

Abstract
Background
Tosedostat is a novel oral aminopeptidase inhibitor with clinical activity in a previous phase I/II study in elderly patients with relapsed/refractory acute myeloid leukaemia (RR AML). We present the results of a randomised phase II study comparing two dosing regimes of tosedostat.
Methods
Patients aged ≥60 years with AML with relapse after a first complete remission (CR) lasting up to 12 months, or no prior CR, were randomised to receive as first salvage tosedostat 120 mg once daily for 6 months or 240 mg once daily for 2 months followed by 120 mg for 4 months. The primary endpoint was the proportion of patients who obtained a Complete Remission or Complete Remission with incomplete Platelet Recovery. The study was analysed on an intention to treat basis. The study was registered on clintrials.gov (NCT00780598) and the final study visit occurred in March 2011.
Findings
Seventy-three patients were treated with tosedostat. Seven patients (10%) achieved CR or a complete remission with incomplete platelet recovery (CRp): 2 of 38 (5%) in the 120 mg group and 5 of 35 (14%) in the 240 mg→120 mg group. The most common adverse events at grade 3 or worse were febrile neutropenia which occurred in 21/73 (29%) patients overall, 11/38 (29%) in the 120 mg group and 10/35 (29%) of the 240 mg→120 mg group, thrombocytopenia (16, 22%; 8, 21% and 8, 23%), fatigue (15, 21%; 7, 18% and 8, 23%), dyspnoea (12, 16%; 5, 13% and 7, 20%), pneumonia (10, 14%; 4, 11% and 6, 17%). There were 5 adverse events with an outcome of death, 3 in the 120 mg group and 2 in the 240 mg→120 mg group. The events were acute hepatitis, respiratory failure, pneumonia, atrial fibrillation and left ventricular dysfunction.
Interpretation
Tosedostat, at either dose schedule, has efficacy in older patients with relapsed or refractory AML, particularly those with prior myelodysplastic syndromes (MDS) or prior hypomethylating agent therapy. Additional studies of tosedostat including combination with hypomethylating agents and low dose cytarabine in patients with high risk MDS and AML are ongoing and/or planned.
Funding
The OPAL study was funded by Chroma Therapeutics Ltd, Abingdon, UK.
Introduction
Treatment outcomes for older patients with acute myeloid leukaemia have not substantially improved since the development of cytarabine and anthracycline-based regimens several decades ago1, 2, 3. This situation may be due to the limited tolerability in this population for the adverse effects of such therapies, as well as a higher prevalence of poor cytogenetic predictors of poor outcome. The problem is even greater in patients who are refractory to, or have relapsed following, first line therapy. The only drug ever approved for this indication, gemtuzumab ozogamycin, was recently withdrawn from the market.
Tosedostat is a novel oral agent that targets aminopeptidases, enzymes that have a key role in the protein cell cycle (see webappendix). Aminopeptidase inhibition results in the Amino Acid Deprivation Response (AADR), a response which occurs selectively in transformed cells and leads to an upregulation of pro-apoptotic factors including CHOP and NOXA4, an activation of stress–related pathways such as NFkB, and an inhibition of mTOR which switches off protein synthesis. Tosedostat induces an AADR in a wide variety of cell lines in vitro, including solid tumour, leukemia and myeloma cell lines and has demonstrated anti-neoplastic activity in a range of in vivo solid tumour models5. Tosedostat is administered as an oral ester moiety with an esterase sensitive motif. Inside the cell the ester is hydrolysed to a polar acid moiety, which is poorly membrane permeable and therefore trapped in the cell. Tosedostat has demonstrated synergy in vitro with a wide range of other drugs used to treat solid and haematological cancers including chemotherapy agents, hypomethylating agents and others5.
Tosedostat has been administered to patients in phase I/II studies in both solid tumours and haematological malignancies. In a phase I/II study in 40 patients with solid tumours treated with tosedostat as a single agent, a Maximum Acceptable Dose (MAD) of 240 mg once daily was determined and 1 durable partial response (PR) and 7 confirmed stable diseases (SD) were observed6. The most common adverse events at any grade were fatigue, diarrhoea and peripheral oedema. In a phase I/II study in 57 patients with haematological malignancies, a MAD of 130 mg once daily was determined and 7 bone marrow complete responses (CR) and 7 PRs were observed among a subset of 51 AML patients, of which 6 bone marrow CRs and 5 PRs occurred in the 35 AML patients who were refractory or relapsed to prior therapy7. The most common adverse events at any grade were thrombocytopenia, fatigue and peripheral oedema. The MAD in the haematological malignancies study was lower than that observed in the solid tumours study, and was primarily due to several cases of thrombocytopenia, a common event in the patient population enrolled. We decided that further evaluation of the optimal doses used in haematological malignancies was appropriate.
The outcomes of these phase I/II studies led to the design of the OPAL study (CHR-2797-038) which was designed to further evaluate the safety and efficacy of two dosing regimens of tosedostat in patients with relapsed or refractory AML.
Methods
Patients
The OPAL study was a phase II multicentre, randomised study conducted in 20 centres in the US (16), Canada (2) and the Netherlands (2). Patients were eligible if aged ≥60 years with AML per WHO classification (excluding APL) which required first salvage treatment following primary induction for AML which resulted in no CR or a CR which lasted <12 months, with an ECOG performance score of 2 or less, adequate hepatic, renal and cardiac function, and an expected life expectancy of at least 3 months. Patients who had received prior first salvage treatment, anti-leukaemia treatment within 2 weeks, or who had significant cardiac disease at baseline were ineligible. The study was approved by Institutional Review Boards or Ethics Committees at participating sites. All patients provided written informed consent and a Drug Safety Monitoring Board was established.
Efficacy Parameters
Primary Efficacy Measure
Complete Remission (CR):
<5% blasts in bone marrow aspirate
No blasts with Auer rods or persistence of extramedullary disease
Absolute neutrophil count (ANC) ≥1000/mm3
Platelet count ≥100,000/mm3
Complete Remission with Incomplete Platelet Recovery (CRp):
<5% blasts in bone marrow aspirate
No blasts with Auer rods or persistence of extramedullary disease
ANC ≥1000/mm3
Platelets <100,000/mm3
Secondary Efficacy Measures
Secondary efficacy parameters included:
- Other levels of clinical response:
-
○Morphological leukemia-free state (MLFS)
-
○Partial Remission (PR)
-
○Stable Disease (SD)
-
○Progression of Disease (PD)
-
○
Overall Survival
Event Free Survival
Duration of Clinical Response
Percentage reduction in blasts in bone marrow
Morphological leukemia free state (MLFS):
<5% blasts in bone marrow aspirate
No blasts with Auer rods or persistence of extramedullary disease
Partial Remission (PR):
5 to 25% blasts in bone marrow aspirate, with at least 50% reduction from baseline
Stable Disease (SD):
Any disease category which does not meet the definition of CR, CRp, MLFS, PR or PD
Progressive Disease (PD):
Any one of:
>50% increase in bone marrow blast percentage from the best value to a value of at least 25%
>50% increase in circulating (peripheral) blasts (with rising trend)
New appearance of circulating (peripheral) blasts (with rising trend on at least 2 consecutive occasions)
Development of extramedullary leukemia
Randomisation
Patients were randomly assigned to receive either 120 mg tosedostat for 6 months or 240 mg tosedostat for 2 months followed by 120 mg tosedostat for 4 months in a 1:1 ratio. Patients who met the eligibility criteria were randomly assigned to treatment by block method via an interactive web response system using a randomisation schedule generated by an external vendor. Sites, sponsor and study personnel had no access to the randomisation codes. There was no stratification performed. This was an open label study.
Procedures
The initial design of the OPAL study had two parts: Part A to assess the efficacy and safety of two dosing regimens of tosedostat followed by Part B, an expansion cohort for the most favorable schedule. Part B was cancelled by the sponsor in favour of proceeding to a future phase III study.
Study visits were scheduled at baseline, day 2, day 15, and then monthly for 6 months, plus an additional visit 28 days after last dose of study drug (Figure 1). Visits at months 1, 2, 3 and 6 included bone marrow aspiration. The protocol allowed the month 1 bone marrow aspiration to be omitted if peripheral blood results showed clearly that the patients was not yet responding, because previous data had shown that at least 2 months treatment was needed for a response and also to reduce unnecessary procedures in patients. During scheduled visits patients had a review of safety events, biochemistry, complete blood counts, pharmacokinetic sampling and ECG. ECG data was transmitted to a central reading facility (Biomedical Systems, Brussels, Belgium). The results reported here represent bone marrow readings at the treating institution. Biochemistry and haematology samples were analysed at a central laboratory (Quintiles Laboratories, Atlanta, US and Edinburgh, UK). Pharmacokinetic samples were analysed at Quotient Bioresearch, Cambridgeshire, UK. Data Monitoring at sites was performed by Quintiles, Durham, US.
Figure 1.
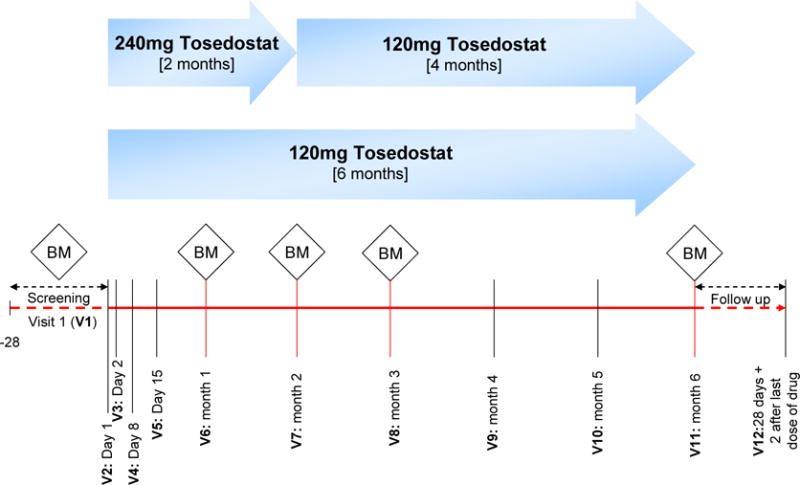
OPAL study schematic
Discontinuation of dosing of tosedostat was allowed for safety reasons, whether the event was considered related to tosedostat or not. The Investigator was asked to document the reasons for discontinuation, and contact the Medical Monitor to discuss the case and possible options. In addition, for subjects receiving 240 mg, the Investigator could reduce the dose of tosedostat to 120 mg once daily for safety reasons for which the Investigator considered temporary or permanent discontinuation was unnecessary. The investigator could also interrupt tosedostat treatment, for a maximum of 2 weeks, in certain circumstances, for example awaiting resolution of a grade 2, 3, or 4 adverse event. If the AE had not resolved after 14 days, the Investigator was asked to discuss the event with the Medical Monitor. If a patient required multiple treatment interruptions, the Medical Monitor was to be consulted for a discussion on the patient’s continued participation in the study. Patients in the 240 mg→120 mg group were allowed to be kept on the 240 mg dose after 2 months, if in the opinion of the Investigator the subject would benefit from remaining on the higher dose.
Patients were allowed to receive hydroxyurea treatment at a daily dose of up to 3g per day during the first 28 days, and leukapheresis, colony stimulating factors, blood transfusions, anticoagulant treatment and other measures of supportive care as prescribed by the investigator. Drug accountability was monitored using a patient diary and pill count returns. An extension study named TOPAZ was set up for patients who completed OPAL and wished to continue to receive tosedostat.
The primary objective of Part A was to evaluate the safety and efficacy of 2 dose regimens of tosedostat to determine an appropriate dose for expansion in Part B. The secondary objectives were to evaluate the safety and tolerability of tosedostat, to further evaluate the efficacy of tosedostat using secondary efficacy parameters and to measure the trough plasma concentrations of tosedostat and its active metabolite CHR-79888. Efficacy endpoints were based on the IWG criteria8, modified with an additional response group of stable disease, defined as any response other than CR, CRp, MLFS, PR or PD. Adverse events were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0). All patients who received at least one dose of tosedostat were included in the safety analysis.
Statistical Analysis
The study had analyses at 3 months (interim, presented at ASCO 20119) and at 6 months (final, reported here). All efficacy and safety analyses were based on study data collected from randomised patients who received at least one oral dose of tosedostat and were performed using SAS 9.2. The planned sample size for Part A of the study was 70 patients. This was based on a sample size of 35 in each group which would give a two-sided 95% CI with limits plus or minus 13·3% from the expected proportion of 20% of subjects with a CR or CRp 30%7 of patients. The study was not formally powered to show a difference between dose groups. Outcomes of subsets are presented descriptively.
Response data was summarized by treatment group. Any patient with no post-baseline bone marrow data was considered a non-responder. Overall survival was measured as the time from the first date of tosedostat dosing until death or last follow-up.
Following the planned interim analysis, a number of post-hoc additional analyses were added. Subgroup analyses of ORR and OS were done for the prognostic factors prior myelodysplastic syndrome (MDS), prior AML therapy, AML type, cytogenetics10, remission experience, and baseline bone marrow blasts.
Limited PK sampling was performed in all patients who agreed to this optional procedure. Analysis was performed on a per protocol basis, i.e. samples only taken from patients who had received tosedostat.
The study was registered on clintrials.gov (NCT00780598).
Role of the Funding Source
The study was sponsored by Chroma Therapeutics. AC, RS and MT are employees of Chroma Therapeutics. The study was designed by the sponsor and Steering Committee members (JC, EF and KY). Data was collected by study investigators and monitored by a contract research organization funded by Chroma Therapeutics. Primary analysis was performed by the contract research organization and subsequent analysis performed by RS and reviewed by MT. The manuscript was written by MT and reviewed, modified and approved by all authors. All authors had access to all raw and processed data from the study. The corresponding author had final responsibility for the integrity of the data and submission of the manuscript.
Results
Patient Characteristics
Seventy-three patients were randomised and received at least one dose of tosedostat (Figure 1). The first patient was randomized on October 21st, 2009 and the last subject on September 16th, 2010. The last study visit was on March 21st, 2011. Patients received a mean of 69·3 days (median 54 days) of tosedostat overall, with little difference between cohorts (Table 1). Twenty-two patients (30%) completed more than 84 days (3 months) of tosedostat. Eighteen patients (25%) completed less than 29 days of dosing.
Table 1.
Extent of exposure
| Duration of exposure (days), N (%) | Tosedostat 120 mg N=38 |
Tosedostat 240 mg→120 mg N=35 |
Overall N=73 |
|---|---|---|---|
|
| |||
| 0 to <29 | 9 (24) | 9 (26) | 18 (25) |
| ≥29 to <57 | 11 (29) | 10 (29) | 21 (29) |
| ≥57 to <85 | 7 (18) | 5 (14) | 12 (16) |
| ≥85 to <113 | 3 (8) | 3 (9) | 6 (8) |
| ≥113 to <141 | 2 (5) | 1 (3) | 3 (4) |
| ≥141 | 6 (16) | 7 (20) | 13 (18) |
| Mean (StD) | 69·1 (51·83) | 69·5 (53·90) | 69·3 (52·46) |
| Median | 56·0 | 53·0 | 54·0 |
| Min, Max | 3, 177 | 5, 175 | 3, 177 |
Max=maximum; Min=minimum; N=number of patients; StD=standard deviation
Baseline demographics and characteristics are presented in Table 2 and previous AML treatment characteristics in Table 3. Patients had a median age of 72 years with 32% aged 75 years or greater, and forty-three (59%) were male. Of the 73 patients, 28 (38%) had AML with multilineage dysplasia, of whom 19 (26% overall) had prior MDS, and 38 (52%) and 26 (36%) had received primary AML induction treatments with conventional chemotherapy regimens, most commonly a ‘3 plus 7’ regime, and hypomethylating agents, respectively. Thirty-eight (52%) had not reached a CR from primary induction, and of the 35 who had reached a CR with prior induction therapy, that CR had lasted less than 6 months for 14 (19% overall) and between 6 and 12 months for 21 (29%). Patients treated with standard chemotherapy were more likely to have had a CR from first induction (30/42, 71%), and a longer remission (214.5 days) than those treated with HMAs (5/26, 19%, 151 days). None of the 5 patients treated with other regimens had a CR. Treatment groups were generally balanced, albeit with small differences. Patients randomised to the 240 →120 mg group were more likely to be female (51% vs 32%), more likely to have been refractory (57% vs 47%), less likely to have had a previous CR lasting between 6 and 12 months (20% vs 37%), and more likely to have a poor karyotype (43% vs 34%).(all patients evaluable for safety and survival) Twenty-five patients received at least one dose of hydroxyurea during the first 28 days of therapy.
Table 2.
Demographics and baseline characteristics
| Parameter | Tosedostat 120 mg N=38 |
Tosedostat 240 mg→120 mg N=35 |
Overall N=73 |
|---|---|---|---|
| Age, (years) | |||
| Median (min, max) | 73 (64, 86) | 71 (65, 86) | 72 (64, 86) |
| Age ≥ 75 years, N (%) | 14 (37) | 9 (26) | 23 (32) |
| Gender, N (%) | |||
| Male | 26 (68) | 17 (49) | 43 (59) |
| Female | 12 (32) | 18 (51) | 30 (41) |
| Baseline AML diagnosis classification | |||
| AML with recurrent genetic abnormalities | 0 | 2 (6) | 2 (3) |
| AML with multilineage dysplasia | 15 (39) | 13 (37) | 28 (38) |
| With prior MDS | 9 (24) | 10 (29) | 19 (26) |
| Without prior MDS | 6 (16) | 3 (9) | 9 (12) |
| AML and MDS, therapy related | 2 (5) | 5 (14) | 7 (10) |
| AML, not otherwise categorised | 21 (55) | 15 (43) | 36 (49) |
| Previous AML Regimen | |||
| Chemotherapy | 22 (58) | 20 (57) | 42 (58) |
| Hypomethylating Agent | 15 (39) | 11 (31) | 26 (36) |
| Other regimens (including LDAC) | 1 (3) | 4 (11) | 5 (7) |
| Prognostic karyotype category, N (%)a | |||
| Better risk | 1 (3) | 0 | 1 (1) |
| Intermediate risk | 23 (61) | 19 (54) | 42 (58) |
| Poor risk | 13 (34) | 15 (43) | 28 (38) |
| AML, N (%) | |||
| Primary | 26 (68) | 21 (60) | 47 (64) |
| Secondary - prior therapy | 2 (5) | 5 (14) | 7 (10) |
| Secondary - prior MDS | 10 (26) | 9 (26) | 19 (26) |
| Bone Marrow Blasts at baseline (%) | n=38 | n=33 | n=71 |
| Median (range) | 32 (6–95) | 37 (4–91) | 32 (4–95) |
| >30% blasts, n (%) | 20 (53%) | 17 (52%) | 37 (52%) |
| White Blood Cell count at baseline (× 109/l) | n=38 | n=34 | n=72 |
| Median (range) | 2.7 (0.3 – 157.4) | 3.1 (0.5 – 153.8 | 2.75 (0.3 – 157.4) |
According to NCCN Practice Guidelines in Oncology – AML v1, 2008, including molecular data when available. Data unavailable for 2 patients
Table 3.
Previous AML treatment
| Parameter | Tosedostat 120 mg N=38 |
Tosedostat 240 mg→120 mg N=35 |
Overall N=73 |
|---|---|---|---|
|
| |||
| Remission experience, N (%) | |||
|
| |||
| Refractory (no prior CR) | 18 (47%) | 20 (57%) | 38 (52%) |
| Prior Chemotherapy | 5 (13%) | 7 (20%) | 13 (34%) |
| Prior Hypomethylating Agent | 12 (32%) | 9 (26%) | 20 (77%) |
| Prior Other (inc. LDAC) | 1 (3%) | 4 (11%) | 6 (66%) |
|
| |||
| Prior CR lasting up to 6 months | 6 (16%) | 8 (23%) | 14 (19%) |
|
| |||
| Prior CR lasting between 6 to 12 months | 14 (37%) | 7 (20%) | 21 (29%) |
|
| |||
| Duration of Prior CR (days) | |||
|
| |||
| Median (IQR) a | |||
| Prior Chemotherapy | 210 (157, 253) | 183 (99, 281) | 208 (117, 267) |
| Prior Hypomethylating Agent | 218 (192, 254) | 183 (99, 277) | 215 (192, 267) |
| Prior Other (inc. LDAC) | 151 (103, 162) | 218 (107, 318) | 151 (107, 162) |
|
| |||
| ECOG Performance Status, N (%) | |||
|
| |||
| 0 | 8 (21) | 5 (14) | 13 (18) |
|
| |||
| 1 | 23 (61) | 20 (57) | 43 (59) |
|
| |||
| 2 | 7 (18) | 10 (29) | 17 (23) |
Patients with no prior CR were excluded from the calculation of median
IQR = Inter Quartile Range
Pharmacokinetics
Within each separate dose group, trough plasma concentration of the active metabolite CHR-79888 was similar at days 2, 8 and 29. However, comparing dose groups across time, mean and median concentrations were higher in those who received 240mg compared to those who received 120 mg at all sampling points. Mean concentrations were 1.4 to 2.7 times higher (in ng/ml; 445 vs 167 at day 2, 448 vs 315 at day 8 and 439 vs 239 at day 29) and median 1.6 to 2.8 times higher (in ng/ml; 412 vs 147 at day 2, 347 vs 217 at day 8 and 329 vs 204 at day 29). These results are consistent with previous monotherapy studies of tosedostat7.
Response Data
The primary outcome for the final analysis was the rate of CR or CRp. There were 7 responders (10%): 2 (5%) in the 120 mg group and 5 (14%) in the 240 →120 mg group (Table 5). For the secondary outcome of PR or better there were 16 responders (22%), 8 (21%) in the 120 mg group and 8 (23%) in the 240 →120 mg group.
Table 5.
Changes in percentage blasts in bone marrow
| Bone Marrow Blasts | Tosedostat 120 mg | Tosedostat 240 mg→120 mg | Overall |
|---|---|---|---|
|
| |||
| Baseline, median (IQR), % | 32 (16.5, 55) | 35 (20. 70) | 33 (18, 60) |
| Month 1 Bone Marrow | n=20 | n=20 | n=40 |
| Median change (IQR), % | 11 (−30, 170) | 14 (−12, 132) | 11 (−18, 165) |
| Patients with 50% reduction, n, % | 3 (15%) | 3 (15%) | 6 (15%) |
| Patients reaching 25% blasts, n | 4 | 4 | 8 |
| Patients reaching 5% blasts, n | 2 | 1 | 3 |
| Month 2 Bone Marrow | n=22 | n=16 | n=38 |
| Median change (IQR), % | 20 (−41, 44) | −15 (−68, 94) | −3 (−65, 71) |
| Patients with 50% reduction, n, % | 4 (18%) | 7 (44%) | 11 (29%) |
| Patients reaching 25% blasts, n | 7 | 8 | 15 |
| Patients reaching 5% blasts, n | 1 | 2 | 3 |
| Month 3 Bone Marrow | n=13 | n=10 | n=23 |
| Median change (IQR), % | 17 (−70, 46) | −40 (−85, −5) | (−17 (−85, 44) |
| Patients with 50% reduction, n, % | 4 (31%) | 5 (50%) | 9 (39%) |
| Patients reaching 25% blasts, n | 5 | 5 | 10 |
| Patients reaching 5% blasts, n | 3 | 2 | 5 |
| Month 6 Bone Marrow | n=5 | n=5 | n=10 |
| Median change (IQR), % | 87.5 (−9, 149) | −89 (−97, −23) | −16 (−93, 88) |
| Patients with 50% reduction, n, % | 1 (20%) | 3 (60%) | 4 (40%) |
| Patients reaching 25% blasts, n | 1 | 4 | 5 |
| Patients reaching 5% blasts, n | 1 | 3 | 4 |
Changes in percentage blasts from baseline include reductions (−) and increases (+)
Patients reaching 25% or 5% includes any patient who achieved a post baseline bone marrow blast percentage of 25 or 5% following a baseline blast percentage of any value higher than these thresholds
Of the 16 responses at PR or better, 1 achieved CR, 6 CRp, 2 Morphologic Leukaemia Free State (MLFS) and 7 PR. A further 21 (29%) had stable disease, into which was classified any patient with bone marrow data post baseline who did not fall into one of the other response categories. Thirteen (18%) had progressive disease and 23 (32%) were non-evaluable for efficacy (all patients evaluable for safety and survival) because they did not have a post-baseline bone marrow sample taken. Of these 23 patients, 9 left the study before 2 weeks and a further 8 between 2 and 4 weeks. Reasons for these patients leaving the study were death (3 patients), disease progression (7 patients), adverse events (9 patients) and withdrawal of consent (4 patients).
Median time to a response of PR or better was 56 days (IQR 30, 62) and was similar in both dose groups (120mg, 51 days, IQR 29, 74; 240 →120mg, 56 days, IQR 34, 59). Median duration of response of PR or better was 39 days (IQR 29, 101); 35 days (IQR 29, 85) in the 120mg group and 62 days (IQR 31, 125) in the 240 →120mg group. This data includes censored data for patients with responses which were ongoing at the completion of OPAL, and this was the case for 8 of the 16 responders in OPAL. Further follow-up of these censored patients in the TOPAZ extension study and outside OPAL showed a median duration of response of PR or better of 84 days (IQR 31. 136); 62 days (IQR 29, 97) in the 120mg group and 102 days (IQR 39, 151) in the 240 →120mg group. Median duration of response for patients who had a CR/CRp/MLFS was longer, with a median of 121 days (IQR 85, 143); 110 days (IQR 78, 62) in the 120mg group and 121 days (112, 143) in the 240 →120mg group.
Changes in the percentage of blasts in bone marrow are given in Table 5. Median blast count at baseline was 33% (32% in the 120mg group, 35% in the 240mg group). At month 1, the median blast count was 11% higher than baseline, (IQR 18% lower to 165% higher), and 6 patients (15%) with a 50% reduction in blast count. At month 2, the median blast count was 3% lower than baseline (IQR -65%, +71%) and 11 patients (29%) with a 50% reduction compared to baseline. At month 3, median blast count was 17% lower (IQR -85%, +44%) and 9 (39%) of patients 50% lower than baseline. At month 6 median blast count was 16% lower (IQR -93%, +88%) and 4 (40%) of patients 50% lower than baseline. Changes in medians appeared to be slightly greater in the 240mg group, although this was not formally statistically tested.
Survival
Median overall survival for the study population was 126 days (interquartile range (IQR) 58, 218, range 4 to 697 days) (Figures 3 and 4 and webappendix). In a landmark analysis which excluded 6 patients who did not survive the first 28 days, survival varied between response groups, with the group of patients with a bone marrow response of CR, CRp or MLFS having a median survival of 322 days (CI 181, 488). Patients with a PR had a median overall survival of 195 days (CI 71, 433), those with stable disease 162 days (CI 103, 216), and the group of patients with progressive disease or who were non-evaluable, 76 days (CI 45, 107). There were some differences between dosing groups, with an overall survival of 181 days (CI 138, 204) in the 120 mg group and 104 days (CI 69, 168) in the 240 →120 mg group. Event free survival (Table 5), defined as time from first dose to death from any cause, disease progression, change or discontinuation or censored at study end (6 months) showed a similar relationship with response, with a median for all patients of 54 days (IQR 28, 85), and a median in days by response category of 142.5 days (IQR 84.5, 169.5) for PR or better, 168 (CI 85, 172) for CR/CRp/MLFS, 84 (CI 30, 142) for PR, 58 (CI 56, 79) for SD and 27.5 (CI 18, 29) for PD/unevaluable.
Figure 3.
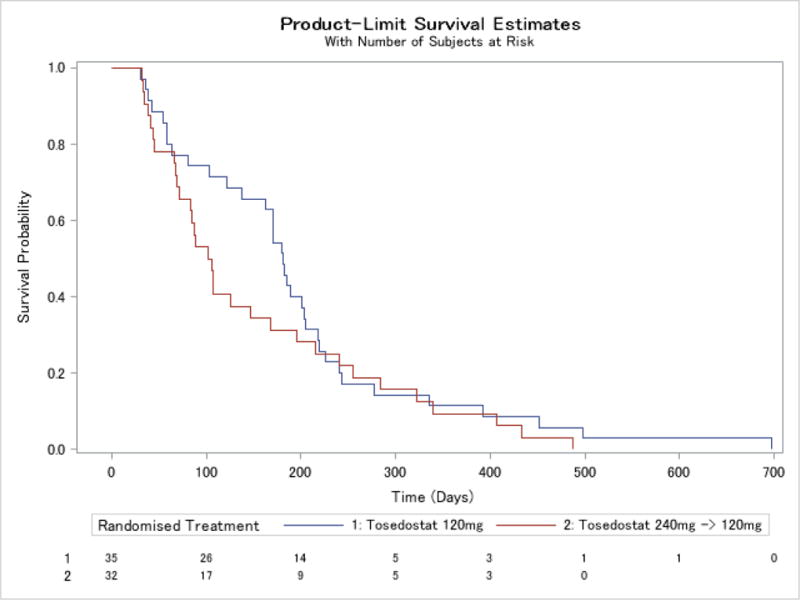
Kaplan-Meier curve for time to death for patients who survived >28 days by treatment group
Figure 4.
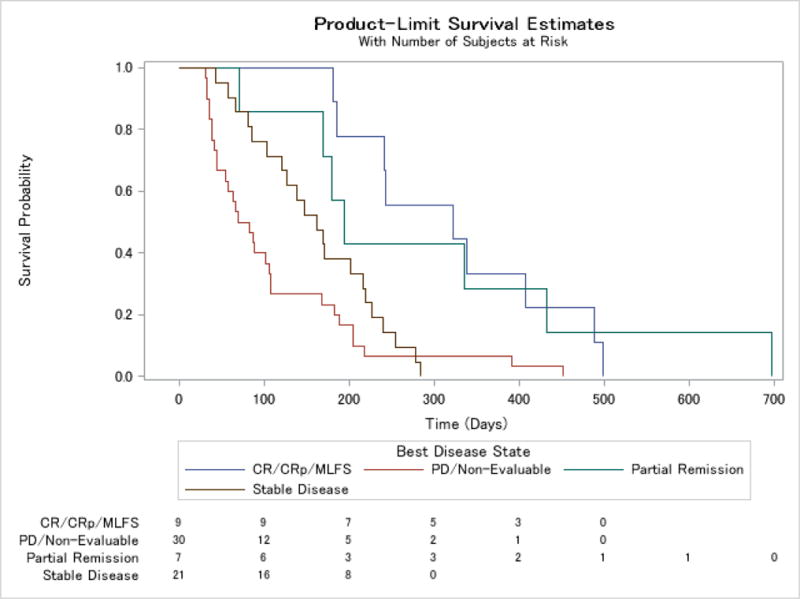
Kaplan-Meier curve for time to death for all patients who survived >28 days by response
Safety
All patients had at least one adverse event, 85% of patients had a serious adverse event and 92% had an adverse event at grade 3 or higher, regardless of causality (Table 7). Rates appeared similar between the dosing groups. Adverse events with an outcome of death were more common in the 240 →120 mg group (49% vs 34%), as were adverse events leading to study termination (31% vs 24%) and adverse events with CTCAE grade 3 or higher (94% vs 90%).
Table 7.
Adverse events summary
| Number of Patients with at least one: | Tosedostat 120 mg N=38 |
Tosedostat 240 mg→120 mg N=35 |
Overall N=73 |
|---|---|---|---|
|
| |||
| AE | 38 (100) | 35 (100) | 73 (100) |
| SAE | 32 (84) | 30 (86) | 62 (85) |
| AE with outcome of death | 13 (34) | 17 (49) | 30 (41) |
| Treatment-relateda AE | 23 (61) | 25 (71) | 48 (66) |
| AE leading to study terminationb | 9 (24) | 11 (31) | 20 (27) |
| AE leading to a dose reduction | 0 | 3 (9) | 3 (4) |
| AE leading to temporary dose interruption | 19 (50) | 17 (49) | 36 (49) |
| CTCAE grade 3, 4 or 5 AE | 34 (90) | 33 (94) | 67 (92) |
CTCAE=Common Terminology Criteria for Adverse Events v3.0
Grade 3=severe AE; Grade 4=life-threatening or disabling AE; Grade 5=Death related to AE
Treatment-related=possibly, probably or definitely related to study drug.
Includes both AEs leading to study termination and AEs leading to permanent discontinuation of study drug.
The most common adverse events observed are listed in Table 8 and serious adverse events in Table 9. The most common adverse events, regardless of causality, at any grade and grade 3+, respectively, were diarrhoea (n=42, 58%;n=3, 4%), peripheral oedema (40, 55%;0 0%), fatigue (36, 49%;15, 21%), dyspnoea (30, 41%:12, 16%), nausea (28, 38%;0, 0%), decreased appetite (27, 37%;2, 3%), hypotension (26, 36%;7,10%), febrile neutropenia (26, 36%;21, 29%), dizziness (25, 34%;0, 0%), pyrexia (23, 32%;2 3%) and cough (23, 32%;1, 1%). The most commonly reported serious adverse events were febrile neutropenia (21, 29%), disease progression (11, 15%), atrial fibrillation (6, 8%), pneumonia (6, 8%), pyrexia (4, 6%) and cardiac failure, cellulitis, dyspnea, multi-organ failure, rectal haemorrhage and respiratory failure (each 3, 4%).
Table 8.
Incidence of Adverse Events Occurring in ≥20% of Patients
| System Organ Class Preferred term CTCAE grade |
Tosedostat 120 mg N=38 |
Tosedostat 240 mg→120 mg N=35 |
Overall N=73 |
|||
|---|---|---|---|---|---|---|
| All | 3+ | All | 3+ | All | 3+ | |
| Percentage of Patients with at least one AE | 100 | 89.5 | 100 | 94.3 | 100 | 91.8 |
| Diarrhoea | 52·6 | 5·3 | 62·9 | 2·9 | 57·5 | 4·1 |
| Oedema peripheral | 47·4 | 0 | 62·9 | 0 | 54·8 | 0 |
| Fatigue | 42·1 | 18·4 | 57·1 | 22·9 | 49·3 | 20·5 |
| Dyspnoea | 42·1 | 13·2 | 40·0 | 20 | 41·1 | 16·4 |
| Nausea | 34·2 | 0 | 42·9 | 0 | 38·4 | 0 |
| Decreased appetite | 31·6 | 2·6 | 42·9 | 2·9 | 37·0 | 2·7 |
| Hypotension | 31·6 | 7·9 | 40·0 | 11·4 | 35·6 | 9·6 |
| Febrile neutropenia | 34·2 | 28·9 | 37·1 | 28·6 | 35·6 | 28·8 |
| Dizziness | 31·6 | 0 | 37·1 | 0 | 34·2 | 0 |
| Cough | 31·6 | 0 | 31·4 | 2·9 | 31·5 | 1·4 |
| Pyrexia | 31·6 | 5·3 | 31·4 | 0 | 31·5 | 2·7 |
| Asthenia | 23·7 | 2·6 | 31·4 | 8·6 | 27·4 | 5·5 |
| Hypokalaemia | 26·3 | 5·3 | 28·6 | 8·6 | 27·4 | 6·8 |
| Constipation | 26·3 | 0 | 20·0 | 0 | 23·3 | 0 |
| Insomnia | 23·7 | 2·6 | 22·9 | 0 | 23·3 | 1·4 |
| Rash | 23·7 | 5·3 | 22·9 | 0 | 23·3 | 2·7 |
| Pneumonia | 21·1 | 10·5 | 25·7 | 17·1 | 23·3 | 13·7 |
| Vomiting | 21·1 | 0 | 22·9 | 0 | 21·9 | 0 |
| Thrombocytopenia | 21·1 | 21·1 | 22·9 | 22·9 | 21·9 | 21·9 |
Treatment-emergent is defined as an AE that started on or after first dose and within 28 days of last dose or an AE that started prior to treatment but worsened during treatment.
Table 9.
Serious adverse events >3% incidence
| System Organ Class | Tosedostat 120 mg N=38 |
Tosedostat 240 mg→120 mg N=35 |
Overall N=73 |
|
|---|---|---|---|---|
| Preferred term | N (%) | |||
|
| ||||
| Number of Patients with at least one SAE | 32 (84·2) | 30 (85·7) | 62 (84·9) | |
| Febrile neutropenia | 9 (23·7) | 12 (34·3) | 21 (28·8) | |
| Disease progression | 6 (15·8) | 5 (14·3) | 11 (15·1) | |
| Atrial fibrillation | 1 (2·6) | 5 (14·3) | 6 (8·2) | |
| Pneumonia | 1 (2·6) | 5 (14·3) | 6 (8·2) | |
| Pyrexia | 3 (7·9) | 1 (2·9) | 4 (5·5) | |
| Cardiac failure congestive | 1 (2·6) | 2 (5·7) | 3 (4·1) | |
| Cellulitis | 2 (5·3) | 1 (2·9) | 3 (4·1) | |
| Dyspnoea | 1 (2·6) | 2 (5·7) | 3 (4·1) | |
| Multi-organ failure | 2 (5·3) | 1 (2·9) | 3 (4·1) | |
| Rectal haemorrhage | 0 | 3 (8·6) | 3 (4·1) | |
| Respiratory Failure | 3 (7·9) | 0 | 3 (4·1) | |
Treatment-emergent is defined as an AE that started on or after first dose and within 28 days of last dose or an AE that started prior to treatment but worsened during treatment.
Adverse events considered to be possibly, probably or definitely related to tosedostat were reported for 48 patients (66%); 23 (61%) in the 120mg group and 25 (71%) in the 240 →120 mg group. The most common events considered by the investigator to be possibly, probably or definitely related to tosedostat were diarrhoea (n=15 (21%) overall; 5 (13%) in the 120 mg group; 10 (29%) in the 240 group), nausea (13 (18%); 7 (18%); 6 (17%)), oedema peripheral (13 (18%); 8 (21%), 5 (14%)), fatigue (14 (19%); 6 (16%); 8 (23%)), decreased appetite (13 (18%); 5 (13%); 8 (23%)), febrile neutropenia (10 (14%); 6 (16%); 4 (11%)) and rash (7 (10%); 3 (8%); 4(11%)).
Dose reductions for toxicity occurred on 16 occasions for 13 patients in the 120 mg group, of which 13 interruptions were for 7 days or less and the maximum was 15 days. There were dose reductions on 14 occasions for 10 patients in the 240 mg→120 mg group, of which 9 interruptions were for 7 days or less and the maximum was 19 days. One patient in the 240 mg→120 mg group had a total of 33 days across several interruptions due to toxicity.
Discontinuations for tosedostat-related toxicity occurred in 14 patients (19%); 9 (24%) in the 120 mg group and 5 (14%) in the240 mg→120 mg group. 13 of these events were considered serious events and there was 1 non-serious event in the 240 mg→120 mg group. The serious adverse events included rash (3 cases) disease progression (2 cases), febrile neutropenia, congestive heart failure, acute respiratory failure, lower GI haemorrhage, shortness of breath associated with pneumonia, diabetic ketoacidosis and paroxysmal atrial fibrillation and left ventricular dysfunction. The non-serious adverse event was pain associated with polymyalgia rheumatica.
There were 5 adverse events with an outcome of death, 3 in the 120 mg group and 2 in the 240 mg→120 mg group. The events were acute hepatitis, respiratory failure, pneumonia, atrial fibrillation and left ventricular dysfunction.
Predictors of response
These post-hoc analyses are listed in the webappendix and were intended to explore the relationship between patient and disease characteristics and response rates and survival in OPAL, and were performed on the pooled population with both dosing groups combined. Higher response rates and/or survival (days) were observed in patients who had previously received hypomethylating agents (5-azacytidine or decitabine) as primary induction compared with those treated with other regimes (10/26, 38% PR or better, median survival 170.5 days (IQR 58, 255) vs 6/47 13%, 106 days (IQR 54, 205), respectively); AML with multilineage dysplasia or prior MDS compared with those with AML not otherwise categorised (10/28, 36%, 182.5 days (IQR 106.5, 242), 7/19, 37%, 185 days (IQR 103, 240) vs 5/26, 14%, 67·5 days(IQR 34.5, 202.5)); and patients who had been refractory (i.e. had not obtained a CR) from previous primary induction compared to those who had a previous CR (11/38, 29%, 169 days (IQR 67, 240) vs 5/35, 14%, 103 days(IQR 145, 195)). In contrast, there were no differences in outcome for patients who had baseline bone marrow blasts of ≤30% or >30% (8/34, 24%, 136·5 days (IQR 44, 278) vs 7/37, 19%, 121 days (IQR 66, 195)), and with poor versus intermediate or better prognostic karyotype (6/28, 21%, 158.5 days (76, 240.5) vs 10/43, 23%, 107 days (IQR 44, 216)).
Discussion
In the OPAL study, 73 patients were treated with tosedostat. Seven patients (10%) achieved CR or a complete remission with incomplete platelet recovery (CRp): 2 in the 120 mg group (5%) and 5 (14%) in the 240 mg→120 mg group. The most common adverse events at grade 3 or worse were febrile neutropenia which occurred in 21 (29%) overall, 11 (29%) of the 120 mg group and 10 (29%) of the 240 mg→120 mg group, thrombocytopenia (16, 22%; 8, 21% and 8, 23%), fatigue (15, 21%; 7, 18% and 8, 23%), dyspnoea (12, 16%; 5, 13% and 7, 20%), pneumonia (10, 14%; 4, 11% and 6, 17%). No clear differences were observed in efficacy or safety between the two dose schedules, but a promising overall response rate (PR or better) of 22% was observed. There was a suggestion for a survival benefit for patients who achieved a PR or better. A further 29% of patients achieved stable disease (SD). Additional analyses suggested that a trend for a higher response rate and survival was observed in patients with prior MDS or who had received hypomethylating agents for previous first induction, as well as patients who had multilineage AML compared to other AML types.
A clear unmet medical need exists for an agent to treat elderly patients who have failed to adequately respond to first-line treatment. Treatment options are limited for this patient population. Similarly, limited therapeutic options are available for patients who, after receiving hypomethylating therapy, experience AML progression from MDS. In a retrospective analysis of 74 such patients, the median overall survival was 3.4 months, with a 1-year survival probability of 8%11. Median survival was 2 months for those treated with best supportive care or palliative chemotherapy (i.e. hydroxyurea or 6-mercaptopurine), while only 1 of 13 patients treated with “active regimens” (i.e. intensive AML-like chemotherapy, other epigenetic therapies, or SCT) was alive after 14 months. Patients on cytarabine-based intensive AML therapy had a response rate of 14% and survival of 8·9 months. Only intensive therapy, allogeneic SCT, and experimental therapies appeared to improve survival over supportive care. Unfortunately, the majority of patients with relapsed AML are elderly and frail and ineligible for intensive therapy or SCT.
Tosedostat is a chronically administered oral agent previously evaluated in patients with solid tumours that has shown anti-leukaemia activity in phase I-II trials in elderly patients with RR AML7. Complete responses were observed and responses proved durable in some patients. The OPAL study was designed to further evaluate the efficacy of tosedostat in patients with RR AML and to determine if there are important differences in efficacy or toxicity between daily dosing at 120 mg for six months and daily dosing at 240 mg for two months followed by 120 mg for four months.
The magnitude of clinical effect observed in OPAL was dampened by a higher than expected early drop-out rate, with 25% of patients having received tosedostat for 28 days or fewer. The PK and mechanism of action of tosedostat suggests that patients need to be on treatment for at least 4 weeks and perhaps longer for full therapeutic effect to be reached. Future studies will focus on patients without highly proliferative disease that will be able to remain on therapy for longer to allow a full treatment effect to occur. This will help establish whether tosedostat may influence the survival of patients in this category, particularly when compared to what could be expected with standard therapy. Good quality data on hospitalization rates and transfusion requirements was not available for this study. These are important outcomes for patients with AML, and not having these data is a potential limitation of the study.
The finding that patients with prior therapy with hypomethylating agents and those with prior MDS had a trend for a better response may be of particular importance since none of the existing agents has been shown to be effective in this setting. Patients with prior MDS are more likely to have less proliferative AML, and this type of patient is more likely to be selected for a low intensity regime such as a hypomethylating agent and may simply be a sign that they have a more indolent disease. However, there might be other factors which make the prior MDS patient more suitable for tosedostat. Although there is no currently available data for the use of tosedostat for patients with MDS who have failed prior hypomethylating agent therapy, based on this preliminary observation findings further studies with tosedostat in this patient population are currently under evaluation. Patients with shorter remissions or with no prior CR had a better response to tosedostat. These patients tend to be less sensitive to standard therapy with cytarabine-based regimens. In a study analyzing the relative benefit of therapy with gemtuzumab ozogamyicin or cytarabine in the first salvage setting, those with a short first CR (less than 10·5 months) benefitted more from gemtuzumab, whereas those with longer remissions benefitted from therapy with cytarabine12. These results were obtained from non-randomised retrospective studies, so should be interpreted with caution, but appear to suggest that patients with short first CRs may be refractory to cytarabine and may require treatment options with alternative mechanisms of action. Further studies with tosedostat will investigate the effect in this particular patient population.
In conclusion, the results of the OPAL study suggest that tosedostat is an active treatment in patients with relapsed or refractory AML. Importantly, most patients were accrued in sites in the United States. Although we have no reason to believe there is bias in the selection of patients between US and other parts of the world, the possibility of differences in patient population and/or healthcare practices may alter the expectations of outcome with this or other interventions. These results form the basis for future studies assessing the activity of tosedostat as a single-agent or in combination therapy in AML and MDS. There are a number of studies ongoing or planned in MDS and AML with tosedostat. Ongoing studies include investigator-sponsored studies of (a) a collaborative group study of tosedostat in combination with conventional chemotherapy (“3 plus 7”) in first line AML, (b) a single centre study of tosedostat in combination with either decitabine or low dose cytarabine in first line AML or high risk MDS, (c) a single centre study of tosedostat in combination with either azacytidine or low dose cytarabine in older patients with first line AML or high risk MDS (d) a single centre study of tosedostat in combination with low dose cytarabine in older patients with first line AML. In addition, there is a multi-centre study ongoing of tosedostat in combination with an investigational histone deacetylase inhibitor agent CHR-3996 in patients with relapsed or refractory myeloma. Planned studies include (a) a collaborative group study of tosedostat in combination with low dose cytarabine in patients with AML and (b) a phase III study of tosedostat in combination with either hypomethylating agents or low dose cytarabine in older patients with first line AML or high risk MDS. The precise design of this study will be informed by the results of ongoing studies.
Figure 2.
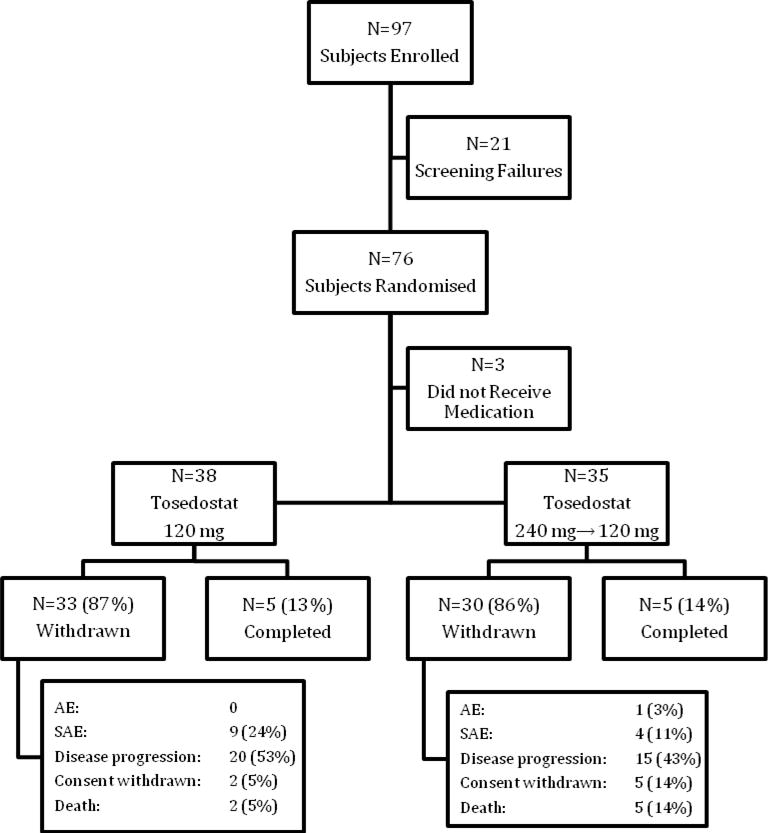
CONSORT diagram for OPAL
Figure 5.
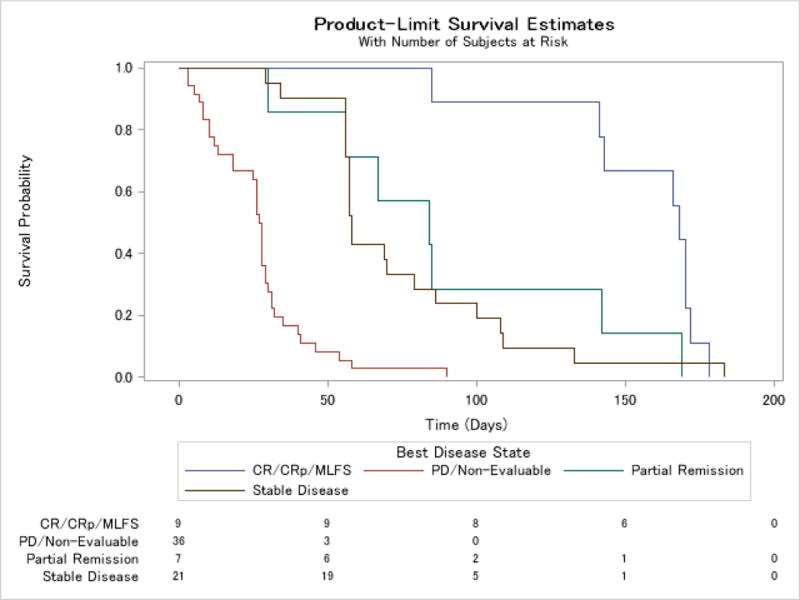
Kaplan-Meier curve for event free survival for all patients by response
Table 4.
Summary of best response to therapy
| Best Response | Tosedostat 120 mgN=38 N (%) |
Tosedostat 240 mg→120 mg N=35 N (%) |
Overall N=73 N (%) |
|---|---|---|---|
|
| |||
| PR or better | 8 (21) | 8 (23) | 16 (22) |
|
| |||
| CR | 0 | 1 (3) | 1 (1) |
| CRp | 2 (5) | 4 (11) | 6 (8) |
| MLFS | 2 (5) | 0 | 2 (3) |
| PR | 4 (11) | 3 (9) | 7 (10) |
| SD | 14 (37) | 7 (20) | 21 (29) |
| PD | 6 (16) | 7 (20) | 13 (18) |
| Non-evaluable | 10 (26) | 13 (37) | 23 (32) |
CR=complete remission; CRp=complete remission with incomplete platelet recovery; MLFS=morphological leukaemia-free state; PR=partial remission; SD=stable disease; PD=progressive disease;
Table 6.
Event-free survival
| Event Free Survival (median number of days; interquartile range)a | Tosedostat 120 mg N=38 |
Tosedostat 240 mg→120 mg N=35 |
Overall N=73 |
|---|---|---|---|
|
| |||
| All patients | 56 (28, 85) | 41 (26, 90) | 54 (28, 85) |
|
|
|||
| PR or Better | 125.5 (84.5, 168.5) n=8 | 142.5 (104, 170) n=8 |
142.5 (84.5,169.5) n=16 |
| CR/CRp/MLFS | 167 (125.5, 173) n=4 |
170 (143, 170) n=5 |
168 (143, 170) n=9 |
| PR | 84.5 (70, 127) n=4 |
67 (30, 142) n=3 |
84 (56, 142) n=7 |
| SD | 63 (56, 86) n=14 |
58 (56, 109) n=7 |
58 (56, 86) n=21 |
| PD/Non-evaluable |
26.5 (9, 28) n=16 |
28·5 (18, 37.5) n=20 |
27.5 (12.5, 31) n=36 |
Event Free Survival is defined as the time (in days) from the date of first dose to the occurrence of an event. An event is defined as the earliest of the following: death (from any cause); disease progression; change and/or discontinuation of therapy for any reason at any time during the study. Subjects who do not have an event are censored at the end of study.
CR=complete remission; CRp=complete remission with incomplete platelet recovery; MLFS=morphological leukaemia-free state; PR=partial remission; SD=stable disease; PD=progressive disease;
Research in Context.
Systematic Review
When the study was being planned in 2007 and 2008, we searched PubMed, clintrials.gov and other online sources for articles published using search terms ‘relapsed AML’, ‘refractory AML’, ‘elderly AML’, and similar terms. We concluded that there was considerable unmet need for this population.
Interpretation
Although OPAL was a small study and not formally powered to show a response benefit, the results suggest that oral tosedostat may be associated with a modest response rate and survival benefit, and seems to be well tolerated by elderly patients with relapsed/refractory AML. This benefit appears to be greater in patients with antecedent MDS or those who have received prior induction treatment with hypomethylating agents. This is a particularly interesting outcome since these patients have a very poor prognosis. Thus, additional studies in this setting are warranted. The future potential for tosedostat when given as a combination agent with currently available agents to patients with MDS and AML is also currently being evaluated.
Acknowledgments
The study was funded by Chroma Therapeutics Ltd, Abingdon, UK. The findings have been presented in part at the 2011 Meeting of the American Society for Clinical Oncology, Chicago, IL, USA; the 6th International Congress On Myeloproliferative Diseases And Myelodysplastic Syndromes November 3–4, 2011, New York, NY, USA; and the 53rd Annual Meeting of the American Society of Hematology, San Diego, CA, December 10–13, 2011. We thank patients and their families who participated in the study, and the investigators and clinical trial sites for their participation in the OPAL study.
Footnotes
Contributors
JC, EF, KY contributed to study design, treatment of patients, data collection and data interpretation. DR, AA and HK contributed to data collection and interpretation. AC, RS and MT contributed to study design and data interpretation. All authors contributed to writing of this report.
Conflicts of Interest
JC, EF and KY were members of the Steering Committee for this study; JC and EF received compensation from Chroma Therapeutics for this role. JC, EF, KY, DR, AA, HK received research funding from Chroma Therapeutics for this study. JC is receiving research funding from Chroma Therapeutics and Cell Therapeutics Inc. for another study with tosedostat. AC, RS and MT are employees of Chroma Therapeutics.
References
- 1.Appelbaum FR, Gundacker H, Head DR, et al. Age and acute myeloid leukemia. Blood. 2006;107:3481–3485. doi: 10.1182/blood-2005-09-3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lang K, Earle CC, Foster T, Dixon D, Van GR, Menzin J. Trends in the treatment of acute myeloid leukemia in the elderly. Drugs Aging. 2005;22:943–955. doi: 10.2165/00002512-200522110-00004. [DOI] [PubMed] [Google Scholar]
- 3.Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 4.Jousse C, Bruhat A, Harding HP, Ferrara M, Ron D, Fafournoux P. Amino acid limitation regulates CHOP expression through a specific pathway independent of the unfolded protein response. FEBS Lett. 1999;448(2–3):211–6. doi: 10.1016/s0014-5793(99)00373-7. [DOI] [PubMed] [Google Scholar]
- 5.Krige D, Needham LA, Bawden LJ, et al. CHR-2797: An Antiproliferative Aminopeptidase Inhibitor that Leads to Amino Acid Deprivation in Human Leukaemic Cells. Cancer Res 2008. 2008;68(16):6669–79. doi: 10.1158/0008-5472.CAN-07-6627. [DOI] [PubMed] [Google Scholar]
- 6.Reid AH, Protheroe A, Attard G, et al. A First-in-Man Phase I and Pharmacokinetic Study on CHR-2797 (Tosedostat), an Inhibitor of M1 Aminopeptidases, in Patients with Advanced Solid Tumors. Clin Can Res. 2009;15:4978–4985. doi: 10.1158/1078-0432.CCR-09-0306. [DOI] [PubMed] [Google Scholar]
- 7.Löwenberg B, Morgan G, Ossenkoppele G, et al. Phase I/II Clinical Study of Tosedostat, an Inhibitor of Aminopeptidases, in Patients With Acute Myeloid Leukemia and Myelodysplasia. J Clin Oncol. 2010;28:4333–4338. doi: 10.1200/JCO.2009.27.6295. [DOI] [PubMed] [Google Scholar]
- 8.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised Recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 9.Feldman EJ, Yee KLW, Rizzieri D, et al. Interim results of OPAL, a study of tosedostat in elderly relapsed/refractory AML. J Clin Oncol. 2011;29(suppl) abstr 6517. [Google Scholar]
- 10.O’Donnell MR, Appelbaum FR, Coutre SE, et al. Acute myeloid leukemia. JNCCN. 2008;6(10):962–992. doi: 10.6004/jnccn.2008.0074. [DOI] [PubMed] [Google Scholar]
- 11.Prebet T, Gore SD, Esterni B, et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J Clin Oncol. 2011;29:3322–3327. doi: 10.1200/JCO.2011.35.8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leopold LH, Berger MS, Cheng SC, Cortes-Franco JE, Giles FJ, Estey EH. Comparative efficacy and safety of gemtuzumab ozogamicin monotherapy and high-dose cytarabine combination therapy in patients with acute myeloid leukemia in first relapse. Clin Adv Hematol Oncol. 2003 Apr;1(4):220–5. [PubMed] [Google Scholar]