

Abstract
Flow cytometry is widely used to measure gene expression and other molecular biological processes with single cell resolution via fluorescent probes. Flow cytometers output data in arbitrary units (a.u.) that vary with the probe, instrument, and settings. Arbitrary units can be converted to the calibrated unit molecules of equivalent fluorophore (MEF) using commercially available calibration particles. However, there is no convenient, non-proprietary tool available to perform this calibration. Consequently, most researchers report data in a.u., limiting interpretation. Here, we report a software tool named FlowCal to overcome current limitations. FlowCal can be run using an intuitive Microsoft Excel interface, or customizable Python scripts. The software accepts Flow Cytometry Standard (FCS) files as inputs and is compatible with different calibration particles, fluorescent probes, and cell types. Additionally, FlowCal automatically gates data, calculates common statistics, and produces publication quality plots. We validate FlowCal by calibrating a.u. measurements of E. coli expressing superfolder GFP (sfGFP) collected at 10 different detector sensitivity (gain) settings to a single MEF value. Additionally, we reduce day-to-day variability in replicate E. coli sfGFP expression measurements due to instrument drift by 33%, and calibrate S. cerevisiae mVenus expression data to MEF units. Finally, we demonstrate a simple method for using FlowCal to calibrate fluorescence units across different cytometers. FlowCal should ease the quantitative analysis of flow cytometry data within and across laboratories and facilitate the adoption of standard fluorescence units in synthetic biology and beyond.
Keywords: flow cytometry, fluorescent protein, molecules of equivalent fluorophore, calibrated gene expression units
Commercial calibration particles are typically mixtures of 4–8 microbead subpopulations, each labeled with a different number of one or more fluorophores. The fluorophore-dependent fluorescence of each subpopulation is given by the manufacturer in molecules of equivalent fluorophore (MEF), the absolute number of fluorophores in solution that gives rise to the same fluorescence as a single microbead (1–3). The MEF values of the subpopulations are approximately evenly (linearly or logarithmically) spaced over 3–4 orders of magnitude, enabling robust standard curves to be generated.
To calibrate flow cytometry data, particles labeled with a similar or identical fluorophore to the cellular probe are used. For example, green fluorescent protein (GFP) expression can be calibrated using fluorescein- or GFP-labeled particles. The cell samples and calibration particles are measured under identical instrument settings. A standard curve is constructed by identifying the individual microbead subpopulations, measuring the a.u. fluorescence of each, and fitting a continuous model to the resulting set of (a.u., MEF) pairs. Then, the standard curve can be used to convert any measured a.u. fluorescence value into its MEF equivalent. If the cell and microbead fluorophores are identical, the same standard curve is obtained with different excitation and emission optics, and the resulting MEF units are instrument independent (4,5). If the two fluorophores have slightly different spectral properties, calibration yields MEF units that are independent of the settings of a single instrument, but may vary on instruments with different optical configurations. In the latter case, data from two instruments can be directly compared by using the same calibration particles and cell samples to determine standard curves for each instrument.
Current software tools for calibrating flow cytometry data to MEF units have not been widely adopted because they require proficiency in a particular computer programming language, are designed for a specific experiment, or are proprietary. Open source tools for the Python (6–8), MATLAB (9–12), and R (13) languages allow the user to read, gate, and plot flow cytometry data from Flow Cytometry Standard (FCS) files, but do not perform the MEF conversion. FlowBeads (14) can identify microbead subpopulations, calculate microbead fluorescence values, generate a standard curve, and calibrate cell fluorescence from a.u. to MEF, but requires knowledge of the R language. Synthetic Biology Tools calibrates from a.u. to MEF units, but is closed source, compatible with only GFP and calibration particles described in molecules of equivalent fluorescein (MEFL), and specifically designed to analyze gene circuits in transiently transfected mammalian cells (15,16). The commercial package FlowJo™ (http://www.flowjo.com/) calibrates data from a.u. to MEF units, but it is costly and closed source.
FlowCal (Figure 1) overcomes the limitations of current tools. FlowCal is a freely-available, open source Python package that enables calibration and processing of FCS files via an intuitive Microsoft Excel user interface (UI) or an underlying Python application programing interface (API). The user simply inputs the manufacturer-provided MEF values and measures calibration particles on a flow cytometer. FlowCal automatically identifies the microbead subpopulations within the FCS file using fluorescence detector channels specified by the user, and quantifies their fluorescence (Notes S1–S3). Accordingly, FlowCal is compatible with different calibration particles and flow cytometers. FlowCal automatically gates events in the densest region of forward scatter/side scatter (FSC/SSC) plots according to user specifications, a feature that eliminates microbead aggregates and other debris and ensures compatibility with diverse cell types (Note S1). The software also calculates common statistics, including mean and coefficient of variation of cell samples, and produces publication quality histogram and density plots. Because FlowCal is open source (Supporting Information), it can be extended by the community for additional applications.
Figure 1.
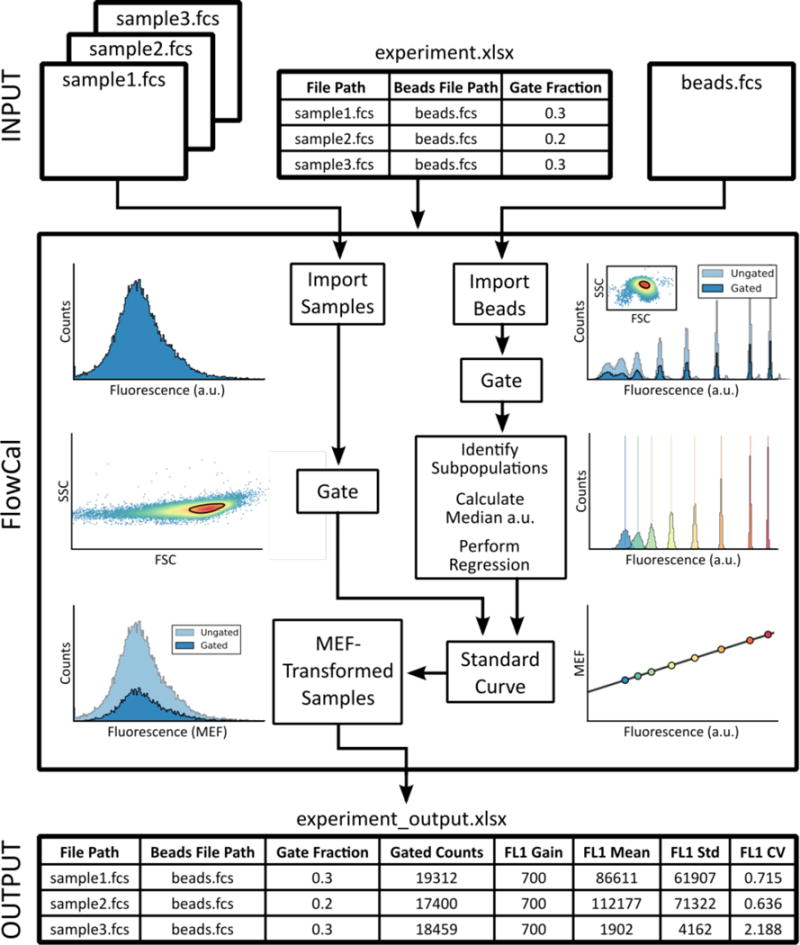
Schematic of the FlowCal package, illustrating the Excel UI workflow. The Excel UI interacts with the underlying Python API and the individual modules involved in calibration particle and cell sample processing. The FlowCal outputs are plots of each step involved in generation of the a.u. to MEF standard curve (density gating, microbead population clustering, and standard curve fitting), scatter plots and histograms for each cell sample in MEF units, and an Excel file containing statistics for each cell sample.
In this study, we validate FlowCal and demonstrate its utility for synthetic biology using inducible E. coli and S. cerevisiae gene expression systems and two different flow cytometers. First, we calibrate measurements of a sample of E. coli expressing sfGFP collected at 10 different detector gain settings. These measurements, which span three orders of magnitude in a.u., are calibrated to MEFL values differing by only 2%. Second, we use MEFL calibration to reduce day-to-day variability in replicate measurements of E. coli sfGFP fluorescence taken over an eight month period by 33%. Third, we demonstrate FlowCal’s compatibility with eukaryotic cells by calibrating mVenus measurements from a light-inducible S. cerevisiae gene expression system to MEFL. Finally, we use FlowCal to determine the a.u. to MEFL standard curves on two separate flow cytometers, and demonstrate a method for cross-calibrating data collected on two different instruments.
Results and Discussion
FlowCal Description
At its core, FlowCal is a Python 2.7 package. It contains separate modules for reading FCS files (versions 2.0, 3.0, and 3.1) (17–19), gating flow cytometry data, calculating statistics, producing histogram and density plots, analyzing calibration particles (identifying subpopulations based on fluorescence and generating standard curves), and running the Excel UI (Figure 1). Complete FlowCal documentation is available on the project website (Procedure S1).
Downloading and Installing FlowCal
Mac and PC users can download and install FlowCal following the instructions in Procedure S1 (additional information in Figures S1–S3).
Using FlowCal
The FlowCal Excel UI can be run following the instructions in Procedure S2. First, the user generates an input spreadsheet in a standard format (Figure S4, Note S4). An example spreadsheet that also serves as a template is provided in the downloadable package. In the spreadsheet, the user enters basic information about the flow cytometer, including the names of the detector channels to be used in the analysis. The names and locations of experimental sample and calibration particle FCS files to be analyzed are entered on individual rows of the spreadsheet. MEF values of the microbead subpopulations, the units (Channel, a.u. or MEF) to be used in calculating statistics and plotting data, and the fraction of events to retain after density-gating are also entered on each row, as appropriate. A single FCS file can be analyzed or plotted in multiple ways simultaneously by entering the file name on multiple rows with different analysis parameters. Multiple calibration particle files can also be specified and applied to different sample files within the same sheet, a useful feature if sample files were acquired on different days or using different instrument settings. After the data are entered, the input spreadsheet file is saved in a desired location. The user then opens FlowCal (Procedure S2) and specifies the location of the input file in an automatically generated prompt window (Figure S5).
First, FlowCal processes the calibration particles (Note S2). Specifically, the FCS file(s) are opened, the list of events in each file is extracted, and the data are density-gated. A FSC/SSC density plot highlighting the gated region and fluorescence histograms of all gated and ungated events (Figure S6) can be automatically produced and saved in the same directory as the input Excel file. Each microbead subpopulation is automatically identified (Figure S7), and a histogram showing each, and its calculated median fluorescence value can also be generated and saved (Figure S8). Finally, the standard curve is generated using a nonlinear regression of a calibration particle fluorescence model, the calculated microbead a.u. values, and the MEF values entered in the spreadsheet (Note S3). A bead autofluorescence term is included as a fit parameter in the model, which allows proper standard curves to be constructed whether or not the manufacturer includes bead autofluorescence in the provided MEF values. The standard curve can also be plotted (Figure S9) and saved to the input file directory.
Next, FlowCal processes the experimental samples. Each FCS file is opened, and cell events are extracted and gated as for the bead samples. The a.u. value of each cell event is then calibrated to MEF using the standard curve. A density plot showing all cell events, the gated region, and fluorescence histograms of ungated and gated data in MEF units are generated for each file (Figure S10). Finally, the arithmetic and geometric mean, median, mode, arithmetic and geometric standard deviation, coefficient of variation, interquartile range, robust coefficient of variation (RCV), and a full histogram are calculated for each fluorescence channel and written to an output Excel file (Figure S11). FSC/SSC density plots and fluorescence histograms can be generated and saved for each file.
Using the Python API, the above steps can be executed in any order, different clustering and gating algorithms can be applied, and more complex plots, such as overlaid cell sample fluorescence histograms (Figure 2B), can be generated. Additionally, the user can explore correlations between fluorescence in two or more fluorescent channels at a single cell level, a useful feature for analyzing cellular noise (20). Example Python scripts that use the API to analyze FCS files are provided in the FlowCal package.
Figure 2.
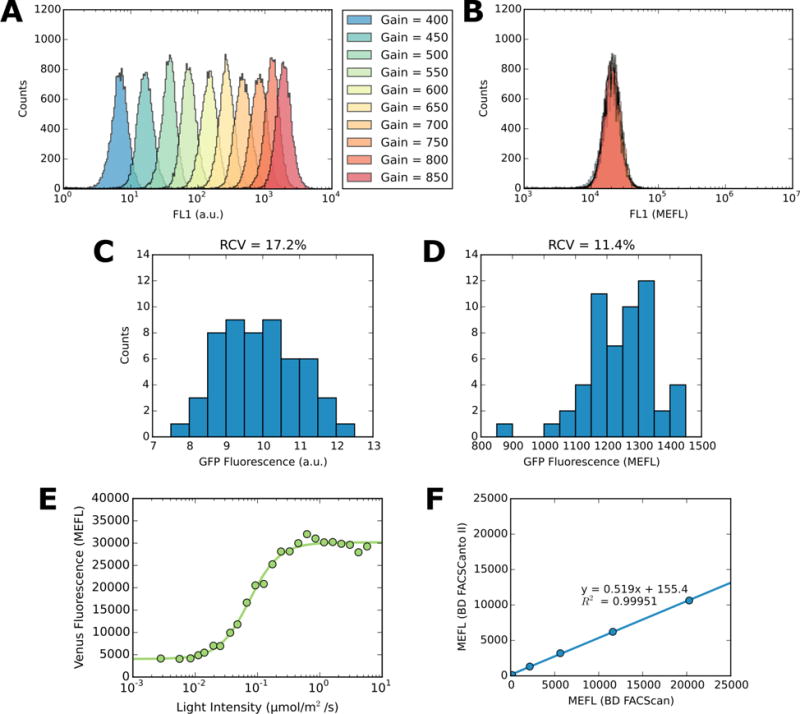
FlowCal histogram plots of a single sample of E. coli BW29655 expressing sfGFP from the IPTG-inducible promoter Ptac (16) when fully induced, measured at 10 different detector gain settings in (A) a.u., and (B) MEFL. Histograms of fluorescence values (geometric mean of each population) of E. coli expressing the CcaS-CcaR v2.0 system (17) in the dark, taken from 54 experimental replicates carried out over eight months in (C) uncalibrated arbitrary units (left) and (D) FlowCal calibrated molecules of equivalent fluorescein (MEFL) values. Robust coefficient of variation (RCV) is shown above each data set. (E) Dose response of the light-responsive PhyB-GBD/PIF3-GAD system in S. cerevisiae (19), where the yellow fluorescent protein Venus is transcribed in response to red (647nm) light. After incubating cells under the specified intensity of light for 18 hours, fluorescence was measured via flow cytometry, and the resulting data was analyzed using FlowCal. (F) E. coli BW29655 expressing sfGFP from the IPTG-inducible promoter Ptac were induced with 0, 81, 161, 318, or 1000 μM IPTG and measured using two different flow cytometers, along with calibration beads. The resulting data was calibrated to MEFL using FlowCal. Geometric mean fluorescence values in MEFL obtained from both instruments are plotted against one another, for each sample. A least-squares linear fit is also shown.
Validating FlowCal by calibrating gene expression data collected at different gain settings
Proper calibration should transform a.u. data collected at different instrument settings to a single MEF value. We validated FlowCal by measuring a single sample of E. coli expressing sfGFP from the IPTG-inducible Ptac promoter (21) in the presence of 1mM IPTG and FITC-labeled calibration particles at 10 different detector gain settings between 400 and 850 V on a single flow cytometer with a logarithmic detector. As expected, the a.u. fluorescence of both the bacteria (Figure 2A) and the calibration particles (Figure S13) increases exponentially with gain. We used FlowCal to calculate a.u.-to-MEF standard curves for each gain setting. As gain changes, the fitted slope parameter of these standard curves is constant and the intercept decreases linearly (Figure S14). Additionally, the 10 E. coli fluorescence histograms, whose geometric means range from 6.7 to 1872 a.u., collapse to a single histogram with mean equal to 19843.3 +/−414.3 MEFL (C.V. = 2.09%) (Figure 2B). These results demonstrate that FlowCal properly performs the MEF calibration.
Using FlowCal calibration to reduce day-to-day variability in gene expression data
In unpublished work characterizing our green/red photoreversible E. coli two component system CcaS-CcaR v2.0 (22), we measured the sfGFP output gene expression levels of 54 replicates of a dark-treated sample alongside calibration particles over an eight month period on a single flow cytometer at the same settings. Despite using a highly reproducible bacterial growth and gene expression measurement protocol (23), we observe relatively large variability in the geometric mean fluorescence of these replicates in uncalibrated a.u. (RCV = 17.2%) (Figure 2C). However, FlowCal calibration reduces the RCV to 11.5% (Figure 2D). Thus, we conclude that 33% of the variation in these data arises from cytometer drift and that FlowCal can automatically calibrate a large number of FCS files to eliminate such errors.
Using FlowCal to calibrate S. cerevisiae gene expression data
We next investigated whether FlowCal can be used to analyze fluorescence data in eukaryotes. To this end, we used an engineered S. cerevisiae red/far-red photoreversible promoter system based on Phytochrome B and Phytochrome Interacting Factor 3 (24) with the yellow fluorescent protein (YFP) gene mVenus as the output. We measured the transfer function of this system by growing the strain under increasing intensities of red light and analyzing mVenus fluorescence with FlowCal. Despite the large difference in size relative to E. coli, FlowCal reliably identifies the S. cerevisiae cells in the densest region of the FSC/SSC diagram (Figure S15). Additionally, mVenus fluorescence is reliably calibrated to MEFL, allowing us to calculate the red light to mVenus transfer function of this system in calibrated units (Figure 2E). We conclude that FlowCal is compatible with bacteria, eukaryotes, and multiple fluorescent proteins.
Cross-instrument calibration of MEF Units
If microbead and cell fluorophores have different spectra, calibrated cell fluorescence data are instrument-dependent (4,5). Given the limited availability of calibration beads labeled with fluorophores used in synthetic biology (e.g. sfGFP, mVenus, mCherry), achieving instrument-independent fluorescence units requires characterizing the dependence of MEF measurements on the instrument. We measured the fluorescence of an IPTG-inducible E. coli sfGFP expression strain at different inducer levels alongside FITC-labeled calibration particles using a customized BD FACScan and a BD FACSCanto II. While both instruments use 488 nm excitation lasers, the emission filters differ (510/21 and 525/50nm, respectively). Accordingly, the FlowCal-calculated MEFL values differ on the two instruments (Figure 2F). However, the values are linearly related (MEFLFACSCanto II = m · MEFLFACScan + b, with m = 0.5194 ± 0.0067 and b = 155.36 ± 53.19) By dropping autofluorescence terms (Note S5), the relationship can be used to convert sfGFP measurements from one instrument to another. This result is expected to be general (4,5), where the value of m depends on the two instruments being cross-calibrated and the identity of both the cell and bead fluorophores.
Outlook
Any two data sets reported in a.u. can be quantitatively compared only if the fluorescent probes, instrument, and acquisition settings used are identical. Even when a single probe and instrument are used, detector sensitivity (gain) settings are often changed to permit analysis of samples with large differences in fluorescence intensities. Consequently, the extent of the analyses that can be performed and conclusions that can be drawn from many flow cytometry studies is limited (25).
FlowCal converts flow cytometry data from a.u. to MEF units that are independent of cytometer gain settings (Figure 2A and 2B), allowing researchers to quantitatively compare cells with widely varying fluorescence values using a single instrument. Additionally, FlowCal calibration reduces day-to-day variability by eliminating the effects of instrument drift (Figure 2C and 2D). FlowCal calibration should thus aid researchers in determining other sources of experimental variability and identify bona fide biological noise, both of which are major needs in synthetic and systems biology. The FlowCal gating and unit conversion algorithms are compatible with diverse cell types and sizes (Figure 2E and S15), enabling our results to be generalized to different experimental systems.
Fluorophores produce detectable fluorescence signals in multiple cytometer detector channels (i.e. bleedthrough). For example, sfGFP is primarily detected in the FACScan FL-1 channel, but bleeds through into the red filtered FL-3 channel commonly used to detect mCherry. Though such bleedthrough can be compensated for automatically (26,27), FlowCal does not currently perform this function. Nonetheless, multiple fluorophores can still be used simultaneously in a single cell with FlowCal when bleedthrough is negligible. If bleedthrough is significant, FlowCal output data can also be manually compensated following established procedures. Due to its open source license, we or others can add automatic bleedthrough compensation or other useful features to FlowCal in the future.
Universal, instrument-independent fluorescence units would allow the synthetic biology community to more directly compare the performance of different engineered biological systems. Beyond these, physical units - describing the absolute number of molecules of fluorophore per cell - would facilitate the development and direct validation of mathematical models of biological circuits, thereby improving the effectiveness of model-based design. Physical gene expression units could be obtained by using FlowCal along with beads labeled with sfGFP, mCherry, and other common fluorescent proteins. Unfortunately, the availability of these is currently limited. Alternatively, calibrated units from different instruments can be compared with the help of a common biological sample (Figure 2F). Universal units could be theoretically obtained with this approach by distributing standard reference strains with annotated fluorescence values in MEF as given by a reference instrument. However, differences in experimental handling, especially across different laboratories, can dramatically affect gene expression and fluorescence, introducing errors in cross-instrument calibrations (28,29). Given current technology, FlowCal allows reporting of gene expression in calibrated units. However, it can, without modifications, report either universal or even physical units pending the availability of beads labeled with fluorescent proteins or a more robust biological reference strain protocol. Such gene expression unit standardization is a major need in synthetic biology and will help the field develop into a mature engineering discipline.
Methods
E. coli Ptac-GFP experimental protocol for Figure 2A and 2B
E. coli strain BW29655 was transformed with plasmid pSR11.2, a ColE1 plasmid containing sfGFP under control of Ptac/LacI, and mCherry under control of PLtet0-1/TetR. After transformation, a single colony was inoculated in LB + 34 μg/mL chloramphenicol, and a −80°C freezer stock was prepared.
To perform the experiment shown in Figure 2A and 2B, the following steps were performed:
One culture with 3mL LB + 34 μg/mL chloramphenicol was started from the −80°C freezer stock and grown at 37°C, 250RPM overnight (~13 hours) to saturation.
A single tube with 3mL M9 media + 34 μg/mL chloramphenicol + 1mM IPTG was inoculated from the overnight culture to an initial OD600 of 0.0001.
This culture tube was incubated at 37°C, 250RPM for 7 hours to a final OD600 of 0.145.
The tube was removed from the shaker and placed on an ice-water bath for approximately 15 minutes.
Rifampicin treatment was performed as previously described (23). In short, 100μL of culture was added to a flow cytometry tube with 1mL PBS + 500μg/mL rifampicin and incubated at 37°C for 1 hour.
The flow cytometry tube was transferred to an ice-water bath, and flow cytometry measurements were performed.
E. coli Ptac-GFP experimental protocol for Figure 2F
E. coli strain BW29655 was transformed with plasmid pSC12, a ColE1 plasmid containing sfGFP under control of Ptac/LacI. After transformation, a single colony was inoculated in LB + 34 μg/mL chloramphenicol, and a −80°C freezer stock was prepared.
To perform the experiment shown in Figure 2F, the following steps were performed:
One culture with 3mL LB + 34 μg/mL chloramphenicol was started from the −80°C freezer stock and grown at 37°C, 250RPM overnight (~13 hours) to saturation.
Five tubes with 3mL M9 media + 34 μg/mL chloramphenicol each were inoculated from the overnight culture to an initial OD600 of 0.000025. IPTG was added to each tube to the following final concentrations: 0, 81, 161, 318, and 1000 μM.
These culture tubes were incubated at 37°C, 250RPM for 6:20 hours to a final OD600 of 0.14.
The tubes were removed from the shaker and placed on an ice-water bath.
100μL of culture was added to a flow cytometry tube with 1mL PBS.
Flow cytometry measurements were performed.
E. coli CcaS/CcaR v2.0 experimental protocol
E.coli strain BW29655 + pSR43.6 + pSR58.6 (22) is the green/red photoreversible CcaS/CcaR v2.0 system used in Figure 2C and 2D. Experiments were performed using Light-Plate Arrays (LPAs).
Prior to the experiments, the following was performed:
One culture with 3mL LB + 34 μg/mL chloramphenicol + 100 μg/mL spectinomycin was started from a −80°C freezer stock and grown at 37°C, 250RPM up to an OD600 < 0.3. The exact final OD600 was measured.
Culture was mixed with 60% glycerol to a 70:30 volume ratio. The mixture was distributed into 50μL in PCR tubes and frozen at −80°C. A value consisting of 70% of the OD600 measured in the previous step was written down. These cultures were used to start experiments.
For the experiments, the following was performed:
M9 media with 34 μg/mL chloramphenicol + 100 μg/mL spectinomycin was prepared.
A single starter culture was removed from the −80°C freezer, thawed, and inoculated in M9 such that the initial OD600 is 5×10−5.
Inoculated M9 media was distributed among the wells of a 24-well plate with 500μL per well. The plate was then sealed with adhesive foil.
The plate was installed in one of the LPAs and incubated at 37°C, 250 RPM for 8 hours.
At the end of the experiment, the plates were removed from the LPA and placed on an ice-water bath for around 15 minutes.
Rifampicin treatment was performed as previously described (23). In short, 100μL of culture from each well was added to a flow cytometry tube with 1mL PBS + 500μg/mL rifampicin and incubated at 37°C for 1 hour.
Flow cytometry tubes were transferred to an ice-water bath, and flow cytometry measurements were performed.
S. cerevisiae PhyB/PIF3/Venus experimental protocol
Strain 160712 [W303, MATa, leu2∷prADH1-GADPIF3, trp1∷prADH1-PHYBGDBD, his3∷ prGAL1-Venus, ade2∷ADE2, can1, ura3] was used. Experiments were performed using Light-Plate Arrays (LPAs).
For the experiments, the following was performed:
A culture tube containing 3mL SD (-His, -Trp, -Leu) was inoculated from a −80°C freezer stock and grown at 30°C, 250 RPM until the OD600 reached approximately 2.0 (around 16 hours).
SD (-His, -Trp, -Leu) containing 10 μM PCB was inoculated from the starter culture to an initial OD600 of 0.00015.
Inoculated SD media was distributed among the wells of a 24-well plate with 500uL per well. The plate was then sealed with adhesive foil.
The plate was installed in an LPA outfitted with 647 nm (centroid) LEDs (L2-0-R5TH50-1, LED Supply) and incubated at 30°C, 250 RPM for 18 hours, with the indicated intensity of 647 nm light.
At the end of the experiment, the plate was removed from the LPA and chilled in an ice-water bath for 15 minutes.
200μL of culture from each well was added to 1mL PBS, and flow cytometry measurements were performed.
Flow Cytometry Data Acquisition and Analysis
Cytometry acquisition was performed using a BD FACScan flow cytometer with the original laser system replaced by blue (488 nm, 30 mW) and yellow (561 nm, 50 mW) solid-state lasers (Cytek). The FL1 (sfGFP) acquisition channel emission filter was also replaced with a 510/21-nm filter. The FL3 (mCherry) acquisition channel emission filter (650 nm long pass) is original to the instrument.
For Figure 2F, an additional set of measurements were carried out using a BD FACSCanto II Flow Cytometer. The excitation laser relevant for the FITC channel is a Coherent Saphire Solid State, 488nm, and the emission filter is a 525/50 band-pass.
Acquisition of cell samples is performed with typical count rates of 1,000–2,000 events/s. Approximately 30,000–50,000 events are stored for each sample. A SSC threshold is used to eliminate instrument noise events that are clearly not due to cell scattering.
The cytometer is calibrated using Spherotech cat. #RCP-30-5A beads, measured either at the beginning or at the end of every measurement round. A single drop is added to 250 or 500μL PBS, which results in count rates of 300–600 events/s. The cytometer settings used for cells and calibration beads have been listed in Table S1 and Table S2.
Analysis has been performed using FlowCal 1.0.0. The scripts used to generate the plots shown in Figures 2, S13, S14 and S15 are included in Supporting Information.
Supplementary Material
Acknowledgments
We thank Andreas Milias-Argeitis and Mustafa Khammash for the S. cerevisiae strain, Karl Gerhardt for performing the yeast experiment, Felix Ekness for helping with FlowCal installation in OSX, and Tabor lab members for beta testing and helpful comments. This work was supported by the National Science Foundation (EFRI-1137266 and MCB-1244135), The Office of Naval Research (MURI N000141310074, YIP N000141410487) The National Institutes of Health (1R21AI115014-01A1), and the Welch Foundation (C-1856). JS is supported by an NSF Graduate Research Fellowship (DGE-0940902). BL is supported by an NDSEG Fellowship.
Footnotes
Supporting Information
Additional notes, procedures, figures, tables, a copy of FlowCal, and scripts used to generate this document’s figures using FlowCal.
References
- 1.Schwartz A, Flow Cytometry Standards Corporation Calibration Method for Flow Cytometry Using Fluorescent Microbeads and Synthesis Thereof. 4,767,206 US Patent. 1988
- 2.Schwartz A, Gaigalas AK, Wang L, Marti GE, Vogt RF, Fernandez-Repollet E. Formalization of the MESF unit of fluorescence intensity. Cytometry B Clin Cytom. 2004;57:1–6. doi: 10.1002/cyto.b.10066. [DOI] [PubMed] [Google Scholar]
- 3.Vogt RF, Jr, Marti GE, Zenger V. Standardization and Quality Assurance in Fluorescence Measurements I. Berlin: Springer; 2008. Quantitative fluorescence calibration: a tool for assessing the quality of data obtained by fluorescence measurements; pp. 3–31. [Google Scholar]
- 4.Schwartz A, Fernandez Repollet E, Vogt R, Gratama JW. Standardizing flow cytometry: Construction of a Standardized Fluorescence Calibration Plot Using Matching Spectral Calibrators. Cytometry. 1996;26(1):22–31. doi: 10.1002/(SICI)1097-0320(19960315)26:1<22::AID-CYTO4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman RA, Wang L, Bigos M, Nolan JP. NIST/ISAC standardization study: variability in assignment of intensity values to fluorescence standard beads and in cross calibration of standard beads to hard dyed beads. Cytometry A. 2012;81:785–796. doi: 10.1002/cyto.a.22086. [DOI] [PubMed] [Google Scholar]
- 6.Yurtsev E, Friedman J. FlowCytometryTools. http://eyurtsev.github.io/FlowCytometryTools/
- 7.Frelinger J. FCM: A python library for working with flow cytometry data. https://github.com/jfrelinger/fcm.
- 8.Kumar RS, Mehta T, Bose B. FlowPy: A Python tool for Flow Cytometry Data. http://flowpy.wikidot.com/
- 9.Balkay L. FCS data reader. http://www.mathworks.com/matlabcentral/fileexchange/9608-fcs-data-reader.
- 10.Balkay L. FCA: Flow Cytometry Analysis. http://www.mathworks.com/matlabcentral/fileexchange/8480-fca-2-2.
- 11.Steinberg N. Flow Cytometry GUI for Matlab. http://www.mathworks.com/matlabcentral/fileexchange/38080-flow-cytometry-gui-for-matlab.
- 12.Henson R. Flow Cytometry Data Reader and Visualization. http://www.mathworks.com/matlabcentral/fileexchange/8430-flow-cytometry-data-reader-and-visualization.
- 13.Ellis B, Haaland P, Hahne F, Le Meur N, Dopalakrishnan N, Spidlen J, Jiang M. flowCore: Basic structures for flow cytometry data. https://bioconductor.org/packages/devel/bioc/html/flowCore.html.
- 14.Pontikos N. flowBeads: Analysis of flow bead data. https://bioconductor.org/packages/devel/bioc/html/flowBeads.html.
- 15.Beal J, Weiss R, Yaman F, Davidsohn N, Adler A. A Method for Fast, High-Precision Characterization of Synthetic Biology Devices. 2012 http://web.mit.edu/jakebeal/www/Publications/MIT-CSAIL-TR-2012-008.pdf.
- 16.Davidsohn N, Beal J, Kiani S, Adler A, Yaman F, Li Y, et al. Accurate predictions of genetic circuit behavior from part characterization and modular composition. ACS Synth Biol. 2014;4(6):673–681. doi: 10.1021/sb500263b. [DOI] [PubMed] [Google Scholar]
- 17.Dean PN, Bagwell CB, Lindmo T, Murphy RF, Salzman GC. Data file standard for flow cytometry. Data File Standards of the Society for Analytical Cytology. Cytometry. 1990;11:323–332. [Google Scholar]
- 18.Seamer LC, Bagwell CB, Barden L, Redelman D, Salzman GC, Wood JC, Murphy RF. Proposed new data file standard for flow cytometry, version FCS 3.0. Cytometry. 1997;28:118–122. doi: 10.1002/(sici)1097-0320(19970601)28:2<118::aid-cyto3>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 19.Spidlen J, et al. Data File Standard for Flow Cytometry, version FCS 3.1. Cytometry A. 2009;77A:97–100. doi: 10.1002/cyto.a.20825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic Gene Expression in a Single Cell. Science. 2002;297(5584):1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 21.De Boer HA, Comstock LJ, Vasser M. The tac promoter: a functional hybrid derived from the trp and lac promoters. Proc Natl Acad Sci USA. 1983;80(1):21–25. doi: 10.1073/pnas.80.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidl SR, Sheth RU, Wu A, Tabor JJ. Refactoring and optimization of light-switchable Escherichia coli two-component systems. ACS Synth Biol. 2014;3(11):820–831. doi: 10.1021/sb500273n. [DOI] [PubMed] [Google Scholar]
- 23.Olson E, Hartsough LA, Landry B, Shroff R, Tabor JJ. Characterizing bacterial gene circuit dynamics with optically programmed gene expression signals. Nat Methods. 2014;11:449–455. doi: 10.1038/nmeth.2884. [DOI] [PubMed] [Google Scholar]
- 24.Milias-Argeitis A, Summers S, Stewart-Ornstein J, Zuleta I, Pincus D, El-Samad H, Khammash M, Lygeros M. In silico feedback for in vivo regulation of a gene expression circuit. Nat Biotechnol. 2011;29(12):1114–16. doi: 10.1038/nbt.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beal J. Bridging the gap: a roadmap to breaking the biological design barrier. Front Bioeng Biotechnol. 2014;2:87. doi: 10.3389/fbioe.2014.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roederer M. Spectral compensation for flow cytometry: visualization artifacts, limitations, and caveats. Cytometry. 2001;45:194–205. doi: 10.1002/1097-0320(20011101)45:3<194::aid-cyto1163>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Gaigalas AK, Marti G, Abbasi F, Hoffman RA. Toward quantitative fluorescence measurements with multicolor flow cytometry. Cytometry A. 2008;73:279–288. doi: 10.1002/cyto.a.20507. [DOI] [PubMed] [Google Scholar]
- 28.Kelly JR, Rubin AJ, Davis JH, Ajo-Franklin CM, Cumbers J, Czar MJ, et al. Measuring the activity of BioBrick promoters using an in vivo reference standard. J Biol Eng. 2009;3:4. doi: 10.1186/1754-1611-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beal J, Haddock-Angeli T, Gershater M, de More K, Lizarazo M, Holenhorst J, Rettberg R. Reproducibility of Fluorescent Expression from Engineered Biological Constructs in E coli. PLoS ONE. 2016;11(3):e0150182. doi: 10.1371/journal.pone.0150182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.