

Abstract
The presence of macromolecules in cells geometrically restricts the available space for poplypeptide chains. To study the effects of macromolecular crowding on folding thermodynamics and kinetics, we used an off-lattice model of the all-β-sheet WW domain in the presence of large spherical particles whose interaction with the polypeptide chain is purely repulsive. At all volume fractions, ϕc, of the crowding agents the stability of the native state is enhanced. Remarkably, the refolding rates, which are larger than the value at ϕc = 0, increase nonmonotonically as ϕc increases, reaching a maximum at 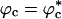 . At high values of ϕc, the depletion-induced intramolecular attraction produces compact structures with considerable structure in the denatured state. Changes in native state stability and folding kinetics at ϕc can be quantitatively mapped onto confinement in a volume-fraction-dependent spherical pore with radius Rs ≈ (4π/3ϕc)1/3 Rc (Rc is the radius of the crowding particles) as long as
. At high values of ϕc, the depletion-induced intramolecular attraction produces compact structures with considerable structure in the denatured state. Changes in native state stability and folding kinetics at ϕc can be quantitatively mapped onto confinement in a volume-fraction-dependent spherical pore with radius Rs ≈ (4π/3ϕc)1/3 Rc (Rc is the radius of the crowding particles) as long as 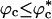 . We show that the extent of native state stabilization at finite ϕc is comparable with that in a spherical pore. In both situations, rate enhancement is due to destabilization of the denatured states with respect to ϕc = 0.
. We show that the extent of native state stabilization at finite ϕc is comparable with that in a spherical pore. In both situations, rate enhancement is due to destabilization of the denatured states with respect to ϕc = 0.
Keywords: confinement effects, protein folding, depletion-interaction
The interior of cells contains several kinds of macromolecules like lipids, sugars, nucleic acids, and proteins, along with large organized macromolecular arrays, such as cytoskeleton fibers. The volume fraction ϕc, occupied by the macromolecular crowding agents in Escherichia coli, can be as large as 0.4 (1, 2). The large volume occupied by crowding agents can have profound effect on a number of processes of biological importance. For example, biochemical reactions, stability of actin, and amyloid formation are profoundly altered in a crowded environment (3, 4). Folding of globular proteins, which is the focus of this study, in such a crowded environment can be significantly different from that at ϕc = 0. Although systematic studies of the effect of crowding on folding kinetics of well characterized proteins are lacking, several experiments have shown that refolding at finite ϕc is greatly affected. The oxidative refolding of hen lysozyme is changed when crowding agents are present (5). The rate for the molecules that reach the native basin of attraction (NBA) rapidly increases by a factor of ≈2-5, whereas the rate for the slow track molecules (i.e., those that are trapped in misfolded states) increases only by ≈80% (5). These studies and the need to systematically assess crowding effects on folding of proteins have motivated this work.
Because of the complexity of interactions, Epc, between the crowding agents and the protein of interest, it is difficult to predict the effect of macromolecular crowding on refolding of proteins. If Epc is short-ranged and repulsive, then excluded volume interactions, which prevent the polypeptide chain from accessing regions occupied by the crowding particles, are the most important. Typically, the range of the site-site excluded interactions between amino acid residues and small molecules (water, urea, GdmHCl, etc.) is on the order of a few angstroms. In contrast, the range of macromolecular excluded volume interactions can be on the order of 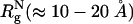 , where
, where 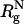 is the size of the protein in the native state. As a result, even inert macromolecules alter folding of proteins, especially at large volume fractions of the crowding agents.
is the size of the protein in the native state. As a result, even inert macromolecules alter folding of proteins, especially at large volume fractions of the crowding agents.
By modeling the crowding particles as hard spheres, we can predict the changes in the folding of two-state folders at finite ϕc by using arguments that have been used (6) to describe collapse of homopolymers in random media. The polypeptide chain would prefer to be localized in a region that is free of the macromolecular objects. Because the probability of finding such regions decreases as exp(-3ϕcRg3/4πRc3), where Rg is the typical dimension of the protein and Rc is the size of the crowding agent, we expect the polypeptide chain to be a compact globule at high values of ϕc. If the polypeptide is compact at large ϕc values, then the entropy change ΔS = S(ϕc) - S(0) < 0 because conformations with large Rg are suppressed. Thus, the stability of the native state, provided that it is not dramatically altered, is predicted to increase as ϕc increases (Fig. 1). Because of the loss in conformational entropy of the denatured states at finite ϕc values, the effective free-energy barrier to folding decreases (Fig. 1). As a result, the ratio kf(ϕc)/kf(0) must exceed unity, where kf(ϕc) is the refolding rate at ϕc.
Fig. 1.

Schematic free-energy profile for WW (PDB ID code 1PIN; sequence, KLPPGWEKRMSRSSGRVYYFNHITNASQWERPSG) domain folding. The simulated structures corresponding to the NBA and representative structures in the unfolded states are shown. The enhanced stability at ϕc ≠ 0 is due to destabilization of the unfolded states.
Similar arguments were used to rationalize the observed enhancement in stability (7-9) and increase in folding rates (8-11) for proteins encapsulated in nanopores (8). Indeed, it has been advocated that folding in a crowded environment can be mimicked by confinement. In this study, we studied the effects of crowding as a function of ϕc as well as confinement in spherical pores of different radii to probe the effects on the stability and kinetics of folding of WW domain. We show that for 0 < ϕc ≤ 0.25, the stability of the native state is enhanced. Surprisingly, whereas kf(ϕc) > kf(0) at all ϕc, the dependence of kf(ϕc) on ϕc is nonmonotonic. The rates kf(ϕc) increase until a critical value 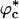 and decrease for
and decrease for 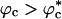 . Crowding effects can be quantitatively mimicked by confinement of the protein in a volume fraction-dependent spherical pore as long as
. Crowding effects can be quantitatively mimicked by confinement of the protein in a volume fraction-dependent spherical pore as long as 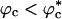 . The differences between the two situations, at high volume fractions, arise because of variations in the initial compact structures. In the crowded environment, density fluctuations can lead to expanded structures in the denatured state ensemble (DSE), whereas for proteins encapsulated in pores, poplypeptide conformations with Rg > Rs, the pore radius, are strictly forbidden. This difference results in variations in the nature of DSE, which in turn affect the folding kinetics at
. The differences between the two situations, at high volume fractions, arise because of variations in the initial compact structures. In the crowded environment, density fluctuations can lead to expanded structures in the denatured state ensemble (DSE), whereas for proteins encapsulated in pores, poplypeptide conformations with Rg > Rs, the pore radius, are strictly forbidden. This difference results in variations in the nature of DSE, which in turn affect the folding kinetics at 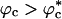 .
.
Methods
Model for the WW Domain. We used a coarse-grained Cα side-chain model (SCM) of the all-β-sheet WW domain (PDB ID code 1PIN) in which only the positions of the α-carbon and the centers of mass of side chains are retained (12, 13). Numerous experimental (14, 15) and computational (16) studies at ϕc = 0 of the WW domain make it an attractive system for our purposes. If Ep and Ec are the potential energies of the protein and the crowding agents, respectively, then the energy of the combined system is 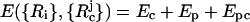 , where {Ri} refers to the coordinates of the polypeptide chain, {
, where {Ri} refers to the coordinates of the polypeptide chain, {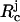 ; j = 1, 2,..., N} are the coordinates of the N spherical crowding agents, and Epc is the interaction energy between the crowding agents and the protein. Following earlier studies (12, 13), we assume that Ep is the sum of bond-length potential, side chain-backbone connectivity potential, bond-angle potential, dihedral potential, hydrogen bond (HB), and nonbonded interactions. A simple energetic bias is used to favor the l-form of the side chains (13). The nonbonded (residues that are separated by at least one residue along the sequence) potential between two side chains at a distance r is as follows:
; j = 1, 2,..., N} are the coordinates of the N spherical crowding agents, and Epc is the interaction energy between the crowding agents and the protein. Following earlier studies (12, 13), we assume that Ep is the sum of bond-length potential, side chain-backbone connectivity potential, bond-angle potential, dihedral potential, hydrogen bond (HB), and nonbonded interactions. A simple energetic bias is used to favor the l-form of the side chains (13). The nonbonded (residues that are separated by at least one residue along the sequence) potential between two side chains at a distance r is as follows:
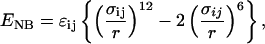 |
[1] |
where σij = f(σi + σj), and σi and σj are the van der Waals radii of the side chains. To avoid clashes between bulky side chains, we use f = 0.9. For the native contact energies εij, we use the Betancourt-Thirumalai statistical interactions potential (17). Because we are mainly interested in the physical consequences of crowding as a function of ϕc, we use a modified Go-like (18) model in which only the native interactions are retained and nonbonded interactions depend on the residue type. The repulsive nonnative interactions depend explicitly on the size of the side chains.
The purely repulsive interactions between the Cα atoms, side chain, and the spherical crowding agents with radius Rc are assumed to be pairwise additive. The repulsive potential energy between two particles i and j at a distance r is as follows:
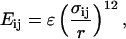 |
[2] |
where σij = σi + σj, σi and σj are the sizes of the interaction sites, and ε is 0.6 kcal/mol (1 cal = 4.18 J).
Simulation Details. The radius of the macromolecular crowding agents is 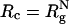 , where
, where 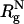 is the radius of gyration of the folded protein at infinite dilution. To minimize finite size effects, we use periodic boundary conditions (PBC), which take into account interactions between images of the crowding agents with themselves and the protein of interest (19). To carry out the simulations, we use the topology files and the PBC implementations in the amber6 program (20). The size of the cubic simulation box is >2L, where L = (N - 1) σ is the maximum length of the polypeptide chain. The unit of length σ ≈ 3.8 Å is the typical distance between α-carbon atoms. To ensure that there are no errors due to truncation of the interactions, we choose L = 75σ, which not only can accommodate an expanded polypeptide chain but also allows us to explore a range of volume fractions
is the radius of gyration of the folded protein at infinite dilution. To minimize finite size effects, we use periodic boundary conditions (PBC), which take into account interactions between images of the crowding agents with themselves and the protein of interest (19). To carry out the simulations, we use the topology files and the PBC implementations in the amber6 program (20). The size of the cubic simulation box is >2L, where L = (N - 1) σ is the maximum length of the polypeptide chain. The unit of length σ ≈ 3.8 Å is the typical distance between α-carbon atoms. To ensure that there are no errors due to truncation of the interactions, we choose L = 75σ, which not only can accommodate an expanded polypeptide chain but also allows us to explore a range of volume fractions
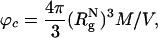 |
where M is the number of crowding agents, and V = L3. The simulations, for ϕc in the range 0 ≤ ϕc ≤ 0.25, are performed by initially equilibrating only the system of spherical crowding agents at the desired ϕc. Subsequently, the polypeptide chain is inserted and the interactions between the protein and the crowding agents are increased incrementally to avoid generating sudden changes in forces.
Calculations of Thermodynamic and Kinetic Properties. To compute the thermodynamic quantities, we integrated the Langevin equation of motion in the low-friction limit by using a Verlet-like algorithm (21, 22). The integration time step is 10-4τL, where τL = (mσ2/ε)1/2. To achieve efficient sampling of the allowed conformational space, we used the replica exchange method (REM) (23-25), covering a wide temperature range (245-425 K). The initial structures were equilibrated from independent molecular dynamics simulations. At certain times (e.g., 4τL), configurations with “neighboring” temperatures are exchanged simultaneously with the probability that is determined by the Metropolis criterion. Only pairs of replicas with temperatures that are close to each other are exchanged, and they are alternatively switched from one of two possible choices: (T1, T2), (T3, T4),....and (T2, T3), (T4, T5)..., given that T1 < T2 < T3 < T4 < T5... We used the weighted-histogram-analysis method (WHAM) (26) to compute the temperature dependence of the thermodynamic properties. The density of states was calculated by using only the energy of the protein. The ϕc-dependent folding temperature Tf(ϕc) are calculated from the fluctuations of the structural overlap function χ (27).
To probe crowding-dependent folding kinetics, we used Brownian dynamics by choosing the friction coefficient that is appropriate for water (27), for which the natural unit of time, τH, is 4.2 ns. The initial denatured configurations of the protein and crowders are prepared at 1.18Tf(ϕc = 0) (Tf(0) = 342 K). Folding kinetics is obtained at a fixed T = 0.83Tf(0). For each ϕc, we obtained 200 folding trajectories. The folding rates are computed from the distribution of first passage times. For the ith trajectory, the first passage time corresponds to the first time a molecule reaches a conformation with overlap 〈χ〉 ≈ 0.19, where the angular brackets indicate equilibrium averages.
Results and Discussions
Native State Is Not Significantly Perturbed in the Range of 0 ≤ ϕc ≤ 0.25. To assess the effects of crowding agents on the folding of the WW domain, it is necessary to ensure that the native state is not greatly altered. The number of native contacts and χ do not vary much as ϕc is increased from 0 to 0.25. For example, χ measured with respect to the coarse-grained version of the PDB structure, changes from 0.06 at ϕc = 0 to 0.04 at ϕc = 0.25. The repulsive interactions with the crowding agents make only a minor contribution to the total energy of the WW domain in the native state. The average repulsive energy of interaction between the crowding agents and the WW domain, 〈Epc〉, varies from 0.03 <〈Epc〉/ε < 0.6 (ε = 0.6 Kcal/mol) as ϕc changes from 0.02 to 0.25. Thus, the native structure of the WW domain does not change significantly, even at ϕc = 0.25.
Crowding Enhances the Stability of the Native State. The WW domain folds in an all-or-none manner as the temperature is changed from high temperature to T < Tf(ϕc) for 0 < ϕc ≤ 0.25 (M.S.C. and D.T., unpublished data). The collapse temperature Tθ at ϕc = 0 is nearly coincident with Tf(ϕc = 0), which is also a characteristic of sequences that fold in an apparent two-state manner (28, 29).
Because only excluded volume interactions between the polypeptide chain and the crowding particles are taken into account, it is clear the WW domain would prefer to be localized in a region that is free of the crowding agents (6, 30). Such regions, which arise because of density fluctuations in the crowding particles, can easily be found with high probability, especially when ϕc is low. Given that the native state is not greatly perturbed, it follows that localization of the polypeptide chain results in the destabilization of the ensemble of unfolded states with respect to the case when ϕc = 0. If Fu(ϕc) - Fu(0) > 0[Fu(ϕc) and Fu(0) are the unfolded state free energies at ϕc and ϕc = 0, respectively], then the change in the folding temperature ΔTf = Tf(ϕc) - Tf(0) > 0. These arguments suggest that crowding should increase the folding temperature and, hence, promote the stability of the native state as long as interactions between the crowding agents and the polypeptide chain are repulsive.
The folding-transition temperatures calculated from the temperature dependence of Δχ (Fig. 2A), Tf(ϕc), at all values of ϕc, are >Tf(0). The change in ΔTf is ≈7% as ϕc is increased from 0 to 0.25. This shift, which is significant, compares well with recent measurements of folding temperature changes of proteins in nanopores (31, 32). The dependence of ΔTf on ϕc can be rationalized by using changes in the free energy due to localization in a volume (33). By assuming that the native state energy is not altered because of crowding, we expect that ΔTf ≈ C ϕcα, where α ≈ 3ν [where ν (≈0.6) is the Flory exponent for the random coil]. A plot of lnΔTf as a function of ln(Cϕc) (Fig. 2B) indeed shows that α = 1.8. The nonlinear dependence of ΔTf on ϕc is in accord with experiments (34).
Fig. 2.

Crowding effect on folding thermodynamics. (A) Fluctuations in the overlap function (Δχ = 〈χ2〉 - 〈χ〉2) as a function of temperature at different values of ϕc. The fraction of conformations in the NBA (fNBA) at ϕc = 0 (□) and ϕc = 0.15 (•) as a function of temperature. (B) Crowding-dependent change in the folding temperature ΔT(ϕc) = Tf(ϕc) - Tf(0) as a function of ϕc. The solid line is a theoretical fit using ΔT(ϕc) = Cϕc3ν, with ν = 0.6. (C) Radius of gyration (Rg), at various volume fractions of the crowding particles, as a function of temperature.
The entropically stabilized native state due to crowding is also reflected in the free energy of stability ΔF(ϕc) = -kBTsln (PNBA(ϕc)/1 - PNBA(ϕc)), where PNBA(ϕc) is the thermal probability of occupying the NBA at ϕ = ϕc and Ts is the simulation temperature. The NBA is the set of conformations for which χ ≤ 〈χ (Tf)〉 = 0.19. In infinite dilution, ΔF(0) = -2.8 Kcal/mol. The value of ΔF(ϕc) increases with ϕc and at ϕc = 0.25, ΔF(0.25) is -3.9 Kcal/mol. The increases in stability of the native state and ΔTf are in accord with the Minton argument that the enhanced stability of the native state is due to crowding-induced, entropic destabilization of the DSE.
Collapse of the WW Domain Is Driven by the Depletion Effect. When ϕc = 0, the denatured poplypeptide d chains are expanded random coils with mean dimension 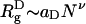 , with ν ≈ 0.6 (35), whereas in the folded states, they are near by maximally compact (
, with ν ≈ 0.6 (35), whereas in the folded states, they are near by maximally compact (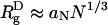 ) (36). Upon reducing T from high temperatures, proteins undergo coil-to-globule transition at the Flory θ temperature, Tθ. Thus, the global characterization of the DSE at ϕc = 0 are in accord with the expectations from polymer theory (33). As ϕc increases, we expect WW to become compact even at high temperatures (T > Tθ at ϕc = 0). The temperature dependence of Rg (T) (Fig. 2C) shows that, at ϕc = 0.25, the WW domain is only ≈25% larger than its value in the native state at T ≈ 375 K.
) (36). Upon reducing T from high temperatures, proteins undergo coil-to-globule transition at the Flory θ temperature, Tθ. Thus, the global characterization of the DSE at ϕc = 0 are in accord with the expectations from polymer theory (33). As ϕc increases, we expect WW to become compact even at high temperatures (T > Tθ at ϕc = 0). The temperature dependence of Rg (T) (Fig. 2C) shows that, at ϕc = 0.25, the WW domain is only ≈25% larger than its value in the native state at T ≈ 375 K.
The substantial reduction in Rg at large values of ϕc at high temperature is due to a coil-to-globule transition in the WW domain at a critical value of 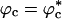 because of volume excluded by the crowding agents. The collapse of WW domain at
because of volume excluded by the crowding agents. The collapse of WW domain at 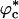 is due to two opposing contributions to the free energy (30). The protein prefers to be in a crowding-free region, which is difficult to find at high ϕc. However, localization in a confined volume decreases chain entropy. As a result, at a critical volume fraction
is due to two opposing contributions to the free energy (30). The protein prefers to be in a crowding-free region, which is difficult to find at high ϕc. However, localization in a confined volume decreases chain entropy. As a result, at a critical volume fraction 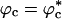 , the WW domain becomes compact even at relatively high temperatures. From this physical reasoning, which was made precise by Shaw and D.T. (ST) (37), it follows that when the mean spacing between the crowding agents,
, the WW domain becomes compact even at relatively high temperatures. From this physical reasoning, which was made precise by Shaw and D.T. (ST) (37), it follows that when the mean spacing between the crowding agents, 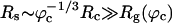 , then the crowding agents do not perturb the folding of the polypeptide chain. In the opposite limit, the probability of finding a region free of the crowding agents in which the polypeptide chain can be accommodated becomes small. The localization results in the reduction of the conformational entropy of the chain. As a result, there is an entropically generated intramolecular effective attractive interaction that drives the collapse transition even when T > Tθ (ϕc = 0). This interaction is the depletion-induced attraction between colloidal particles in polymer solution (38).
, then the crowding agents do not perturb the folding of the polypeptide chain. In the opposite limit, the probability of finding a region free of the crowding agents in which the polypeptide chain can be accommodated becomes small. The localization results in the reduction of the conformational entropy of the chain. As a result, there is an entropically generated intramolecular effective attractive interaction that drives the collapse transition even when T > Tθ (ϕc = 0). This interaction is the depletion-induced attraction between colloidal particles in polymer solution (38).
The depletion effect is most clearly evident in the radial distribution functions between the crowding agents and the polypeptide chain gpc(r) (Fig. 3 A and B). At all values of ϕc, there is clear depletion in the density of crowding particles near the WW domain. In particular, for 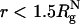 , the probability of finding the crowding agents is zero, which shows that the WW domain is localized in a region that is essentially devoid of the crowding particles. Because large conformational fluctuations are not probable because of the volume occupied by crowding agents, the WW chain adopts a compact conformation even at relatively high temperatures (Fig. 3C). The striking similarity between the gpc(r) shown here and the ST theory for homopolymer (see figure 1 in ref. 37) in a colloidal solution further reinforces the importance of depletion-induced collapse for proteins because of macromolecular crowding.
, the probability of finding the crowding agents is zero, which shows that the WW domain is localized in a region that is essentially devoid of the crowding particles. Because large conformational fluctuations are not probable because of the volume occupied by crowding agents, the WW chain adopts a compact conformation even at relatively high temperatures (Fig. 3C). The striking similarity between the gpc(r) shown here and the ST theory for homopolymer (see figure 1 in ref. 37) in a colloidal solution further reinforces the importance of depletion-induced collapse for proteins because of macromolecular crowding.
Fig. 3.

Depletion-induced attraction. (A and B) Radial distribution functions between the protein residues and crowding particles as a function of ϕc at two temperatures. At both temperatures, there is a depletion layer (shaded area) surrounding the protein, which effectively excludes the crowding agents (see also C). (C) A snapshot of the WW protein and crowding particles in the simulation. There is a rim of depletion layer surrounding the protein in which the crowding particles are absent. Sizes of side chains are highlighted in red.
Folding Rates Vary Nonmonotonically with ϕc. The refolding rates at various ϕc values were calculated by a temperature quench from T = 1.18Tf(0) to Ts = 0.83 Tf(0). The distributions of first passage times, from which kf(ϕ) were computed, were obtained for 200 folding trajectories.
Based on the entropic destabilization of the DSE, we expect that kf(ϕc) should increase as ϕc increases, provided that the free-energy barrier does not change (Fig. 1). When ϕc exceeds 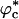 , the volume fraction above which the WW chain adopts a globular conformation even at high temperatures, we expect kf(ϕc) to decrease. For
, the volume fraction above which the WW chain adopts a globular conformation even at high temperatures, we expect kf(ϕc) to decrease. For 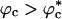 , the polypeptide chain is localized in a small enough volume that the conformational fluctuations that may be required to cross the free-energy barrier are restricted. The expected nonmonotonic dependence of kf(ϕc) on ϕc is borne out in the simulations (Fig. 4). The rate initially increases as ϕc increases. At ϕc ≈ 0.1, kf(ϕc) is nearly three times as large as in ϕc = 0. Surprisingly, kf(0.15) is nearly 80% larger than kf(0), which shows that the search among compact conformations is faster than transitions to the NBA from the expanded random coil conformations. Thus, even high density of crowding can lead to modest increases in folding rates.
, the polypeptide chain is localized in a small enough volume that the conformational fluctuations that may be required to cross the free-energy barrier are restricted. The expected nonmonotonic dependence of kf(ϕc) on ϕc is borne out in the simulations (Fig. 4). The rate initially increases as ϕc increases. At ϕc ≈ 0.1, kf(ϕc) is nearly three times as large as in ϕc = 0. Surprisingly, kf(0.15) is nearly 80% larger than kf(0), which shows that the search among compact conformations is faster than transitions to the NBA from the expanded random coil conformations. Thus, even high density of crowding can lead to modest increases in folding rates.
Fig. 4.

Dependence of the folding rates as a function of ϕc and the radius of the volume-fraction-dependent confining sphere,  . This figure shows that crowding effects can be mimicked by encapsulation in a spherical pore with radius Rs.
. This figure shows that crowding effects can be mimicked by encapsulation in a spherical pore with radius Rs.
Crowding Can Be Mimicked by Confinement. Encapsulation of polypeptide chains in nanopores has been used to mimic macromolecular crowding. The ST arguments suggest that, at a volume fraction ϕc, the polypeptide chain should be localized in a spherical region with the most probable radius
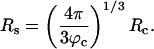 |
If this mapping between crowding and effective confinement is qualitatively correct, then we expect thermodynamic and kinetic properties at ϕc and an encapsulated protein in a spherical pore with radius Rs to be similar. To validate this proposal, we computed changes in Tf and folding rates for the WW chain confined to spherical pores by using the procedures described in ref. 8.
As shown in refs. 8 and 10, ΔTf(Rs) = Tf(Rs) - Tf(Rs → ∞) [Tf(Rs → ∞) ≡ Tf(ϕc = 0)] increases as Rs increases. The ST mapping between confinement and crowding is quantitatively accurate for ϕc ≤ 0.1. For example, when 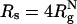 , ϕc ≈ 0.065, for which
, ϕc ≈ 0.065, for which 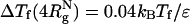 , whereas ΔTf(ϕc = 0.06) = 0.03kBTf/ε. Similarly, we find that ΔΔG = (ΔG(ϕc) - ΔG(Rs))/kBTs is small, ranging from 0.16 to 0.32 in the range of 0 < ϕc ≤ 0.1. As ϕc exceeds 0.1, encapsulation in an equivalent nanopore results in marginally greater stability. At higher volume fraction, the optimal cavity in which the WW domain is localized is likely to be cylindrical. Thus, the mapping onto an equivalent spherical pore is only qualitatively valid. These results and a recent analysis (39) show that crowding and confinement affect the stability of the native state to the same extent.
, whereas ΔTf(ϕc = 0.06) = 0.03kBTf/ε. Similarly, we find that ΔΔG = (ΔG(ϕc) - ΔG(Rs))/kBTs is small, ranging from 0.16 to 0.32 in the range of 0 < ϕc ≤ 0.1. As ϕc exceeds 0.1, encapsulation in an equivalent nanopore results in marginally greater stability. At higher volume fraction, the optimal cavity in which the WW domain is localized is likely to be cylindrical. Thus, the mapping onto an equivalent spherical pore is only qualitatively valid. These results and a recent analysis (39) show that crowding and confinement affect the stability of the native state to the same extent.
Strikingly, the folding rates kf(Rs) and kf(ϕc) exhibit similar behavior. By the proposed mapping, the folding rates kf(ϕc) can be quantitatively predicted by using kf(Rs) as long as 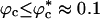 (Fig. 4). When
(Fig. 4). When 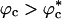 , decrease in kf(ϕc) is somewhat greater than kf(Rs) (Fig. 4). This difference arises because of destabilization of the DSE due to crowding does not occur to the same extent as it does in nanopores in which conformations with R > Rs are strictly disallowed. Even at high volume fractions, density fluctuations in the crowding particles can accommodate polypeptide conformations with Rg > Rs. These conformational fluctuations may be required in the transition from compact nonnative states to the folded structure.
, decrease in kf(ϕc) is somewhat greater than kf(Rs) (Fig. 4). This difference arises because of destabilization of the DSE due to crowding does not occur to the same extent as it does in nanopores in which conformations with R > Rs are strictly disallowed. Even at high volume fractions, density fluctuations in the crowding particles can accommodate polypeptide conformations with Rg > Rs. These conformational fluctuations may be required in the transition from compact nonnative states to the folded structure.
Confinement Suppresses Large-Scale Shape Fluctuations. In the transition from denatured to folded states, the WW domain undergoes substantial changes in the overall shape, which we characterize as Δ (Δ = 0 for spheres), the asphericity parameter, and S, the shape parameter (S > 0 for prolate ellipsoid, and S < 0 for oblate ellipsoid) (36). Both Δ and S are calculated by using the inertia tensor (36). From the 200 folding trajectories, we computed the profiles of Δ and S under various conditions (Fig. 5). Although on an average WW domain is compact (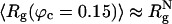 ) at ϕc ≈ 0.15, the distribution of Δ and S are qualitatively similar to what is found at ϕc = 0 (Fig. 5 A and B). In going from the unfolded states to the NBA, S changes substantially from ≈1.5 to the native value of 0.07, which shows large fluctuations in the shape. Similarly, substantial changes in Δ also occur in the transition from the DSE to the NBA (Fig. 5 A and B).
) at ϕc ≈ 0.15, the distribution of Δ and S are qualitatively similar to what is found at ϕc = 0 (Fig. 5 A and B). In going from the unfolded states to the NBA, S changes substantially from ≈1.5 to the native value of 0.07, which shows large fluctuations in the shape. Similarly, substantial changes in Δ also occur in the transition from the DSE to the NBA (Fig. 5 A and B).
Fig. 5.

Crowding effects and confinement on folding kinetics. Histograms of 200 folding kinetic trajectories are plotted at Ts as a function of shape parameters, Δ and S, at several different conditions: ϕc = 0 (A); ϕc = 0.15 (B); 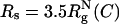 ; and
; and 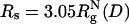 . The color bar is scaled by kBTs. The green arrows indicate the NBA. The dashed circle in C and D is a visual guide to highlight the suppression of the extended configurations as the radius of the spherical pore decreases. In contrast, both at ϕc = 0 (A) and ϕc = 0.15 (B), density fluctuations of the crowding agents allow large fluctuations in Δ and S. When the spherical pore is small enough,
. The color bar is scaled by kBTs. The green arrows indicate the NBA. The dashed circle in C and D is a visual guide to highlight the suppression of the extended configurations as the radius of the spherical pore decreases. In contrast, both at ϕc = 0 (A) and ϕc = 0.15 (B), density fluctuations of the crowding agents allow large fluctuations in Δ and S. When the spherical pore is small enough, 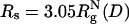 , which mapped to ϕc ≈ 0.15, folding occurs by a kinetic partitioning mechanism as a result of being trapped in a structure with an inaccurate turn (highlighted in a purple β-strand in D). There is no such barrier caused at ϕc = 0.15 (B).
, which mapped to ϕc ≈ 0.15, folding occurs by a kinetic partitioning mechanism as a result of being trapped in a structure with an inaccurate turn (highlighted in a purple β-strand in D). There is no such barrier caused at ϕc = 0.15 (B).
In contrast to crowding, encapsulation greatly restricts fluctuations in Δ and S (Fig. 5 C and D). Regions with S > 1 and Δ > 0.6 are strictly forbidden when 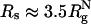 , and the are allowed conformations further restricted when Rs is decreased to
, and the are allowed conformations further restricted when Rs is decreased to 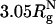 . When
. When 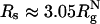 , a maximally compact state with both Δ≈ 0 and S ≈ 0 is populated (dark blue in Fig. 5D). The structure in this region has a nonnative turn (purple in Fig. 5D). The formation of this low-energy kinetic trap results in a decrease in kf(Rs) when
, a maximally compact state with both Δ≈ 0 and S ≈ 0 is populated (dark blue in Fig. 5D). The structure in this region has a nonnative turn (purple in Fig. 5D). The formation of this low-energy kinetic trap results in a decrease in kf(Rs) when 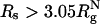 . Because density fluctuations in the crowding agents can always allow polypeptide chains to sample different Rg values, we find that at high-volume fractions, highly spherical structures (Δ≈ 0 and S ≈ 0) are also explored. However, these structures are more mobile and can rearrange readily, which prevents them from being kinetic traps. The differences in the nature of DSE as ϕc increases and Rs decreases lead to variations in the folding mechanisms between the two situations.
. Because density fluctuations in the crowding agents can always allow polypeptide chains to sample different Rg values, we find that at high-volume fractions, highly spherical structures (Δ≈ 0 and S ≈ 0) are also explored. However, these structures are more mobile and can rearrange readily, which prevents them from being kinetic traps. The differences in the nature of DSE as ϕc increases and Rs decreases lead to variations in the folding mechanisms between the two situations.
Confinement-Induced Unfolded States Are Highly Structured. To assess the nature of the structures in the denatured states, we generated an ensemble of conformations at T = 1.18Tf(0). The clustering procedure (40) shows that the DSE ensemble can be classified into two dominant clusters at ϕc = 0 and ϕc = 0.15, and in the spherical pore with 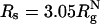 (Fig. 6). The structures in the two clusters are remarkably similar at both volume fractions (compare Fig. 6 B and C). Structures in DSI are expanded, whereas in DSII there is evidence for formation of native fold (Fig. 6 B and C). However, at ϕc = 0.15, there is a suppression of chain conformations with large Rg, which leads to destabilization of the DSE. In contrast to the behavior at ϕc = 0.15, the DSE structures in the spherical pore are dramatically different (compare Fig. 6 C and D). In the nanopore, the DSE is more compact and highly structured, which shows that confinement leads to substantial long-range interactions even at elevated temperature. The structures in DSII in the encapsulated WW domain can have incorrect topologies (Fig. 6D) that serve as kinetic traps.
(Fig. 6). The structures in the two clusters are remarkably similar at both volume fractions (compare Fig. 6 B and C). Structures in DSI are expanded, whereas in DSII there is evidence for formation of native fold (Fig. 6 B and C). However, at ϕc = 0.15, there is a suppression of chain conformations with large Rg, which leads to destabilization of the DSE. In contrast to the behavior at ϕc = 0.15, the DSE structures in the spherical pore are dramatically different (compare Fig. 6 C and D). In the nanopore, the DSE is more compact and highly structured, which shows that confinement leads to substantial long-range interactions even at elevated temperature. The structures in DSII in the encapsulated WW domain can have incorrect topologies (Fig. 6D) that serve as kinetic traps.
Fig. 6.

Distributions of denatured states at 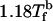 under high volume fractions of crowding agents and in spherical pore with encapsulation
under high volume fractions of crowding agents and in spherical pore with encapsulation 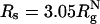 . (A) The native structure of WW domain (PDB ID code 1P1N) in ribbon representation as a reference to show the relative positions of Trp-11 (green) and Trp-34 (yellow) along the backbone. (B)ϕc = 0. (C)ϕc = 0.15. (D)
. (A) The native structure of WW domain (PDB ID code 1P1N) in ribbon representation as a reference to show the relative positions of Trp-11 (green) and Trp-34 (yellow) along the backbone. (B)ϕc = 0. (C)ϕc = 0.15. (D) 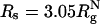 . The dominant cluster, at ϕc = 0 (B) in the DSE (DSI) corresponds to expanded structures. DSI has few contacts between side chains and between backbone α-carbon atoms. DSII contain a significant amount of side-chain contacts, but backbone structures are not aligned properly for hydrogen bonding. A DSE structure represents an averaged conformation over a given cluster. Percentages of DSI decrease as ϕc increases (C), whereas those of DSII increase, implying that compact structures are favored at high volume fraction. Interestingly, under stringent spherical encapsulation (D), there are trapped structures in DSII because the bulky Trp side chain (dashed circle) is unable to flip over to form correct contacts as a result of a hindrance by the spherical boundary condition.
. The dominant cluster, at ϕc = 0 (B) in the DSE (DSI) corresponds to expanded structures. DSI has few contacts between side chains and between backbone α-carbon atoms. DSII contain a significant amount of side-chain contacts, but backbone structures are not aligned properly for hydrogen bonding. A DSE structure represents an averaged conformation over a given cluster. Percentages of DSI decrease as ϕc increases (C), whereas those of DSII increase, implying that compact structures are favored at high volume fraction. Interestingly, under stringent spherical encapsulation (D), there are trapped structures in DSII because the bulky Trp side chain (dashed circle) is unable to flip over to form correct contacts as a result of a hindrance by the spherical boundary condition.
Conclusions
We have made experimentally testable predictions for crowding-induced changes in the native stability and kinetics (1). Excluded volume effects due to macromolecular crowding enhance native state stability at all values of ϕc. The stabilization is linked directly to crowding-induced destabilization of the unfolded states (39). This interpretation is reaffirmed further by comparing unfolding rates at ϕc = 0 and ϕc ≠ 0. If the free energy landscape changes, as indicated in Fig. 1 at ϕc ≠ 0, then we expect that unfolding rates ku(ϕc) should not vary much with ϕc. Indeed, unfolding simulations show that ku(0) and ku(0.15) at T = 395 K are nearly identical (M.S.C. and D.T., unpublished data) (2). Surprisingly, we find that the refolding rates change nonmonotonically with ϕc. Even at relatively high ϕc, kf(ϕc) > ku(0) (3). Crowding effects can be mimicked quantitatively by confinement in a spherical pore with a ϕc-dependent pore size for 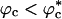 . Even when
. Even when 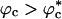 , we find that the rates in the two cases do not differ significantly. Although calculations have been done for the WW domain, the general results should be applicable to other proteins as well.
, we find that the rates in the two cases do not differ significantly. Although calculations have been done for the WW domain, the general results should be applicable to other proteins as well.
There are two factors that were not considered in our model that could potentially affect our conclusions. (i) It could be argued that, in the presence of crowding agents, the neglected nonnative interactions could be enhanced, thus increasing the roughness of the energy landscape. However, because of the repulsive nature of the interactions between the crowding agents and the polypeptide chain, such an interaction is unlikely. Thus, the sole effect of the crowding particles is to create a large depletion zone around the polypeptide chain. (ii) It is possible that the intrapeptide interactions are modified by the interstitial water. Because the WW domain is localized in a region whose volume, at the highest volume fraction, is nearly 27 times as large as that of the folded protein (see Fig. 3), we suspect that interactions between the amino acids are the same as in the bulk. Thus, we expect the qualitative nature of the predicted results to be valid even for more realistic models.
We have only considered only one value for the size of the crowding particles, namely, 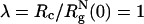 . Besides temperature, the effective binary system of proteins and molecular crowding effects is described by three variables (namely, λ, ϕc, and ϕp, the volume fraction of the proteins). At a fixed temperature, variations of the three variables can produce a complex phase diagram. Several scenarios can be anticipated by using results familiar in the colloid literature (see ST for an example) and by modeling the poplypeptide chains as approximately spherical particles. When λ < 1, the smaller crowding agents promote protein-protein association because of a depletion effect. In this limit, we propose that folding would be unaffected as long as
. Besides temperature, the effective binary system of proteins and molecular crowding effects is described by three variables (namely, λ, ϕc, and ϕp, the volume fraction of the proteins). At a fixed temperature, variations of the three variables can produce a complex phase diagram. Several scenarios can be anticipated by using results familiar in the colloid literature (see ST for an example) and by modeling the poplypeptide chains as approximately spherical particles. When λ < 1, the smaller crowding agents promote protein-protein association because of a depletion effect. In this limit, we propose that folding would be unaffected as long as 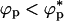 , the overlap concentration. However, if
, the overlap concentration. However, if 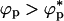 , crowding particles should promote aggregation of proteins. In the limit λ > 1, attractive interactions between the crowding agents is enhanced by the polypeptide chains. This attraction would lead to a microphase separation containing protein-rich phase and crowding agent rich phase. In this limit, folding would be unaffected when ϕp → 0 and aggregation would result if
, crowding particles should promote aggregation of proteins. In the limit λ > 1, attractive interactions between the crowding agents is enhanced by the polypeptide chains. This attraction would lead to a microphase separation containing protein-rich phase and crowding agent rich phase. In this limit, folding would be unaffected when ϕp → 0 and aggregation would result if 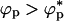 . Thus, in both limits (λ < 1 or λ > 1) and
. Thus, in both limits (λ < 1 or λ > 1) and 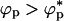 , we predict that crowding agents enhance protein aggregation.
, we predict that crowding agents enhance protein aggregation.
Acknowledgments
This work was supported by a postdoctoral fellowship from the Alfred P. Sloan Foundation (to M.S.C.) and by the National Science Foundation (to D.T.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NBA, native basin of attraction; DSE, denatured state ensemble; ST, Shaw and D.T.
References
- 1.Ellis, R. J. & Minton, A. P. (2003) Nature 425, 27-28. [DOI] [PubMed] [Google Scholar]
- 2.Ellis, R. J. (2001) Trends Biochem. Sci. 26, 597-604. [DOI] [PubMed] [Google Scholar]
- 3.Minton, A. P. (2000) Curr. Opin. Struct. Biol. 10, 34-39. [DOI] [PubMed] [Google Scholar]
- 4.Ellis, R. J. (2001) Curr. Opin. Struct. Biol. 11, 114-119. [DOI] [PubMed] [Google Scholar]
- 5.van den Berg, B., Wain, R., Dobson, C. M. & Ellis, R. J. (2000) EMBO J. 19, 3870-3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thirumalai, D. (1988) Phys. Rev. A 37, 269-276. [DOI] [PubMed] [Google Scholar]
- 7.Zhou, H. X. & Dill, K. A. (2001) Biochemistry 40, 11289-11293. [DOI] [PubMed] [Google Scholar]
- 8.Klimov, D. K., Newfield, D. & Thirumalai, D. (2002) Proc. Natl. Acad. Sci. USA 99, 8019-8024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Betancourt, M. R. & Thirumalai, D. (1999) J. Mol. Biol. 287, 627-644. [DOI] [PubMed] [Google Scholar]
- 10.Takagi, F., Koga, N. & Takada, S. (2003) Proc. Natl. Acad. Sci. USA 100, 11367-11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedel, M., Sheeler, D. J. & Shea, J.-E. (2003) J. Chem. Phys. 118, 8106-8113. [Google Scholar]
- 12.Klimov, D. K. & Thirumalai, D. (2000) Proc. Natl. Acad. Sci. USA 97, 2544-2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung, M. S., Finke, J. M., Callahan, B. & Onuchic, J. N. (2003) J. Phys. Chem. B 107, 11193-11200. [Google Scholar]
- 14.Jager, M., Nguyen, H., Crane, J. C., Kelly, J. W. & Gruebele, M. (2001) J. Mol. Biol. 311, 373-393. [DOI] [PubMed] [Google Scholar]
- 15.Ferguson, N., Berriman, J., Petrovich, M., Sharpe, T. D., Finch, J. T. & Fersht, A. R. (2003) Proc. Natl. Acad. Sci. USA 100, 9814-9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karanicolas, J. & Brooks, C. L., III, (2003) Proc. Natl. Acad. Sci. USA 100, 3954-3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Betancourt, M. R. & Thirumalai, D. (1999) Protein Sci. 8, 361-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Udea, Y., Taketomi, H. & Go, N. (1978) Biopolymers 17, 1531-1548. [Google Scholar]
- 19.Allen, M. P. & Tildesley, D. J. (1987) Computer Simulation of Liquids (Oxford Univ. Press, New York).
- 20.Case, D. A., Pearlman, D. A., Caldwell, J. W., Cheatham, T. E., Ross, W. S., Simmerling, C. L., Darden, T. A., Merz, K. M., Stanton, R.V., Cheng, A. L., et al. (1999) AMBER6 (University of California, San Francisco).
- 21.Guo, C., Cheung, M. S., Levine, H. & Kessler, D. A. (2002) J. Chem. Phys. 116, 4353-4365. [Google Scholar]
- 22.Honeycutt, J. D. & Thirumalai, D. (1992) Biopolymers 32, 695-709. [DOI] [PubMed] [Google Scholar]
- 23.Sanbonmatsu, K. Y. & Garcia, A. E. (2002) Proteins 46, 225-234. [DOI] [PubMed] [Google Scholar]
- 24.Sugita, Y. & Okamoto, Y. (1999) Chem. Phys. Lett. 314, 141-151. [Google Scholar]
- 25.Lin, C. Y., Hu, C. K. & Hansmann, U. H. (2003) Proteins 52, 436-445. [DOI] [PubMed] [Google Scholar]
- 26.Kumar, S., Bouzida, D., Swendsen, R. H., Kollman, P. A. & Rosenberg, J. M. (1992) J. Comput. Chem. 13, 1011-1021. [Google Scholar]
- 27.Veitshans, T., Klimov, D. & Thirumalai, D. (1997) Folding Des. 2, 1-22. [DOI] [PubMed] [Google Scholar]
- 28.Thirumalai, D. & Klimov, D. K. (1999) Curr. Opin. Struct. Biol. 9, 197-207. [DOI] [PubMed] [Google Scholar]
- 29.Cheung, M. S. & Chavez, L. L., Onuchic, J. N. (2004) Polymer 45, 547-555. [Google Scholar]
- 30.Honeycutt, J. D. & Thirumalai, D. (1990) J. Chem. Phys. 93, 6851-6858. [Google Scholar]
- 31.Eggers, D. K. & Valentine, J. S. (2001) J. Mol. Biol. 314, 911-922. [DOI] [PubMed] [Google Scholar]
- 32.Bolis, D., Politou, A. S., Kelly, G., Pastore, A. & Temussi, P. A. (2004) J. Mol. Biol. 336, 203-212. [DOI] [PubMed] [Google Scholar]
- 33.Gennes, P.-G. d. (1979) Scaling Concepts in Polymer Physics (Cornell Univ. Press, New York).
- 34.Sasahara, K., McPhie, P. & Minton, A. P. (2003) J. Mol. Biol. 326, 1227-1237. [DOI] [PubMed] [Google Scholar]
- 35.Kohn, J. E., Millett, I. S., Jacob, J., Zagrovic, B., Dillon, T. M., Cingel, N., Dothager, R. S., Seifert, S., Thiyagarajan, P., Sosnick, T. R., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 12491-12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dima, R. I. & Thirumalai, D. (2004) J. Phys. Chem. B 108, 6564-6570. [Google Scholar]
- 37.Shaw, M. R. & Thirumalai, D. (1991) Phys. Rev. A 44, R4797-R4800. [DOI] [PubMed] [Google Scholar]
- 38.Asakura, S. & Oosawa, F. (1954) J. Chem. Phys. 22, 1255-1256. [Google Scholar]
- 39.Minton, A. P. (2005) Biophys J. 88, 971-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klimov, D. K. & Thirumalai, D. (1998) J. Mol. Biol. 282, 471-492. [DOI] [PubMed] [Google Scholar]