

Abstract
The apicomplexan parasite Cryptosporidium is a leading cause of diarrheal disease and an important contributor to overall global child mortality. We currently lack effective treatment and immune prophylaxis. Recent advances now permit genetic modification of this important pathogen. We expect this to produce rapid advances in fundamental as well as translational research on cryptosporidiosis. Here we outline genetic engineering for Cryptosporidium in sufficient detail to establish this technology in any laboratory that requires access to this key technology. This chapter details the conceptual design consideration, as well as all the experimental steps required to transfect, select, and isolate transgenic parasites. We also provide detail on key in vitro and in vivo assays to detect, validate, and quantify genetically modified Cryptosporidium parasites.
Keywords: (3–5) CRISPR, Cryptosporidium, transfection, genetics, diarrheal disease
INTRODUCTION
Cryptosporidium parvum and Cryptosporidium hominis are apicomplexan parasites that can cause severe diarrhea in children and immunocompromised adults. These two agents have also been linked to several large-scale waterborne disease outbreaks worldwide (Striepen, 2013). Lack of continuous culture, genetic tools, and facile animal models has hampered research efforts to understand the parasite’s biology and to develop efficacious drugs or vaccines for this important infectious disease. However, driven by the tremendous public health impact of the disease, Cryptosporidium research has experienced a remarkable recent surge. Leveraging new technologies from stem cell derived organ culture to CRISPR genome engineering, several laboratories have begun to push at these boundaries. Here we describe, in technical detail, how Cryptosporidium can be engineered genetically.
Using newly available molecular tools for transfection and genetic engineering (Vinayak et al., 2015), it is now possible to design and construct reporter strains and to interrogate specific parasite genes by knockout or modification to unravel their role in Cryptosporidium biology. Cryptosporidium is transmitted by ingestion of food or water contaminated with oocysts, the environmentally stable form of the parasite (Fayer R, 1997; Mac Kenzie et al., 1994). The oocyst is remarkably resistant to environmental stresses due to a chemically inert wall that shelters the parasite e.g. from the sterilizing effects of water chlorination (Bushkin et al., 2013; Korich, Mead, Madore, Sinclair, & Sterling, 1990). Oocysts are stable for months with refrigeration and are the best starting point for genetic engineering (see Fig. 1 for outline). Large numbers of oocysts are available from academic and commercial sources that typically propagate C. parvum in calves. To generate a stable transgenic parasite, C. parvum oocysts are “excysted” to release the 4 sporozoites harbored inside. Reporter and selection constructs (usually an Nluc-NeoR fusion cassette) are introduced into sporozoites by electroporation followed by infection of highly susceptible mice (Griffiths, Theodos, Paris, & Tzipori, 1998; Mead, Arrowood, Sidwell, & Healey, 1991). Because transfected sporozoites are no longer encased in the protective oocyst wall, their ability to survive the acidic environment of the stomach is reduced if infected orally. Therefore, we surgically inject transfected sporozoites directly at the site of infection—the small intestine. Surgery is only required when transfected sporozoites are initially used to infect mice. After transgenic oocysts have been recovered, subsequent mice can be orally infected by simple gavage with oocysts—similarly transgenic oocysts can be used to infect tissue culture for in vitro experiments.
Figure 1. Strategy for generating stable transgenic Cryptosporidium parvum.
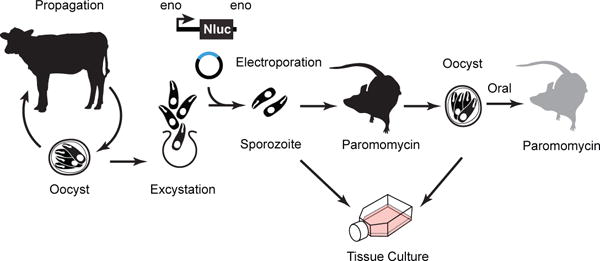
C. parvum mutants are generated by electroporation of sporozoites that are then used to infect IFN-γ KO mice followed by in vivo drug selection for stable transgenic parasites. Transfected sporozoites can also be cultured in vitro for a shorter amount of time (up to 72 hrs). Modified from Vinayak et al., 2015.
Recent work has employed transfection to produce transgenic C. parvum expressing a chimeric Nanoluciferase reporter-neomycin resistance marker (Nluc-NeoR) (Hall et al., 2012). Stable maintenance of this marker is selected for by growth in the presence of paromomycin (Gueiros-Filho & Beverley, 1994; Mochizuki, 2008), which is administered to mice in the drinking water. Following infection, oocysts are shed with the feces and transgenic oocysts can be appreciated and quantified by measuring Nluc activity directly in the fecal material collected from infected mice. Similarly, successful transfection is evident by Nluc activity from tissue culture. In a typical experiment, transgenic organisms emerge over the course of two to four weeks and oocysts can be isolated from fecal material for downstream experiments in tissue culture or animals.
This protocol describes all aspects of generating a stable transgenic C. parvum strain, including the design of DNA constructs, in vitro culture, transfection, mouse handling and surgical infection, Nluc luciferase assays, and oocyst purification from fecal material. Protocols may be used individually, or executed in succession as described here.
Safety Concerns
Cryptosporidium parvum and Cryptosporidium hominis are human pathogens that cause mild to severe diarrhea in young children, naïve adults, and chronic diseases in the immunosuppressed. Because transmission is fecal to oral, it is advised to conduct experiments in a designated area of the laboratory with minimal traffic, and with equipment (cooling, pipettes, centrifuges, etc.) dedicated to use with Cryptosporidium only. Equipment and surfaces that come into contact with the pathogen should be decontaminated with 3–6% hydrogen peroxide (prepared in water; we also use commercial products like Diversey Oxyvir Tb), since bleach and ethanol do not neutralize Cryptosporidium oocysts. To decontaminate a work area, douse surfaces with excess hydrogen peroxide solution, and allow to air dry for 20–30 minutes. Always wear lab coats, eye protection and gloves (dispose regularly). All trash, including liquid wastes, should be autoclaved before disposal. We have found collecting liquid waste in 10-liter carboys (autoclaved when waste reaches 5 liters) to be efficient. Note that our safety and animal protocols were reviewed by University of Georgia oversight committees and officials. Other institutions may require modified safety measures for handling, storing, and decontaminating Cryptosporidium and Cryptosporidium infected animals reflecting specific local regulatory requirements.
Handling Cryptosporidium Oocysts
Oocysts are the hardy, transmissible form of the pathogen. The oocyst is comprised of a protective wall, impenetrable to common disinfectants, and contains 4 sporozoites. When ingested, oocysts naturally excyst and release sporozoites in the gastrointestinal tract in response to acid, bile salts, and an increase in temperature. To reduce unintended excystation, oocysts should be stored at 4 °C and maintained on ice at all times. We have found oocysts to be most viable within 2 months of isolation from an infected animal. After this length of time, both transfection and infection rates decrease rapidly. Oocysts may be stored in PBS with antibiotics or in 2.5% potassium dichromate (prepared in deionized water). Iowa strain II oocysts are commercially available from Sterling Parasitology Laboratory at the University of Arizona in Tuscon, AZ (Tel. +1-520-621-4580); Bunchgrass Farms, Idaho (+1-208-877-1276); and Waterborne Inc. New Orleans, Louisiana (+1-504-895-3338). We have found oocysts from all sources to be suitable for the protocols described herein. While all sources provide C. parvum Iowa II strain they are not identical and some differences in virulence and drug susceptibility (and genome sequence) have been noted.
BASIC PROTOCOL 1: CRYPTOSPORIDIUM TISSUE CULTURE
C. parvum parasites can be maintained for up to 72 hours in a simple co-culture with human intestinal epithelial cells (Upton, Tilley, & Brillhart, 1994; Upton, Tilley, Nesterenko, & Brillhart, 1994). Although they cannot be continuously maintained, invasion of host cells and intracellular development of several stages can be observed during this time frame. Continuous in vitro culture for Cryptosporidium have been reported (DeCicco RePass et al., 2017; Morada et al., 2016); however, at this point they require enclosed microfiber cartridges or specialized silk scaffolds, yield only moderate amplification of parasites, and are not immediately amenable to the assays described below.
Materials
HCT-8, human ileocecal colorectal adenocarcinoma cells, (ATCC catalog #CCL-244)
HCT-8 Media (see recipe)
15 ml centrifugation tubes
Tissue culture treated flasks and plates
Sterile 12 mm glass coverslips
Trypsin (0.25%)
RPMI 1460 with L-glutamine (Corning, catalog #10-040-CV)
FBS
Sodium Pyruvate (10 mM stock)
Penicillin/Streptomycin (100x stock)
Amphotericin B
1X PBS pH 7.5
Cryptosporidium oocysts (available from Sterling Labs or Bunchgrass Farms; or produced in-house)
Clorox bleach
Cryptosporidium Infection Media (see recipe)
1:4 bleach solution (see recipe)
37 °C water bath
Centrifuge with adaptors for 15 ml tubes
37 °C incubator maintained at 5% CO2
Microcentrifuge at 4 °C
All steps to be carried out in a biosafety cabinet using aseptic technique. Cryptosporidium oocysts should be maintained on ice or at 4 °C at all times.
Thaw a vial of HCT-8 cells in a 37 °C water bath for 1–2 minutes.
Add 10 ml HCT-8 media into a 15 ml centrifugation tube. Transfer HCT-8 cells from cryovial and resuspend in HCT-8 media.
Centrifuge at 500 × g for 5 minutes.
Remove supernatant and resuspend cells in 10 ml HCT-8 media.
Add cells to a T-25 flask and culture in an incubator at 37 °C with 5% CO2 until confluent (typically 2–3 days).
Once confluent, remove the culture media and rinse cells twice with PBS.
Add 1 ml trypsin to flask, swirl liquid until it covers the surface of the flask, and place in incubator for 5–10 minutes or until HCT-8 cells have become detached.
Resuspend cells in 10 ml HCT-8 media to neutralize trypsin.
Seed HCT-8 cells at 10–20% confluency into 24 well plates (with or without sterile 12 mm glass coverslips), 96 well plates, or flasks.
-
Culture HCT-8 cells until ~40–60% confluent.
HCT-8 cells grow quickly and overgrowth of these cells should be avoided, especially when preparing samples for microscopy. Infect cultures with Cryptosporidium at 60% confluency when preparing a 0–24 hour co-culture sample, or at 40% confluency for longer co-culture.
- Transfer appropriate number of oocysts to a 1.5 ml centrifuge tube. Depending on the experimental procedure, the below gives an estimate of the optimal number of starting oocysts to be used for each assay condition.
- Microscopy of coverslips in 24-well plate: 100,000 oocysts per well
- Nluc assay of 24-well plate: 1,000–10,000 oocysts per well
- Nluc assay of 96-well plate: 1,000–5,000 oocysts per well
Centrifuge at 16,000 ×g for 3 minutes at 4 °C. Remove supernatant.
If oocysts were stored in 2.5% potassium dichromate, resuspend cells in 0.5 ml PBS, and repeat step 12 before continuing. If not skip to step 14.
Resuspend oocysts in 100–400 μl 1:4 bleach solution.
Incubate on ice for 5 minutes. This step serves to kill residual bacteria from the preparation of Cryptosporidium and enhances excystation.
Centrifuge at 16,000 ×g for 3 minutes at 4 °C. Remove supernatant.
Resuspend oocysts in 0.5 ml PBS.
-
Repeat steps 16 and 17 for a total of 3 washes with PBS. This serves to thoroughly wash out the bleach before adding the oocysts to tissue culture.
Oocysts will naturally excyst over the first 24 hours in culture, but if you prefer to use sporozoites, refer to the excystation protocol described in steps 1–14 of Basic Protocol 2 (Transient Transfection of Cryptosporidium).
Resuspend oocysts in appropriate volume of Cryptosporidium Infection Media (1 ml for 24-well plates, 200 μl for 96-well plates).
-
Remove media from plates/flasks containing HCT-8 cells at 40–60% confluency and replace with oocysts in media from step 19.
You may wash the culture anytime from 3 to 24 hours post infection and refresh the media to remove unexcysted oocysts if desired.
SUPPORT PROTOCOL 1: IMMUNOFLUORESCENCE ASSAY OF CRYPTOSPORIDUM INFECTED CULTURES
Immunofluorescence is a convenient way to observe parasites and monitor expression of transgenes. Coverslip cultures of HCT-8 cells infected with Cryptosporidium may be fixed at any time after infection for immunofluorescence assay (IFA) and microscopy. Various intracellular stages are observed during this time frame: trophozoites (1 nucleus) are observed 0–24 hours post infection, merozoites (4–8 nuclei) from 12–48 hours post infection, and male and female gametes (>12 condensed or 1 decondensed nucleus, respectively) 48–72 hours post infection.
Materials
PBS pH 7.4
3% paraformaldehyde, prepared in PBS pH 7.4
125 mM glycine, prepared in PBS pH 7.4
0.25% Triton X-100, prepared in PBS pH 7.4
3% BSA, prepared in PBS pH 7.4
24-well plate
parafilm
Fluoro-Gel with TES Buffer (Electron Microscopy Sciences, catalog #17985-30)
Microscope Slides (Fisher Scientific, catalog #12-544-1)
- Antibodies of choice
- We have found several antibodies to work well in IFA. Listed below are commercially available antibodies for detecting both wild type and transgenic Cryptosporidium mutants.
- Sporo-Glo (Waterborne, Inc. Catalog #A600FLR)
- Vicia Villosa Lectin, VVL (Vector Laboratories, Fluorescein labeled, Catalog # FL-1231)
- Anti-Human Neomycin Phosphotransferase II, NPII (Alpha Diagnostics International; only stains in transgenic parasites.)
DAPI
Forceps
Paper towels
In a new 24-well plate, fill wells corresponding to your samples with PBS.
Carefully transfer glass coverslips from HCT-8 culture infected with Cryptosporidium to wells of the second plate.
Remove PBS and add 1 ml 3% paraformaldehyde to each well. Incubate at room temperature for 10 to 20 minutes.
-
Optional: Replace paraformaldehyde with 1 ml 125 mM glycine to each well. Incubate at room temperature for 5 minutes.
Glycine quenches excess paraformaldehyde and enhances visual contrast.
Replace glycine with 1 ml 0.25% Triton X-100 to each well. Incubate at room temperature 10 minutes.
Wash wells with PBS. Add 1 ml 3% BSA to each well and incubate at room temperature for 30 minutes, or overnight at 4 °C.
Prepare the primary antibodies (can use multiple as long as they were generated in different animal species) in 3% BSA in PBS, 100 μl for each coverslip. We have confirmed the use of a mouse monoclonal anti neomycin phosphotransferase II (NPII) from Alpha Diagnostic International (used at 1:1000 dilution) to detect transgenic C. parvum with the Nluc-NeoR casette.
On a flattened piece of parafilm, aliquot 100 μl of the primary antibody solution spaced evenly apart.
Remove each coverslip from the 24-well plate using forceps; blot the edge of the coverslip on a thin paper towel, and place upside down on top of the drop of primary antibody solution. The antibody solution should be in contact with the side of the coverslip with attached cells. Orient the placement of coverslips in parallel with the original 24-well plate configuration to avoid confusion.
Incubate at room temperature for 1 hour.
Transfer coverslips to 24-well plate, cell-side up, and add 1 ml PBS to each well.
Incubate samples for 5 minutes. Remove PBS and replace with 1 ml PBS.
Repeat Step 12 for a total of 3 washes. Be careful not to detach cells from coverslip.
Prepare the secondary antibody in 3% BSA in PBS, 200 μl for each coverslip.
After the final PBS wash, remove the PBS and add 200 μl secondary antibody solution.
Incubate at room temperature for 1 hour, protected from light.
Remove secondary antibody solution from each well and add 1 ml PBS.
Incubate 5 minutes. Remove PBS and replace with 1 ml PBS.
Repeat Step 18 for a total of 3 washes.
Prepare DAPI in PBS. Use DAPI at a final concentration of 2 μg/ml.
Incubate at room temperature protected from light for 10 minutes.
Pipette 20 μl of Fluoro-Gel on a clean glass slide. Remove each coverslip from the 24-well plate using forceps; blot the edge of the coverslip on a thin paper towel, and place upside down on top of the drop of mounting gel.
Optional: You may seal the edges of the coverslip with clear nail polish to protect for future imaging.
BASIC PROTOCOL 2: TRANSFECTION OF CRYPTOSPORIDUM (AMAXA 4D NUCLEOFECTION DEVICE)
Cryptosporidium is transmitted via an oocyst, and traditional electroporation or lipofection protocols cannot deliver DNA through the oocyst wall to the sporozoites; therefore, the oocysts must first excyst to release their sporozoites. Excystation occurs during the course of natural infection and can be mimicked in the laboratory by incubating oocysts with either bile salts or sodium deoxytaurocholate (chief ingredient of bile) at 37 °C (Gut & Nelson, 1999). After the sporozoites are released, they can be electroporated with various reporter constructs and co-cultured with HCT-8 cells for up to 72 hours. We found this transient assay to be helpful in testing Nluc luciferase reporter constructs prior to generating stable transgenic parasite lines. However, note that the transfection rate is low (<10−5) rendering transient experiments with less sensitive reporters, e.g. microscopy-based fluorescent protein assays, impractical.
Materials
On day of transfection a 24- or 48-well plate of HCT-8 cells at 40–60% confluency should be ready. Typically, parasites from a single cuvette (ie each DNA construct or experimental condition to be tested) are split to three wells.)
Cryptosporidium oocysts
Clorox bleach
PBS pH 7.4
0.2 mM sodium deoxytaurocholate in PBS, pH 7.4
parafilm
Disposable hemacytometer (Kova Glasstic Slide 10 with Grids, catalog #87144)
DNA desired for transfection (DNA should be prepared at a minimum of 2 μg/μl if transfecting a single plasmid, or 4 μg/μl if transfecting two plasmids. Transfection of more than two plasmids is not recommended. DNA should be prepared in Tris-EDTA (pH 8.0) or nuclease free water.
Cryptosporidium Infection Media (see recipe)
SF Cell Line 4D-Nucleofector X Kit Small cuvettes (Lonza, catalog #V4XC-2032)
4D AMAXA Nucleofector (Lonza, Cologne)
Microcentrifuge at 4 °C
Water bath maintained at 15 °C (we have a dedicated water bath placed in a cold room)
-
Resuspend oocysts and aliquot 5×107 oocysts into a new microcentrifuge tube. If needed, add PBS to bring total volume up to 600 μl.
These instructions are for 5×107 oocysts, enough to test at least five different DNA constructs or five different experimental conditions. We generally excyst 25% more oocysts than desired as the rate of excystation is usually 60–80%. Scale up/down as needed. Keep oocysts and solutions on ice or at 4 °C unless otherwise described. Perform all steps in a biosafety cabinet.
Centrifuge at 16,000 ×g for 3 minutes at 4 °C. Remove supernatant.
Resuspend in 600 μl cold PBS.
Centrifuge at 16,000 ×g for 3 minutes at 4 °C. Remove supernatant.
Resuspend oocysts in 100–400 μl cold 1:4 bleach solution.
Incubate on ice for 5 minutes.
Centrifuge at 16,000 ×g for 3 minutes at 4 °C. Remove supernatant.
Resuspend oocysts in 600 μl cold PBS.
Repeat steps 7 and 8 for a total of 3 washes with PBS. This serves to thoroughly wash out the bleach before oocyst excystation.
Finally, resuspend oocysts in 400 of μl 0.2 mM sodium deoxytaurocholate, wrap microcentrifuge tube lid with parafilm, and incubate for 10 minutes at 15 °C.
Next, transfer oocysts to a 37 °C water bath and incubate for 1 hour.
-
After an hour, bring the total volume up to 1 ml using PBS and count the number of sporozoites using a disposable hemocytometer.
If desired, the sample can be filtered using a 3 μm polycarbonate membrane (Whatman catalog # 110612) to remove empty oocyst shells and unexcysted oocysts. In our experience, the presence of oocyst shells or unexcysted oocysts in the sample does not affect electroporation. Please note that filtration of the sample may lower the overall number of sporozoites recovered.
-
Determine total number of sporozoites (1×107 sporozoites are required for each transfection).
It is ok if the sample contains oocysts, just disregard them in the count. They will not affect electroporation.
-
Aliquot the total number of sporozoites desired into a new microcentrifuge tube.
If after counting, the total sporozoite number determined is less than desired, the sample can be incubated at 37 °C for an additional hour. Alternatively, if you have more sporozoites than needed we often use these sporozoites for genomic DNA extraction or other purposes. Sporozoites are only infective for a few hours; unused parasites cannot be saved for infection at a later time.
Centrifuge sporozoites at 16,000 ×g for 3 minutes at 4 °C. Remove supernatant.
Resuspend in 600 μl cold PBS.
Centrifuge sporozoites at 16,000 ×g for 3 minutes at 4 °C. Remove supernatant.
-
Resuspend sporozoites at 1×107 sporozoites per 15 μl Complete SF Buffer.
Buffer and cuvettes are purchased together for use with the 4D Nucleofector. We recommend mixing SF Buffer with Supplement #1 fresh on the day of the transfection to make “Complete SF Buffer”. These should be added at a ratio of 4.5 to 1, i.e. 16.4 μl Buffer SF with 3.6 μl Supplement #1 = 20 μl Complete SF Buffer. Scale up as needed.
Aliquot 15 μl sporozoites into new microcentrifuge tubes and label with plasmid name.
-
Add plasmid DNA to corresponding microcentrifuge tube. For 4D Nucleofector, the total volume of DNA should not exceed 5 μl per small cuvette. Use 10–30 μg of DNA (applies to each plasmid if transfecting with more than one).
In our experience, use of a larger volume of DNA than described disrupts the conduction of electricity through the cuvette and results in unsuccessful transfection.
DNA should be prepared at a minimum of 2 μg/μl if transfecting a single plasmid, or 4 μg/μl if transfecting two plasmids. Transfection of more than two plasmids is not recommended. DNA should be prepared in Tris-EDTA (pH 8.0).
Pipette parasite and DNA mixture up and down gently to mix, and transfer entire 20 μl volume to a single, small cuvette in the cuvette strip (strip contains a 2 by 8 layout of cuvettes).
Repeat until all samples have been prepared and transferred to the cuvettes. Make sure the lid of the cuvette strip is tightly closed.
Electroporate each cuvette using program EH 100.
Add 80 μl Cryptosporidium Infection Media to each small cuvette. Pipette up and down gently to mix, and transfer the 100 μl volume to a 15 ml conical containing 3ml Cryptosporidium Infection Media. Repeat for each cuvette.
Remove HCT-8 media from 24- or 48-well plate.
Replace with Crytosporidium Infection Media containing sporozoites, 1 ml per well, 3 wells per cuvette.
Incubate sporozoite infected HCT-8 culture for 24 to 48 hours at 37 °C, 5% CO2.
ALTERNATE PROTOCOL 2: TRANSFECTION OF CRYPTOSPORIDIUM USING LARGE 4D NUCLEOFECTION CUVETTES
Amaxa Nucleofection cuvettes are available in small and large sizes, and scale well. The small cuvettes hold 20 μl of sample and are used to transfect 1×107 sporozoites; the large cuvettes hold 100 μl of sample and are used to transfect 5×107 sporozoites. The large cuvettes are convenient for mouse-related transfections as a single large cuvette generates enough transfected parasites to infect an entire cage of mice (5 mice).
Materials
SF Cell Line 4D-Nucleofector X Kit Large cuvettes (Lonza, catalog #V4XC-2012)
Follow steps 1–17 of Basic Protocol 2 (Transfection of Cryptosporidium: Amaxa 4D Nucleofection Device), and then continue below as described:
Resuspend 5×107 sporozoites in 75 μl Complete SF Buffer and add 25μl DNA (50 – 150 μg total).
Pipette parasite and DNA mixture up and down gently to mix, and transfer entire 100 μl volume to a single, large cuvette.
Electroporate each cuvette using program EH 100.
Use disposable Pasteur pipette that comes with cuvette to transfer parasites to microcentrifuge tube.
ALTERNATE PROTOCOL 3: TRANSFECTION OF CRYPTOSPORIDIUM (BTX ELECTROPORATION DEVICE)
Transfection of sporozoites may be accomplished using a variety of electroporation devices. We have found transfection using the Amaxa 4D Nucleofection system to have a ten-fold higher transfection rate than the BTX electroporation device, but include instructions here for those without access to an Amaxa 4D Nucleofector. While the excystation process is the same, the final steps to prepare parasites for electroporation differ for each device.
Materials
2 mm BTX Electroporation Cuvettes
1X Cytomix (see recipe)
BTX Electroporation System (ECM 630, Harvard Apparatus)
Follow steps 1–17 of Basic Protocol 2 (Transfection of Cryptosporidium: Amaxa 4D Nucleofection Device), and then continue below as described:
-
Resuspend sporozoites at 1×107 sporozoites per 100 μl Complete Cytomix.
Cytomix should be prepared fresh, filtered using a 0.22 μm membrane, and stored at 4 °C. On the day of transfection, remove 10 ml of cytomix, supplement with ATP and L-glutathione, and refilter (after supplementation, it is referred to as “Complete Cytomix”).
Aliquot 100 μl sporozoites into new microcentrifuge tubes and label with plasmid name.
-
Add plasmid DNA to corresponding microcentrifuge tube. For BTX, the total volume of DNA should not exceed 30 μl per cuvette. Use 10–30 μg of DNA (applies to each plasmid if transfecting with more than one).
In our experience, use of a larger volume of DNA than described disrupts the conduction of electricity through the cuvette and results in unsuccessful transfection.
DNA should be prepared at a minimum of 2 μg/μl if transfecting a single plasmid, or 4 μg/μl if transfecting two plasmids. Transfection of more than two plasmids is not recommended. DNA should be prepared in Tris-EDTA (pH 8.0) or nuclease free water.
Pipette parasite and DNA mixture up and down gently to mix, and transfer entire volume to labeled 2mm BTX cuvette.
Repeat until all samples have been prepared and transferred to BTX cuvettes.
Electroporate each cuvette using a single 1,500V pulse; resistance of 25Ω; and a capacitance of 25 μF.
Resuspend the contents of each cuvette in 3 ml Cryptosporidium Infection Media.
Remove HCT-8 media from 24- or 48-well plate.
Replace with Crytosporidium Infection Media containing sporozoites, 1 ml per well, 3 wells per cuvette.
Incubate sporozoite infected HCT-8 culture for 24 to 48 hours at 37 °C, 5% CO2.
SUPPORT PROTOCOL 2: DESIGNING AND CREATING DNA CONSTRUCTS FOR GENETIC MODIFICATION OF CRYPTOSPORIDIUM
It is now feasible to genetically modify C. parvum to generate transgenic strains where a gene is deleted or tagged. To generate a transgenic strain, sporozoites are transfected with 1. a CRISPR/Cas9 plasmid containing a guide RNA sequence that targets the gene of interest (Jinek et al., 2012; Sidik, Hackett, Tran, Westwood, & Lourido, 2014) and 2. a repair cassette that contains the Nluc-NeoR selection marker. Integration of the repair cassette can replace and thus delete the gene of interest, or modify it by inserting for example an epitope tag at the C-terminus of the encoded protein (to allow for expression and localization studies). To direct integration to the desired locus, the repair cassette is flanked by sequences homologous to the desired crossover sites. In this section, we explain how to select and clone guide RNAs for knockout or tagging, and how to design the repair cassette for targeted integration. We use an experimentally validated example, the thymidine kinase knockout (described below and in Vinayak et al., 2015), as a detailed example. There are several resources available for first time CRISPR/Cas9 users that offer excellent explanations and tips (Newman & Ausubel, 2016; https://www.addgene.org/crispr/guide/).
STRATEGIC PLANNING
When planning to engineer a C. parvum transgenic strain note that this parasite completes its entire life cycle in a single host; deletion of a gene or other genome feature essential at any step of the life cycle will prevent successful recovery of mutants. Only a single drug marker is currently available. This necessitates a ‘single hit’ construct that places all transgenes into a single locus. Fortunately, because C. parvum is haploid for most of its lifecycle, a single targeting event is typically sufficient.
When selecting the guide RNA sequence consider selecting a guide within the open reading frame (if tolerated). C. parvum intergenic regions are small and regulatory sequences of adjacent genes may overlap; careful planning may help to avoid unintended disruption of these regions. Additionally, intergenic regions may have even lower GC content and less sequence complexity than coding regions, making it more challenging to choose unique and non-repetitive sequences. Pick guides that are close to the repair flanks you chose. When tagging a gene at the C-terminus it is best to identify a guide RNA sequence as close to the stop codon as possible. If an appropriate guide cannot be found in the open reading frame, expand your search to include regions just beyond the open reading frame.
The Eukaryotic Pathogen Database (http://www.eupathdb.org and http://www.cryptodb.org) provides a critical resource to access the genome of Cryptosporidium and associated datasets (Abrahamsen et al., 2004; Harb & Roos, 2015) and this includes tools to select guide RNA sequences. We have found the Eukaryotic Pathogen CRISPR guide RNA Design Tool (Brooks et al., 2010; Peng & Tarleton, 2015) to be simple and straightforward as the C. parvum genome is preloaded (http://grna.ctegd.uga.edu). A ‘G’ is needed to aide appropriate transcription from the U6 promoter. To increase the number of available guides in the AT-rich Cryptosporidium genome consider to simply add an artificial ‘G’ to the 5’ end of guide RNA sequences, as described for predicted guide RNAs via EuPaGDT. We found guides generated in this manner on par with perfect matches.
There are several convenient approaches to clone the short guide RNA sequence taking advantage of artificial oligonucleotides. We typically design two oligonucleotides complementary for 20 bp; their orientation such that the protospacer adjacent motif (PAM, not included in the oligonucleotides but present in the genomic target) is 3’ of the guide sequence. To the 5’ end of the oligonucleotide pair, add an artificial ‘GTTG’ to the forward oligonucleotide and an ‘AAAC’ to the reverse oligonucleotide. Once the oligonucleotides are annealed, these additional sequences will generate ‘sticky’ overhangs compatible to those generated by BbsI treatment of the plasmid.
Next, select the region of the locus where you intend to insert the repair cassette containing Nluc-NeoR. The genome of Cryptosporidium does not encode enzymes required for non-homologous end joining found e.g. in the related apicomplexan Toxoplasma gondii (Fox, Ristuccia, Gigley, & Bzik, 2009; Huynh & Carruthers, 2009). The transgene must thus be targeted for insertion at the locus using homology-directed repair. We found constructs that rely on homologous recombination at both ends to be more efficient in modifying the genome of C. parvum than single crossover plasmids (unpublished observations). Therefore, we suggest flanking the Nluc-NeoR cassette at the 5’ and 3’ end with sequences homologous to the regions flanking the desired insertion site with the CRISPR site in between and removed by recombination.
For gene knockouts, this is straightforward: homology regions consist of untranslated regions directly upstream of the start codon and directly downstream of the stop codon, and the guide RNA targeting site should be contained within the open reading frame and should be as close to one of the homology regions as possible. In this way, the gene of interest (including the guide sequence and PAM) is replaced with Nluc-NeoR rendering the transgenic locus resistant to further cleavage by Cas9 (Figure 2A).
Figure 2. Genetic Strategy to Knockout or Tag Cryptosporidium Thymidine Kinase.
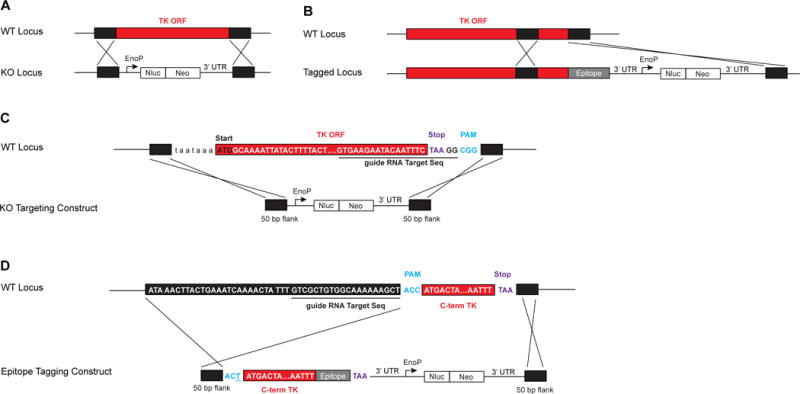
The thymidine locus (tk) can be targeted for knockout or C-terminal epitope tagging. A) Flanks of 50 bp of homology from directly upstream and downstream of the gene target the repair cassette (black) containing Nluc-NeoR (white) to replace the entire tk open reading frame (ORF, red). B) To epitope-tag TK, a repair cassette containing the C-terminus of the gene (red) is cloned fused to the epitope tag (grey). The Nluc-NeoR cassette (white) is expressed under independent regulatory sequences. Flanks of 50 bp of homology (black) target the repair cassette for correct integration and produce a direct fusion of TK with an epitope tag (grey). C) Homology flanks of 50 bp were designed such that the entire TK ORF (red), including the guide RNA sequence (underlined) and the PAM (blue), were replaced with the Nluc-NeoR cassette. D) To fuse TK at the C-terminus with an epitope tag, the C-terminal region of TK (C-term TK, red) was cloned without a stop codon (purple) in frame with an epitope tag (grey). This TK-epitope fusion was cloned upstream of the Nluc-NeoR cassette (white) to generate the repair epitope tagging construct. The epitope tagging construct is flanked by 50 bp of homology (black) from directly upstream of the PAM (blue) and directly downstream of the stop codon (purple). To avoid further targeting of the repair cassette after integration, the PAM is mutated (underlined).
When a gene is tagged rather than replaced, the guide RNA sequence may need to remain after the repair cassette is introduced. In that case, we suggest mutation of the PAM to prevent further targeting of the locus by Cas9. We use the human codon optimized Streptococcus pyogenes Cas9 that recognizes a PAM motif sequence of ‘NGG’ located directly adjacent to the 20 bp guide sequence. Because the PAM sequence is required for endonuclease activity, mutation of this sequence inhibits further Cas9 activity. It is often simple to mutate one of the bases of the PAM to disrupt the motif without altering the codon and resulting amino acid. Rather than displace the 3’ UTR of the gene of interest, we often design the tagging repair cassette to be inserted between the endogenous gene’s stop codon and its 3’ UTR. This serves to avoid disrupting any possible regulatory sequences located in the 3’ UTR. Tagging constructs contain in order—an upstream homology region, a region of the open reading frame containing the guide RNA and mutated PAM, an epitope tag (with stop codon), 3’ UTR, the Nluc-NeoR cassette, and a downstream homology region (Figure 2B). In this way, the repair cassette will generate a fusion of the gene of interest and the epitope tag, express Nluc-NeoR, and will be resistant to future targeting by Cas9. Traditional restriction cloning or Gibson assembly may be used to flank the marker with homology regions specific to the gene of interest.
In Fig. 2 we use the C. parvum thymidine kinase (tk) gene as example for gene replacement or tagging. We found the thymidine kinase gene to be non-essential (Vinayak et al., 2015) and reliably amenable to modification making it a convenient locus to introduce transgenes, and a good positive control for a first C. parvum transgensis experiment.
To knockout tk, the guide RNA sequence recognizes the last base pairs of the tk coding sequence with a PAM after the stop codon (Figure 2C). We selected homology regions directly upstream of the tk open reading frame (for the 5’ homology region) and directly downstream of the guide RNA site (for the 3’ homology region). Our initial transfection experiments used flanks of ~1000 bp, as longer regions of homology typically enhance the recombination frequency (Brooks et al., 2010). However, we found that aided by Cas9, shorter flanks of 50 bp, but not 20 bp, suffice (Pawlowic and Striepen, unpublished). We now routinely employ 50 bp flanks, which is most convenient as synthetic oligonucleotides can be used to flank the targeting cassette in a simple PCR amplification. This greatly simplifies preparation and provides a linear molecule.
Similarly, to fuse the TK protein to a sequence encoding an epitope tag, we use a guide RNA within the open reading frame, but as close as possible to the stop codon (Figure 2D).
We replace the C-terminal region of the gene with a tagged version that contains the Nluc-NeoR cassette with its own regulatory sequences. To avoid further targeting of the repaired locus, the PAM is mutated. The change of a single base pair maintains the correct amino acid of the TK protein but renders the sequence no longer susceptible to Cas9 activity.
Materials
Luria-Bertani (LB) broth
Kanamycin
CpAldo_Cas9_Ribo + U6 Plasmid (Figure 3, Striepen Lab plasmid #185, University of Georgia (Vinayak et al., 2015))
Forward and Reverse Oligonucleotide pair corresponding to your guide RNA sequence (see strategic planning section for instructions on design) To the 5’ end of the oligonucleotide pair corresponding to the guide RNA sequence, add an artificial ‘GTTG’ to the forward oligo and an ‘AAAC’ to the reverse oligo. Once the oligos are annealed, these additional sequences will generate sticky ends compatible to the BbsI cloning site on the plasmid.
Figure 3.
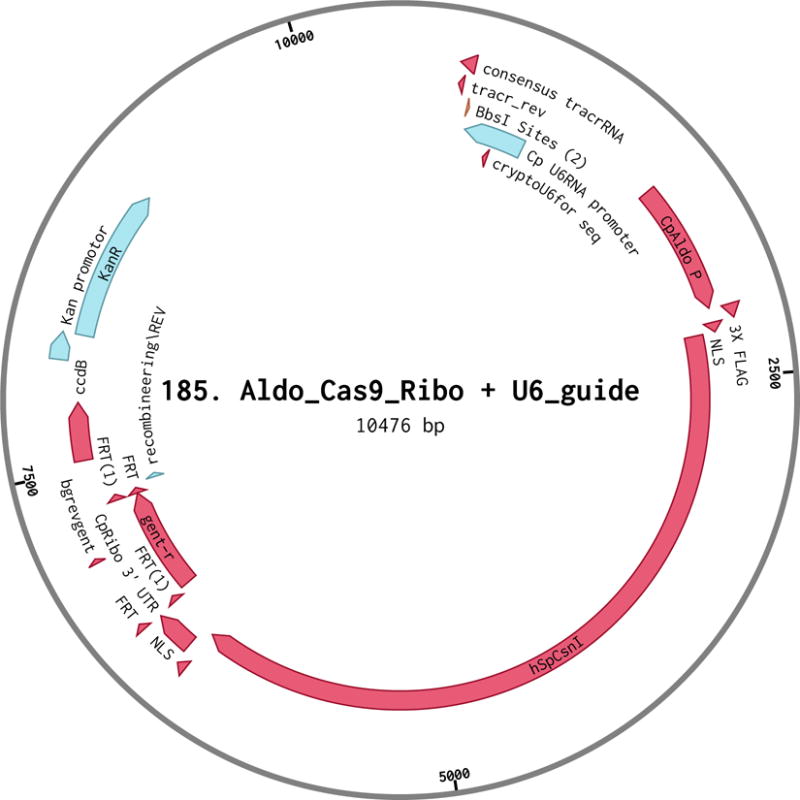
Plasmid map of 185. Aldo_Cas9_Ribo + U6.
AccuPrep Nano-Plus Plasmid Mini Extraction Kit (Bioneer, catalog #K-3111)
AccuPrep Purification Kit (Bioneer, catalog #K-3035)
BbsI (NEB catalog #R0539, store at −80 °C)
Calf Intestinal Alkaline Phosphatase, CIP (NEB, catalog #M0290, store at −20 °C)
T4 Polynucleotide Kinase, T4PNK (NEB, catalog #M0201, store at −20 °C)
T4 DNA Ligase Reaction Buffer, (NEB, catalog #B0202, store in 10 μl aliquots at -20 °C and do not freeze-thaw more than twice)
Transformation competent bacterial cells
Agar for bacterial culture plates
Petri dishes
Low-fidelity DNA polymerase master mix for PCR (Promega GoTaq or similar)
Forward Screening Primer: 5′-CTTTACTATTTATTCCGCTTCCACATGC-3′
Heated, shaking incubator, set at 250 rpm and 37 °C
37 °C water bath
Thermocycler
DNA gel electrophoresis apparatus
37 °C incubator for bacterial cultures
Preparation of the CpAldo_Cas9-Ribo + U6 plasmid:
-
1
Inoculate up to 10 ml of Luria broth (LB) with kanamycin (50 μg/ml) with bacteria containing the CpAldo_Cas9_Ribo + U6 plasmid. Culture in a heated shaking incubator at 37 °C overnight until dense.
-
2
Isolate and purify at least 10 μg of plasmid from bacterial culture.
-
3
Digest CpAldo_Cas9_Ribo + U6 plasmid with BbsI at 37 °C in water bath until completely digested.
-
4
Run 5% of the volume of the digested DNA on an agarose gel to confirm the plasmid is fully cut.
-
5
Use Gel extraction/PCR purification clean up kit to purify the linearized DNA.
-
6
Dephosphorylate the DNA with CIP in a 37 °C in water bath for 1 hour.
-
7
Dilute with ultra-pure water to a DNA concentration of 50 ng/μl.
Preparation of the guide RNA insert:
-
8Combine the following in a PCR tube on ice, in the order listed:
- 6.5 μl ultra-pure water
- 1 μl 100 μM Forward Guide Oligo
- 1 μl 100 μM Reverse Guide Oligo
- 1 μl T4 DNA Ligase Reaction Buffer
- 0.5 μl T4 PNK Enzyme
-
9Mix well and incubate in a thermocycler as described below:
- 37°C for 30 minutes
- 95°C for 5 minutes. Repeat this step 14 times, each time decreasing by 5°C.
- 4°C hold.
-
10
Use immediately or store at −20 °C.
The forward and reverse oligos have now been annealed and phosphorylated to generate an ~20 bp double-stranded DNA. This DNA has sticky ends that match the overhangs created when cutting the plasmid with BbsI
Ligation of guide RNA insert into Cas9 plasmid:
-
15
In separate microcentrifuge tubes, dilute the Guide RNA insert at a ratio of 1:200 and 1:500 using ultra-pure water.
-
16Combine the following in a microcentrifuge tube on ice, in the order listed. Make one reaction using the 1:200 Guide RNA dilution and a second reaction using the 1:500 Guide RNA dilution.
- 6.5 μl ultra-pure water (adjust to final volume of 10 μl as needed)
- 50 ng CpAldo_Cas9_Ribo + U6 plasmid (should be 1 μl)
- 1 μl diluted Guide RNA insert
- 1 μl T4 DNA Ligase Reaction Buffer
- 0.5 μl T4 DNA Ligase Enzyme
-
17
Incubate at room temperature for 1–3 hours, or at 4 °C overnight.
-
18
Transform 50 μl competent bacterial cells with 2 μl of the ligation reaction.
-
19
Plate transformed cells on LB agar with kanamycin and incubate at 37 °C overnight.
Screen for positive clones:
-
20
Pick 5–10 colonies and individually inoculate cultures of 3 ml LB with kanamycin (50 μg/ml) and incubate in a heated, shaking incubator at 37 °C overnight.
-
21
To screen each colony, use a small amount of bacterial culture (2 μl is sufficient) from the previous step to perform PCR, using the Reverse Guide Oligo from Step 8 and the Forward Screening Primer (5′-CTTTACTATTTATTCCGCTTCCACATGC-3′).
-
22
Analyze PCR reactions on an agarose gel. An ~200 bp product indicates correct ligation.
Miniprep positive cultures and sequence DNA using Forward Screening Primer described in Step 21.
BASIC PROTOCOL 3: SURGICAL INFECTION OF MICE WITH TRANSFECTED SPOROZOITES
Mice are not the natural host for Cryptosporidium parvum, but they are a valuable model to study infection in the lab. We have adapted a mouse model of infection as a means to generate and propagate C. parvum genetically modified parasite strains. Infection of interferon-gamma knockout mice (IFN-γ KO; Jackson Laboratory #B6.129S7-Ifngtm1Ts/J)(Griffiths et al., 1998) generates a robust infection with C. parvum: it is acute, characterized by a weeklong peak of oocyst shedding, followed by gradual decrease and eventual resolution of the infection within a month. Therefore, to propagate transgenic parasites we serially infect groups of co-housed mice, with each mouse infection passage lasting approximately a month. We have found infectivity of engineered C. parvum to increase with mouse passage, as the parasites apparently adapt to the host switch from cow to mouse, and eventually mice may become overly susceptible to infection. At this point (usually three to four passages in IFN-γ KO mice) the infection may be transferred to NOD scid gamma mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; Jackson Laboratory Catalog #005557) (Mead et al., 1991). NOD scid gamma mice are less susceptible to death from cryptosporidiosis; they shed oocysts at lower levels than IFN-γ KO mice, but maintain the infection for up to two months. Typically, younger mice are more susceptible to Cryptosporidium infection. For surgical infections, we use mice 5–6 weeks old. For passaging Cryptosporidium transgenics we use 6–10 weeks old mice to compensate for their usual increase in virulence. We find variation in the ability to infect mice with Cryptosporidium between laboratories, a phenomenon likely linked to microbiome differences among other factors. Optimization may thus be required to adapt the animal procedures described here for best use in different local facilities.
To generate a stable transgenic parasite, C. parvum sporozoites are transfected and immediately used to infect mice. Transfected sporozoites are no longer protected by the oocyst wall and show reduced capacity to orally infect mice when compared to oocysts, which may be due to passage through the acidic stomach. Therefore, we surgically inject transfected sporozoites directly into the small intestine —the natural site of infection. Note that surgery is only required in this first passage following transfection. Subsequent passage of established transgenic parasites is achieved by simple oral infection of mice with oocysts.
The protocols described below were approved by the Institutional Animal Care and Use Committee at the University of Georgia. These protocols represent best practices for animal handling to minimize animal discomfort and to promote well-being and were developed in continued discussion with local veterinarians and animal technicians. All animal procedures require institutional approval and oversight and the protocols described may require modification to comply with local rules and regulations. Working with animals requires specialized training on animal handling and use typically provided by the local animal facilities; while the surgery procedure used here is straightforward, personnel performing this procedure may wish to seek additional hands on training from the local veterinary staff.
Materials
4 female interferon-gamma knockout mice per each desired strain (B6.129S7-Ifngtm1Ts/J; JAX 002287) Mice should be visually examined for good health post transport and allowed to acclimate to new environment and food for 7 days prior to antibiotic pre-treatment.
Ampicilin
Streptomycin
Vancomycin
Mouse Antibiotic Pre-Treatment Solution (see recipe)
Paromomycin sulfate salt (Sigma Aldrich catalog #P5057, or similar)
Microcentrifuge tube storage boxes (1 per cage)
Blue food coloring dye
Sterile PBS
Ophthalmic ointment
Betadine surgical scrub, 7.5% povidone-iodine
Sterile draping
70% ethanol
Surgical latex gloves (fresh pair for each mouse)
Sterile gauze pads
Permanent markers (black, red, green)
1ml tuberculin syringe, 27G detachable needle (Covidien, catalog #8881501368)
PDS*II (polydioxanone) suture, 4–0 violet monofilament (Ethicon, catalog #Z304H)
VetBond tissue adhesive (3M, catalog #1469SB)
Meloxicam 5 mg/ml solution for injection (Eloxiject, Henry Schein Animal Health, catalog #049755)
DietGel® Boost, a purified high calorie dietary supplement (Clear H2O; catalog #72-04-5022)
Surgical instrument cleaner solution (Roboz catalog #IC-1000)
Phillips Norelco GoStyler precision trimmer (Style #NT9145)
Isoflurane anaesthesia set up (induction box, nose cone, oxygen tank, and isoflurane scavenging system)
Far infrared warming pad
Surgical grade scissors and blunt forceps (we generally use one set per cage and sterilize using a hot glass bead sterilizer between animals)
Sterilization packs for autoclaving surgical tools
Hot glass bead sterilizer
One week before surgery:
-
1
A week before surgery, the regular drinking water is exchanged for Mouse Antibiotic Pre-Treatment Solution. We find that removing the intestinal bacterial flora significantly increases the susceptibility of mice to C. parvum infection. We recommend this treatment for surgical infections, or for strains and mutants that show reduced infectivity.
One day before surgery:
-
2
Exchange drinking water containing antibiotic pretreatment solution with regular, sterile water.
Mice are given regular water 24 hours before surgery and until the first day post-surgery.
-
3
Shave surgical area of upper abdomen of mice, just below sternum to mid-abdomen.
Doing this the day before surgery reduces the handling of mice prior to surgery and reduces their stress.
-
4
Prepare an empty “fecal collection cage” dedicated for each C. parvum strain. Cage should contain 2–3 food pellets and a mouse house, but should be devoid of absorbent burrowing material.
-
5
Prepare boxes to store collected fecal material collected in microcentrifuge tubes, and place box at 4 °C.
-
6
Prepare 3–6% hydrogen peroxide solution used to decontaminate surfaces used in transfection and surgery.
-
7
Surgical tools should be treated in a dry sterilizer or autoclaved in surgical packs.
Day of surgery:
Preparation and electroporation of sporozoites is carried out as previously described in Basic Protocol 2 (Transfection of Cryptosporidium). We refer to C. parvum carrying a single, specific stable transgene or mutation as a “strain.” To increase the likelihood of recovering the desired strain of Cryptosporidium, we prepare enough transfected sporozoites to infect a cage of four mice. Each mouse is infected with 1×107 sporozoites; we recommend preparing slightly more—5×107 transfected sporozoites—per desired strain to account for loss during the excystation process.
See Alternative Protocol 3 for transfection of 5×107 sporozoites using large 4D Nucleofector cuvettes.
Preparation of transfected sporozoites:
-
8
Dilute 5×107 transfected sporozoites to 500 μl final volume using PBS containing sterile blue coloring food dye. The dye must be sterile as some can drip or leak out into peritoneum from needle or intestine.
The dye helps visualize the sample during injection.
-
9
Place the parasites on ice and take directly to the surgical facility Plan electroporation to minimize time from excystation to infection to avoid loss of viability of parasites.
Parasites should be kept on ice during the time between transfection and surgical injection. We try to minimize this time to less than 2 hours. Depending on personnel and access to surgical equipment, three cages of mice (12 mice total) is a reasonable number of mice to infect in a single day.
Immediately before surgery:
Visually examine mice to ensure health (weight, appearance, respiration, etc) of each animal prior to surgery. Prepare a sterile surgical field first by placing a warming pad in the center and cover with a sterile draping. Place surgical tools, hot glass bead sterilizer, suture material, antiseptics, and sharps disposal container within close reach. If possible, the surgical area should be set up inside a biosafety cabinet. Sterile gloves, hair restraint, surgical mask, and a gown should be worn throughout surgery and monitoring period. Traffic flow should be minimized and procedure should be performed under aseptic technique with change of gloves as needed. Working in a team of two can increase efficiency and enhance monitoring of animals pre- and post-surgery. Typically, one person performs the surgery while an assistant prepares needles for injection, fills out surgery log and other records, transfers animals in and out of the surgical field, and monitors mice pre- and post-surgery.
Instructions described for a surgical procedure on a single animal from start to finish:
Typically we can complete a single surgery in approximately 15 minutes/mouse. Therefore, surgery for a cage of four mice can be completed in approximately an hour.
-
10
Place mouse in isoflurane (3–5%) anesthesia induction chamber with a 2L/minute oxygen flow rate and monitor until non-responsive to toe pinch. This may take up to 5 minutes depending on the size of the chamber.
-
11
Once the mouse is non-responsive, place mouse to the sterile surgical field, lying on its back with its head in the nosecone (1–3% isoflurane as needed), and tail closest to you. Isoflurane should be diverted from the anesthesia induction chamber to the nose cone. Ophthalmic ointment should be applied as per manufacturer instructions to prevent drying of eyes.
Respiration and response to stimulation (toe pinch or touch to medial corner of eye) should be monitored during procedure and vaporizer adjusted as needed (i.e. increase level of anesthetic if mouse is responsive to stimulation and wait to continue procedure until the mouse is no longer responsive). Mucous membranes and footpads should remain a normal color indicating that the animal’s perfusion is adequate.
-
12
The surgeon should clean hands and wrists by scrubbing for 3 minutes with povidone iodine or chlorhexidine (or other suitable disinfectant) and then rinsing with water and drying with a clean towel.
-
13
Wash the shaved area of the mouse’s abdomen three times with betadine and once with 70% ethanol (apply using sterile gauze pads).
-
14
Color the portion of the mouse’s tail (near the body) with a permanent marker to allow for identification of individual mice.
As an alternative to tattooing, we use tail colors to identify individual mice. We use permanent markers to mark the tails of three mice (black, red, and green work well) and leave one unmarked. Recoloring every third day may be necessary.
-
15
Replace gloves with new pair of sterile gloves.
During this time, the assistant may load the needle with the sample of Cryptosporidium and set to the side safely in preparation for injection.
-
16. Using sterile surgical instruments (directly removed from packaging or from hot bead sterilizer) incise skin vertically for no more than 1.5 cm in length (Figure 4A–C). The incision should be made midline in the abdominal region below the sternum with microsurgical scissors.
Make the incision as small as possible. A small incision, we can usually close with a single suture. This improves the time and ease of the healing process.
-
17
Gently, using blunt forceps, expose a small loop of the ileum of the small intestine (Figure 4F–G).
-
18
Inject 100 μl of transfected Cryptosporidium prepared in PBS with sterile blue food coloring (Figure 4H). Inject slowly and allow to absorb into the lumen of the intestine.
-
19
Gently replace the loop of the ileum back into the abdominal cavity.
-
20
Suture the peritoneum closed with the PDS (Figure 4I–K).
-
21
Suture the skin closed with the PDS. Make two tight knots as close to the skin as possible (Figure 4L). The quality of the sutures has significant impact on the healing of the incision. Therefore, we suggest that all scientists performing this survival surgery contact a local animal surgical technician or veterinarian for training. A strong suture that closes the incision without irritation is paramount to the animal’s health and the success of the experiment.
-
22
Turn off the vaporizer and allow the mouse to breathe the oxygen supply gas until it begins to wake. Remove the mouse from the surgical area to recovery area (clean cage) with thermal support until ambulatory and exhibiting normal respiration. If there are obvious problems with full recovery such as continued unconsciousness, inability to maintain normal body position, or abnormal physiological function (e.g. breathing), then the mouse should be euthanized. If the mouse resumes normal activity, proceed and repeat the surgical procedure for the next animal.
Wipe surgical instruments clean with sterile saline to remove blood and tissue particles and place in a hot bead sterilizer between animals. If the instruments become contaminated by contact with a non-sterile surface or non-sterile portions of the body (such as fur) use a new sterile set of instruments on subsequent animals.
-
23
Four mice per strain should be infected with transfected sporozoites to increase likelihood of obtaining the desired strain.
Figure 4. Surgical technique for direct infection of the small intestine.

Transfected sporozoites are injected directly into the small intestine of IFN-γ KO mice. A-C) Once the anesthetized mouse is non-responsive, make a small incision in the skin and the peritoneum. E-G). Using blunt forceps, pull a small loop of the small intestine out of the abdominal cavity. Slowly inject solution containing transfected parasites and sterile blue food coloring into the lumen of the small intestine and allow to absorb. Gently replace the small intestine back into the cavity. I-K) Suture the opening of the peritoneum closed. L) Suture the skin closed using two knots.
Monitoring animal health after surgery:
-
24
Immediately after surgery and prior to recovery, meloxicam (1.0 mg/kg subcutaneous) is administered once to minimize post-operative pain.
-
25
Throughout the surgery session, the assistant should document the activity of recovering mice every 15 minutes as they regain consciousness, move purposefully, right themselves, and maintain balance and do not exhibit any adverse effects from the surgery. Mice usually generally regain consciousness within 5 minutes and exhibit mobility in less than half an hour.
-
26
Monitor surgical site and overall animal health 2 hours post-surgery. At this time we apply VetBond to the skin to further protect the surgery site. Should peritoneal sutures be damaged, mice must be anesthetized again to replace suture, as VetBond is not indicated for internal use.
Paromomycin selection (1–30 days post infection):
-
28
Prepare ~200 ml paromomycin (in ultra-pure water at 16 g/L) per cage, and replace regular drinking water. Excess paromomycin solution may be stored at 4 °C for up to 2 weeks.
-
29
Continue to monitor mice daily until the incision is completely healed and sutures are resorbed or removed (7–10 days).
-
30
Collect fecal material from infected mice every three days, starting six days post infection.
We have found transferring mice from their usual cage to a “fecal collection cage” (see Materials) for 1–4 hours encourages them to defecate. We combine all the fecal material from a cage into a single microcentrifuge tube and store at 4 °C. Fecal collection every three days is sufficient for most experiments; however fecal samples may be collected more often if desired.
ALTERNATE PROTOCOL 3: ORAL INFECTION OF MICE WITH CRYPTOSPORIDIUM OOCYSTS
Transgenic parasites emerging from selection may be used to infect a second cage of mice in order to amplify their numbers. This may be done by purifying oocysts from fecal material followed by gavage of uninfected mice with 1,000–100,000 oocysts. A lower dose is recommended for younger mice (6–7 weeks old) or for strains of Cryptosporidium adapted to mice that show increased virulence. We found infection levels (as measured by Nanoluciferase activity in the fecal material) to vary due to mouse strain and source, age, food, microbiota, etc. Monitor infection closely and proceed according to the measurements obtained. In practice, it is best to dose at lower numbers of oocysts and incrementally increase as needed to generate a robust infection (up to 100,000 transgenic oocysts).
Materials
4–6 female mice: IFN-γ KO (Jackson Laboratory Catalog # 002287) or NOD scid gamma (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; Jackson Laboratory Catalog #005557), depending on virulence of the Cryptosporidium strain)
sterile gavage needles (see Table 1 below)
Table 1.
Gavage needle specifications based on mouse size.
| Weight of mouse (grams) | Gauge (G) | Length of gavage need (inches) | Ball Diameter (mm) | Shape |
|---|---|---|---|---|
| <14 | 24 | 1 | 1.25 | Straight |
| 15 – 20 | 22 | 1, 1.5 | 1.25 | Straight |
| 20 – 25 | 20 | 1, 1.5, 3 | 2.25 | Straight, Curved |
| 25 – 30 | 18 | 1, 1.5, 2 | 2.25 | Straight, Curved |
| 30 – 35 | 18 | 2, 3 | 2.25 | Straight, Curved |
Cryptosporidium oocysts diluted in PBS
Oocysts should be prepared in PBS at a dose of 1,000–100,000 oocysts per mouse. Gavage volumes should not exceed the maximum volume (10ml/kg of body weight =0.1ml/10g of body weight), and are typically 100–200 μl per mouse.
Before using gavage to infect mice, seek training in proper technique. Also note that appropriately sized and type of gavage needles (Table 1) must be selected. Gavage needles can either be stainless steel and reusable or disposable and made of flexible plastic. Stainless steel needles are more durable against chewing, but plastic needles are less likely to cause injury. Stainless steel needles can be straight or curved and plastic needles bend and flex to follow the esophagus of mice. (A curved needle is often easier to use and less traumatic as are needles with barrel vs ball.)
Load gavage need with the dose of oocysts needed for a single mouse.
Measure the mouse from the mouth to the last rib. This length is the appropriate needle length to reach the stomach. Do not pass the needle beyond this point to avoid injury.
Gently scruff the animal so that the forelegs are extended outward and away from the body and the head is pulled back creating a straight line from the neck through the esophagus. Hold vertically.
Gently insert the gavage needle into the mouth behind the front teeth and allow needle to pass downward as if falling by gravity, do not force. If resistance is encountered, do not push the needle, but rather pull the gavage needle out slowly and try again.
Once placed, inject slowly and avoid air bubbles. Then pull straight out gently and steadily.
Repeat steps 1–5 for each additional mouse. Gavage needle may be reused for infection of mice with the same transgenic strain of Cryptosporidium.
BASIC PROTOCOL 4: NANOLUCIFERASE ASSAY TO MEASURE INFECTION IN IN VITRO CULTURES
The Nanoluciferase reporter is ATP-independent, with glow-type luminescence, emits in the blue-light spectra, and is highly sensitive (Hall et al., 2012). It is the only reporter to date that we can detect in transient transfection assays. Nanoluciferase is also stable in unpurified fecal material, meaning fecal samples are used directly to determine infection levels in mice. Nanoluciferase activity, as a measurement of oocysts in the feces, is comparable in pattern and amplitude to infection levels as determined by qPCR (Mary et al., 2013), but is faster and requires less fecal material. We have observed variation in expression of Nanoluciferase dependent on the targeted locus. Therefore, the specific relationship between Nanoluciferase activity and oocyst number has to be determined empirically for each strain.
Materials
NanoGlo Luciferase Assay Kit (Promega, catalog #N1110)
CoStar Assay Plate, 96 well, no lid, round bottom white polyprolyene (catalog #3355)
Plate reader that measures luminescence (e.g. Promega GloMax)
To be carried out in a biosafety cabinet with the light off:
Combine NanoGlo substrate and lysis buffer immediately before use at a ratio of 1:50, substrate to lysis buffer. Prepare enough for 200 μl per well and keep protected from light.
Remove media from each well and replace with 200 μl NanoGlo mixture.
Using a pipette tip, scratch the surface of each well to dislodge and lyse HCT-8 cells. After 20 seconds of agitation, transfer volume to opaque, white 96-well plate.
Repeat Step 3 for all samples.
Measure luminescence using a plate reader. For Promega GloMax we use quick luminescence protocol with an integration time of 0.3 seconds. Activity degrades over time, so measure immediately upon mixing sample and substrate.
ALTERNATE PROTOCOL 4: NANOLUCIFERASE ASSAY TO MEASURE INFECTION IN FECAL SAMPLES
Materials
Microcentrifuge tubes
3 mm glass beads (Fisher Scientific catalog #11-312A)
Fecal Lysis Buffer (see recipe)
thin metal or wooden spatula (one per fecal sample)
Using a thin metal spatula, mash and mix the fecal sample.
-
Weigh out 20 mg fecal material and transfer to new microcentrifuge tube.
We have found 20 mg to produce strong reading while consuming the least amount of the sample and reagent. Using a set mass allows for comparison of NanoLuc activity over time.
Add 10–15 glass beads and 1 ml Fecal Lysis Buffer.
Repeat steps 1 through 3 for each sample using a new metal or wooden spatula.
- Incubate microcentrifuge tubes (containing 20 mg fecal material, glass beads, and Fecal Lysis Buffer) for 20–30 minutes at 4 °C. This softens the sample but will not reduce Nluc activity.
- During the last five minutes of the incubation time, prepare the substrate.
- Combine NanoGlo substrate and lysis buffer (from the NanoGlo kit) at a ratio of 1:50, substrate to lysis buffer. Prepare enough for 300 μl per fecal sample and keep protected from light.
Vortex each microcentrifuge tube until there are no solid pieces visible (usually 1 minute is sufficient).
Centrifuge for 1 minute at 16,000 xg to pellet debris and improve ease of pipetting.
In a white, opaque CoStar 96-well plate, load 100 μl NanoGlo substrate (prepared as described in Step 5b) to each well. To reduce spillover luminescence between samples, load every other well.
Each fecal sample is measured in triplicate. Transfer 100 μl of each fecal sample to three corresponding wells.
Measure luminescence using a plate reader. For the Promega GloMax reader we use the quick luminescence protocol with an integration time of 0.3 seconds. Activity degrades over time, so measure immediately upon mixing sample and substrate.
SUPPORTING PROTOCOL 4: PCR SCREEN OF FECAL DNA CONTAINING TRANSGENIC CRYPTOSPORIDIUM TO MAP GENETIC MODIFICATION
Once fecal samples register significant Nluc activity (at least five times above background), DNA may be extracted to map the target locus in a PCR screen to confirm successful genetic modification. We use the ZR Fecal DNA MicroPrep kit (Zymo Research). In addition to the kit’s instructions, we recommend five cycles of freeze and thaws before processing to disrupt oocysts.
Materials
Zymo Fecal DNA MicroPrep Kit (Zymo Research catalog #D6012)
Liquid Nitrogen
Fecal DNA (including an unmodified wild type control sample)
Upstream Forward Primer
Internal Reverse Primer
Internal Forward Primer
Downstream Reverse Primer
Low-Fidelity DNA polymerase master mix for PCR (Promega GoTaq)
Thermocycler
DNA Gel Electrophoresis Apparatus
DNA Extraction
Fill a rubber ice bucket with 1 inch of liquid nitrogen. Keep covered.
Set a heat block to 100 °C.
Weigh out 50–100 mg fecal material and transfer to bashing bead tube. Prepare one bashing bead tube for each sample.
Add 750 μl Lysis Soution (from Zymo kit) to each sample. Vortex each microcentrifuge tube until there are no solid pieces (usually 1 minute is sufficient).
Place bashing bead tubes in liquid nitrogen until the sample is frozen (approximately 15 seconds).
Place bashing bead tubes in heat block until fully melted (approximately 2 minutes).
Repeat Steps 5 and 6 for a total of five freeze-thaw cycles.
Continue with ZR Fecal DNA MicroPrep instructions.
PCR Screening of fecal DNA to confirm transgenic mutants
Perform PCR with primers designed to detect 5’ and 3’ integration. Additional primers to demonstrate loss of a gene or as a general positive control may also be used. See Support Protocol 2A for an example.
BASIC PROTOCOL 5: PURIFICATION OF OOCYSTS FROM FECAL MATERIAL
Collect fecal material containing your transgenic Cryptosporidium for several days, and proceed to oocyst isolation (Upton, 1997). Oocysts are physically separated from fecal material (food particles, other microorganisms, etc.) by extensive washings followed by two floatations. Because this procedure involves large quantities of infectious oocysts, pay particular attention to biosafety, lab coats and eye goggles must be worn, and gloves should be changed frequently. A specific area of the laboratory and specific equipment (e.g. centrifuges) dedicated to this procedure may further minimize exposure and cross contamination risk.
To avoid excystation of oocysts, all equipment, solutions, and samples should be maintained at 4°C or on ice. We use several ice buckets or fill a large autoclave tray with ice to hold all samples and solutions. All liquids generated from oocyst purification, including the ice used to chill samples, must be autoclaved before disposal.
Materials
Ice-cold tap water
Conical tubes (sized 15 ml; 50 ml; and 500 ml Corning catalog #431123)
Cell scraper, length 300 mm, blade 20 mm (TPP catalog #99003)
Disposable hemocytometer (Kova Glasstic Slide 10 with Grids; catalog #87144)
Ice-cold Sucrose Floatation Solution (see recipe)
Ice-cold 0.85% NaCl (see recipe)
Ice-cold 1.25 M cesium chloride
2.5% potassium dichromate, prepared in ultra-pure water
PBS with antibiotics (1X Pen/Strep)
Microcentrifuge at 4 °C
Large centrifuge 4 °C
Cole Parmer LabGEN 125 Homogenizer with autoclavable Omni Tip plastic tip generator probes
250 and 850 μm mesh filters and PVC fittings for mesh filters (Bel-Art 378451000 Mini-Sieve Micro Sieve Set)
small plastic funnels
Large autoclave tray, or several buckets filled with ice
10 L carboy for autoclaving liquid waste
-
To a 50 ml conical, add approximately 35 ml cold water and fecal material collected from several days post infection.
Typically, we use up to a month of fecal collections (3–10 microcentrifuge tube tubes’ worth of fecal material.
Attach a sterile, plastic tip generator probe to an upright, immersion blender (LabGEN 125).
While the blender is off, submerge homogenizer in contents of conical tube.
Blend sample on medium speed until all clumps are broken and a slurry texture is achieved. This may take up to five minutes. Take care to avoid aerosolizing the sample. Place sample on ice and carefully dismantle the blender.
Assemble the filter apparatus: first insert a 850 μm mesh filter into PVC fitting; place this in the wide side of a funnel, then place the entire apparatus over a 500 ml conical on ice.
Slowly pour the homogenized sample over the mesh filter, using a cell scraper to assist the sample to drain through the filter into the conical below.
Add approximately 35 ml of cold tap water to the now empty 50 ml conical. Use the cell scraper to gather the solid pieces on top of the mesh, and transfer them back into the conical tube. Shake vigorously.
Repeat Steps 6 and 7 for a total of 5–6 times.
Remove the mesh filter from the apparatus and replace with a 250 μm mesh filter. We find it helpful to use the cell scraper to push the filter out of the PVC fitting from the underside. Reassemble the filter apparatus.
Slowly pour the homogenized sample over the mesh filter, using a cell scraper to assist the sample to drain through the filter into the conical below.
Add approximately 35 ml of cold tap water to the now empty 50 ml conical. Use the cell scraper to gather the solid pieces on top of the mesh, and transfer them back into the conical tube. Shake vigorously.
Repeat Steps 10 and 11 for a total of 3–4 times.
Disassemble filter apparatus. Take care to place all components in an autoclavable bin for decontamination at the end of this procedure.
Centrifuge 500 ml conical for 10 minutes at 1000×g at 4 °C.
Remove supernatant by carefully decanting into 10 L carboy. Resuspend pellet in 50mL of cold tap water and transfer to a 50 ml conical.
-
Centrifuge sample for 10 minutes at 1000×g at 4 °C.
During this spin, remove 1.33 specific gravity sucrose from 4 °C and place on a stir plate. Stir until immediately before use. Sucrose is heavy and will settle over time, disrupting the specific gravity.
Decant supernatant into liquid waste container. Resuspend pellet in 50 ml cold tap water. Split sample into two 50 ml conical tubes, 25 ml in each.
Add 25mL Sucrose Floatation Solution (1.33 specific gravity) to each conical and mix gently by inverting several times.
Immediately centrifuge for 5 minutes at 1000×g at 4 °C.
After this spin, oocysts are floating in the sucrose solution. Carefully decant entire supernatant from both conicals into a new 500mL conical to collect oocysts. Add 300 ml cold tap water to the 500 ml conical (should be 400 ml total).
Centrifuge 15 minutes at 1500×g at 4 °C.
- Decant supernatant into liquid waste container.
-
At this point, oocysts (in pellet) can be resuspended in PBS with antibiotics and stored overnight at 4 °C. Protocol can be continued the following day.-OR-
- Oocysts (in pellet) are resuspended in 5 ml 0.85% NaCl. It is important to keep this volume as close to 5 ml as possible.
-
Add 0.8 ml cold 1.25 M CsCl2 solution to 10 microcentrifuge tube tubes.
Slowly overlay 0.5 ml oocysts from Step 22 (resuspended in 0.85% NaCl) on top of CsCl2 solution in each microcentrifuge tube. Dispense slowly so that two distinct layers are created. Do not disrupt microcentrifuge tubes or allow solutions to mix! Oocysts are floated for a second time during this step and separation with CsCl2 layer is critical.
Centrifuge for 3 minutes at 16,000×g at 4 °C.
Remove the top 1 ml of the solution from each microcentrifuge tube and transfer to a new microcentrifuge tube. Oocysts are floating in the supernatant and bacterial contamination and debris will be pelleted. Discard original microcentrifuge tubes with pelleted debris.
To microcentrifuge tubes containing floating oocysts, add 0.5 ml 0.85% NaCl.
Centrifuge for 3 minutes at 16,000×g at 4 °C.
-
Oocysts are now contained in the pelleted material. Discard the supernatant.
Check with your local safety department to see if cesium chloride needs its own separate waste collection container.
Use 1 ml 0.85% NaCl to resuspend the pellets from each microcentrifuge tube and combine into a single microcentrifuge tube tube.
Centrifuge for 3 minutes at 16,000×g at 4 °C.
Discard supernatant and resuspend in 1 ml PBS with antibiotics or in 2.5% potassium dichromate (prepared in ultra-pure water).
Remove an aliquot of the purified oocysts, dilute 1:10 in PBS and count on a disposable hemacytometer.
Autoclave all metal and plastic equipment to decontaminate (PVC fittings, metal mesh filters, etc), liquid waste, and contents of ice buckets used to chill solutions and samples.
REAGENTS AND SOLUTIONS
HCT-8 Media: RPMI with glutamine, supplemented at a final concentration of 10% heat-inactivated FBS, 1 mM sodium pyruvate, 1X Pen/Strep, and 1X fungizone. Store at 4 °C for up to 2 months. Warm in 37°C water bath directly before use.
Cryptosporidium Infection Media: DMEM supplemented at a final concentration of 2% heat-inactivated FBS, 0.2 mM L-glutamine, 1× Pen/Strep, and 1× fungizone. Store at 4 °C for up to 2 months. Warm in 37 °C water bath directly before use.
1:4 bleach solution: 100 μl Clorox bleach mixed with 300 μl deionized or milli-Q water.
1X Cytomix (prepared with ultra-pure water at a final pH 7.6): 120mM KCl; 0.15mM CaCl2; 10mM K2HPO4/KH2PO4, pH 7.6; 25mM HEPES, pH 7.6; 2mM EGTA; 5mM MgCl2. Store 1X Cytomix at 4 °C for up to 4 months. On day of use, prepare 10 ml “Complete Cytomix” by supplementing 1X Cytomix with 2mM ATP and 5mM glutathione (final concentrations). Make Complete Cytomix fresh immediately before use, keep on ice during protocol, and use only once.
Mouse Antibiotic Pre-Treatment Solution: 1 mg/ml ampicillin, 1 mg/ml streptomycin, 0.5 mg/ml vancomycin final concentrations prepared in ultra-pure water. Store at 4 °C for up to 1 month.
Fecal Lysis Buffer: 50 mM Tris pH 7.6, 2 mM DTT, 2 mM EDTA pH 8.0, 10 % glycerol, 1% Triton X-100 (final concentrations) prepared in water. Store at 4 °C (make 100 ml at a time).
Sucrose Floatation Solution (1.33 specific gravity): 756 g sucrose dissolved in 483 mL deionized water will make approximately 1 liter (3 mL phenol optional). Sucrose at this concentration may take up to 2 hours to dissolve; we suggest this solution be prepared at least a day ahead. Store at 4 °C for up to 6 weeks.
0.85% NaCl: 0.85 g sodium chloride in 100 ml deionized water. Store at 4 °C for up to 2 months.
COMMENTARY
Diarrheal diseases are the cause of 9% of overall mortality in children under the age of five worldwide (Liu et al., 2015). Cryptosporidium is the second leading cause of diarrheal disease in children (Kotloff et al., 2013); the disease is protracted and life threatening in particular among malnourished children (Guerrant et al., 1999). Cryptosporidiosis is also of great concern in immunocompromised individuals and was one of the early-recognized AIDS defining opportunistic infections (Clifford et al., 1990; Manabe et al., 1998). There are no vaccines available, and nitazoxanide the only drug approved to treat cryptosporidiosis is not effective in immunocompromised patients and has diminished activity in malnourished children—the groups that need drug therapy the most (Amadi et al., 2002; Rossignol, Kabil, el-Gohary, & Younis, 2006). Compared to the well-studied apicomplexan parasites Plasmodium and Toxoplasma, we know little about Cryptosporidium. This lack of knowledge extends from fundamental questions about the biology of the parasite and the disease to translational insights into how to design and test effective treatments and vaccines (Checkley et al., 2015). This in part is due to the significant technical challenges of working with the parasite that have stifled progress. There is currently no system to continuously propagate Cryptosporidium in tissue culture; therefore Cryptosporidium must be maintained in infected animals. Work with animals largely relies on newborn large animal models (calves and gnotobiotic piglets) or immunosuppressed mice. Long-term maintenance of parasite strains is complicated by the lack of freezing protocols (Fayer, Nerad, Rall, Lindsay, & Blagburn, 1991). These hurdles have conspired to keep this parasite genetically intractable and effectively cut off cryptosporidiosis research from the molecular biology that revolutionized parasitology over the last 25 years (Striepen, 2013).
Critical Parameters
Vigor of parasites is paramount at each step in the process of generating and maintaining a stable transgenic strain. Oocysts have a “shelf life” and excystation, transfection, and mouse infection rates will significantly decrease beginning two months after initial isolation. It is critical that oocysts are stored and handled properly (4 °C or on ice) as an increase in temperature is a trigger for natural excystation. Once sporozoites are free from the oocyst, they have a narrow window of time in which they are infective, generally less than 6 hours. Therefore, proper handling of oocysts and a consideration of timing are critical to both in vitro and in vivo infections.
Trouble Shooting
It is important to remember that if the gene or genomic region being targeted is essential in any stage of the Cryptosporidium life cycle, the transgenic will not be recovered. We suggest that you gain proficiency with individual protocols i.e. transient transfection, mouse surgery, etc using a positive control like the thymidine kinase locus. While most protocols described herein are robust (immunofluorescence microscopy slide preparation, DNA cloning, Nluc assays, oocyst purification), transfection and mouse infection may require optimization.
Common reasons for transfection failure are:
DNA is not concentrated enough. Cryptosporidium sporozoites are resuspended in transfection buffer and then combined with DNA. If the DNA is not concentrated enough, upon addition to sporozoites, the transfection buffer becomes diluted and the electrical pulse can no longer be conducted through the sample.
Incorrect number of sporozoites used for transfection. Because the transfection efficiency is so low, a significant number of parasites are required in order to guarantee transfection. Additionally, transfection parameters are optimized for a specific number of organisms. Even if all other conditions are correct, we have found using an incorrect number of organisms to cause the transfection to fail.
Possible reasons for inability to recover transgenic:
This may happen if your transfection was unsuccessful. If the 4D Nucleofector fails to give your sample a “green” success code after delivering the pulse, it is very unlikely that the transfection was successful, and we do not recommend using this sample to infect mice. We suggest that you perform a transient transfection assay with a verified positive control using the same materials intended for generating a stable transgenic (i.e. same batch of oocysts, same transfection buffer stock, etc).
It is possible the gene is essential or cannot be fused to an epitope tag. Therefore, you may be unable to recover the desired strain.
The targeting strategy may have caused disruption of the expression of an adjacent gene. If possible, create a new strategy by identifying a new guide RNA sequence and new homology sequences and minimize alterations to the genome.
Inability to infect subsequent cages of mice with transgenic:
The dose required to infect the following cage of mice may be too low. Consider infecting several cages at different doses per animal to optimize infection dose.
Disruption of your gene of interest may result in decreased parasite viability. To confirm such a defect, in vitro invasion and growth should be compared to wild type Cryptosporidium. If the strain produces only weak infections, you may need to pre-treat mice with antibiotics for a week before infecting (as described in Basic Protocol 3: Surgical Infection of Mice with Transfected Sporozoites) or infect at a higher dose.
What to do if mice become overly susceptible to Cryptosporidium infection:
Consider lowering the infection dose. With more virulent strains, 1,000 – 10,000 oocysts/mouse are often enough to generate a robust infection.
Older mice are less susceptible to Cryptosporidium infection. Consider infecting mice aged 8–10 weeks with virulent strains.
Compared to IFN-γ KO mice, NOD scid gamma mice become infected at lower levels but maintain a chronic infection. Consider passaging more virulent strains through NOD scid gamma, especially after 2–3 passages in IFN-γ KO mice.
Anticipated Results
For an example of the process and results involved generating a stable transgenic C. parvum strain, see Support Protocol 2A.
Time Considerations
Typically, isolation of a transgenic strain takes a month; initial efforts may require additional planning, experimentation and optimization. Before beginning animal experiments, approval from local institutional animal care and use committees should be obtained; and before beginning C. parvum transgenesis experiments, institutional offices of biosafety should be consulted. Designing a strategy to target a gene of interest and preparing the necessary DNA constructs takes anywhere from one week to a month depending on molecular biology experience. Transfection of C. parvum sporozoites and surgical infection of mice must be completed in a single day. Finally, fecal samples are collected regularly and assayed for up to a month after the surgical procedure is performed. Because of the significant amount of time required and the involved nature of each step, we suggest that groups start by reproducing the thymidine kinase knockout strain. We suggest readers to read an entire section for more detailed time considerations before beginning an experiment.
Acknowledgments
Work on the genetic analysis and modification of Cryptosporidium was supported by multiple grants from the National Institutes of Health (5R01AI112427-03), the Bill and Melinda Gates Foundation, the Wellcome Trust, and the UGARF-CDC collaborative program to BS. MCP is supported by training grant NIH T32AI060546 and AS is supported by NIH fellowship F32AI124518. BS is a Georgia Research Alliance Distinguished Investigator.
Footnotes
LITERATURE CITED
- Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Kapur V. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304(5669):441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- Amadi B, Mwiya M, Musuku J, Watuka A, Sianongo S, Ayoub A, Kelly P. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet. 2002;360(9343):1375–1380. doi: 10.1016/S0140-6736(02)11401-2. doi:S0140-6736(02)11401-2 [pii] [DOI] [PubMed] [Google Scholar]
- Brooks CF, Johnsen H, van Dooren GG, Muthalagi M, Lin SS, Bohne W, Striepen B. The toxoplasma apicoplast phosphate translocator links cytosolic and apicoplast metabolism and is essential for parasite survival. Cell Host Microbe. 2010;7(1):62–73. doi: 10.1016/j.chom.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushkin GG, Motari E, Carpentieri A, Dubey JP, Costello CE, Robbins PW, Samuelson J. Evidence for a structural role for acid-fast lipids in oocyst walls of Cryptosporidium, Toxoplasma, and Eimeria. MBio. 2013;4(5):e00387–00313. doi: 10.1128/mBio.00387-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley W, White AC, Jr, Jaganath D, Arrowood MJ, Chalmers RM, Chen XM, Houpt ER. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect Dis. 2015;15(1):85–94. doi: 10.1016/S1473-3099(14)70772-8. doi:S1473-3099(14)70772-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford CP, Crook DW, Conlon CP, Fraise AP, Day DG, Peto TE. Impact of waterborne outbreak of cryptosporidiosis on AIDS and renal transplant patients. Lancet. 1990;335(8703):1455–1456. doi: 10.1016/0140-6736(90)91478-s. doi: 0140-6736(90)91478-S [pii] [DOI] [PubMed] [Google Scholar]
- DeCicco RePass MA, Chen Y, Lin Y, Zhou W, Kaplan DL, Ward HD. A Novel Bioengineered 3D Human Intestinal Model for Long-Term Infection of Cryptosporidium parvum. Infect Immun. 2017 doi: 10.1128/IAI.00731-16. doi:IAI.00731-16 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayer R, Nerad T, Rall W, Lindsay DS, Blagburn BL. Studies on cryopreservation of Cryptosporidium parvum. J Parasitol. 1991;77(3):357–361. [PubMed] [Google Scholar]
- Fayer R, S CA, Dubey JP. The general biology of Cryptosporidium. Boca Rotaon, Florida: CRC Press Inc; 1997. [Google Scholar]
- Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot Cell. 2009;8(4):520–529. doi: 10.1128/EC.00357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths JK, Theodos C, Paris M, Tzipori S. The gamma interferon gene knockout mouse: a highly sensitive model for evaluation of therapeutic agents against Cryptosporidium parvum. J Clin Microbiol. 1998;36(9):2503–2508. doi: 10.1128/jcm.36.9.2503-2508.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueiros-Filho FJ, Beverley SM. On the introduction of genetically modified Leishmania outside the laboratory. Exp Parasitol. 1994;78(4):425–428. doi: 10.1006/expr.1994.1048. [DOI] [PubMed] [Google Scholar]
- Guerrant DI, Moore SR, Lima AA, Patrick PD, Schorling JB, Guerrant RL. Association of early childhood diarrhea and cryptosporidiosis with impaired physical fitness and cognitive function four-seven years later in a poor urban community in northeast Brazil. Am J Trop Med Hyg. 1999;61(5):707–713. doi: 10.4269/ajtmh.1999.61.707. [DOI] [PubMed] [Google Scholar]
- Gut J, Nelson RG. Cryptosporidium parvum: synchronized excystation in vitro and evaluation of sporozoite infectivity with a new lectin-based assay. J Eukaryot Microbiol. 1999;46(5):56S–57S. [PubMed] [Google Scholar]
- Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Wood KV. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7(11):1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harb OS, Roos DS. The Eukaryotic Pathogen Databases: a functional genomic resource integrating data from human and veterinary parasites. Methods Mol Biol. 2015;1201:1–18. doi: 10.1007/978-1-4939-1438-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh MH, Carruthers VB. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot Cell. 2009;8(4):530–539. doi: 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korich DG, Mead JR, Madore MS, Sinclair NA, Sterling CR. Effects of ozone, chlorine dioxide, chlorine, and monochloramine on Cryptosporidium parvum oocyst viability. Appl Environ Microbiol. 1990;56(5):1423–1428. doi: 10.1128/aem.56.5.1423-1428.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Levine MM. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382(9888):209–222. doi: 10.1016/S0140-6736(13)60844-2. doi:S0140-6736(13)60844-2 [pii] [DOI] [PubMed] [Google Scholar]
- Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, Black RE. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. 2015;385(9966):430–440. doi: 10.1016/S0140-6736(14)61698-6. doi:S0140-6736(14)61698-6 [pii] [DOI] [PubMed] [Google Scholar]
- Mac Kenzie WR, Hoxie NJ, Proctor ME, Gradus MS, Blair KA, Peterson DE, et al. A massive outbreak in Milwaukee of cryptosporidium infection transmitted through the public water supply. N Engl J Med. 1994;331(3):161–167. doi: 10.1056/NEJM199407213310304. [DOI] [PubMed] [Google Scholar]
- Manabe YC, Clark DP, Moore RD, Lumadue JA, Dahlman HR, Belitsos PC, Sears CL. Cryptosporidiosis in patients with AIDS: correlates of disease and survival. Clin Infect Dis. 1998;27(3):536–542. doi: 10.1086/514701. [DOI] [PubMed] [Google Scholar]
- Mary C, Chapey E, Dutoit E, Guyot K, Hasseine L, Jeddi F, Network A. C. N. Multicentric evaluation of a new real-time PCR assay for quantification of Cryptosporidium spp. and identification of Cryptosporidium parvum and Cryptosporidium hominis. J Clin Microbiol. 2013;51(8):2556–2563. doi: 10.1128/JCM.03458-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead JR, Arrowood MJ, Sidwell RW, Healey MC. Chronic Cryptosporidium parvum infections in congenitally immunodeficient SCID and nude mice. J Infect Dis. 1991;163(6):1297–1304. doi: 10.1093/infdis/163.6.1297. [DOI] [PubMed] [Google Scholar]
- Mochizuki K. High efficiency transformation of Tetrahymena using a codon-optimized neomycin resistance gene. Gene. 2008;425(1–2):79–83. doi: 10.1016/j.gene.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Morada M, Lee S, Gunther-Cummins L, Weiss LM, Widmer G, Tzipori S, Yarlett N. Continuous culture of Cryptosporidium parvum using hollow fiber technology. Int J Parasitol. 2016;46(1):21–29. doi: 10.1016/j.ijpara.2015.07.006. doi:S0020-7519(15)00220-9 [pii] [DOI] [PubMed] [Google Scholar]
- Newman M, Ausubel FM. Introduction to Gene Editing and Manipulation Using CRISPR/Cas9 Technology. Curr Protoc Mol Biol. 2016;115:31, 34, 31–36. doi: 10.1002/cpmb.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Tarleton R. EuPaGDT: a web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microbial Genomics. 2015;1(4) doi: 10.1099/mgen.0.000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol JF, Kabil SM, el-Gohary Y, Younis AM. Effect of nitazoxanide in diarrhea and enteritis caused by Cryptosporidium species. Clin Gastroenterol Hepatol. 2006;4(3):320–324. doi: 10.1016/j.cgh.2005.12.020. S1542-3565(05)01191-2 [pii] [DOI] [PubMed] [Google Scholar]
- Sidik SM, Hackett CG, Tran F, Westwood NJ, Lourido S. Efficient genome engineering of Toxoplasma gondii using CRISPR/Cas9. PLoS One. 2014;9(6):e100450. doi: 10.1371/journal.pone.0100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striepen B. Parasitic infections: Time to tackle cryptosporidiosis. Nature. 2013;503(7475):189–191. doi: 10.1038/503189a. [DOI] [PubMed] [Google Scholar]
- Upton SJ. In vitro cultivation. Boca Raton, Florida: CRC Press Inco.; 1997. [Google Scholar]
- Upton SJ, Tilley M, Brillhart DB. Comparative development of Cryptosporidium parvum (Apicomplexa) in 11 continuous host cell lines. FEMS Microbiol Lett. 1994;118(3):233–236. doi: 10.1111/j.1574-6968.1994.tb06833.x. [DOI] [PubMed] [Google Scholar]
- Upton SJ, Tilley M, Nesterenko MV, Brillhart DB. A simple and reliable method of producing in vitro infections of Cryptosporidium parvum (Apicomplexa) FEMS Microbiol Lett. 1994;118(1–2):45–49. doi: 10.1111/j.1574-6968.1994.tb06801.x. [DOI] [PubMed] [Google Scholar]
- Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, Striepen B. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature. 2015;523(7561):477–480. doi: 10.1038/nature14651. [DOI] [PMC free article] [PubMed] [Google Scholar]