

Abstract
The genus Conus sensu lato consists of 500–700 species. However, the mitochondrial genomes of only few species have been fully sequenced and reported so far. In this study, the complete mitochondrial genome of Conus tribblei, a member of the poorly known subgenus Splinoconus is sequenced with the mean coverage of 604x. The mitochondrial genome is 15,570 bp long and consists of genes encoding for 13 respiratory chain proteins, 22 tRNA, and 2 rRNA. The gene organization is highly conserved among the Conus species. The longest intergenic region between tRNA-Phe and cytochrome c oxidase subunit III (cox3), which in C. tribblei is 169 bp long and contains a 112-bp long segment of inverted repeat, may represent the putative control region. The control regions of Conus species exhibited variability in the length and position of the inverted repeats. Therefore, this region may have the potential to be used as a genetic marker for species discrimination.
Keywords: Mitogenome, control region, conoidea
Introduction
The genus Conus is a large genus, with ~500–700 species. To date, the complete mitochondrial genome sequences have been reported for only few Conus species (Bandyopadhyay et al. 2008; Cunha et al. 2009; Brauer et al. 2012). Although the previous studies indicate a high degree of conservation of organization and sequence of the mitochondrial genomes, interesting differences were noted, such as the novel putative control region (CR) in Conus consors, apparently absent in the mitochondrial DNA of other Conus species (Brauer et al. 2012). In this study, the complete mitochondrial genome of Conus tribblei as the first representative of subgenus Splinoconus was sequenced and annotated.
Methods
DNA was extracted from the foot tissue of C. tribblei specimen collected in the Philippines using DNeasy blood and tissue kit (Qiagen, USA). Three libraries with different insert sizes (170, 500 and 800 bp) were constructed, and each library was sequenced on one lane of Illumina HiSeq2000. The 100-bp long paired-end (PE) reads were filtered by FASTX (Pearson et al. 1997) and error corrected by SOAPec (Luo et al. 2012). The reads were mapped to the mitochondrial genomes of Conus textile, Conus borgesi, and C. consors using Bowtie2 (Langmead and Salzberg 2012). The aligned reads (184,061 PE and 30,037 single-end) were used for de novo assembly using ABySS-1.3.7 (Simpson et al. 2009). Open reading frames were identified using ORF finder (Stothard 2000), and tRNAscan (Lowe and Eddy 1997) was used for the identification of tRNAs. Annotation of 12S and 16S rRNA was performed by alignment of the C. tribblei mitochondrial genome with the reference genomes using MUMmer3 (Kurtz et al. 2004).
Results
The complete mitochondrial genome of C. tribblei, 15,570-bp long, was assembled with the mean coverage of 604x using k-mer 57. Thirteen genes encoding proteins of the respiratory chain, 22 tRNA genes and two rRNA genes were identified. The gene organization in the mitochondrial genome of C. tribblei and other Conus species are highly conserved (Fig. 1). The mitochondrial genome contained two large intergenic spacers: one between cox1 and cox2 (138 bp), and another between tRNA-Phe and cox3 (169 bp). The region between tRNA-Phe and cox3 in C. tribblei is longer than this region in C. textile (126 bp) and C. borgesi (127 bp) but shorter than C. consors (698 bp). This region in C. tribblei consists of a segment of 112 bp with 100% match inverted repeat sequence of 55 bp. This inverted repeat in C. tribblei is longer than the inverted repeat in C. textile (41 bp), C. borgesi (39bp) and the first inverted repeat in C. consors (21 bp); but it’s shorter than the second inverted repeat in C. consors (455bp). This region may represent the putative control region (CR) (Brauer et al. 2012) suggested to play a role in the initiation of replication and transcription (Boore 1999). Our results show that the length of the inverted repeat in CR is variable among Conus species. Further survey of this region in other species of Conus may reveal the suitability of using this region as a genetic marker for species discrimination.
Fig. 1.
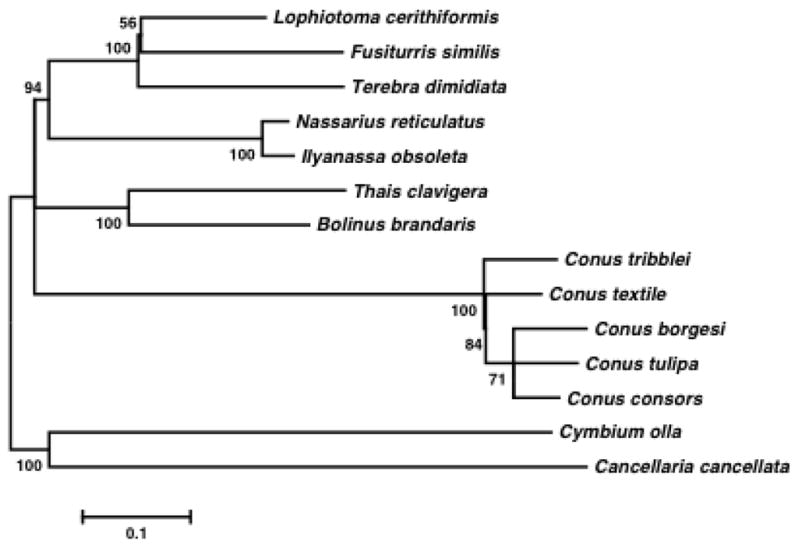
Phylogenetic relationship within Neogastropods. The evolutionary history was inferred using the Maximum likelihood method based on the General Reversible Mitochondrial+Freq model from a single concatenated data set of deduced amino acid sequences of 13 mitochondrial protein-coding genes. A discrete Gamma distribution was used to model evolutionary rate differences among cites. The rate variation model allowed for some sites to be evolutionary invariable. The bootstrap values are indicated on the nodes. All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted in MEGA5 (Tamura et al. 2011). Conus tulipa (KR006970), Conus borgesi (EU827198), Conus consors (KF887950), Conus textile (DQ862058), Terebra dimidiata (EU827196), Fusiturris similis (EU827197), Lophiotoma cerithiformis (DQ284754), Cancellaria cancellata (EU827195), Cymbium olla (EU827199), Bolinus brandaris (EU827194), Thais clavigera (DQ159954), Ilyanassa obsoleta (DQ238598) and Nassarius reticulatus (EU827201).
Footnotes
Declaration of interest
The specimen used in this study was obtained in conjunction with a collection trip supported in part by ICBG grant #1U01TW008163. This study was supported by a grant to AOL from the Emerging Interdisciplinary Research Program of the University of the Philippines System through the Philippine Genome Center. The data analysis was carried out using the high performance computing systems at the Philippine Genome Center. The mitochondrial genome sequence of C. tribblei is deposited in the GenBank database under the accession number KT199301. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Contributor Information
Neda Barghi, Marine Science Institute, University of the Philippines-Diliman, Quezon City 1101, Philippines.
Gisela P. Concepcion, Philippine Genome Center, University of the Philippines, Quezon City 1101, Philippines, Marine Science Institute, University of the Philippines-Diliman, Quezon City 1101, Philippines
Baldomero M. Olivera, Department of Biology, University of Utah, Salt Lake City, UT-USA
Arturo O. Lluisma, Philippine Genome Center, University of the Philippines, Quezon City 1101, Philippines, Marine Science Institute, University of the Philippines-Diliman, Quezon City 1101, Philippines
References
- Alikhan N, Petty NK, Zakour NLB, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay PK, Stevenson BJ, Ownby J, Cady MT, Watkins M, Olivera MB. The mitochondrial genome of Conus textile, coxI–coxII intergenic sequences and Conoidean evolution. Mol Phylogenet Evol. 2008;46:215–223. doi: 10.1016/j.ympev.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer A, Kurz A, Stockwell T, Baden-Tillson H, Heidler J, Wittig I, Kauferstein S, Mebs D, Stöcklin, Remm M. The Mitochondrial genome of the venomous cone snail Conus consors. PLoSOne. 2012;7(12):e51528. doi: 10.1371/journal.pone.0051528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RL, Grande C, Zardoya R. Neogastropod phylogenetic relationships based on entire mitochondrial genomes. BMC Evol Biol. 2009;9:210. doi: 10.1186/1471-2148-9-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–360. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu S, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam T, Wang J. SOAPdenovo2: an empirically improved memory-efficient short-read de Novo assembler. GigaScience. 2012;1(18):1–6. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Wood T, Zhang Z, Miller W. Comparison of DNA sequences with protein sequences. Genomics. 1997;46:24–36. doi: 10.1006/geno.1997.4995. [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, Birol İ. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19(6):1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P. The Sequence Manipulation Suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques. 2000;28:1102–1104. doi: 10.2144/00286ir01. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]