compared with K16 WT strains, and 67 of them showed >2-fold change in gene expression in at least one mutant (Fig. 2B). These results clearly demonstrate a distinct role for K16 in gene expression and provide further evidence against specific gene expression changes associated with lysines 5, 8, or 12. We note that many of the K16R-down-regulated genes are involved in the pheromone response [16 of 81 down-regulated K16R-specific genes had a Gene Ontology (GO) annotation of “mating,” P = 1.1 × 10–13], because of derepression of the silent mating type locus and subsequent diploid-like repression of the mating pathway (30). The only nonmating GO or MIPS (Munich Information Center for Protein Sequences) annotation that was enriched among the K16 discriminators was pyridoxine metabolism (P = 1.4 × 10–6), because of misregulated expression of the subtelomeric genes SNO2, SNO3, SNZ2, and SNZ3.
Mutation of K5, K8, and K12 Causes Cumulative Transcriptional Changes. The specific gene expression changes found in K16R mutants were consistent with a reading mechanism involving a K16-specific binding protein. Both Sir3, a component of the repressive Sir complex, and Bdf1, a yeast homolog of the C-terminal domain of mammalian TAF(II)250, bind preferentially to H4 deacetylated at K16 (12, 13, 15), although these proteins differ in their requirements for acetylation on the remaining lysines in the H4 tail (14, 31). However, the lack of residue-specific gene expression effects of the other lysines implies that there is no equivalent protein specifically binding to H4 acetylated at K5, K8, or K12. This finding is somewhat surprising in light of data showing a specific requirement for K8 acetylation in hSwi/Snf recruitment to the IFN-β promoter (19).
Given the near-identical incremental gene expression differences for strains bearing mutations in K5, K8, and K12, as well as the clustering of histone tails according to the number of mutations, we wondered whether we might find genes that acted as “charge counters” (i.e., genes whose expression levels incrementally changed as the number of K → R mutations increased). We note that an interaction between a histone tail-binding protein and the histone tail that involves all three lysines in question (each residue contributing roughly equally to the thermodynamics of binding), could also produce the results we see here. Thus, we refer to genes whose expression changes incrementally with the number of lysines mutated as charge counters out of convenience and note that this class of genes supports a previously suggested mechanism (9) for the reading of H4 tail acetylation based on higher-order chromatin decompaction. Nevertheless, we urge caution in this interpretation and emphasize its speculative nature, given the chemical constraints of genetically encoded mutations.
Gene expression data for K5R, K8R, and K12R were averaged to produce a value for gene expression changes given a single mutation. Data for the three relevant double mutants were averaged to give a value for double mutants, and the data from the K5,8,12R mutant were used for the triple mutant. The rank sum test was used to identify genes whose expression changed monotonically with the total number of K → R mutations. As shown in Fig. 3A, 682 genes are up-regulated in a graded manner with the number of K → R mutations, whereas 585 genes are down-regulated. The up-regulated class was enriched for genes involved in energy generation, with the most highly enriched Gene Ontology (GO) category being “energy derivation by oxidation of organic compounds” (P = 1.1 × 10–10). For example, 35 of the 69 genes annotated in the mitochondrial ribosome were up-regulated charge counters (P < 10–14). Down-regulated charge counters were enriched for annotations associated with ribosome synthesis, with the highest enrichment for the GO biological process of “ribosome biogenesis” (P = 1.7 × 10–13). All together, ≈22% of the 5,745 genes studied act as charge counters for lysines 5, 8, and 12 on histone H4.
Fig. 3.

Charge counters. (A) The number of ORFs showing monotone, nonspecific response to mutations in K5, K8, and K12 was 1,267. ORFs that monotonically increase in mean expression level as a function of the number of lysine mutations (in K5, K8, and K12) are shown for confidence parameter α = 0.05 (see Materials and Methods). In total, we found 682 (+) counters and 585 (–) counters. ORFs are sorted by the geometric mean of the mean expression differences. x axis is in increasing order of number of mutations, from 1 to 3. (B) Location of chromosomal regions enriched for charge-regulated domains. We chose the 1,267 counters (using α = 0.05) and aligned them to chromosomal coordinates. Mean log ratio expression values for the average single mutant, average double mutant, and triple K5,8,12R mutant counters are shown. x axis is proportional to base-pair distance, and ORF widths are drawn to scale. The complete genomic view is shown in Fig. 9. (C) K16R has counter effects, in addition to discriminator effects, on gene expression. Gene expression data from the K5,8,12R mutant is plotted on the x axis against gene expression data from the K5,12,16R mutant on the y axis. Pink points indicate genes identified as K16R discriminators; blue points indicate the remainder of genes.
K5/8/12-Regulated Genes Occur in Chromosomal Clusters. The proposed mechanism to “read” charge counters is stabilization of 30-nm fiber by electrostatic interactions between the H4 tail and an acidic patch on the H2A/H2B dimer of an adjacent nucleosome (11). We hypothesized that charge counters should cluster near each other in the genome, because there is clearly a minimum length of DNA required to fold into 30-nm fiber. Charge counters were mapped onto their chromosomal locations and were indeed found to occur in clusters (see Fig. 3B for examples; see Fig. 9, which is published as supporting information on the PNAS web site, for a whole genomic view). A sliding window of 11 ORFs was tested for enrichment of charge-counter genes relative to 5,000 randomized data sets. Statistically significant clusters were found throughout the genome (Fig. 10, which is published as supporting information on the PNAS web site), in contrast to the previously described Htz-associated domains (32) and Hda1-associated subtelomeric domains (33), both located within 40 kb of telomeres.
Nonspecific Transcriptional Changes Due to Mutation of K16. Although K16 is clearly distinct from the other lysines, with its unique role in the regulation of a set of genes, K16 also has a general role in gene expression. In the K16 comparisons in Fig. 2 A, we observe that although multiple genes fall off a straight line (the discriminators), a great number of genes change similarly in K16R strains and in any of the other K → R strains. In addition, examination of the cluster in Fig. 1 reveals close correlation between the K5,12,16R mutant and the K5,8,12R mutant (which differ only by the incremental effect of K8R or K16R on the K5,12R background). Without the histone H3 N-terminal tail, mutation of K5 and K12 reveals a requirement for K8 (but not K16) (17). From this observation, it might be expected that there would be great differences between gene expression in K5,8,12R mutants and K5,12,16R mutants, because H4-dependent chromatin assembly in the K5,12,16R mutant would be intact because of the WT K8; however, chromatin assembly in the K5,8,12R mutant would be expected to be entirely H3-dependent (34). To test this hypothesis, we plotted the gene expression data for the K5,8,12R mutant against the data for the K5,12,16R mutant (Fig. 3C). Genes identified as K16R discriminators (Fig. 2B) were plotted separately, and the remaining nondiscriminator genes show a good correlation between gene expression changes in the two triple mutants (r2 = 0.85). These results demonstrate that K16 has nonspecific effects on gene expression as well as the specific effects presumably mediated by Bdf1 and Sir3.
Discussion
Understanding the role of multiple covalent modifications in any biological process requires knowledge of both the modifications that occur combinatorially in vivo and what modifications are functionally redundant. For lysine acetylation in the histone tails in yeast, the former has recently been described by Kurdistani et al. (15). In this study, we have used H4 tail mutations to investigate the latter. We demonstrated that K16 was unique among the H4 N-terminal lysines because it alone caused a distinct transcriptional phenotype when mutated. This observation is consistent with a reading mechanism for lysine acetylation in which the effectors are proteins that bind histone H4 in a K16 acetylation-regulated manner. Conversely, we failed to find any specific gene expression phenotypes caused by mutation of the other three lysines. Instead, K5, K8, and K12 are partially redundant, and a large fraction of the genome changes gene expression monotonically with the number of lysine residues mutated to arginine.
The existence of charge counters is consistent with the hypothesis that 30-nm chromatin fiber is stabilized by charge-dependent interactions between histone tails and acidic patches on adjacent nucleosomes. Furthermore, these coregulated genes occur in clusters in the genome. However, the charge counters could be the result of a number of other reading mechanisms. First, these results are consistent with any number of mechanisms involving acetylation-dependent histone tail-binding proteins that bind more strongly as the number of acetyl groups increases. However, we have found no significant correlation between gene expression in the K5,8,12R mutant and the binding of Bdf1 (15, 31), ruling out one significant candidate for this protein (data not shown). Second, histone tail binding to adjacent linker DNA could prevent binding of regulatory proteins in a charge-dependent manner (6, 35, 36). Third, H4-dependent histone deposition requires at least one of K5, K8, or K12 (17), suggesting the possibility that up-regulated charge-counter clusters corresponded to domains where repressive chromatin structures were improperly established (because of exclusively H3-dependent chromatin deposition) during replication in the K5,8,12R mutant. We failed to find significant colocalization of up-regulated charge counters with origins of replication (37), suggesting that replication defects do not account for the up-regulated charge-counter clusters (data not shown). Future studies to determine chromatin structure changes in the K5,8,12R mutant will be necessary to further explore the mechanistic basis for the incremental gene expression caused by progressive mutation of these lysines and to distinguish between the changes in chromatin structure at up-regulated and down-regulated charge counters.
Finally, we note that this work provides insight into the functional complexity of the histone H4 acetylation code. We found that the four H4 lysines give rise to eight transcriptional states (corresponding to zero, one, two, or three of the K5/8/12 mutations with or without the K16 mutation) rather than the full complement of 16 possible states. In addition, the combinatorial effects in the histone H4 acetylation code are cumulative in nature and thus show that insofar as these histone modifications form a “code,” it is a simple one. This lack of combinatorial complexity is consistent with results from higher organisms, including a recent study showing that histone modifications in Drosophila are highly correlated with each other, which the authors argue demonstrates a “binary” pattern of histone modifications in which modifications do not occur in complex combinations but occur together as a group (38). Taken together, we feel these results shed significant light on the histone code hypothesis (1–4) as it concerns the case of histone acetylation. These results demonstrate that insofar as combinations of histone H4 acetylation have functional consequences, these consequences are simple and cumulative and do not reveal a “complex, multimark code.” (2) It will be interesting to see whether other types of histone modification behave in a similar fashion.
Supplementary Material
Acknowledgments
We thank J. Oshiro, T. Maniatis, A. Murray, K. Thorn, M. Laub, R. Kishony, A. Regev, and S. Kurdistani for critical reading of the manuscript and suggestions and B. Bernstein (Harvard University) and S. Schreiber for the pHHT2HHF2 plasmid. This work was supported by the Bauer Center for Genomics Research and the National Institute of General Medical Sciences.
Author contributions: O.J.R. designed research; M.F.D. performed research; S.J.A., L.F.W., and O.J.R. analyzed data; and O.J.R. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
See Commentary on page 5308.
References
- 1.Turner, B. M. (2000) Bioessays 22, 836–845. [DOI] [PubMed] [Google Scholar]
- 2.Strahl, B. D. & Allis, C. D. (2000) Nature 403, 41–45. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber, S. L. & Bernstein, B. E. (2002) Cell 111, 771–778. [DOI] [PubMed] [Google Scholar]
- 4.Kurdistani, S. K. & Grunstein, M. (2003) Nat. Rev. Mol. Cell. Biol. 4, 276–284. [DOI] [PubMed] [Google Scholar]
- 5.Dhalluin, C., Carlson, J. E., Zeng, L., He, C., Aggarwal, A. K. & Zhou, M. M. (1999) Nature 399, 491–496. [DOI] [PubMed] [Google Scholar]
- 6.Vettese-Dadey, M., Grant, P. A., Hebbes, T. R., Crane-Robinson, C., Allis, C. D. & Workman, J. L. (1996) EMBO J. 15, 2508–2518. [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson, J. D., Lowary, P. T. & Widom, J. (2001) J. Mol. Biol. 307, 977–985. [DOI] [PubMed] [Google Scholar]
- 8.Wolffe, A. P. & Hayes, J. J. (1999) Nucleic Acids Res. 27, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tse, C., Sera, T., Wolffe, A. P. & Hansen, J. C. (1998) Mol. Cell. Biol. 18, 4629–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong, L., Schroth, G. P., Matthews, H. R., Yau, P. & Bradbury, E. M. (1993) J. Biol. Chem. 268, 305–314. [PubMed] [Google Scholar]
- 11.Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. (1997) Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 12.Kimura, A., Umehara, T. & Horikoshi, M. (2002) Nat. Genet. 32, 370–377. [DOI] [PubMed] [Google Scholar]
- 13.Suka, N., Luo, K. & Grunstein, M. (2002) Nat. Genet. 32, 378–383. [DOI] [PubMed] [Google Scholar]
- 14.Carmen, A. A., Milne, L. & Grunstein, M. (2002) J. Biol. Chem. 277, 4778–4781. [DOI] [PubMed] [Google Scholar]
- 15.Kurdistani, S. K., Tavazoie, S. & Grunstein, M. (2004) Cell 117, 721–733. [DOI] [PubMed] [Google Scholar]
- 16.Sobel, R. E., Cook, R. G., Perry, C. A., Annunziato, A. T. & Allis, C. D. (1995) Proc. Natl. Acad. Sci. USA 92, 1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma, X. J., Wu, J., Altheim, B. A., Schultz, M. C. & Grunstein, M. (1998) Proc. Natl. Acad. Sci. USA 95, 6693–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanno, T., Kanno, Y., Siegel, R. M., Jang, M. K., Lenardo, M. J. & Ozato, K. (2004) Mol. Cell 13, 33–43. [DOI] [PubMed] [Google Scholar]
- 19.Agalioti, T., Chen, G. & Thanos, D. (2002) Cell 111, 381–392. [DOI] [PubMed] [Google Scholar]
- 20.Megee, P. C., Morgan, B. A. & Smith, M. M. (1995) Genes Dev. 9, 1716–1727. [DOI] [PubMed] [Google Scholar]
- 21.Baudin, A., Ozier-Kalogeropoulos, O., Denouel, A., Lacroute, F. & Cullin, C. (1993) Nucleic Acids Res. 21, 3329–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts, C. J., Nelson, B., Marton, M. J., Stoughton, R., Meyer, M. R., Bennett, H. A., He, Y. D., Dai, H., Walker, W. L., Hughes, T. R., et al. (2000) Science 287, 873–880. [DOI] [PubMed] [Google Scholar]
- 23.Eisen, M. B., Spellman, P. T., Brown, P. O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Megee, P. C., Morgan, B. A., Mittman, B. A. & Smith, M. M. (1990) Science 247, 841–845. [DOI] [PubMed] [Google Scholar]
- 25.Hecht, A., Laroche, T., Strahl-Bolsinger, S., Gasser, S. M. & Grunstein, M. (1995) Cell 80, 583–592. [DOI] [PubMed] [Google Scholar]
- 26.Johnson, L. M., Kayne, P. S., Kahn, E. S. & Grunstein, M. (1990) Proc. Natl. Acad. Sci. USA 87, 6286–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin, A. M., Pouchnik, D. J., Walker, J. L. & Wyrick, J. J. (2004) Genetics 167, 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basehoar, A. D., Zanton, S. J. & Pugh, B. F. (2004) Cell 116, 699–709. [DOI] [PubMed] [Google Scholar]
- 29.Wang, A., Kurdistani, S. K. & Grunstein, M. (2002) Science 298, 1412–1414. [DOI] [PubMed] [Google Scholar]
- 30.Wyrick, J. J., Holstege, F. C., Jennings, E. G., Causton, H. C., Shore, D., Grunstein, M., Lander, E. S. & Young, R. A. (1999) Nature 402, 418–421. [DOI] [PubMed] [Google Scholar]
- 31.Matangkasombut, O. & Buratowski, S. (2003) Mol. Cell 11, 353–363. [DOI] [PubMed] [Google Scholar]
- 32.Meneghini, M. D., Wu, M. & Madhani, H. D. (2003) Cell 112, 725–736. [DOI] [PubMed] [Google Scholar]
- 33.Robyr, D., Suka, Y., Xenarios, I., Kurdistani, S. K., Wang, A., Suka, N. & Grunstein, M. (2002) Cell 109, 437–446. [DOI] [PubMed] [Google Scholar]
- 34.Ling, X., Harkness, T. A., Schultz, M. C., Fisher-Adams, G. & Grunstein, M. (1996) Genes Dev. 10, 686–699. [DOI] [PubMed] [Google Scholar]
- 35.Yang, Z., Zheng, C., Thiriet, C. & Hayes, J. J. (2005) Mol. Cell. Biol. 25, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vitolo, J. M., Thiriet, C. & Hayes, J. J. (2000) Mol. Cell. Biol. 20, 2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raghuraman, M. K., Winzeler, E. A., Collingwood, D., Hunt, S., Wodicka, L., Conway, A., Lockhart, D. J., Davis, R. W., Brewer, B. J. & Fangman, W. L. (2001) Science 294, 115–121. [DOI] [PubMed] [Google Scholar]
- 38.Schubeler, D., MacAlpine, D. M., Scalzo, D., Wirbelauer, C., Kooperberg, C., van Leeuwen, F., Gottschling, D. E., O'Neill, L. P., Turner, B. M., Delrow, J., et al. (2004) Genes Dev. 18, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}