

Abstract
Translation in the mitochondria is regulated by mechanisms distinct from those acting in the cytosol and in bacteria, yet precise methods for investigating it has lagged behind. This unit describes an approach, mitochondrial ribosome (mitoribosome) profiling, to quantitatively monitor mitochondrial translation with high temporal and spatial resolution in Saccharomyces cerevisiae. Mitoribosomes are immunoprecipitated from whole cell lysate and the protected mRNA fragments are isolated. These fragments are then converted to sequencing libraries or analyzed by northern blot hybridization to reveal the distribution of mitoribosomes across the mitochondrial transcriptome. As information about RNA abundance is required to resolve translational from RNA effects, we also present an RNA sequencing approach that can be performed in parallel. Accurately capturing the biologically relevant distribution of mitoribosome positions depends on several critical parameters that are discussed. Application of mitoribosome profiling can reveal mechanisms of mitochondrial translational control that were not previously possible to uncover.
Keywords: mitochondria, translation, ribosome profiling, ribosome immunoprecipitation, RNA-seq, northern blot
INTRODUCTION
Mitochondria have their own genome that is expressed by a dedicated mitochondrial RNA polymerase and mitochondrial ribosome (mitoribosome). This unit presents a set of protocols for monitoring mitochondrial gene expression in Saccharomyces cerevisiae with high temporal resolution and in any condition (Figure 1). Basic Protocols 1 and 2 describe mitoribosome profiling: a method to monitor mitoribosome positions with codon resolution across the mitochondrial transcriptome. The method is adapted from the ribosome profiling technique developed to monitor cytosolic ribosome (cytoribosome) positions by deep sequencing ribosome-protected fragments (footprints) (Ingolia, 2010; Ingolia et al., 2009). Here, to ensure enrichment of mitoribosomes (74S) from the similar-sized but more abundant cytoribosomes (80S), they are immunoprecipitated via a 3X-FLAG epitope tag added endogenously to a mitoribosomal protein. Two critical modifications required to capture mitoribosome footprints are (1) buffer conditions that do not induce subunit dissociation, and (2) size selection that accounts for the larger footprint of mitoribosomes (Couvillion et al., 2016). Once isolated, mitoribosome footprints are used to generate a library to be sequenced on the Illumina platform.
Figure 1.
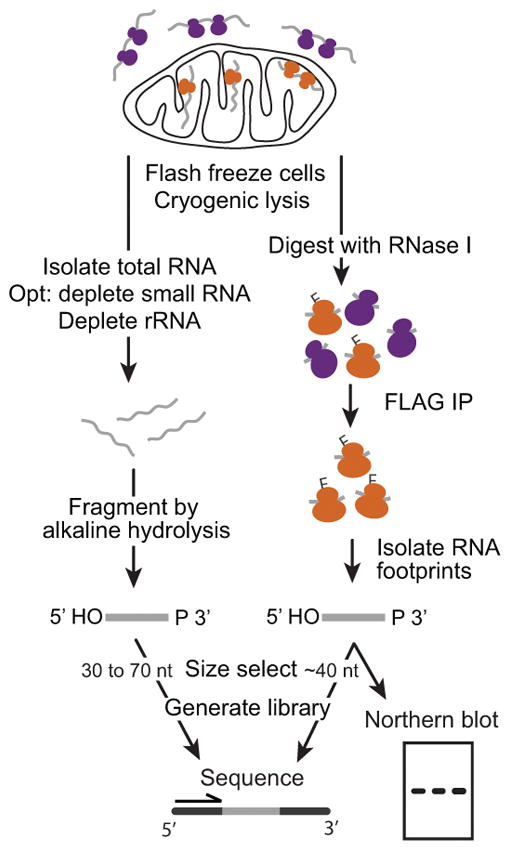
Flowchart of the protocols presented in this unit. mRNA isolation and fragmentation (left, Support Protocol), and mitoribosome footprint isolation (right, Basic Protocol 1) result in chemically similar RNA species that are then size selected and converted to libraries for sequencing (Basic Protocol 2). Alternatively, mitoribosome footprints can be analyzed by northern blot hybridization (Alternate Protocol). IP: immunoprecipitation.
Two additional protocols expand the potential of mitoribosome profiling (Support Protocol and Alternate Protocol). One, RNA sequencing (RNA-seq), performed in parallel allows the deconvolution of RNA-level and translational effects. Two, detection of mitoribosome footprints by northern blot hybridization, can be used instead of or in addition to library generation and sequencing. It provides a rapid way to verify the success of the experiment and the size range of footprints. It can also be used to compare ribosome occupancy on a transcript across conditions or strains.
Here, steps to isolate mitoribosome footprints from the yeast Saccharomyces cerevisiae are presented (Basic Protocol 1), as well as how to isolate total mRNA fragments in parallel for RNA sequencing (Support Protocol). Mitoribosome footprints (and total mRNA) can then be converted into libraries for deep sequencing (Basic Protocol 2), or footprints can be analyzed by northern blotting (Alternate Protocol).
BASIC PROTOCOL 1. ISOLATE MITORIBOSOME PROTECTED FRAGMENTS BY IMMUNOPRECIPITATION
This protocol describes the isolation of mitochondrial ribosome footprints. A yeast strain with 3X-FLAG tagged mitoribosomes is rapidly harvested and lysed under cryogenic conditions to preserve ribosome positions. Endogenously tagged mitoribosome small subunit protein MrpS17 (MrpS17-FLAG) is used here, but tagged large subunit proteins Mrp20 (Mrp20-FLAG) and Img1 (Img1-FLAG) yield similar results. The lysate is thawed and treated with RNase I to digest unprotected RNA. Next, FLAG antibody-conjugated agarose beads are used for batch purification, the beads are washed, and mitoribosomes are eluted by competition with 3X-FLAG peptide. The RNA is then extracted from the immunoprecipitate using phenol (Gallagher & Wiley, 2008).
Materials
Solutions and reagents:
Yeast strain expressing a C-terminal epitope tagged (3X-FLAG) MrpS17 grown in medium of choice
RNase-free water
Lysis buffer (including protease inhibitors and lauryl maltoside; see recipe)
50 U/μl RNase I (RNase If, NEB M0243)
5X SDS-PAGE sample buffer (see recipe)
20 U/μl SUPERase-In
Agarose slurry conjugated with FLAG antibody (Sigma)
Wash buffer (including TritonX-100; see recipe)
5 mg/ml 3X-FLAG peptide in TBS (aliquot and store at −80°C, avoid freeze-thaw; Sigma F4799)
125:24:1 (v/v/v) Acid-phenol:chloroform:isoamyl alcohol, pH 4.5
3 M sodium acetate, pH 5.5 (RNase-free)
25 mg/ml linear polyacrylamide (LPA)
Isopropanol
70% (v/v) ethanol
10 mM Tris, pH 7.0 (RNase-free)
Anti-FLAG M2 antibody (Sigma F3165)
Special equipment:
-
Microfiltration assembly (90-mm, ULTRA-WARE):
4-liter side-arm flask with a fritted glass support base
Glass funnel
Anodized aluminum clamp
No. 8 silicone stopper
Nitrocellulose (0.45-um, 90-mm diameter membranes; Whatman)
Mixer mill, 50-ml chambers and 25-mm stainless steel ball (Retsch)
100°C heat block
Floor centrifuge with rotor able to reach 20,000 x g (e.g. Sorvall with SS-34 rotor and Oak Ridge tubes)
End over end rotator
Costar Spin-X centrifuge tube filter (0.45-um cellulose acetate in 2-ml tube; Corning)
Other equipment:
Spectrophotometer
Vacuum source
Liquid nitrogen
Vacuum source
Styrofoam container
Forceps
50-ml conical tubes
15-ml conical tubes
20-G needles
Metal spatulas with one flat end and one curved end
Tongs
Cryo-gloves
Graduated cylinder
Kimwipes
−80°C freezer
Refrigerated centrifuge
Vortex
CAUTION: Phenol/chloroform is harmful if swallowed or comes in contact with skin, causes severe skin burns and eye damage, is fatal if inhaled, and is potentially carcinogenic. It should be used with appropriate safety measures such as protective gloves, glasses, clothing, and sufficient ventilation. All waste should be handled according to hazardous waste regulations.
Culture yeast cells and harvest by filtration
-
1
Culture cells (Sherman, 2002) as desired for experiment. For analysis by northern blotting, collect ~60 OD600 equivalents (small scale, ~100 ml). For analysis by sequencing, collect ~400 OD600 equivalents (large scale, ~700 ml).
Keep cell number equivalent across samples to be compared. See Treco and Winston (2008) for a discussion on relating optical density and cell number. -
2
Set up filtration apparatus and prepare 50-ml conical tubes submerged in and filled with liquid nitrogen. Additionally, use a needle to make a hole in tube caps for removal of liquid nitrogen later.
Prepare all materials prior to retrieving culture from shaker in order to minimize time between removing cells from condition of interest and freezing. -
3
Measure culture volume for desired cell number and harvest cells by filtration.
-
4
Using a room temperature flat metal spatula, scrape cells from filter and transfer to liquid nitrogen-filled tube. Do this in 3–4 iterations, being sure to remove all cells from filter.
To remove cell paste from spatula after freezing, press into wall of tube, or use a second, pre-chilled spatula to scrape it off. To quickly warm spatula after plunging into liquid nitrogen, have a beaker of room temperature water nearby, and a Kimwipe for drying. -
5
Cap tubes and remove liquid nitrogen through hole in cap.
Tubes with cell pellets can be stored for 6 months at −80°C.
Prepare frozen lysis buffer
Alternatively, lysis buffer can be prepared and frozen first and cells collected into the same tube.
-
6
Prepare 2.6 ml (small scale) or 4 ml (large scale) of 1× lysis buffer per sample. Make 2.6 or 4 ml aliquots, respectively, and chill on ice 10 min.
-
7
Prepare 50-ml conical tubes submerged in and filled with liquid nitrogen, with holes in caps as in step 2.
Additional liquid nitrogen may have to be poured on top as it is boiled off during the subsequent step. -
8
Drip lysis buffer 150 μl at a time into liquid nitrogen-filled tubes.
Wait several seconds after each drop until the buffer is completely frozen before adding more. If many samples are being prepared at once, move on to the next tubes, cycling through to reduce waiting time. -
9
Cap tubes and remove liquid nitrogen through hole in cap.
-
10
Transfer one tube of frozen buffer into each tube of frozen cells, cap, and shake to combine.
Tap tubes to remove material from cap before proceeding or storing. Samples can be stored for 6 months at −80°C.
Cryogenically lyse by mixer mill
-
11
Prechill mixer mill chamber, disassembled except for the plastic o-ring, and stainless steel ball in liquid nitrogen.
Wait for vigorous boiling to stop – this may take several minutes. -
12
Remove chamber and place frozen yeast/buffer mix, then ball, inside. Screw chamber together and place into liquid nitrogen.
-
13
Load chambers and run mixer mill 3 min at 15 Hz. Place chambers back into liquid nitrogen until vigorous boiling stops. Repeat for a total of 6 rounds.
-
14
Prepare 50-ml conical tubes submerged in liquid nitrogen, but do not fill.
-
15
Prechill rounded side of metal spatula.
-
16
Remove chamber from liquid nitrogen, open, and scrape grindate out, transferring to prepared 50-ml tube.
Remove as much of grindate as possible, transferring the steel ball a couple times and scraping the walls of both sides of the chamber. Samples can be stored for 6 months at −80°C.
Digest RNA for ribosome footprinting
-
17
For a large scale prep only, freshly prepare additional 1× lysis buffer to dilute thawed sample (see below for amount needed).
-
18
Thaw grindate in a bath of room temperature water.
As grindate thaws, gently swirl to maintain more homogenous temperatures throughout the sample. As soon as ice is melted, move immediately to the next step. -
19
For large scale prep only, add 1× lysis buffer to dilute lysate to a concentration of 25 OD600/ml. Starting volume is approximately 4 ml, so e.g., if you collected 400 OD600 cell equivalents, add 12 ml to bring volume to 16 ml. Mix gently by inverting several times.
-
20
Remove aliquot for RNA sequencing.
Yield is at least 100 ng total RNA/μl lysate. Typically 200 μl is sufficient for downstream applications. For best quality, proceed directly to isolate RNA (see Support Protocol) or alternatively, flash freeze and store for up to 6 months at −80°C. -
21
Add RNase I to 500 U/ml lysate, swirl thoroughly to mix, incubate 30 min in room temperature (25°C) water bath. Swirl to mix again after 15 min.
Optional: save 40 μl of this input sample for analysis by immunoblotting: combine with 10μ 5X SDS-PAGE sample buffer, vortex 5 sec, boil 5 min, and store −20°C. -
22
To stop digestion add Superase-In to 100 U/ml. Mix by swirling, and ice.
-
23
Centrifuge 20,000 × g at 4°C for 15 min.
For small scale prep, lysate can be transferred to microfuge tubes for centrifugation in a microcentrifuge. For large scale prep, transfer to Oak Ridge centrifuge tube and use Sorvall floor centrifuge with SS-34 rotor or equivalent.
Immunoprecipitate mitoribosomes
-
24
Prepare agarose slurry (12 μl per ml lysate) by washing 3 times in at least 40 volumes of wash buffer. To wash, invert tube several times, centrifuge 1 min at 1000 × g to pellet beads, and discard supernatant. Resuspend beads by adding 4.5 volumes wash buffer.
To avoid crushing the agarose beads, do not centrifuge at high speed. -
25
Transfer cleared lysate from step 23 to fresh tube at 4°C.
Optional: save 40 μl of this soluble sample for analysis by immunoblotting: combine with 10 μl 5X SDS-PAGE sample buffer, vortex 5 sec, boil 5 min, and store −20°C. -
26
To cleared lysate, add 60 μl of washed, diluted agarose slurry (from step 24) per ml of lysate.
Agarose beads settle quickly – pipette up and down before removing each aliquot. -
27
Incubate lysate with beads for 3 hr at 4°C, rotating end over end.
-
28
Centrifuge 2 min at 1000 × g, 4°C, to pellet beads.
Optional: save 40 μl of the supernatant as unbound sample for analysis by immunoblotting: combine with 10 μl 5X SDS-PAGE sample buffer, vortex 5 sec, boil 5 min, and store −20°C. -
29
Remove and discard supernatant
-
30
Wash beads in 50 volumes wash buffer 3 times, rotating end over end for 10 min at room temperature each time. To pellet beads between washes, centrifuge 1 min at 1000 × g.
-
31
Elute 3X -FLAG-tagged mitoribosomes from beads: After final wash, pellet beads and resuspend in 6 bead slurry volumes wash buffer containing 200 μg/ml 3X FLAG peptide.
For example, if the original lysate volume was 15 ml, 180 μl agarose slurry was used and the elution volume added should be 1080 μl. -
32
Incubate for 40 min rotating end over end at room temperature.
-
33
To remove beads, apply mixture to 0.45 μm filter column (Costar Spin-X) and centrifuge 1 min at 16,000 × g. Transfer flow-through (eluate) to a 1.5-ml microcentrifuge tube.
Optional: save 20 μl of the eluate for analysis by immunoblotting: combine with 5 μl 5X SDS-PAGE sample buffer, vortex 5 sec, boil 5 min, and store −20°C.
Extract RNA
-
34
If eluate volume is >600 μl, split sample into two 1.5-ml tubes. Add equal volume of acid-phenol:chloroform:isoamyl alcohol (pH 4.5).
Avoid upper aqueous buffer layer when removing phenol:chloroform:isoamyl alcohol from stock bottle. -
35
Shake vigorously by hand 10 sec, then vortex top speed for 10 sec. Centrifuge 5 min at 10,000 × g, room temperature.
-
36
Transfer aqueous (top) layer to a fresh 1.5-ml tube, being careful to leave behind any debris, interphase, or organic material.
Collect organic (bottom) phase for appropriate disposal in phenol/chloroform waste. -
37
To reserved aqueous phase, add one-tenth volume 3M sodium acetate and 0.7 μl LPA. Vortex 10 sec.
Isopropanol precipitate RNA
-
38
Add 1.25 volumes room temperature isopropanol. Vortex 10 sec. Incubate 20 min at −80°C.
Alternatively, incubate 10 to 15 min in ethanol/dry ice bath, or overnight at −20°C.RNA can be stored at this step for 6 months at −80°C. -
39
Let thaw, mix, and centrifuge 15 min at 20,000 × g at 4°C to pellet RNA.
Note orientation of tube in centrifuge. -
40
Remove most of supernatant, leaving 50 – 100 ul. Wash pellet by adding 800 μl 70% ethanol at room temperature, vortex 5 sec. Centrifuge 1 min at 20,000 × g, room temperature.
-
41
Remove supernatant, being careful not to disturb pellet.
To remove as much supernatant as possible, centrifuge again briefly to collect ethanol wash from the walls and aspirate liquid using a fine tip. Best results are obtained if all ethanol is removed instead of allowed to dry off, but take great care to not disturb pellet. -
42
Resuspend pellet in 11 μl 10 mM Tris, pH 7.0.
Optional: check quality of lysis and immunoprecipitation by immunoblot
-
43
Perform immunoblot (Gallagher & Wiley, 2008) on input, soluble, unbound, and eluate samples using anti-FLAG antibody.
Solubility and immunoprecipitation efficiency should be at least 60%.
BASIC PROTOCOL 2. CONSTRUCT DNA SEQUENCING LIBRARIES FROM MITORIBOSOME FOOTPRINTS AND/OR FRAGMENTED TOTAL mRNA
RNase I treatment to generate footprints (Basic Protocol 1) and fragmentation of total mRNA by alkaline hydrolysis (Support Protocol) result in chemically similar RNA species with 5′ hydroxyl and 3′ phosphate ends, facilitating identical library generation methods. To minimize bias enzymatic steps should be as efficient as possible. Here we follow, with minor modifications, the method devised for cytosolic ribosome profiling (Ingolia et al., 2012). First the ~40 nt footprints are size selected from mitoribosome-associated RNA. For fragmented mRNA that has already been size selected, begin at Step 15, in which the 3′ phosphate group is removed and a pre-adenylated linker (linker-1) is ligated using a modified RNA ligase. Reverse transcription is then performed with a primer containing carbon spacers to increase flexibility. The cDNA product is then circularized to create a template with known flanking sequences for PCR. PCR is performed with primers containing unique indexing sequences (Table 1) so that samples can be pooled for sequencing and sorted in silico. The resulting product is compatible for sequencing on Illumina platforms. Typically a single MiSeq run (~30 million reads) produces sufficient coverage across 5 indexed mitoribosome profiling libraries, and a single NextSeq run (~400 million reads) produces sufficient coverage across 5 indexed RNA-seq libraries.
Table 1.
DNA oligonucleotides
| Library preparation | |
| Linker-1a | 5′ /5rApp/CTGTAGGCACCATCAAT/3ddC/ 3′ |
| oSMD-RT1b,c | 5′ /5Phos/GATCGTCGGACTGTAGAACTCTGAACCTGTC/iSp18/CACTCA/iSp18/C AAGCAGAAGACGGCATACGAGATATTGATGGTGCCTACAG 3′ |
| oSMD2b | 5′ CAAGCAGAAGACGGCATACGAGATATTGATGG 3′ |
| oSMD1b,d | 5′ AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTG AACTCCAGTCACATCACGCGACAGGTTCAGAGTTC 3′ |
| oMTC1b,d | 5′ AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTG AACTCCAGTCACTGACCACGACAGGTTCAGAGTTC 3′ |
| oMTC2b,d | 5′ AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTG AACTCCAGTCACACATGTCGACAGGTTCAGAGTTC 3′ |
| oMTC3b,d | 5′ AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTG AACTCCAGTCACGCCAATCGACAGGTTCAGAGTTC 3′ |
| oMTC4b,d | 5′ AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTG AACTCCAGTCACCAGATCCGACAGGTTCAGAGTTC 3′ |
| oMTC5b,d | 5′ AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTG AACTCCAGTCACACTTGACGACAGGTTCAGAGTTC 3′ |
| oNTI202b | 5′ CGACAGGTTCAGAGTTCTACAGTCCGACGATC 3′ |
|
| |
| DNA template generation for northern probes | |
| COB(+) | 5′ GTACCTGAATGATACTTATTACCATTCTATGC 3′ |
| COB(−) | 5′ TACCGATATAGAATAAAACATTTTCAATAGTAGAG 3′ |
| COX1(+) | 5′ ATGGTACAAAGATGATTATATTCAACAAATGC |
| COX1(−) | 5′ TGTAATCCTGATAAAGACTTGTAAGAAGTTCG 3′ |
| COX2(+) | 5′ ATGTTAGATTTATTAAGATTACAATTAACAAC 3′ |
| COX2(−) | 5′ CAGAAACTTGATTTAATCTACCAGGAG 3′ |
| COX3(+) | 5′ CCATTTCATATGGTTATGCCTTCACCATGAC 3′ |
| COX3(−) | 5′ GAAATGTAATCCTGTACCAGCATAGAATACTG 3′ |
| ATP6(+) | 5′ TCACCATTAGATCAATTTGAGATTAGAC 3′ |
| ATP6(−) | 5′ ACCTAATCTTAAACCTAATGAAATAGCTC 3′ |
| ATP9(+) | 5′ GATTAAATATTTAATAAATAAATATTATGC 3′ |
| ATP9(−) | 5′ TTATACACCGAATAATAATAAGAATGAAACC 3′ |
Available as IDT part# 11-04-03-05.
Order as PAGE purified.
iSp18 are 18-carbon spacers.
Used when indexing on the Illumina platform is desired. The bolded sequence is the index read during sequencing.
Materials
Solutions and reagents:
Mitoribosome-associated RNA (from Basic Protocol 1) and/or fragmented and size-selected mRNA (from Support Protocol)
2X urea RNA loading buffer (see recipe)
10-bp DNA ladder
15%, 10% TBE-urea, 8% TBE polyacrylamide gels
SYBR gold (10,000X concentrate)
RNase-free water
3 M sodium acetate, pH 5.5 (RNase-free)
25 mg/ml linear polyacrylamide (LPA)
Isopropanol
70% (v/v) ethanol
10X T4 polynucleotide kinase (PNK) buffer
10 U/μl T4 PNK
Oligonucleotide linker and primers (see Table)
50% (w/v) PEG 8000 (RNase-free)
10X T4 RNA ligase reaction buffer (NEB, B0216L)
200 U/μl T4 RNA ligase 2, truncated (NEB)
10 mM Tris, pH 8.0 (RNase-free)
5X First-strand (FS) RT reaction buffer (Invitrogen, 18080-993)
10 mM dNTPs
0.1 M DTT
20 U/μl SUPERase-In
200 U/μl Superscript III
1 N sodium hydroxide
3 M sodium chloride
10X CircLigase reaction buffer (Epicentre, CL4111K)
1 mM ATP
50 mM manganese chloride
100 U/μl CircLigase ssDNA ligase
5X Phusion HF reaction buffer (NEB, M0530S)
2 U/μl Phusion HF DNA polymerase
10X DNA loading buffer (see recipe)
100-bp DNA ladder
DNA gel extraction buffer (see recipe)
10 mM Tris, pH 8.5
Qubit dsDNA HS Assay kit
Special equipment:
Heat blocks: 80°C, 70°C, 37°C, 25°C
Typhoon fluorescent/phosphorescent image scanner, or UV transilluminator with camera
Blue light transilluminator (or UV transilluminator) box
End over end rotator
Costar Spin-X centrifuge tube filter (0.45-um cellulose acetate in 2-ml tube; Corning)
Qubit Fluorometer
Bioanalyzer DNA high sensitivity chip and reagents
Other equipment:
Polyacrylamide electrophoresis apparatus and power source
0.5-ml and 1.5-ml RNase-free non-stick microcentrifuge tubes
Plastic sheet protector
Razor blades or scalpels
Thermocycler
Plastic sheet protector
Size select mitoribosome footprints
-
1
Combine 10 μl RNA from large scale mitoribosome purification with 10 μl 2X urea RNA loading buffer.
-
2
Prepare 0.5 μg of 10-bp ladder in 20 μl (final volume) of 1X urea RNA loading buffer.
-
3
Denature samples and ladder for 90 sec at 80°C, immediately followed by at least 90 sec on ice.
-
4
Pre-run a 15% TBE-urea polyacrylamide gel in 1X TBE for 15 min at 200 V.
Clear urea from wells with a syringe and needle prior to pre-run and again immediately before loading samples. -
5
Load gel and run for 1 hr at 200 V.
For a 1 mm thick, 12-lane gel, load each sample in two wells. Alternatively, use a 1.5 mm thick 10-lane gel and load each sample in one well. -
6
Stain gel with SYBR gold (3 μl in 30 ml TBE) for 3 min with shaking at room temperature. Rinse gel in water, place in a sheet protector, and image on Typhoon scanner with 520 BP emission filter, blue (488) laser, and PMT voltage setting 400–600, depending on the power of the laser. Alternatively, image using UV transilluminator.
Perform rapid gel extraction
-
7
Pierce a 0.5-ml RNase-free, non-stick mircrocentrifuge tube with a 20-G needle and place it inside a 1.5-ml RNase-free, non-stick tube with the cap cut off.
-
8
Place gel on a blue light or UV light box and excise fragments in the range of 37 – 42 nt using the 10-bp ladder as a guide (Figure 2). Place each gel slice into a pierced 0.5-ml RNase-free microcentrifuge tube of the nested tubes from step 7.
It is recommended to perform northern blotting (see Alternate Protocol) on a fraction of the sample prior to gel extraction to verify size range of footprints. -
9
Centrifuge nested tubes 2 min at 20,000 × g, room temperature, to force the gel through the needle hole. Transfer any residual gel from the small tube into the larger tube.
-
10
Add 360 μl RNase-free water to gel pieces and incubate 10 min at 70°C.
-
11
Vortex gel slurry for 30 sec and transfer gel mixture to a Costar Spin-X column using a wide-bore tip.
To make a wide-bore tip, cut the end of a tip with a clean razor blade. -
12
Centrifuge 2 min at 20,000 × g, room temperature to recover the elution mixture free of gel debris. Transfer flow-through to a 1.5-ml microcentrifuge tube.
-
13
Add 40 μl 3M sodium acetate and 0.7 μl LPA. Vortex 10 sec.
-
14
Add 500 μl room temperature isopropanol. Vortex 10 sec. Precipitate RNA as described above.
Figure 2.
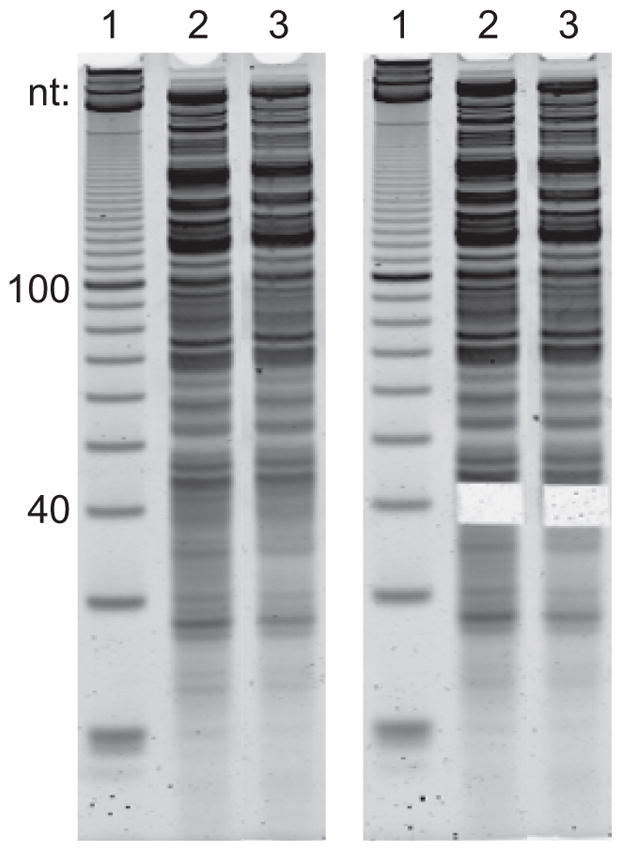
Representative example of gel excision to size select mitoribosome footprints. Left and right panels are before and after size selection, respectively. Lane 1 shows a denatured 10-bp DNA ladder, and lanes 2 and 3 show two different samples of RNA extracted after mitoribosome IP. The majority of RNA is fragmented mito- and cytoribosomal RNA.
Dephosphorylate 3′ ends
-
15
Resuspend pellet in 5 μl RNase-free water.
-
16
Denature for 90 sec at 80°C then immediately place on ice for at least 90 sec.
-
17
Add 4.5 μl PNK mix: 0.95 μl 10X T4 PNK buffer, 2.55 μl RNase-free water, 1 μl T4 PNK.
-
18
Incubate at 37°C for 1 hr.
-
19
Heat kill T4 PNK by incubating 70°C for 10 min.
After this step, centrifuge tube before opening to collect any condensate from walls and cap.
Ligate 3′ adaptor
-
20
To dephosphorylated sample, add 0.5 μl 1 μg/μl Linker-1 (Table 1) (0.5 μg, 85 pmol).
Linker-1 should be present in at least 5-fold molar excess compared to substrate RNA. -
21
Denature for 90 sec at 80°C then immediately place on ice for at least 90 sec.
-
22
Add 10 μl ligation mix: 8 μl 50% PEG 8000, 1 μl 10X T4 RNA ligase buffer, 1 μl T4 RNL2, truncated.
PEG 8000 is viscous and care must be taken in preparing this reaction. Use a wide-bore tip to pipette PEG 8000, and thoroughly mix the master mix solution before aliquoting to samples. -
23
Incubate at 25°C for 3 hr.
-
24
Add 340 μl RNase-free water, 40 μl 3M sodium acetate, 0.7 μl LPA, vortex 10 sec.
-
25
Add 500 μl isopropanol, vortex 10 sec. Precipitate RNA as described above.
-
26
Resuspend pellet in 10 μl 1X urea RNA load buffer.
-
27
Prepare 0.3 μg 10-bp ladder in a final volume of 10 μl 1X urea RNA load buffer.
-
28
Prepare and run samples and ladder on 15% TBE-urea polyacrylamide gel, stain and image as described above.
Each sample can now be loaded in one well of a 1-mm thick 12-lane gel. -
29
Excise ligated product at approximately 60 nt (50 to 90 nt for RNA-seq) and perform rapid gel extraction and isopropanol precipitation.
Perform reverse transcription
-
30
Resuspend pellet in 10 μl of 10 mM Tris, pH 8.0 and transfer to a PCR tube. All subsequent incubation steps should be performed in a thermocycler.
-
31
Add 2 μl of 1.25 μM oSMD-RT1 (Table 1) reverse transcription primer.
-
32
Denature for 90 sec at 80°C then immediately place on ice for at least 90 sec.
-
33
Add 8 μl of reverse transcription mix: 4 μl 5X FS buffer, 1 μl 10 mM dNTPs, 1 μl 0.1 M DTT, 1 μl Superase-In, 1 μl Superscript III. Mix well.
-
34
Incubate for 30 min at 48°C.
-
35
Add 2.2 μl 1 N sodium hydroxide and incubate for 20 min at 98°C to degrade RNA.
-
36
Transfer to 1.5 ml microcentrifuge tube, and add 157.1 μl water, 20 μl 3M sodium acetate, and 0.7 μl LPA. Vortex 10 sec.
-
37
Add 300 μl isopropanol, vortex 10 sec. Precipitate RNA as described above.
-
38
Resuspend pellet in 10 μl 1X urea RNA load buffer.
-
39
Prepare 0.3 μg 10-bp ladder in a final volume of 10 μl 1X urea RNA load buffer.
-
40
Prepare and run samples and ladder, as described above, on 10% TBE-urea polyacrylamide gel at 200V for 65 min, stain and image as described above.
Load each sample in one well of a 1-mm thick 12-lane gel. -
41
Excise cDNA product, which should be >100 nt (Figure 3), and perform rapid gel extraction and isopropanol precipitation with 3 M sodium chloride in place of 3M sodium acetate.
For best results, blot excess buffer from gel after staining and work rapidly. Diffusion of the unextended reverse transcription primer into excess buffer can contaminate desired product and result in PCR amplification of unextended reverse transcription primer in subsequent steps.
Figure 3.
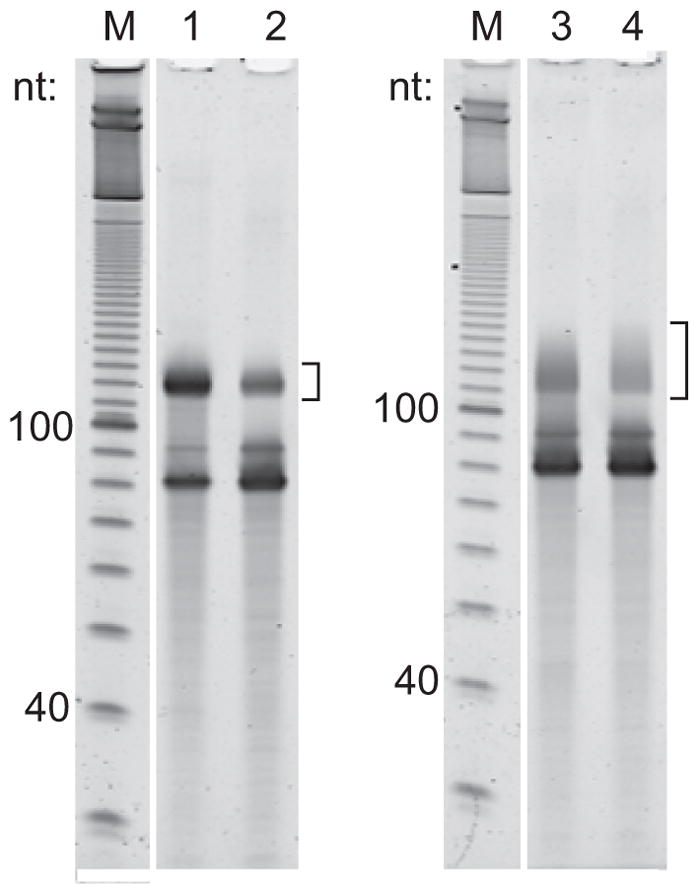
Representative examples of reverse transcription products. The brackets indicate the region to be excised for cDNA produced from mitoribosome footprints (lanes 1 and 2), or from fragmented RNA that has been rRNA-depleted (lane 3) or rRNA- and small RNA-depleted (lane 4). The ~80-nt species is unextended reverse transcription primer.
Circularize cDNA
-
42
Resuspend pellet in 5 μl water.
-
43
Add 15 μl circularization mix: 10 μl water, 2 μl 10X Circularization buffer, 1 μl 1 mM ATP, 1 μl 50 mM manganese chloride, 1 μl Circ ligase.
-
44
Incubate for 1 hr at 60°C.
-
45
Heat kill Circ ligase by incubating for 10 min 80°C.
Circles can be stored for 6 months at −20°C.
Determine cycle number to be used for PCR amplification
-
46
Combine 5 μl of circularization reaction with 78.5 μl PCR mix: 56.3 μl water, 16.7 μl 5X Phusion HF buffer, 1.7 μl 10 mM dNTPs, 1.5 μl 20 μM oSMD2 (Table 1), 1.5 μl 20 μM indexed primer (oSMD1, oMTC1, oMTC2, etc; Table 1), 0.8 μl 2 U/μl Phusion polymerase.
Perform this PCR test with the index primer that will be used for each library. -
47
Aliquot 16.7 μl of reaction mixture into each of 5 PCR tubes.
-
48
Program thermocycler for 30 sec at 98°C and 14 cycles of [15 sec at 98°C, 15 sec at 65°C, 7 sec at 72°C].
-
49
Remove samples during elongation step (72°C) after 6, 8, 10, 12, and 14 cycles.
-
50
Add 1.8 μl 10X DNA loading buffer.
-
51
Prepare 0.3 μg 100 bp in a final volume of 20 μl 1X DNA load buffer.
-
52
Resolve on 8% TBE non-denaturing polyacrylamide gel for 45 min at 180V. Do not heat samples prior to loading.
Pre-running the non-denaturing gel is not necessary. -
53
Stain and image the gel as previously described.
Choose cycle number that produces product in the linear range of amplification and does not produce the larger products that result from reannealed partial duplexes (Figure 4).
Figure 4.
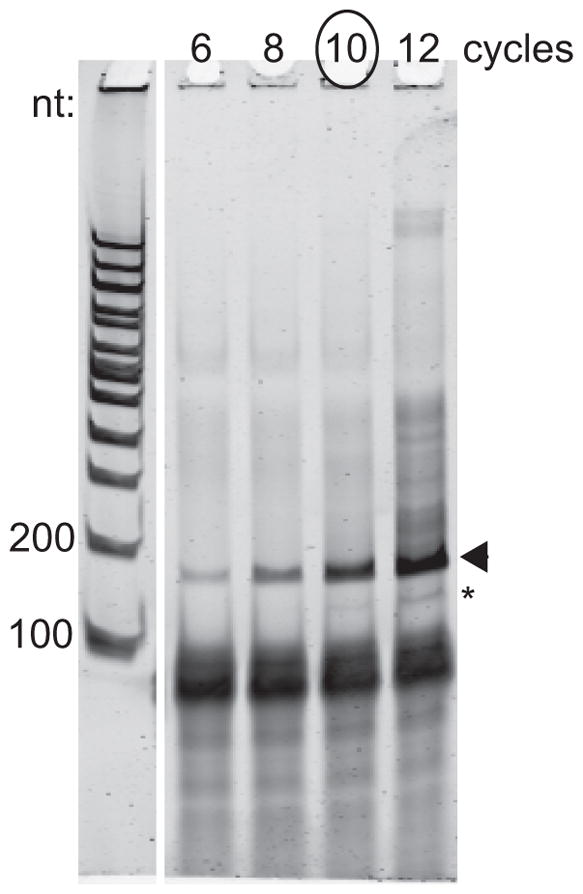
PCR cycle number determination. The arrowhead indicates the ~180-nt product to be purified. The asterisk indicates the product derived from unextended RT primer and should be avoided. In this example, 10 PCR cycles was chosen because the larger products that result after 12 cycles indicate the PCR amplification has approached saturation.
Perform PCR amplification
-
54
Prepare PCR reaction mix as described in step 46.
This reaction can be scaled down to use 3 μl circles depending on yield. -
55
Aliquot 16.7 μl reaction mixture into PCR tubes.
-
56
Program thermocycler for 30 sec at 98°C and N cycles of [15 sec at 98°C, 15 sec at 65°C, 7 sec at 72°C]. N is determined by the PCR test above.
-
57
Resolve samples as described above.
-
58
Excise the amplified PCR product, avoiding any lower product band resulting from unextended reverse transcription primer.
-
59
Perform overnight gel extraction: To gel pieces, add 600 μl of DNA gel extraction buffer. Incubate overnight at room temperature, rotating end over end.
Do not heat. -
60
Remove gel pieces by applying to Costar Spin-X column as described, add 0.7 μl LPA, and precipitate with 750 μl isopropanol as described.
-
61
Resuspend pellet in 12 μl 10 mM Tris, pH 8.5.
-
62
Assess library concentration and quality using Qubit Fluorometric Quantitation dsDNA kit (high sensitivity), and Bioanalyzer DNA high-sensitivity chip according to manufacturer’s instructions.
-
63
When submitting samples for sequencing, supply custom “Read 1” sequencing primer: oNTI202 (Table 1).
ALTERNATE PROTOCOL 1. QUANTIFY MITORIBOSOME PROTECTED FRAGMENTS BY NORTHERN BLOTTING
Analysis of mitoribosome footprints by northern blotting can provide valuable information when ribosome occupancy on a message needs to be directly compared across samples, codon resolution is not required, or sequencing facilities are not readily available. Because of the limited number of mitochondrial mRNAs, virtually all of them can be probed in a matter of days. As of writing the authors have successfully probed for COB, COX1, COX2, COX3, ATP6, and ATP9, but not VAR1 or ATP8 transcripts. The following northern blotting method has been optimized for detection of small RNA species. RNA is blotted to a nylon membrane after polyacrylamide gel electrophoresis. A probe is prepared from a template amplified from the region of interest by internally labeling random-primed polymers. The double-stranded probe mix is then denatured and hybridized to the RNA blot at low (room) temperature. Non-specific interactions are washed away by incubating with decreasing concentrations of salt, and specific probe hybridizations are visualized by exposing to a storage phosphor screen.
Additional materials
Solutions and reagents:
Mitoribosome-associated RNA (Basic Protocol 1)
300–500 bp dsDNA template for probe generation (produced using standard PCR and agarose gel extraction methods)
3 μg/μl random hexamers
Nuclease-free water
10X NEB2 reaction buffer (NEB)
90 μM dATP
0.3 mM dG, dC, 0.9 mM dT mix
10 mCi/ml α32P-dATP
5 U/μl DNA Polymerase I Klenow fragment (3′ to 5′ exo-)
1X TE (see recipe)
Church’s hybridization buffer with formamide (see recipe)
3X blot wash buffer (see recipe)
2X blot wash buffer (see recipe)
Special equipment:
Hybond N+ nylon membrane
Biorad mini trans-blot electrophoretic transfer cell
UV cross-linker
Plexiglass shielding, plexiglass tube racks, and appropriate licensing for handling radioactive material
Geiger counter
100°C heat block
Typhoon fluorescent/phosphorescent image scanner and storage phosphor screen, or autoradiography film
Storage phosphor cassette
NucAway Spin Columns (Invitrogen, AM10070)
Hybridization oven
Hybridization tube
CAUTION: 32P poses a radiological hazard. It should be used with appropriate safety measures such as shielding and personal protective equipment, especially safety glasses. Measures should be taken to reduce exposure time and maximize distance from source. Follow institutional guidelines for handling and disposing of radioactive material.
Prepare RNA blot
-
1
Resolve RNA (1 μl from large scale or 5 μl from small scale mitoribosome purification) on a 15% TBE-urea polyacrylamide gel, along with 0.3 μg 10-bp ladder.
Bring sample volume to 10 μl in 1X urea RNA loading buffer before heating and loading. -
2
Stain and image gel as previously described.
-
3
Transfer to nylon membrane (Hybond N+) in 0.5X TBE using Biorad mini trans-blot electrophoretic transfer cell, 80V for 1 hr at room temperature.
-
4
Disassemble blotting sandwich with gel still on membrane and mark bottoms of wells with a pencil. Remove gel and let membrane dry for 10 min.
-
5
Place blot in UV crosslinker chamber and apply 120,000 microjoules/cm2 at a wavelength of 254 nm.
After crosslinking, the blot can be stored at room temperature indefinitely.
Prepare radioactive probe
-
6
Combine 25 ng double stranded template DNA in 4 μl water with 5 μl 3 μg/μl random hexamers. Primer sequences for probe template amplification are provided in Table 1.
TemplateDNA should be 300–500 bp and gel purified. The ratio of DNA to random hexamers is important for probe quality. -
7
Incubate for 2 min at 100°C and immediately ice for at least 2 min.
-
8
Add 16 μl extension mix:
5 μl water
2.5 μl 10X NEB2 buffer
2.5 μl 90 μM dATP*
2.5 μl 0.3 mM each dG, dC, 0.9 mM dT*
2.5 μl 10 mCi/ml α32P-dATP
1 μl 5 U/μl Klenow fragment (3′ to 5′ exo-)
*Amounts are given to optimize synthesis of AT-rich probes. For templates that are not AT-rich, use 50 μM dATP, 0.5 mM each dG,dC,dT.
-
9
Incubate overnight (or at least 5 hr) at room temperature.
-
10
Remove unincorporated nucleotides with Ambion NucAway spin column according to manufacturer’s instructions.
As a crude check for label incorporation efficiency, use Geiger counter to compare counts per minute of probe to counts per minute of unincorporated nucleotide in spent spin column. Ideally the probe should produce a higher signal. -
11
Add 75 μl 1X TE to probe solution.
Probe can be stored for up to a week at −20°C. Longer storage time is possible, but specific activity will be reduced, as the half-life of 32P is 14 days.
Hybridize probe to RNA blot
-
12
Block non-specific interactions by pre-hybridizing blot without probe in 15 ml Church’s buffer with formamide for at least 1 hr at room temperature.
-
13
Just before adding probe, denature by incubating for 2 min at 100°C and immediately ice for at least 2 min.
This step is critical because hybridization is performed at room temperature. -
14
Add 100 μl of probe to buffer in hybridization tube and swirl by mixing.
Take care to not pipette probe directly onto blot. -
15
Hybridize overnight (at least 8 hr) at room temperature.
-
16
Properly dispose of buffer with probe by pouring into radioactive liquid waste container.
Diluted probe can alternatively be transferred to a 50-ml conical tube and stored at 4°C to be used again. Before use, heat to 50°C and cool to room temperature. -
17
Rinse blot once in 3X blot wash buffer, wash in 3X blot wash buffer for 20 min at 30°C, then wash in 2X blot wash buffer for 20 min at 30°C.
An additional wash in 2X or 1X blot wash buffer may be necessary. -
18
Remove blot from hybridization tube and wrap in plastic wrap.
Be sure no liquid can escape, as it will damage phosphoimager screen. -
19
Expose face up to storage phosphor screen for 5 hr to overnight.
-
20
Image on Typhoon using Storage Phosphor acquisition mode.
-
21
Align ladder from SYBR gold-stained image with northern image using pencil marks at wells (Figure 5).
Figure 5.
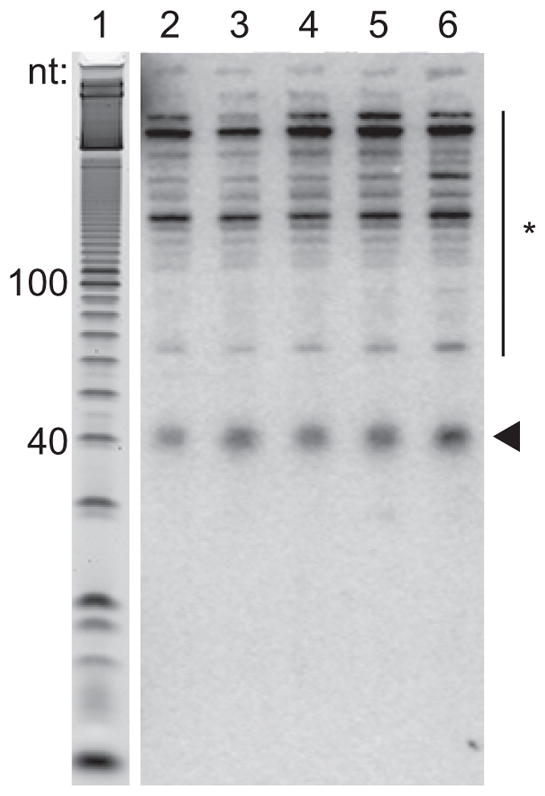
Northern blot hybridization to visualize footprints. Lane 1 shows the denatured 10-bp DNA ladder from the gel stained with SYBR gold before blotting. Lanes 2 – 6 show COX2 probe hybridization to RNA extracted after mitoribosome immunoprecipitation from a series of samples. The arrowhead indicates hybridization to mitoribosome footprints. The asterisk indicates non-specific cross-hybridization.
SUPPORT PROTOCOL 1. ISOLATE mRNA FOR RNA SEQUENCING
This protocol describes how to isolate and process mRNA from mitoribosome profiling lysates to generate RNA fragments that can be used in Basic Protocol 2 to make stranded RNA sequencing libraries. Importantly, ribosomal RNA (rRNA) depletion is used to enrich mRNA in place of poly(A) selection in order to not bias against recovery of mitochondrial mRNAs, which are not polyadenylated in S. cerevisiae. First, lysate is deproteinized and DNA is removed. An optional step is included for small RNA depletion (Nilsen, 2012) to remove transfer RNA (tRNA), which otherwise makes up >50% of the library. This step should be used with caution and only if quality of RNA is carefully monitored. Next, rRNA is depleted and the remaining RNA is fragmented and size selected at which point it is ready for library generation as described in Basic Protocol 2.
Additional materials
Solutions and reagents:
20% (w/v) SDS (RNase-free)
20 μg/ml proteinase K
24:1 Chloroform:isoamyl alcohol
100% ice cold ethanol
10X RQ1 RNase-free DNase reaction buffer (Promega, M6101)
1 U/μl RQ1 RNase-free DNase (Promega, M6101)
Ribo-Zero Gold rRNA Removal Kit for yeast (Illumina, MRZY13)
2X alkaline fragmentation buffer (see recipe)
Special equipment:
Heat blocks: 42°C, 37°C, 70°C, 80°C
NanoDrop UV/Vis Spectrophotometer
CAUTION: Chloroform/isoamyl alcohol is harmful if swallowed, causes skin and eye irritation, and is potentially carcinogenic. It should be used with appropriate safety measures such as protective gloves, glasses, clothing, and sufficient ventilation. All waste should be handled according to hazardous waste regulations.
Isolate total RNA from lysate
-
1
Start with reserved lysate from step 20 in Basic Protocol 1.
-
2
Bring volume to 390 μl with 10 mM Tris, pH 7.0. Allow sample to reach room temperature.
-
3
Add 10 μl of 20% SDS, vortex on medium-high setting 10 sec.
-
4
Add 2 μl of 20 μg/ml proteinase K, vortex on medium-high setting 10 sec, incubate 20 min at 42°C.
-
5
Add 40 μl of 3M sodium acetate. Vortex 10 sec.
-
6
Add 600 μl of 125:24:1 acid-phenol:chloroform:isoamyl alcohol (pH 4.5).
-
7
Shake vigorously by hand 10s, then vortex top speed for 10 sec. Centrifuge 5 min at 10,000 × g, room temperature.
-
8
Transfer aqueous (top) layer to a fresh 1.5-ml tube, being careful to leave behind any debris, interphase, or organic material.
Collect organic (bottom) phase for appropriate disposal in phenol/chloroform waste. -
9
To remove residual phenol, add 500 μl of 24:1 chloroform:isoamyl alcohol to reserved aqueous phase.
-
10
Repeat steps 7 and 8.
-
11
Add 800 μl ice cold 100% ethanol. Vortex 10 sec. Precipitate RNA as described above.
Ethanol is used in place of isopropanol at this step because it may precipitate less phenol. -
12
Resuspend pellet in 40 μl 10 mM Tris, pH 7.0.
Gently tap tube, let sit at room temperature up to 30 min with periodic tapping, and pipette up and down to ensure all pellet is dissolved. -
13
Dilute 1 μl to 1:10 and quantify using NanoDrop, “RNA” settings.
Remove remaining DNA
-
14
To 40 – 60 μg material as quantified by NanoDrop, add 10 mM Tris, pH 7.0 to bring volume to 25 μl.
If the optional small RNA depletion step will be carried out it is ideal to use 60 μg here. -
15
Add 35 μl DNase mix: 26 μl RNase-free water, 6 μl 10X DNase buffer, and 3 μl RQ1 DNase. Mix and incubate for 30 min at 37°C.
-
16
Add 240 μl RNase-free water.
-
17
Add 300 μl of 125:24:1 acid-phenol:chloroform:isoamyl alcohol (pH 4.5). Extract RNA as in steps 7 and 8 above.
-
18
To reserved aqueous phase, add 30 μl 3M sodium acetate and 0.7 μl LPA. Vortex 10 sec.
-
19
Add 600 μl ice cold 100% ethanol. Vortex 10 sec. Precipitate RNA as described above.
-
20
Resuspend pellet in 31 μl 10 mM Tris, pH 7.0.
-
21
Dilute 1 μl to 1:10 and quantify using NanoDrop. Concentration should be 1 – 2 μg/μl.
Deplete small RNAs
Optional: For high-quality RNA this step removes tRNA alone and dramatically increases number of mRNA-mapping reads, without bias against short mRNA transcripts (unpublished data). However, if RNA is fragmented, this step may deplete mRNA fragments <100 nt. Gel visualization of total, small, and large RNA fractions is encouraged for quality control (Figure 6).
Figure 6.
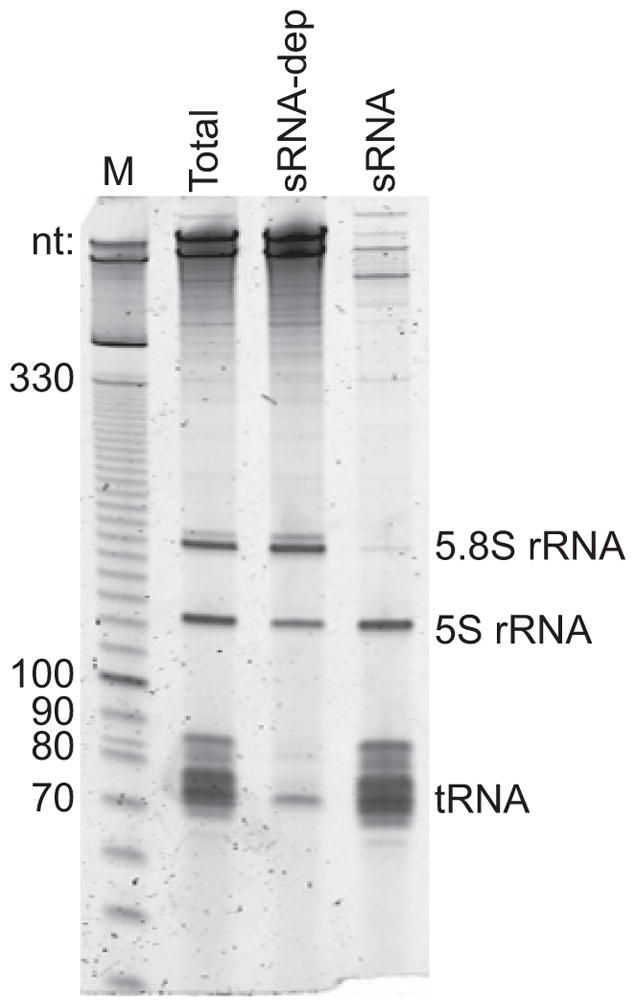
Quality control step to visualize fractions after small RNA depletion. Total RNA (Total; 0.7 μg loaded), large RNA to be carried through library generation (sRNA-dep), and the PEG/NaCl supernatant (sRNA) resolved on a 6% TBE-urea polyacrylamide gel.
-
22
Combine 15 μl RNA (concentration at least 1 μg/μl) with PEG precipitation mix: 9 μl RNase-free water, 3 μl 50% PEG 8000, 3 μl 3M sodium chloride. Mix and incubate for 30 min on ice.
-
23
Centrifuge 30 min at 20,000 × g, 4°C to pellet RNAs >100 nt.
-
24
Discard supernatant containing tRNA and other small RNAs.
Alternatively, ethanol precipitate small RNAs for analysis as in Figure 6. -
25
Add 800 μl 70% ethanol at room temperature, vortex 5 sec. Centrifuge 1 min at 20,000 × g, room temperature.
-
26
Remove supernatant, being careful not to disturb pellet.
To remove as much supernatant as possible, centrifuge again briefly to collect ethanol wash from the walls and aspirate liquid using a fine tip. -
27
Resuspend pellet in 21 μl 10 mM Tris, pH 7.0.
-
28
Dilute 1 μl to 1:5 and quantify using NanoDrop.
Deplete ribosomal RNA
-
29
Apply 5 μg RNA (total or small RNA-depleted) to Ribo-zero kit according to manufacturer’s instructions.
-
30
After depleting rRNA, bring volume to 180 μl with RNase-free water.
-
31
Add 20 μl 3M sodium acetate and 0.7 μl LPA. Vortex 10 sec.
-
32
Add 600 μl ice cold 100% ethanol. Vortex 10 sec. Precipitate RNA as described above.
-
33
Resuspend pellet in 16 μl 10 mM Tris, pH 8.0.
Create fragmentation curve to determine fragmentation time
If multiple samples are being prepared in parallel, it is only necessary to perform the following fragmentation test with a single sample. Perform fragmentation in a thermocycler with heated lid.
-
34
Combine 4 μl RNA and 36 μl RNase-free water. Add 40 μl 2X alkaline fragmentation buffer, mix and aliquot into 4 PCR tubes, 20 μl each.
-
35
Incubate 0, 10, 20, or 30 min at 95°C in PCR machine.
-
36
Transfer each sample to a 1.5-ml tube, add 340 μl water, 40 μl 3M sodium acetate, and 0.7 μl LPA. Vortex 10 sec.
-
37
Add 500 μl room temperature isopropanol. Vortex 10 sec. Precipitate RNA as described above.
Perform gel electrophoresis to visualize fragmentation curve
-
38
Resuspend pellet in 10 μl 1X urea RNA loading buffer.
-
39
Prepare 0.2 μg of 10-bp ladder in a final volume of 10 μl 1X urea RNA load buffer.
-
40
Prepare and run samples and ladder, as described above, on 15% TBE-urea polyacrylamide gel at 200V for 1 hr.
Load each sample in one well of a 1-mm thick 12-lane gel. -
41
Stain and image the gel as previously described.
Fragment RNA
Choose fragmentation time based on the incubation time in which most fragments lie in the range of 30–70 nt (Figure 7).
Figure 7.
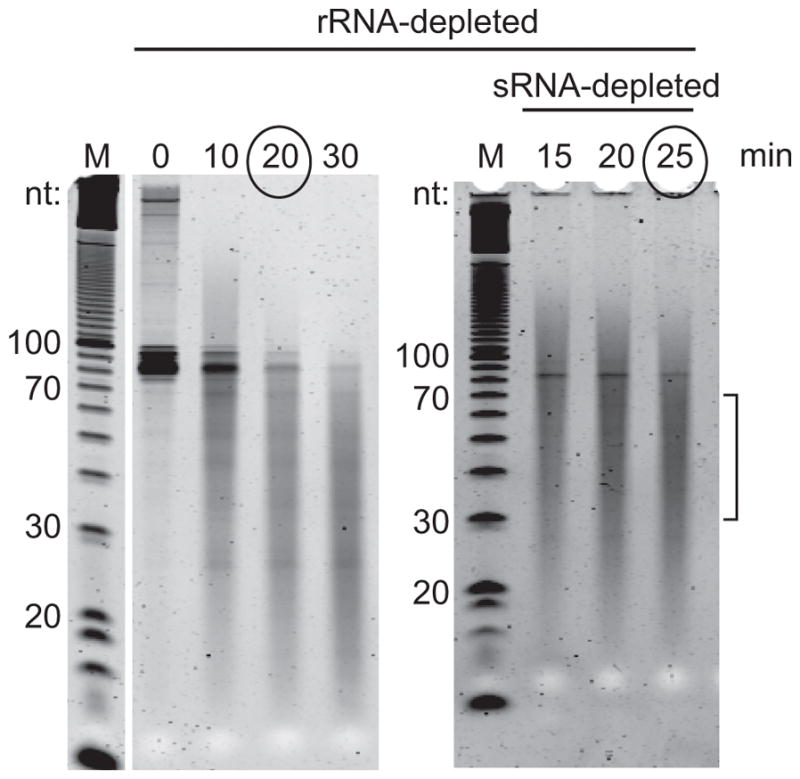
Representative examples of fragmentation curves. Purified mRNA, with or without small RNA depletion was incubated in alkaline fragmentation buffer for the indicated time at 95°C. The incubation time chosen is indicated. Bracket indicates target region for fragments.
-
42
Combine 8 μl RNA and 72 μl RNase-free water. Add 80 μl 2X alkaline fragmentation buffer, mix and aliquot into 8 PCR tubes, 20 μl each.
Conditions should be kept identical to those used in the fragmentation curve. -
43
Incubate in thermocycler for time determined above at 95°C.
-
44
Pool all 8 aliquots per sample to a 1.5-ml tube, add 335 μl water, 55 μl 3M sodium acetate, and 0.7 μl LPA. Vortex 10 sec.
-
45
Add 690 μl room temperature isopropanol. Vortex 10 sec. Precipitate RNA as described above.
Size select fragmented RNA and perform rapid gel extraction
-
46
Resuspend RNA, run, stain, and image gel as described above.
-
47
Excise fragment in the range of 30 – 70 nt using the 10-bp ladder as a guide.
-
48
Perform rapid gel extraction as described, except resuspend gel pieces in 495 μl RNase-free water and after elution add 55 μl 3M sodium acetate and 0.7 μl LPA. Precipitate with 690 μl room temperature isopropanol.
A larger elution volume is used to accommodate the extra gel mass. -
49
Prepare sequencing libraries identically to procedure described in Basic Protocol 2 for ribosome footprints, beginning with dephosphorylation of 3′ ends (step 15).
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes unless noted as RNase-free in which case DEPC-treated or commercial RNase-free water and RNase-free reagents.
Alkaline fragmentation buffer, 2X (RNase-free)
100 mM sodium carbonate-bicarbonate buffer, pH 9.2 (12% 100 mM Na2CO3 and 88% 100 mM NaHCO3)
Store in 200 μl aliquots, tightly capped, for 1 year at room temperature
Blot wash buffer, 2X
2X SSC
0.1% SDS
Store 6 to 12 months at room temperature
Blot wash buffer, 3X
3X SSC
0.1% SDS
Store 6 to 12 months at room temperature
Church’s hybridization buffer with formamide
0.5 M Phosphate buffer, pH 7.2 (68.4% 1 M Na2HPO4 [dibasic] and 31% 1 M NaH2PO4 [monobasic])
1 mM EDTA
7% SDS
1% BSA
Heat and stir to dissolve
Add formamide to 15%
Store in 15 ml aliquots for 2 or more years at −20°C
DNA gel extraction buffer
10 mM Tris, pH 8.0
0.3 M NaCl
1 mM EDTA
Store, tightly capped, 2 or more years at room temperature
DNA loading buffer, 10X
10 mM Tris, pH 7.0
50% glycerol
100 mM EDTA
Bromophenol blue (avoid measuring – transfer powder with pipette tip until desired color is achieved)
Store 2 or more years at 4°C
Lysis buffer (RNase-free)
10 mM Tris, pH 8.0
50 mM NH4Cl
10 mM MgCl2
0.5% lauryl maltoside (n-Dodecyl β-D-maltoside, Sigma, cat. no. D4641)
0.25 mM DTT
1.5X EDTA-free protease inhibitor cocktail (Sigma, cat. no. 11873580001)
Prepare fresh on day of use
SDS-PAGE sample buffer, 5X
300 mM Tris, pH 7.0
10% SDS
20% β-mercaptoethanol
50% glycerol
Bromophenol blue (avoid measuring – transfer powder with pipette tip until desired color is achieved)
Store in 1-ml aliquots 2 or more years at −20°C
Urea RNA loading buffer, 2X (RNase-free)
8 M urea
30 mM EDTA
Bromophenol blue (avoid measuring – transfer powder with pipette tip until desired color is achieved)
Store 6 months at −20°C
Wash buffer (RNase-free)
10 mM Tris, pH 8.0
50 mM NH4Cl
10 mM MgCl2
0.1% TritonX-100
Store 6 to 12 months at 4°C
COMMENTARY
Background Information
Ribosome profiling relies on deep sequencing of ribosome footprints to provide codon-resolution information about ribosome positions and a quantitative readout of expression (Ingolia et al., 2009). Mitochondrial ribosomes (mitoribosomes) are less abundant and more fragile than cytosolic ribosomes (cytoribosomes), and protect a longer stretch of mRNA (~40 nt; (Couvillion et al., 2016), necessitating a dedicated procedure to examine mitochondrial translation. The method traditionally used to monitor mitochondrial translation is metabolic labeling, which relies on incorporation of labeled amino acids under conditions that inhibit cytosolic translation (Fox et al., 1991). While mitoribosome profiling is more labor-intensive, it overcomes many limitations of metabolic labeling. First, it provides high temporal and positional resolution, which is critical for many applications as translational responses can occur on a rapid time scale. Second, it can be performed on cells in any condition and is not confounded by labeled amino acid uptake efficiency. Finally, it does not rely on inhibition of cytosolic translation, which we have found affects mitochondrial translation (Couvillion et al., 2016).
The Basic Protocol in which mitoribosome footprints are converted to a sequencing library yields codon-resolution data that reports on the distribution of mitoribosomes across mitochondrial mRNAs, allowing within-sample comparisons of protein synthesis. While distributions among the messages can be compared across samples, absolute increase or decrease of ribosome occupancy must be evaluated using the Alternate Protocol in which mitoribosome footprints are quantified by northern blotting, with normalization to cell number. Both the Basic and Alternate Protocols report on the combined effects of RNA levels and translation. To isolate translation regulation, translation efficiency must be calculated by normalizing to RNA abundance. We also provide a Support Protocol for isolating and preparing mRNA for RNA sequencing. Combining these protocols provides a powerful means to investigate S. cerevisiae mitochondrial translation in stress, aging, mutants, and mitochondrial disease models.
Critical Parameters
Culture growth
S. cerevisiae metabolism, and thus potentially mitochondrial translation, is highly sensitive to growth conditions. It is important to be consistent by using cells fresh from the freezer (within one week), to note culture inoculation and collection density, and flask to culture volume. Additionally, we found it necessary to pH culture media (pH 5.0) to maintain consistent growth rate, especially in glycerol-containing media.
Cell collection
Because translational responses can be rapid, it is critical to move cells from the condition of interest to liquid nitrogen as quickly as possible. This requirement precludes mitochondrial purification, during which cells must be incubated for long periods to break down the cell wall. Filtration is the collection method of choice for its speed. The cell number collected should be kept constant between samples to be compared.
Solubilization of mitoribosomes
Yeast mitoribosomes are substantially more fragile than cytoribosomes and their subunits are prone to dissociation in typical yeast lysis conditions (Couvillion et al., 2016). At the same time, translating mitoribosomes are membrane associated (Ott et al., 2016), so solubility and subunit association must be balanced. The critical parameter in subunit association is a low ratio of monovalent (NH4+, Tris+) to divalent (Mg2+) cations in the buffer (Vignais et al., 1972).
RNase digestion/gel excision
Footprint capture during gel excision as well as data quality depend on precise nuclease digestion. If any parameters or conditions are modified nuclease must be titrated and footprints visualized by northern blotting prior to gel excision and library generation. When size selecting footprints from the gel, it is ideal to excise as small a region as possible without leaving behind any footprint fragments. The larger the region excised, the greater the rRNA contamination will be in the final sequenced library. It is also possible to deplete rRNA fragments if more footprint reads are needed (Ingolia et al., 2012), but typically this is not necessary. In contrast, rRNA depletion should always be performed in preparation for RNA-seq, as described in the Support Protocol.
Quantification of northern blot signal
Digital images of northern blots can be quantified by densitometry using free software such as ImageJ (https://imagej.nih.gov/ij/). Relative signal should only be compared between bands on the same blot from a single probe, and only when equal cell numbers were collected for each sample and each step in the protocol yielded similar efficiency, including RNA precipitation in the final step. One indicator of success is equal intensity of rRNA fragments visible in the gel after SYBR gold staining (e.g. see Figure 2). However, it is possible that different strains or cells in different states may yield more or less of these rRNA fragments for biologically relevant reasons.
Troubleshooting
Troubleshooting problems and their possible solutions may be found in Table 2.
Table 2.
Troubleshooting guide for mitoribosome profiling and related protocols
| Problem | Possible Cause | Solution |
|---|---|---|
| Low recovery of footprints | Low starting cell number | Start with more cells; some strains or mutants may have less mitochondrial translation. |
| Low solubility | Increase buffer volume in cryogenic lysis | |
| Mitoribosome dissociation | Check lysis buffer conditions for ratio of monovalent to divalent cations. Do not vigorously shake (aerate) lysate. | |
| RNA degradation | Be sure all reagents are RNase-free and work area is free of dust; use barrier tips | |
| Poor separation or smearing of RNA in gels | Incomplete ethanol/salt removal | Wash pellet thoroughly with vortexing in 70% ethanol; remove all trace of ethanol wash |
| Incomplete resuspension of pellet | Mark position of pellet in tube before it dries | |
| Urea accumulation in gel wells prior to loading | Use a syringe to clear wells of urea within 2 minutes of loading | |
| RNA not fully denatured | Heat RNA in urea load buffer and place immediately on wet ice | |
| High fraction of rRNA reads in mitoribosome library | Gel slice too big in initial size selection | Excise a more narrow range |
| Mitoribosome dissociation | Check lysis buffer conditions for ratio of monovalent to divalent cations. Do not vigorously shake (aerate) lysate. | |
| Poor signal on northern blot | Probe specific activity too low | Check labeling protocol and 32P stock |
| Probe not full denatured | Boil probe for at least two minutes and place immediately on wet ice just before use | |
| High background | Repeat wash step with 2X or even 1X blot wash buffer | |
| Poor/inconsistent fragmentation for RNA-seq | RNA is in too high a concentration of buffer | Tris concentration should be 1 mM before addition of 2X alkaline fragmentation buffer |
| Condensation is forming during heating | Perform fragmentation in thermocycler with heated lid |
Anticipated Results
The vast majority of the RNA extracted after mitoribosome immunoprecipitation is RNase-digested rRNA (mitochondrial and cytosolic), which appears in a characteristic banding pattern in the gel (see Figure 2). The mobility of the footprint fragments cannot be visualized without northern blotting (see Figure 5). Because of this, recovered RNA mass is not a useful indicator of footprint abundance. Instead, success should be gauged using immunoblotting by efficiencies of MrpS17-FLAG solubility and immunoprecipitation, which are expected to be >60%. Mitoribosome profiling libraries typically have 15–25% footprint reads, 40–60% cytosolic rRNA, 20–30% mitochondrial rRNA, and <1% tRNA and nuclear mRNA-mapping reads. Mitoribosome footprint reads should map precisely to open reading frames with their 5′ ends beginning ~18 nt upstream of start codons. RNA sequencing libraries made without small RNA depletion typically have 20% mRNA-mapping reads, 65% tRNA mapping reads, and 1% rRNA reads. When small RNA depletion is included libraries are 40–50% mRNA-mapping reads, 15–20% tRNA, and 20–25% rRNA.
Time Considerations
All steps for the protocols presented in this unit can be completed in 8 days if only one or two samples are being processed. Day 1: Culture, collect, and cryogenically lyse cells. Day 2: Immunoprecipitate mitoribosomes and associated RNA; extract total RNA. Days 3–4: Perform immunoblot and northern blot to verify quality of immunoprecipitation. Day 5: Fragment total RNA; size select footprints and total RNA. Days 6–8: Library generation.
The protocols are flexible with many stopping points, and not every experiment will require every protocol in this unit. For an experienced investigator up to 6 samples can be processed in parallel, but more than 8 days will be required to complete all steps.
Acknowledgments
The authors thank Iliana Soto, Andreas Mayer, and Aaron Aker for critical comments on the protocol. This research was supported by the National Institutes of Health (F32GM106531), the Burroughs Wellcome Fund (Career Award at the Scientific Interface), and the Ellison Medical Foundation (New Scholar in Aging Award).
LITERATURE CITED
- Couvillion MT, Soto IC, Shipkovenska G, Churchman LS. Synchronized mitochondrial and cytosolic translation programs. Nature. 2016;533(7604):499–503. doi: 10.1038/nature18015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AP, Patel SS. Mechanism of transcription initiation by the yeast mitochondrial RNA polymerase. Biochim Biophys Acta. 2012;1819(9–10):930–938. doi: 10.1016/j.bbagrm.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox TD, Folley LS, Mulero JJ, McMullin TW, Thorsness PE, Hedin LO, Costanzo MC. Analysis and manipulation of yeast mitochondrial genes. Methods Enzymol. 1991;194:149–165. doi: 10.1016/0076-6879(91)94013-3. [DOI] [PubMed] [Google Scholar]
- Gallagher SR, Wiley EA. Current protocols essential laboratory techniques. Hoboken: John Wiley & Sons; 2008. [Google Scholar]
- Ingolia NT. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010;470:119–142. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7(8):1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrein K, Bonnefoy N, Ott M. Mitochondrial protein synthesis: efficiency and accuracy. Antioxid Redox Signal. 2013;19(16):1928–1939. doi: 10.1089/ars.2012.4896. [DOI] [PubMed] [Google Scholar]
- Nilsen TW. Selective precipitation of large RNAs. Cold Spring Harb Protoc. 2012;2012(12) doi: 10.1101/pdb.prot072322. [DOI] [PubMed] [Google Scholar]
- Ott M, Amunts A, Brown A. Organization and Regulation of Mitochondrial Protein Synthesis. Annu Rev Biochem. 2016;85:77–101. doi: 10.1146/annurev-biochem-060815-014334. [DOI] [PubMed] [Google Scholar]
- Sherman F. Getting started with yeast. Methods Enzymol. 2002;350:3–41. doi: 10.1016/s0076-6879(02)50954-x. [DOI] [PubMed] [Google Scholar]
- Treco DA, Winston F. Growth and manipulation of yeast. Curr Protoc Mol Biol. 2008;Chapter 13(Unit 13):12. doi: 10.1002/0471142727.mb1302s82. [DOI] [PubMed] [Google Scholar]
- Vignais PV, Stevens BJ, Huet J, Andre J. Mitoribosomes from Candida utilis. Morphological, physical, and chemical characterization of the monomer form and of its subunits. J Cell Biol. 1972;54(3):468–492. doi: 10.1083/jcb.54.3.468. [DOI] [PMC free article] [PubMed] [Google Scholar]