

Abstract
A long-standing question of human immunodeficiency virus (HIV) genetic variation and evolution has been whether differences exist in mutation rate and/or mutation spectra among HIV types (i.e., HIV-1 versus HIV-2) as well as among HIV groups (i.e., HIV-1 groups M-P and HIV-2 groups A-H) and HIV-1 Group M subtypes (i.e., subtypes A-D, F-H, and J-K). To address this, a new single-strand consensus sequencing assay was developed for the determination of HIV mutation frequencies and spectra using the Illumina sequencing platform. This assay enables parallel and standardized comparison of HIV mutagenesis among various viral vectors with lower background error than traditional methods of Illumina library preparation. We found significant differences in viral mutagenesis between HIV types but intriguingly no significant differences among HIV-1 Group M subtypes. More specifically, HIV-1 exhibited higher transition frequencies than HIV-2, due mostly to single G-to-A mutations and (to a lesser extent) G-to-A hypermutation. These data suggest that HIV-2 RT exhibits higher fidelity during viral replication, and, taken together, these findings demonstrate that HIV type but not subtype significantly affects viral mutation frequencies and spectra. These differences may inform antiviral and vaccine strategies.
Graphical abstract
Introduction
Human immunodeficiency virus type-1 (HIV-1) has typically been found to mutate on the order of 10−4–10−5 (average of ~3.6×10−5) mutations/base pair/cycle (m/bp/c) in cells [1–4], a rate approximately 10,000–100,000 times faster than eukaryotic genomic DNA [5]. The fidelity of HIV-1 replication can be modulated by a variety of viral and cellular factors. HIV-1 reverse transcriptase (RT) is thought to be a key driver of viral mutagenesis, primarily due to its high error rates in vitro (typically ~10−4 m/bp) [6]. Further, the intermediates of reverse transcription (RNA-DNA hybrids in the cytosol) are thought to avoid host repair processes. RNA polymerase II can also generate mutations when transcribing viral genomic RNA from integrated proviral DNA, but limited evidence suggests this is a lesser source of error than RT [2]. Cellular DNA polymerases could theoretically introduce mutations when replicating the integrated provirus during cell division, but the high fidelity of cellular DNA replication (≤10−9 m/bp/c) argues that this is a relatively minor source of virus variation.
In addition to RNA and DNA polymerases, the APOBEC3 family of DNA deaminases can induce mutations in HIV-1 by performing C-to-U editing of minus strand viral DNA during reverse transcription [7, 8]. Ultimately, APOBEC3 editing leads to fixation of multiple G-to-A mutations on the plus strand viral DNA, a process referred to as G-to-A hypermutation. Although the viral accessory protein Vif counteracts most APOBEC3 activity, multiple lines of evidence demonstrate that Vif-mediated protection is not absolute. In cell culture, expression of Vif often does not fully restore the infectivity of virus produced in the presence APOBEC3 proteins [9–11]. Further, Vif alleles from different virus isolates have been shown to vary widely in their abilities to counteract various APOBEC3 proteins, which appears to at least partially depend on the HIV-1 Vif subtype [10, 12]. Vif variants that are partially or fully defective against specific APOBEC3 proteins can be readily identified in patient samples [11]. In addition, G-to-A hypermutants have often been observed in proviral DNA from patient samples [11, 13–19], which provides strong evidence that APOBEC3-mediated editing occurs in vivo. Some evidence suggests that APOBEC3 proteins may promote HIV-1 variation by sub-lethal mutation or by recombination between hypermutated and non-hypermutated viruses [20–23]. However, a recent study demonstrated that APOBEC3-mediated hypermutation likely contributes substantially less to HIV-1 variation than RT [24]. Lastly, there is some evidence that adenosine deaminases acting on RNA (ADARs) perform A-to-I editing of HIV-1 RNAs, although the frequency and functional consequences of such editing events remain unclear [4, 25–30].
While HIV-1 is the major cause of the worldwide pandemic, human immunodeficiency virus type-2 (HIV-2) infects ~1–2 million individuals globally, primarily in West Africa [31]. HIV-2 is likely less prevalent due to its reduced transmissibility, which has been demonstrated for both sexual transmission and mother to child transmission [32–34]. Further, HIV-2 is generally less pathogenic than HIV-1, leading to AIDS in only ~25% of untreated individuals (compared to >95% for HIV-1) [35, 36]. The reduced transmissibility and attenuated pathogenicity of HIV-2 are associated with much lower viral loads for HIV-2 than HIV-1, both in plasma and in genital secretions [34, 37–39]. We previously hypothesized that HIV-2 would exhibit a lower mutation frequency than HIV-1, which might limit its ability to diversify and thus reduce viral fitness and/or pathogenicity [40]. We found that HIV-2 exhibited lower total, substitution, and transition mutation frequencies than HIV-1, primarily due to reduced levels of G-to-A hypermutants indicative of APOBEC3 activity. However, the previous work was limited by the relatively high levels of background errors due to PCR and/or sequencing. Further, replication fidelity was only compared between a single molecular clone of HIV-1 and HIV-2, which may not be truly representative of their respective viral types. Most importantly, replication fidelity was not compared between HIV-1 Group M subtypes (i.e., A–D, F–H, and J–K), even though the extent to which mutation frequencies and spectra differ between subtypes has remained a longstanding question within the field of HIV genetic variation and evolution.
To overcome these limitations, we developed a new standardized assay, called single-strand consensus sequencing (SSCS), for comparison of HIV mutation frequencies and spectra in a high-throughput and parallel manner. We found that HIV-1 exhibited an ~1.8-fold higher transition frequency than HIV-2. For both HIV-1 and HIV-2, rare G-to-A and A-to-G hypermutants were observed, likely due to APOBEC3 or ADAR-mediated editing, respectively. The lower transition frequency of HIV-2 was primarily due to fewer single G-to-A mutations (rather than hypermutation), arguing that differences in viral mutation frequencies were primarily caused by greater fidelity of HIV-2 RTs. Intriguingly, there were no significant differences between HIV-1 subtypes in terms of mutation frequencies or spectra. Taken together, these findings demonstrate that HIV type but not subtype significantly impacts viral mutation frequencies and spectra. These differences may affect the relative abilities of HIV-1 and HIV-2 to diversify or evolve within infected individuals and inform strategies for vaccines and antiviral treatments.
Results
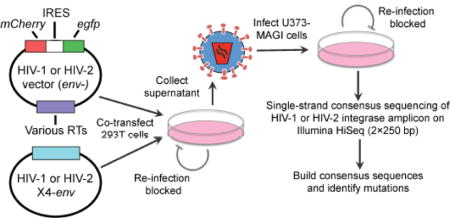
Development of an assay to measure HIV-1 and HIV-2 mutation frequencies and spectra in a comparative and parallel manner
To compare viral mutagenesis in a standardized manner, multiple HIV-1 and HIV-2 RTs were cloned into parental HIV-1 (pNL4-3 MIG) or HIV-2 (pROD14 MIG) vectors, respectively. The HIV-2 ROD14 RT was also swapped into pNL4-3 MIG (or vice versa), but the resulting HIV-1/HIV-2 chimeric viruses were non-infectious. RT swapping was performed in a near-isogenic manner, with only the last 20–25 amino acids of protease and the first amino acid of integrase exchanged in addition to RT. This experimental strategy helped standardize the assay (within HIV-1 or HIV-2) by eliminating differences in accessory genes (e.g. vif) and template sequences. In total, 10 HIV-1 vectors and 6 HIV-2 vectors were constructed (Table 1). On average, HIV-2 RTs shared ~62% amino acid identity with HIV-1 NL4-3 RT, whereas HIV-1 RTs from non-B subtypes shared ~90–93% identity with NL4-3 RT and other subtype B RTs shared ~97% identity with NL4-3 RT. Viral stocks were produced by co-transfecting the HIV-1 and HIV-2 vectors with pNL4-3 Env or pROD10 Env, respectively, into 293T cells (Figure 1). Viruses were then DNaseI-treated, titered, and used to infect 100,000 U373-MAGI cells at an MOI of 1.0. In this assay, the producer cells and target cells cannot be re-infected (due to a lack of CD4 or Env expression, respectively), and thus viruses were limited to a single round of replication. Genomic DNA was purified 72 h later and subjected to single-strand consensus sequencing (SSCS), a method previously described by several other research groups that allows identification and exclusion of most background errors due to PCR and sequencing [41–44]. Notably, an SSCS-related method, termed Primer ID, has already been developed for sequencing of HIV-1 viral RNA [44], but a similar method for consensus sequencing of proviral DNA has not yet been reported. Briefly, SSCS was performed by assigning unique identifier tags (UIDs, strings of 14 degenerate bases) to starting templates by linear extension (Figure S1). Linear extensions were then bead-purified, reverse- extended, bead-purified again, and amplified by PCR. Illumina Nextera XT libraries were prepared from the PCR products, pooled in an equimolar manner, and redundantly sequenced on the Illumina HiSeq 2500 using 2×250 paired-end sequencing. Consensus sequences were constructed by aligning reads with identical UIDs (Figure S1). Consensus families were required to contain at least three reads, and bases (whether wild-type or mutations) had to be present in at least 75% of reads to be represented in the consensus sequence. Ambiguous positions were converted to uncalled bases and were not included in subsequent analysis of mutational events. Consensus sequences were then mapped to the appropriate reference sequence, and mutation frequencies and spectra were calculated as described before [40]. Note that mutations detected by this method should have originated from the non-coding (i.e. antisense) strand of proviral DNA, as the original primer used in linear extension was in the forward (i.e. sense) orientation. SSCS was performed on homologous regions of HIV-1 and HIV-2 integrase (200 bp in length, not including UIDs or primer sequences), which share ~71% nucleotide identity. SSCS was also performed using purified plasmids (diluted into genomic DNA from uninfected cells) to measure the background mutation frequency of the assay.
Table 1.
HIV-1 and HIV-2 vectors for comparative parallel analyses of viral mutagenesis by single-strand consensus sequencing.a
| Virus | Reference | HIV Type | RT Subtype or Groupb | Identity to NL4-3 RTc | Identity to ROD14 RTc |
|---|---|---|---|---|---|
| 94CY017.41 | [79] | HIV-1 | A | 90% | 62% |
| NL4-3 | [77] | HIV-1 | B | — | 62% |
| YU2 | [80, 81] | HIV-1 | B | 97% | 61% |
| BH10 | [82] | HIV-1 | B | 97% | 61% |
| 93BR029.4 | [83] | HIV-1 | B | 97% | 62% |
| MJ4 | [84] | HIV-1 | C | 93% | 61% |
| 96ZM651.8 | [85] | HIV-1 | C | 93% | 61% |
| 98IS002.5 | [85] | HIV-1 | C | 93% | 61% |
| 94UG114.1.6 | [83] | HIV-1 | D | 92% | 61% |
| 92NG003.1 | [83] | HIV-1 | G | 90% | 62% |
| ROD14 | [78] | HIV-2 | A | 62% | — |
| ST | [86, 87] | HIV-2 | A | 61% | 93% |
| ISY | [92] | HIV-2 | A | 62% | 93% |
| D194 | [88, 89] | HIV-2 | A | 61% | 90% |
| CBL-20 | [90] | HIV-2 | A | 62% | 93% |
| GH123 | [93] | HIV-2 | A | 63% | 93% |
A panel of 10 HIV-1 and 6 HIV-2 vectors was created by sub-cloning reverse transcriptase (RT) genes from other HIV-1 vectors into pNL4-3 MIG or from other HIV-2 vectors (or from genomic DNA purified from cells infected with HIV-2) into pROD14 MIG in a near-isogenic fashion (see Methods).
HIV-1 vectors were engineered with RTs from multiple subtypes of Group M, especially subtypes B (the predominant subtype in the U.S.) and C (the predominant subtype globally). All HIV-2 vectors were engineered with RTs from Group A.
The columns indicate the extent of identity between RT sequences at the amino acid level.
Figure 1. Experimental strategy to measure HIV-1 and HIV-2 mutation frequencies and spectra by single-strand consensus sequencing.
Viral stocks were produced by co-transfecting 293T cells with Env-deficient HIV-1 or HIV-2 vectors containing various HIV-1 or HIV-2 RT genes (Table 1), respectively, and HIV-1 or HIV-2 CXCR4-tropic Env expression constructs. Virus stocks were concentrated, DNase I-treated to minimize plasmid carryover, and titered in U373-MAGI-CXCR4 cells. Next, 100,000 U373-MAGI cells/sample were infected at an MOI of 1.0, and genomic DNA was purified from cells collected 72 h post-infection. Producer cells and target cells cannot be re-infected due to a lack of receptor or Env expression, respectively, such that viruses were only able to perform a single cycle of replication in this assay. Using the genomic DNAs, single-strand consensus sequencing (SSCS) was performed of homologous HIV-1 and HIV-2 integrase amplicons by uniquely tagging, exponentially amplifying, and redundantly sequencing starting templates on the Illumina HiSeq 2500, as described in Methods and Figure S1. The resulting data were used to build consensus families and determine consensus sequences, allowing identification and exclusion of PCR and sequencing errors.
Comparison of mutation frequencies and spectra between HIV-1 and HIV-2
After performing SSCS, HIV-1 and HIV-2 mutation frequencies were calculated for each possible type of mutation and compared to the corresponding plasmid controls to determine the types of mutations that could be detected at levels significantly higher than the background. Unfortunately, even using SSCS, for both viruses transversions, insertions, and deletions were not detected at significantly higher levels in the biological samples than in the plasmid controls. However, transitions (and larger categories of mutations including transitions, i.e., substitutions and total mutations) were detected at significantly higher levels in the biological samples than in the plasmid controls (p = 0.024 [HIV-1] or 0.014 [HIV-2]), and many previous studies have demonstrated that transitions comprise the majority of mutations during HIV-1 replication [1, 2, 4, 45–48]. Thus, further mutational analyses focused exclusively on transition mutations. Overall, HIV-1 demonstrated an ~1.8-fold higher transition frequency than HIV-2 (4.6×10−5 vs. 2.6×10−5 m/bp) (p = 0.020; Figure 2A), but transition frequencies varied depending on the specific RT and in some cases biological replicate (i.e., NL4-3 and 92NG003.1). Although NL4- 3 exhibited a particularly high transition frequency, HIV-1 still demonstrated an ~1.5-fold higher transition frequency than HIV-2 in the absence of NL4-3 (p = 0.009). Transition frequencies did not vary significantly among HIV-1 Group M subtypes (p = 0.41). Next, transition spectra were compared between HIV-1 and HIV-2 by determining the relative percentage of each transition type (G-to-A, A-to-G, C-to-T, or T-to-C). HIV-1 exhibited a transition spectrum much more biased toward G-to-A transitions than HIV-2 (58% vs. 29%; Figure 2B), as we observed previously for HIV-1 NL4-3 and HIV-2 ROD14 [40]. In contrast, HIV-2 exhibited higher relative levels of A-to-G and T-to-C transitions, while the relative level of C-to-T transitions was similar between HIV-1 and HIV-2.
Figure 2. Comparison of transition frequencies and spectra between HIV-1 and HIV-2.
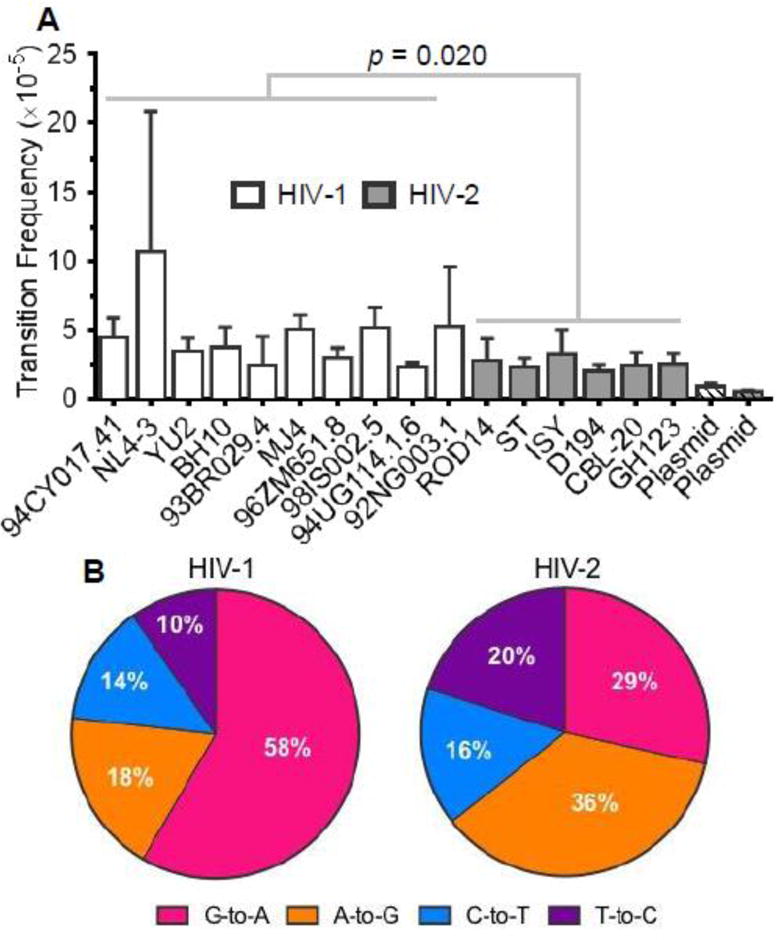
A. Transition frequency analysis. Transition frequencies were calculated by dividing the number of transition mutations by the number of reference bases (mutations + wild-type bases) and are expressed as mutations/bp. Transition frequencies were determined for 10 HIV-1 vectors and 6 HIV-2 vectors, as well as for plasmid controls, which indicate the level of residual background error. The data represent the average of three independent biological replicates, with error bars indicating standard deviation. HIV-1 and HIV-2 transition frequencies were compared statistically using generalized linear mixed effects models (see Methods). In these tests, data were not pooled across viral isolates or biological replicates. B. Transition spectra analysis. Transition spectra were determined by dividing the frequency of each type of transition by the total transition frequency, with the results expressed as a percentage of total transitions. The data represent the average transition spectra across all HIV-1 or HIV-2 vectors.
HIV-1 exhibits higher levels of G-to-A transitions than HIV-2
To compare viral mutagenesis across HIV-1 and HIV-2 RTs in more detail, transition frequencies were calculated for each type of transition and each specific HIV-1 or HIV-2 vector. On average, HIV-1 and HIV-2 exhibited G-to-A transition frequencies of 2.4×10−5 and 7.1×10−6 m/bp, respectively, so that HIV-1 demonstrated a 3.4-fold higher frequency of G-to-A transitions than HIV-2 (p = 0.0006; Figure 3). Importantly, this trend was observed fairly consistently across the different HIV-1 and HIV-2 vectors. HIV-1 and HIV-2 exhibited less than 2-fold differences in frequencies of the other three types of transitions, and none of these differences were statistically significant (C-to-T: p = 0.52; A-to-G: p = 0.30; T-to-C: p = 0.18). Additionally, these differences were not observed as consistently across the various HIV-1 or HIV-2 RTs. NL4-3 exhibited a very high (but variable) A-to-G frequency, which was later found to be due to several extensively A-to-G hypermutated sequences in one particular experimental replicate for NL4-3. HIV-1 RTs from different subtypes did not exhibit significantly different transition frequencies. Overall, the most pronounced difference in viral mutagenesis across HIVs was the 3.4-fold lower frequency of G- to-A transitions for HIV-2, which could be due to reduced G-to-A hypermutation (resulting from the activity of APOBEC3 proteins) or reduced fidelity of HIV-1 RTs.
Figure 3. HIV-1 exhibits higher levels of G-to-A transition mutations than HIV-2.
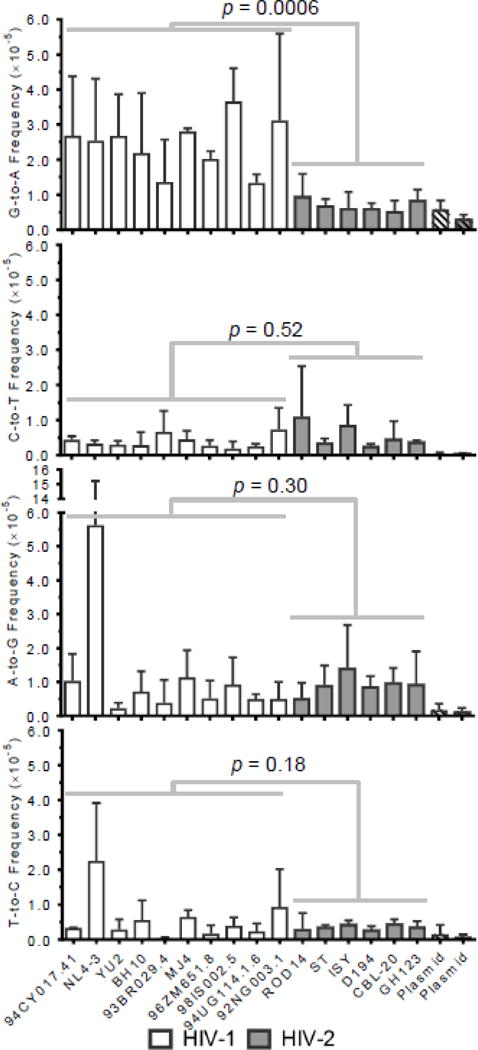
To compare the mutagenesis of HIV-1 and HIV-2 in greater detail, mutation frequencies were calculated for each type of transition for all HIV-1 and HIV-2 viral vectors. Mutation frequencies were calculated by dividing the number of mutations (of the indicated type) by the number of reference bases (mutations + wild-type bases) and are expressed as mutations/bp. The data represent the average of three independent biological replicates, with error bars indicating standard deviation. Individual transition frequencies were compared between HIV-1 and HIV-2 using generalized linear mixed effects models (see Methods). Data were not pooled across viral isolates or biological replicates in these analyses.
HIV-1 and HIV-2 exhibit G-to-A hypermutation consistent with APOBEC3 editing
Previously, HIV-1 and HIV-2 were found to exhibit G-to-A hypermutants consistent with the activity of APOBEC3 proteins, although little APOBEC3 activity was expected due to the presence of Vif and the low expression levels of restrictive APOBEC3 proteins in 293T cells [40]. G-to-A hypermutation was mostly (but not completely) responsible for the higher G-to-A frequency for HIV-1. To address whether the higher G-to-A frequency observed for HIV-1 in this study was also due to hypermutation, we determined G-to-A mutation frequencies due to G-to-A hypermutants for HIV-1 and HIV-2. G-to-A hypermutants were defined as individual consensus reads (200 bp in length after processing) containing two or more G-to-A transitions. G-to-A mutations from hypermutants were observed for most viral vectors (13/16) and were detected at levels significantly higher than background (p < 0.0001 for both HIV-1 and HIV-2; Fisher’s exact test) but were extremely rare events (HIV-1: 2.4×10−4, or ~2.4 in 10,000 reads; HIV-2: 4.4×10−5, or ~0.4 in 10,000 reads), consistent with the expectation of little APOBEC3 activity in this particular experimental system. On average, HIV-1 exhibited a 5.2-fold higher G-to-A mutation frequency from hypermutants than HIV-2 (6.0×10−6 vs. 1.2×10−6, p = 0.012) (Figure 4A), demonstrating that G-to-A hypermutants were at least partially responsible for the higher frequency of G-to-A transitions observed for HIV-1. Although HIV-1 92NG003 exhibited a particularly high frequency of G-to-A mutations from hypermutants, HIV-1 still displayed an ~3.7-fold higher frequency of G-to-A mutations from hypermutants than HIV-2 even in the absence of 92NG003 (4.3×10−6 vs. 1.2×10−6, p = 0.038). HIV-1 G-to-A hypermutants contained between 2 and 5 G-to-A transitions (mean of 2.6), while HIV-2 G-to-A hypermutants contained between 2 and 8 G-to-A transitions (mean of 3.8) (Figure 4B). For HIV-1, G-to-A mutations within hypermutants were heavily biased toward GA and, to a lesser extent, GG dinucleotides, whereas for HIV-2 G-to-A mutations within hypermutants were almost exclusively located at GA dinucleotides (Figure 4C). These patterns are consistent with the known editing preferences of APOBEC3 proteins [9, 49–52]. In contrast, G-to-A mutations in single mutants (i.e., nonhypermutants) were not strongly biased toward GA dinucleotides (Figure 4C), ruling out the possibility that G-to-A mutations were generally biased toward GA dinucleotides in this particular integrase amplicon. These biases were not significantly altered by normalization of the dinucleotide context frequencies to the dinucleotide counts within the integrase amplicon sequences (Figure S2), demonstrating that these biases were not simply due to more GA and/or GG dinucleotides (relative to GT/GC) within the integrase amplicon sequences. Because the extent of APOBEC3 editing has been shown to depend on the speed of reverse transcription [53], we also compared the relative rates of HIV-1 and HIV-2 reverse transcription using a time- of-addition assay with two different reverse transcription inhibitors (Figure S3). After infection, HIV-1 and HIV-2 progressively lost susceptibility to reverse transcription inhibitors in a time- dependent manner that was not significantly different between the two viruses, suggesting that the timing of reverse transcription is similar for HIV-1 and HIV-2. Further, we performed qPCR to measure levels of three different RT products at various time points post-infection, and we did not detect any significant difference between HIV-1 and HIV-2 in the rate of RT product accumulation (Figure S4). In sum, the data from these two experiments indicate that HIV-1 and HIV-2 reverse transcription proceed at similar rates in this particular experimental system and, thus, that the speed of reverse transcription is unlikely to significantly contribute to the observed differences in HIV-1 and HIV-2 G-to-A hypermutation.
Figure 4. HIV-1 and HIV-2 generate G-to-A hypermutants consistent with APOBEC3 editing.
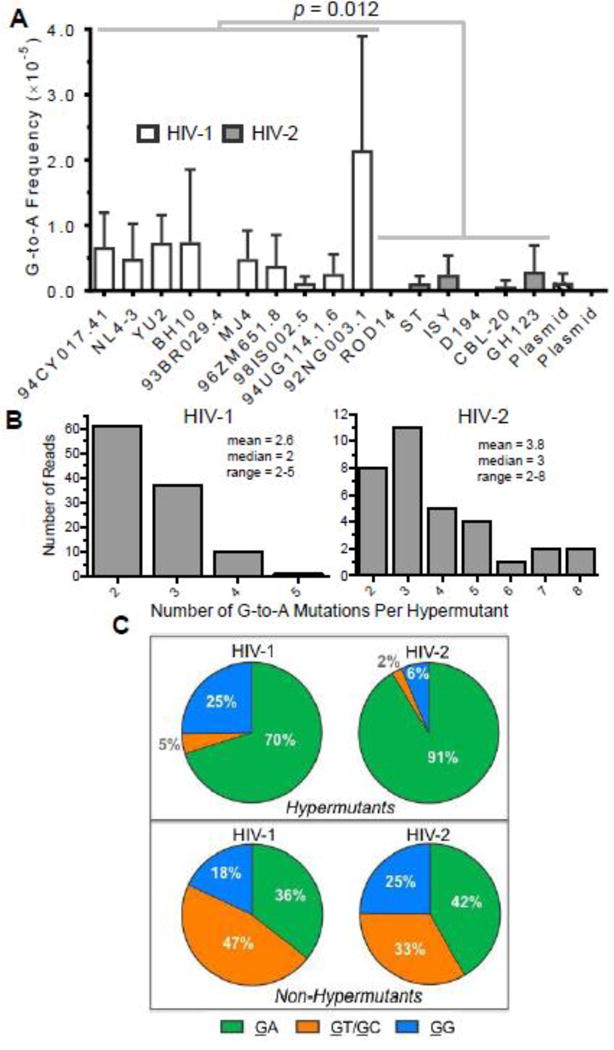
A. G-to-A hypermutation analysis. The frequencies of G-to-A mutations from hypermutants were determined for all HIV-1 and HIV-2 vectors as well as for plasmid controls. For this analysis, hypermutants were defined as consensus reads (200 bp in length after bioinformatics processing) containing two or more G-to-A mutations. G-to-A mutation frequencies from hypermutants were then calculated by dividing the number of G-to-A mutations (in hypermutants only) by all reference bases. HIV-1 and HIV-2 G-to-A mutation frequencies were compared statistically using generalized linear mixed effects models without pooling data across viral isolates or biological replicates (see Methods). Further, the data were analyzed with or without the outlier HIV-1 92NG003.1. B. The degree of G-to-A hypermutation was assessed by determining the numbers of G-to-A mutations within hypermutant reads. C. The 3’ dinucleotide contexts of G-to-A mutations from G-to-A hypermutants or non-hypermutants (i.e., single mutants) was determined and expressed as a percentage of the total. Sites of mutation are underlined (e.g., GG). In panel A, the data represent the average of three independent biological replicates, with error bars indicating standard deviation, while the data in panels B and C represent total (i.e., compiled) data.
HIV-1 and HIV-2 exhibit A-to-G hypermutants potentially induced by ADAR-mediated editing
Several previous groups have reported that the adenosine deaminases acting on RNA (ADAR-1 and 2) proteins perform editing of HIV-1 RNAs, although reports differ about whether editing suppresses or promotes viral infectivity [25–30]. While these groups did not analyze virion RNA (i.e., packaged RNA) or proviral DNA for mutations indicating ADAR editing, another group has observed A-to-G hypermutation within proviral DNA potentially due to ADAR- mediated editing [4]. To determine whether A-to-G hypermutation contributed to HIV-1 or HIV-2 mutagenesis within our experimental system, A-to-G mutation frequencies from A-to-G hypermutants were calculated for all HIV-1 and HIV-2 vectors. Like G-to-A hypermutants, A-to- G hypermutants were defined as consensus reads containing two or more A-to-G mutations. A- to-G hypermutants were observed for most of the viral vectors (13/16), while no A-to-G hypermutants were observed in the plasmid controls (Figure 5A). Both HIV-1 and HIV-2 exhibited levels of A-to-G mutations from hypermutants that were significantly higher than the plasmid control (p < 0.0001; Fisher’s exact test). HIV-1 and HIV-2 both exhibited average A-to- G hypermutant frequencies of 1.1 ×10−4, or ~1 in 10,000 reads. HIV-1 and HIV-2 did not exhibit significantly different frequencies of A-to-G transitions from hypermutants (6.8×10−6 vs. 4.6×10−6, p = 0.245). Although NL4-3 exhibited a very high average A-to-G hypermutant frequency, this was due to a large number of hypermutated sequences in one specific experimental replicate, and HIV-1 and HIV-2 A-to-G transition frequencies from hypermutants did not significantly differ even when this replicate was excluded (p = 0.074). Next, the numbers of A-to-G mutations within hypermutants were examined to determine whether these reads were all simply double or triple mutants (which could occur at low levels due to combinations of RT, RNA polymerase II, and/or background errors) or more extensive hypermutants, which would more strongly implicate ADAR-mediated editing. While some hypermutants contained lower levels of A-to-G mutations (2 to 5 mutations/read), others contained intermediate (7 to 10 mutations/read) or very high levels (15+ mutations/read) of A-to-G mutations (Figure 5B). Further examination of some of the most heavily A-to-G hypermutated reads demonstrated that they cannot simply be attributed to generally error-prone or poor-quality reads, as most of them do not contain any uncalled bases or any mutations besides A-to-G mutations (Figure S5). Further, A-to-G mutations within hypermutants were biased toward TA and AA dinucleotides, more so than A-to- G mutations from single mutants (i.e., non-hypermutants), consistent with the known editing preferences of ADAR-1 and 2 proteins (Figure 5C) [54, 55]. These biases could potentially be due to higher numbers of TA and AA dinucleotides (as opposed to CA and GA) within the integrase amplicon sequences. However, after normalizing the data to the numbers of the various dinucleotides within the amplicon sequences, A-to-G hypermutants were still biased toward TA and AA dinucleotides, with ~91% of all A-to-G mutations occurring in these specific contexts (Figure S6).
Figure 5. HIV-1 and HIV-2 generate A-to-G hypermutation that is consistent with ADAR- mediated editing.
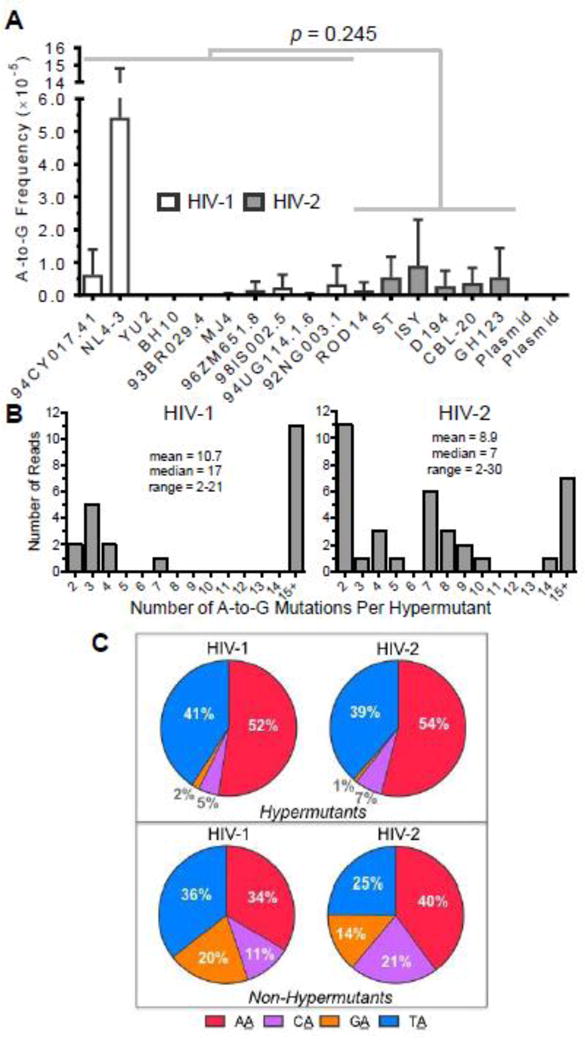
A. A-to-G hypermutation analysis. The frequencies of A-to-G mutations form hypermutants were determined for all HIV-1 and HIV-2 vectors as well as for plasmid controls. For this analysis, hypermutants were defined as consensus reads (200 bp in length after bioinformatics processing) containing two or more A-to-G mutations. A-to-G mutation frequencies from hypermutants were then calculated by dividing the number of A-to-G mutations (in hypermutants only) by all reference bases. HIV-1 and HIV-2 A-to-G mutation frequencies were compared statistically using generalized linear mixed effects models without pooling data across viral isolates or biological replicates (see Methods). Further, the data were analyzed with or without the outlier HIV-1 NL4-3. B. The extent of A-to-G hypermutation was assessed by determining the numbers of A-to-G mutations within hypermutant reads. C. The 5’ dinucleotide contexts of A-to-G mutations from A-to-G hypermutants or non-hypermutants (i.e., single mutants) were determined and expressed as a percentage of the total. Sites of mutation are underlined (e.g., AA). In panel A, the data represent the average of three independent biological replicates, with error bars indicating standard deviation, while the data in panels B and C represent total (i.e. compiled) data.
HIV-1 exhibits a higher frequency of G-to-A transitions than HIV-2 even in the absence of G-to-A hypermutation
To further compare viral mutagenesis between HIV-1 and HIV-2, transition frequencies and spectra were re-examined after excluding all transition hypermutants, defined as reads containing two or more of the same type of transition mutation (G-to-A, A-to-G, C-to-T, or T-to-C). This analysis was performed to compare the general fidelity of viral replication (i.e., due to RNA polymerase II and RT) in the absence of hypermutation likely resulting from the activities of APOBEC3 and ADAR-1 or 2 enzymes. Additionally, hypermutants might greatly skew the mutation frequencies of specific samples, as some contained very large numbers of mutations. In practice, only G-to-A and A-to-G transition frequencies were substantially altered by excluding transition hypermutants, as they comprised 97% (200/206) of all hypermutants observed. After removal of hypermutants, HIV-1 and HIV-2 exhibited transition frequencies of 3.3×10−5 and 2.0×10−5 m/bp, respectively, such that HIV-1 still demonstrated a 1.6-fold higher average transition frequency than HIV-2 (p = 0.006; Figure 6A). As HIV-1 demonstrated only a 1.8-fold higher transition frequency than HIV-2 in the presence of hypermutants (Figure 2A), this analysis indicates that differences in transition frequencies between HIV-1 and HIV-2 are driven primarily by single mutations. Further, HIV-1 still demonstrated a 3.1-fold higher frequency of G-to-A transitions than HIV-2 even after removing hypermutants (Figure 6B, p = 0.016), compared to a 3.4-fold difference in the presence of hypermutants (Figure 3). The frequencies of the other three types of transitions did not differ significantly between HIV-1 and HIV-2 in the absence of hypermutants. Even in the absence of hypermutants, HIV-1 retained a mutation spectrum much more heavily biased toward G-to-A transitions than HIV-2 (Figure 6D). In sum, these findings indicate that HIV-1 exhibits a higher transition frequency than HIV-2 primarily due to greater levels of single G-to-A mutations. As reads with single G-to-A mutations likely arise primarily through RT-mediated errors, these data argue that HIV-2 RT exhibits higher fidelity than HIV-1 RT during viral replication.
Figure 6. HIV-1 exhibits a higher frequency of G-to-A transition mutations than HIV-2 in the absence of G-to-A hypermutants.
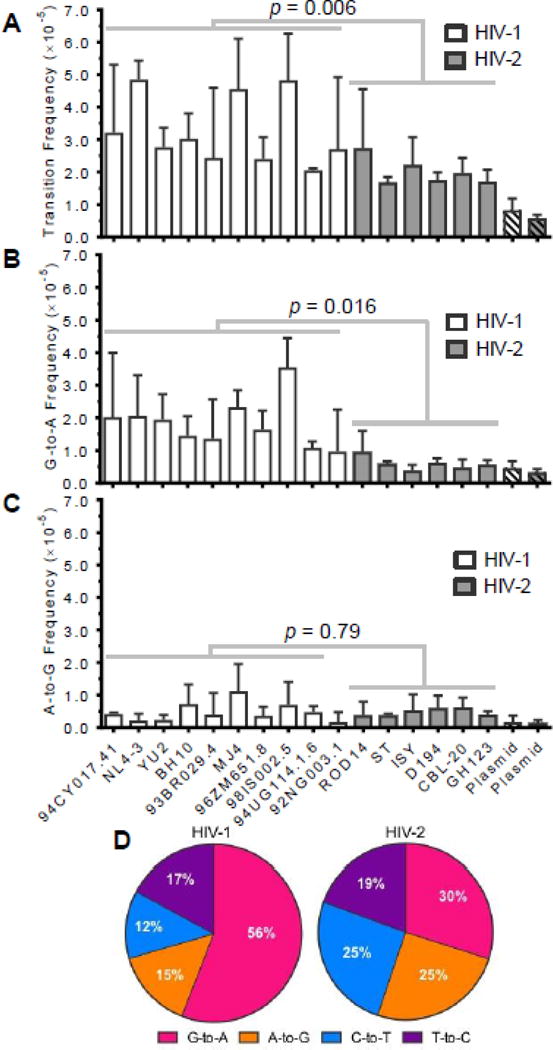
To determine the extent to which transition hypermutants contributed to mutagenesis of HIV-1 and HIV-2 (as well as observed differences between HIV-1 and HIV-2), transition frequencies and spectra were re-calculated after exclusion of all transition hypermutants, defined as reads containing two or more G-to-A, A-to-G, C-to-T, or T-to-C mutations. Only G-to-A and A-to-G transition frequencies were substantially altered by excluding transition hypermutants, as they comprised ~97% (200/206) of all hypermutants. After removing all transition hypermutants, mutation frequencies were re-calculated for all transitions (A), G-to-A transitions (B), and A-to-G transitions (C). Mutation frequencies were determined by dividing the number of the indicated type of mutation by the number of reference bases (mutations + wild-type bases) and are expressed as mutations/bp. The data represent the average of three independent biological replicates, with error bars indicating standard deviation. D. Transition spectra analysis. Transition spectra were re-determined after excluding transition hypermutants by dividing the frequency of each type of transition by the total transition frequency, with the results expressed as a percentage of total transitions. The data represent the average transition spectra across all HIV-1 or HIV-2 vectors.
Discussion
A long-standing question regarding HIV genetic variation and evolution has been whether HIV types, groups, and/or subtypes differ in terms of mutation frequencies or spectra. To address this, we developed a new high-throughput assay based on the concept of single-strand consensus sequencing (SSCS), a method that has previously been reported to greatly reduce background error rates due to PCR and sequencing in other systems [41–44]. During Illumina sequencing experiments, background error rates are typically ~10−2 m/bp for raw sequencing data and ~10−3–10−4 m/bp for stringently filtered data [56–62], although it should be noted that these previous studies were completed using a variety of sample types, PCR conditions, and Illumina sequencing instruments. In this study, the background error frequency (using SSCS) was measured using the plasmid controls as ~2×10−5 m/bp, demonstrating that SSCS significantly reduced levels of background errors. Unfortunately, even using SSCS, transversion frequencies could not be compared between HIV-1 and HIV-2, as the relative levels of background transversion errors were still too high. During SSCS, background errors may persist for several reasons: 1. Errors during the initial linear extension or during the first few cycles of exponential PCR may be frequent enough within consensus families to be represented in consensus sequences; 2. Specific positions may act as hotspots for PCR and/or sequencing errors, so that these errors independently occur often enough to be represented in consensus sequences; and 3. Errors within the unique identifier tags may lead to new consensus families that contain PCR errors [63]. Additional error-correcting methods have more recently been introduced, such as duplex consensus sequencing (DCS) [64], circular resequencing (CirSeq) [65, 66], and cypher sequencing (CypherSeq) [67], which should further reduce background error rates and thus facilitate the use of next-generation sequencing for examining viral variation.
Intriguingly, HIV-1 transition frequencies did not significantly differ between Group M subtypes, regardless of whether hypermutants were included or excluded. The average HIV-1 transition frequency was 4.6×10−5 m/bp, while the HIV-1 plasmid control transition frequency was 9.5×10−6 m/bp, leading to an estimate of 3.7×10−5 m/bp for the true mutation rate of HIV-1. These results are comparable to the mutation rate originally reported for HIV-1 of 2.3×10−5 m/bp for transitions and 3.4×10−5 m/bp for all mutations [1]. Overall, HIV-1 demonstrated an ~1.8-fold higher transition frequency than HIV-2 (4.6×10−5 vs. 2.6×10−5 m/bp) (Figure 2A), due primarily to a higher level of single G-to-A mutations and (to a lesser extent) G-to-A hypermutation. Although HIV-1 and HIV-2 RTs share only ~62% identity at the amino acid level (Table 1), nearly all the residues known to impact HIV-1 RT fidelity are completely conserved across the 16 RTs analyzed in this study (Figure S7). There are two notable exceptions: First, HIV-2 RT contains a highly conserved V75I polymorphism, which is a drug resistance-associated mutation in HIV-1 that has been found to improve HIV-1 RT fidelity 2 to 3-fold in vitro [68, 69]. This polymorphism is found in all 6 HIV-2 RTs analyzed in this study, as well as all 30 HIV-2 RTs in a pre-constructed Pol alignment from the HIV sequence database at Los Alamos National Laboratory (www.hiv.lanl.gov). Second, HIV-2 RT also contains a highly conserved K263V polymorphism, and K263A has been shown to reduce HIV-1 RT fidelity by ~1.5-fold in vitro [70]. Additional studies will be required to determine whether V75I or K263V polymorphisms contribute to the differences we observed here between HIV-1 and HIV-2 mutagenesis. Notably, none of the RT residues known to impact replication fidelity vary among the 10 HIV-1 RTs examined here (Figure S7), consistent with our finding that HIV-1 subtypes did not exhibit significantly different mutation frequencies or spectra.
Unexpectedly, HIV-1 and HIV-2 exhibited rare A-to-G hypermutants (~1 in 10,000 reads; Figure 5A), although these hypermutants did not contribute significantly to the difference in transition frequencies between HIV-1 and HIV-2 (Figure 2A). These A-to-G hypermutants were biased toward AA and TA dinucleotides (Figure 5C), consistent with the editing preferences of ADAR-1 and 2 proteins [54, 55], and in some cases contained more than 15 A-to-G mutations in a single read (Figure 5B, Figure S5). These data implicate editing by ADAR-1 and 2 proteins, but formally testing this hypothesis will require additional studies using viruses produced from cells without ADAR expression. While several groups have examined the effects of ADAR-1 and 2 proteins on HIV-1, some have reported positive effects of ADAR proteins on virus production or infectivity while others have reported antiviral effects [25–30]. Thus, it is still unclear whether ADARs promote or suppress HIV-1 infectivity, whether ADARs edit HIV RNAs in physiologically relevant contexts, or whether the effects of ADAR proteins on HIV-1 are editing-dependent or independent. Although A-to-G hypermutants were very rare in our study, they might be observed at higher frequencies in other experimental systems, such as in primary T-cells or macrophages untreated or treated with interferon (as ADAR-1 is interferon-inducible) [71, 72]. Also, viral RNAs from producer cells would likely exhibit much higher frequencies of A- to-G hypermutation than proviral DNA, as hypermutation could interfere with RNA splicing, export, or packaging. Additional studies are clearly warranted to determine whether ADAR proteins caused the A-to-G hypermutants observed in this study and whether such hypermutants are relevant to variation of HIV-1 and HIV-2.
In a previous study [40], HIV-1 (NL4-3) and HIV-2 (ROD14) mutation frequencies and spectra were compared in U373-MAGI cells using standard PCR and Illumina sequencing of five amplicons: Gag, Vif, HSA (heat stable antigen), EGFP-1 and EGFP-2 (representing nonoverlapping regions of the egfp gene). HIV-1 exhibited a higher mutation frequency than HIV-2, primarily due to a higher frequency (~28-fold, averaged across all amplicons) of G-to-A hypermutants for HIV-1. In the absence of G-to-A hypermutants, HIV-1 and HIV-2 did not exhibit significantly different total mutation frequencies. However, HIV-1 still exhibited an ~1.8- fold higher frequency of G-to-A transitions than HIV-2 in the absence of hypermutants, as well as a higher relative percentage of G-to-A transitions [40]. In the present study, HIV-1 exhibited an ~1.8-fold higher frequency of transition mutations than HIV-2 (Figure 2A), consistent with an ~3.4-fold higher frequency of G-to-A transitions (Figure 3) and an ~5.2-fold higher frequency of G-to-A transitions from hypermutants (Figure 4A). However, even in the absence of transition hypermutants, HIV-1 displayed ~1.6-fold and ~3.1 -fold higher frequencies of all transitions and G-to-A transitions, respectively, and a mutation spectrum much more heavily biased toward G- to-A transitions (Figure 6). Thus, relative to the previous study [40], the differences between HIV-1 and HIV-2 in viral mutagenesis reported here are more attributable to single G-to-A mutants than to G-to-A hypermutants. These differences may be due to the reduced G-to-A hypermutant frequencies observed in the present study, which were ~33-fold lower for HIV-1 (7.9×10−3 vs. 2.4×10−4) and ~6.4-fold lower for HIV-2 (2.8×10−4 vs. 4.4×10−5). Notably, even in the previous study, G-to-A hypermutant frequencies varied significantly depending upon the specific amplicon analyzed, ranging from 7.6×10−4 (Gag) to 2.4×10−2 (EGFP-1) for HIV-1 and from 8.0×10−5 (Gag) to 5.7×10−4 (EGFP-1) for HIV-2 [40]. Additional studies will be required to determine whether the lower hypermutant frequencies observed in the present study were due to the specific region of the genome that was examined, ideally using near full-genome sequencing to compare HIV-1 and HIV-2 mutagenesis across the viral genome. Furthermore, studies should be performed to compare HIV-1 and HIV-2 mutagenesis in primary CD4+ T-cells and macrophages, as these cell types express more physiologically relevant levels of host factors (APOBEC3, ADARs, SAMHD1) that might influence viral mutagenesis.
In sum, these data demonstrate that HIV-1 exhibits an ~1.8-fold higher transition frequency and an ~3.4-fold higher frequency of G-to-A transitions than HIV-2. Low levels of G- to-A and A-to-G hypermutants were observed for both HIV-1 and HIV-2, indicative of rare editing by host immune proteins. However, differences in HIV-1 and HIV-2 replication fidelity were primarily driven by fewer single G-to-A mutations (rather than hypermutation) for HIV-2. These data imply that HIV-2 reverse transcription is less error-prone than HIV-1 reverse transcription, as reads with single G-to-A mutations most likely result from error-prone reverse transcription. This could be due in part to the aforementioned conserved V75I polymorphism in HIV-2 RT. Nonetheless, additional studies should be performed to compare HIV-1 and HIV-2 mutagenesis in the complete absence of APOBEC3 proteins, which would enable more direct comparison of reverse transcription fidelities. While HIV-1 exhibited only an ~1.6-fold higher transition frequency than HIV-2 in the absence of hypermutants, other studies have demonstrated that RNA virus variants with even modest increases in replication fidelity (i.e. 1.4 to 2.5-fold) can significantly impair viral infection, replication, and/or pathogenicity in vivo [73–76]. For example, a chikungunya virus variant with a single mutation (C483Y) in its RNA-dependent RNA-polymerase (RdRp) exhibited a 1.4-fold lower mutation frequency and did not affect viral fitness in cell culture, but led to significantly lower infection and dissemination titers in mosquitos as well as abbreviated viremia and reduced organ titers in mice [73]. Similar findings have been observed with RdRp mutants of other RNA viruses, including West Nile virus, foot- and-mouth disease virus, and enterovirus 71 [74–76]. Thus, the difference in HIV-1 and HIV-2 fidelity observed here could plausibly contribute to the reduced fitness and pathogenicity of HIV- 2 observed clinically. On the other hand, we did not observe significant differences between HIV-1 Group M subtypes in terms of mutation frequencies or spectra. Taken together, these findings reveal an association between phylogenetic differences and differences in mutation frequency and spectra only when comparing HIV types (i.e., HIV-1 versus HIV-2). These differences may help explain the unique features of HIV-2 infection in vivo and inform antiviral and vaccine strategies.
Materials and Methods
Plasmids, cell lines, and reagents
The HIV-1 vector, pNL4-3 MIG, and the HIV-2 vector, pROD14 MIG, have already been previously described [77, 78]. These vectors contain cassettes inserted within nef that encode mCherry, an internal ribosomal entry site (IRES), and enhanced green fluorescent protein (EGFP). Both HIV-1 and HIV-2 vectors contain intact open reading frames for all genes except for env and nef. HIV-1 and HIV-2 vectors with swapped RTs were created by first amplifying RTs from a variety of plasmids and infected cell lines. The following plasmids and viruses were obtained from the NIH AIDS Reagent Program, NIAID, NIH: p94CY017.41 from Drs. Stanley Trask, Feng Gao, Beatrice Hahn, and the Aaron Diamond AIDS Research Center [79]; pYU-2 and pBH10 from Drs. Beatrice Hahn and George Shaw [80–82]; p93BR029.4, p94UG114.1.6, and p92NG003.1 from Drs. Beatrice Hahn, Feng Gao, and the UNAIDS Network for HIV Isolation and Characterization [83]; pMJ4 from Drs. Thumbi Ndung’u, Boris Renjifo, and Max Essex [84]; p96ZM651.8 and p98IS002.5 from Drs. Cynthia Rodenburg, Beatrice Hahn, Feng Gao, and the Zambian-UAB HIV Research Project [85]; pHIV- 2/ST from Drs. Beatrice Hahn and George Shaw [86, 87]; HIV-2D194/HUT-78 from Dr. Hagen von Briesen [88, 89]; HIV-2 CBL-20/H9 from Dr. Robin Weiss [90]. HIV-2 ST and D194 cell-free virus stocks from the AIDS Reagent Program were used to infect CEM-GFP cells (provided to the AIDS Reagent Program by Dr. Jacques Corbeil) [91] followed by purification of genomic DNA (~1 week later) and PCR. The HIV-2 ISY molecular clone was a gift from Dr. Genoveffa Franchini (Animal Models & Retroviral Vaccines Section, National Cancer Institute, Bethesda, MD) [92]. The HIV-2 GH123 molecular clone was a gift from Dr. Akio Adachi (The University of Tokushima Graduate School, Tokushima, Japan) [93]. All HIV-1 viral stocks were produced by co-transfection with pNL4-3 Env, a gift from Dr. Eric Freed (HIV Dynamics and Replication Program, Center for Cancer Research, National Cancer Institute, Frederick, MD). HIV-2 viral stocks were produced by co-transfection with pROD10-Env [94], a gift from Dr. Paula Cannon (University of Southern California, Los Angeles, CA). The human embryonic kidney (HEK 293T) cells were purchased from American Type Culture Collection (Manassas, VA) and maintained in Dulbecco's Modified Eagle's Medium (DMEM) from Cellgro (Manassas, VA) with 10% HyClone FetalClone III (FC3) from Thermo Scientific (Waltham, MA) and 1% penicillin/streptomycin from Life Technologies (Grand Island, NY). U373-MAGI-CXCR4CEM cells were obtained from Dr. Michael Emerman through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH [95]. U373-MAGI cells were maintained similarly to 293T cells but with addition of 1.0 μg/mL puromycin, 0.1 mg/mL hygromycin B, and 0.2 mg/mL G418 to the medium. For transfections, poly-L-lysine was from Newcomer Supply (Middleton, WI) and polyethylenimine (PEI) was from Polysciences, Inc. (Warrington, PA).
Construction of HIV-1 and HIV-2 vectors with swapped RTs
To compare viral mutagenesis in a standardized manner, HIV-1 RTs were swapped with the NL4-3 RT in pNL4-3 MIG and HIV-2 RTs were swapped with the ROD14 RT in pROD14 MIG in a near-isogenic manner. Briefly, new restriction sites (RsrII and XbaI for HIV-1, BssHII and XbaI for HIV-2) were engineered flanking the RT regions by site-directed mutagenesis using the QuikChange II XL kit (Agilent Technologies, Inc.; Santa Clara, CA). These restriction sites did not alter the protein sequence or any important known RNA structures, and these viruses exhibited infectivities similar to the parental viruses. Next, HIV-1 and HIV-2 RTs were amplified by PCR from a variety of molecular clones or from genomic DNA purified from infected cells. Both the PCR products and vectors were digested with the indicated restriction enzymes, and the appropriate fragments were ligated using T4 DNA ligase. The resulting vectors were verified by DNA sequencing to confirm that the correct RT had been amplified and ligated and to confirm that additional mutations had not been introduced into the parental vector or into RT. None of the viruses described in this manuscript exhibited major infectivity defects relative to the parental viruses. In HIV-1, this cloning strategy swapped the last 20 amino acids of protease, all of RT (560 amino acids), and the first amino acid of integrase. In HIV-2, this cloning strategy swapped the last 26 amino acids of protease, all of RT (559 amino acids), and the first amino acid of integrase.
Preparation of HIV-1 and HIV-2 proviral DNA
HIV-1 and HIV-2 viral stocks were produced, concentrated, DNaseI-treated, and titered in U373-MAGI cells as described previously [40], but without column concentration. Next, virus stocks were used to infect 100,000 U373-MAGI cells at a multiplicity of infection (MOI) of 1.0. Uninfected cells were included as negative controls. All infections were performed three times, using independently produced viral stocks. Cells were collected at 72 h post-infection, and genomic DNA was purified using the High Pure PCR Template Preparation Kit (Roche; Basel, Switzerland), eluting in 50 μL buffer. The level of plasmid carryover in genomic DNA was assessed by a previously described method [40]. Briefly, quantitative PCR was used to measure the starting quantities of the ampicillin resistance gene and of HIV-1 or HIV-2 integrase (using untailed primers to amplify the same integrase region that was later subjected to Illumina sequencing). The level of plasmid carryover was calculated by dividing the resulting starting quantities and was found to range from ~0.5–5.0%, a level that would not significantly alter mutation frequencies or spectra.
Assignment of unique identifiers (UIDs) by linear extension
To prepare samples for SSCS, 50 μL linear extensions were performed using 26 μL water, 10 μL genomic DNA, 10 μL 5X HF buffer, 1 μL dNTPs (10 mM), 2.5 μL forward primer (10 μM), and 0.5 μL Phusion Hot Start II High-Fidelity DNA Polymerase (Thermo Fisher Scientific; Waltham, MA). The cycling conditions used were an initial denaturation of 98 °C 30 s, and 10 cycles of 98 °C 10 s/61 °C 30 s/72 °C 30 s. Extensions were performed without template or with plasmid (105 copies pNL4-3 MIG or pROD14 MIG) as negative or positive controls, respectively. Although it would be preferable to use a single cycle of linear extension (to minimize re-extension from the same starting template), extension efficiencies in a single cycle were usually very low (~1–5%), necessitating multiple extension cycles. The forward primers in these reactions contained a region specific to integrase, a UID composed of 14 degenerate bases (Ns), and Illumina adapter sequence (Table S1). HIV-1 and HIV-2 primers were homologous in sequence.
The newly synthesized and tagged strand was purified to remove free primers. Free primers could lead to further extension from proviral DNA and also lead to the formation of primer heterodimers that would be difficult to remove, a common problem when using long primers containing Illumina adapter sequences (due to partial complementarity between the adapter sequences). The tagged strand was purified using AMPure XP beads (Beckman Coulter; Brea, CA) at a 0.8:1 (beads:DNA) ratio and eluted in 42 μL water, followed by transfer of 36 μL/sample to a new 96-well plate. Next, the complementary strand was synthesized using a reverse primer specific to integrase by mixing 36 μL purified DNA, 10 μL 5X HF buffer, 1 μL dNTPs, 2.5 μL primer, and 0.5 μL Phusion. The reaction was incubated at 98 °C 40 s, 58 °C 30 s, and 72 °C 30 s. During this step, the reverse primer added part of the Illumina adapter sequence as well. Lastly, the DNA was again purified using AMPure XP beads at a 0.8:1 ratio to remove free primer, eluting in 41 μL water. In principle, reverse extension could be omitted, and forward extensions could be used directly for exponential PCR with an extremely long reverse primer containing adapter and integrase sequences. However, we empirically found that reverse extension followed by exponential PCR with shorter primers was more efficient, leading to amplification of a higher proportion of the starting template (as assessed by qPCR).
Exponential PCR of tagged DNA
Tagged DNA was next subjected to exponential PCR using primers specific to the tails of earlier primers, which represent Illumina adapter sequences (Table S1). Thus, templates lacking one or both tags could not be exponentially amplified. First, the two extensions (~35 μL each) from the same genomic DNA sample were pooled together (~70 μL in total). Next, quadruplicate exponential PCR reactions were performed by mixing 15 μL water, 16 μL template, 10 μL 5X HF buffer, 1 μL dNTPs, 2.5 μL each primer, 2.5 μL SYBR Green I (diluted 1:2000 in DMSO), and 0.5 μL Phusion polymerase. SYBR Green I was included in the exponential PCR reactions in order to measure the starting quantity of tagged extensions, which was used as a predictor of consensus family size. The reactions were performed on a Bio-Rad C1000 thermal cycler attached to a CFX96 real-time PCR detection system, using the following cycling conditions: 98 °C 30 s, 30 cycles of 98 °C 10 s/70 °C 30 s/72 °C 30 s, and a final extension of 72 °C 2 min. PCR reactions were then purified using AMPure XP beads at a 1:1 ratio, and quadruplicate reactions were eluted from the magnetic beads using the same 30 μL of water (in order to avoid excessive dilution of the DNA). The purified DNA was then quantified by the Qubit dsDNA HS assay, normalized to a concentration of 1 ng/μL, and submitted to the University of Minnesota Genomics Center for library preparation and sequencing.
Library preparation and Illumina sequencing
The University of Minnesota Genomics Center performed additional PCR on the purified amplicons in order to add library indices and remaining Illumina adapter sequences. These amplifications were performed using 5 μL template DNA, 1 μL water, 2 μL 5x KAPA HiFi buffer (Kapa Biosystems, Woburn, MA), 0.3 μL dNTPs (10 mM), 0.5 μL DMSO, 0.5 μL each primer (10 μM), and 0.2 μL KAPA HiFi Polymerase (Kapa Biosystems). The cycling conditions were 95 °C for 5 min, followed by 10 cycles of 98 °C for 20 s, 55 °C for 15 s, 72°C for 1 min, and a final extension at 72 °C for 10 min. The primers used were standard Illumina dual-indexing primers (Table S1). PCR products were quantified using the PicoGreen dsDNA Assay Kit (Life Technologies), and the libraries were normalized, pooled, purified with 1.8X AMPure XP beads, and eluted in 20 μL of EB buffer (10 mM Tris-HCl, pH 8.5). The final library pool was again quantified using the PicoGreen dsDNA Assay Kit and further assessed using a Bioanalyzer High Sensitivity chip (Agilent Technologies, Inc. Santa Clara, CA). The library pool was denatured with 0.2N NaOH, diluted to 8 pM, spiked with 15% PhiX to improve sequence diversity and quality, and subjected to 2×250 paired-end sequencing on the Illumina HiSeq 2500.
Bioinformatics processing of sequencing data
Samples were demultiplexed using the standard indices added during library preparation. Illumina adapters were trimmed using cutadapt [96], and paired-end reads were merged using PANDAseq [97]. The amplicons were small enough (~254 bp, not including Illumina adapter sequences) such that forward and reverse reads were almost totally overlapping. If a base did not match between forward and reverse reads, the base with higher quality was chosen by PANDAseq. Next, consensus families were generated using BioPython [98] and a custom Python script. The script was used to create a list of the unique identifier (UID) sequences corresponding to the first 14 bp of each merged read. For each UID that was present at least three times in the dataset, a list of sequences containing the UID was generated and the consensus sequence at each position was determined by a simple pileup. The threshold used for making a consensus call was 75%. In cases where there was not a consensus base, the position was converted to an uncalled base (X) and was not considered during mutational analysis. Consensus reads were mapped to the appropriate reference sequence (pNL4-3 MIG or pROD14 MIG) using GSNAP [99], and a small number of misaligned reads were discarded. Mutation frequencies and hypermutant frequencies were determined using a custom algorithm based on the Genome Analysis Toolkit walker framework [100]. All primer sequences were excluded from mutational analysis, as errors within primers would not represent biologically meaningful mutations. Additionally, plasmid error hotspots (defined as upper outliers within the plasmid control raw data [i.e., prior to consensus-building] based on the 1.5 × interquartile range rule) were masked, a strategy previously used to reduce the level of background error [40]. Rather than masking all mutational types at hotspot positions, only the problematic type(s) (i.e., C-to-A) were masked at error hotspots.
Statistical analyses
Most mutation frequencies were compared between groups using generalized linear mixed effects models (exceptions are described below). For each analysis, the raw count of one kind of mutation (e.g., transitions or G-to-A mutations) was modeled as a Poisson random variable with logarithmic link, as is standard for Poisson outcomes, with the log of the total number of reference bases as the offset. All analyses used penalized quasilikelihood to compute estimates, standard errors, and tests [101]. Computations were done in R (v 3.2.2, R Core Team 2015) using the glmmPQL function in the MASS package v 7.3–43 [102]. For all analyses, variance components (random effects) included variation between viral strains (of a virus type and subtype), variation between biological replicates, and remaining extra-Poisson variation (“error”). Features specific to individual analyses are as follows. For comparing HIV-1 subtypes, the analysis included only HIV-1 subtypes and the fixed effect was HIV-1 subtype. For comparing plasmid controls vs. virus, HIV-1 and HIV-2 were analyzed separately; for each virus type, the fixed effect was plasmid control vs. virus. For comparing HIV-1 vs. HIV-2, plasmid controls were excluded and the fixed effect was HIV-1 vs. HIV-2. For the latter two comparisons, a further random effect was added, describing variation between subtypes within HIV-1. For mutations with very low or zero counts (e.g., G-to-A and A-to-G mutations from hypermutants for virus vs. plasmid control), comparisons used Fisher's exact test rather than generalized linear mixed effects models because the latter were not feasible. All P-values are presented without adjustment for multiple comparisons.
Supplementary Material
Research Highlights.
Studied if differences exist in mutation rate and/or mutation spectra among HIV types/subtypes
A new single-strand consensus sequencing assay was developed
Significant differences found in viral mutagenesis between HlV types but not among subtypes
HlV type but not subtype significantly impacts viral mutation frequencies and spectra
Acknowledgments
We would also like to extend our gratitude to the staff of the University of Minnesota Genomics Center for helpful advice in the design, performance, and analysis of Illumina sequencing experiments. We also thank the Minnesota Supercomputing Institute for providing computing, software, and support with data storage for this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. Journal of virology. 1995;69:5087–94. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O’Neil PK, Sun G, Yu H, Ron Y, Dougherty JP, Preston BD. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. The Journal of biological chemistry. 2002;277:38053–61. doi: 10.1074/jbc.M204774200. [DOI] [PubMed] [Google Scholar]
- 3.Schlub TE, Grimm AJ, Smyth RP, Cromer D, Chopra A, Mallal S, et al. 15–20% of HIV substitution mutations are associated with recombination. Journal of virology. 2014 doi: 10.1128/JVI.03136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. Journal of virology. 2010;84:9864–78. doi: 10.1128/JVI.00915-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell research. 2008;18:148–61. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menendez-Arias L. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses. 2009;1:1137–65. doi: 10.3390/v1031137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Refsland EW, Harris RS. The APOBEC3 family of retroelement restriction factors. Current topics in microbiology and immunology. 2013;371:1–27. doi: 10.1007/978-3-642-37765-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desimmie BA, Delviks-Frankenberrry KA, Burdick RC, Qi D, Izumi T, Pathak VK. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. Journal of molecular biology. 2014;426:1220–45. doi: 10.1016/j.jmb.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Current biology: CB. 2004;14:1392–6. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 10.Binka M, Ooms M, Steward M, Simon V. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. Journal of virology. 2012;86:49–59. doi: 10.1128/JVI.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS pathogens. 2005;1:e6. doi: 10.1371/journal.ppat.0010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwabu Y, Kinomoto M, Tatsumi M, Fujita H, Shimura M, Tanaka Y, et al. Differential anti- APOBEC3G activity of HIV-1 Vif proteins derived from different subtypes. The Journal of biological chemistry. 2010;285:35350–8. doi: 10.1074/jbc.M110.173286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fourati S, Lambert-Niclot S, Soulie C, Wirden M, Malet I, Valantin MA, et al. Differential impact of APOBEC3-driven mutagenesis on HIV evolution in diverse anatomical compartments. Aids. 2014;28:487–91. doi: 10.1097/QAD.0000000000000182. [DOI] [PubMed] [Google Scholar]
- 14.Janini M, Rogers M, Birx DR, McCutchan FE. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4(+) T cells. Journal of virology. 2001;75:7973–86. doi: 10.1128/JVI.75.17.7973-7986.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kieffer TL, Kwon P, Nettles RE, Han Y, Ray SC, Siliciano RF. G—>A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. Journal of virology. 2005;79:1975–80. doi: 10.1128/JVI.79.3.1975-1980.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kijak GH, Janini LM, Tovanabutra S, Sanders-Buell E, Arroyo MA, Robb ML, et al. Variable contexts and levels of hypermutation in HIV-1 proviral genomes recovered from primary peripheral blood mononuclear cells. Virology. 2008;376:101–11. doi: 10.1016/j.virol.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Pace C, Keller J, Nolan D, James I, Gaudieri S, Moore C, et al. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. Journal of virology. 2006;80:9259–69. doi: 10.1128/JVI.00888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei M, Xing H, Hong K, Huang H, Tang H, Qin G, et al. Biased G-to-A hypermutation in HIV-1 proviral DNA from a long-term non-progressor. Aids. 2004;18:1863–5. doi: 10.1097/00002030-200409030-00023. [DOI] [PubMed] [Google Scholar]
- 19.Wood N, Bhattacharya T, Keele BF, Giorgi E, Liu M, Gaschen B, et al. HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS pathogens. 2009;5:e1000414. doi: 10.1371/journal.ppat.1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim EY, Bhattacharya T, Kunstman K, Swantek P, Koning FA, Malim MH, et al. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. Journal of virology. 2010;84:10402–5. doi: 10.1128/JVI.01223-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mulder LC, Harari A, Simon V. Cytidine deamination induced HIV-1 drug resistance. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5501–6. doi: 10.1073/pnas.0710190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell RA, Moore MD, Hu WS, Pathak VK. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology. 2009;6:16. doi: 10.1186/1742-4690-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadler HA, Stenglein MD, Harris RS, Mansky LM. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. Journal of virology. 2010;84:7396–404. doi: 10.1128/JVI.00056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delviks-Frankenberry KA, Nikolaitchik OA, Burdick RC, Gorelick RJ, Keele BF, Hu WS, et al. Minimal Contribution of APOBEC3-Induced G-to-A Hypermutation to HIV-1 Recombination and Genetic Variation. PLoS pathogens. 2016;12:e1005646. doi: 10.1371/journal.ppat.1005646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biswas N, Wang T, Ding M, Tumne A, Chen Y, Wang Q, et al. ADAR1 is a novel multi targeted anti-HIV-1 cellular protein. Virology. 2012;422:265–77. doi: 10.1016/j.virol.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doria M, Neri F, Gallo A, Farace MG, Michienzi A. Editing of HIV-1 RNA by the double-stranded RNA deaminase ADAR1 stimulates viral infection. Nucleic acids research. 2009;37:5848–58. doi: 10.1093/nar/gkp604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doria M, Tomaselli S, Neri F, Ciafre SA, Farace MG, Michienzi A, et al. ADAR2 editing enzyme is a novel human immunodeficiency virus-1 proviral factor. The Journal of general virology. 2011;92:1228–32. doi: 10.1099/vir.0.028043-0. [DOI] [PubMed] [Google Scholar]
- 28.Phuphuakrat A, Kraiwong R, Boonarkart C, Lauhakirti D, Lee TH, Auewarakul P. Double-stranded RNA adenosine deaminases enhance expression of human immunodeficiency virus type 1 proteins. Journal of virology. 2008;82:10864–72. doi: 10.1128/JVI.00238-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuadrado E, Booiman T, van Hamme JL, Jansen MH, van Dort KA, Vanderver A, et al. ADAR1 Facilitates HIV-1 Replication in Primary CD4+ T Cells. PLoS One. 2015;10:e0143613. doi: 10.1371/journal.pone.0143613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiden MD, Hoshino S, Levy DN, Li Y, Kumar R, Burke SA, et al. Adenosine deaminase acting on RNA-1 (ADAR1) inhibits HIV-1 replication in human alveolar macrophages. PLoS One. 2014;9:e108476. doi: 10.1371/journal.pone.0108476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gottlieb GS, Eholie SP, Nkengasong JN, Jallow S, Rowland-Jones S, Whittle HC, et al. A call for randomized controlled trials of antiretroviral therapy for HIV-2 infection in West Africa. Aids. 2008;22:2069–72. doi: 10.1097/QAD.0b013e32830edd44. discussion 73–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adjorlolo-Johnson G, De Cock KM, Ekpini E, Vetter KM, Sibailly T, Brattegaard K, et al. Prospective comparison of mother-to-child transmission of HIV-1 and HIV-2 in Abidjan, Ivory Coast. Jama. 1994;272:462–6. [PubMed] [Google Scholar]
- 33.Kanki PJ, Travers KU, S MB, Hsieh CC, Marlink RG, Gueye NA, et al. Slower heterosexual spread of HIV-2 than HIV-1. Lancet. 1994;343:943–6. doi: 10.1016/s0140-6736(94)90065-5. [DOI] [PubMed] [Google Scholar]
- 34.O’Donovan D, Ariyoshi K, Milligan P, Ota M, Yamuah L, Sarge-Njie R, et al. Maternal plasma viral RNA levels determine marked differences in mother-to-child transmission rates of HIV-1 and HIV-2 in The Gambia. MRC/Gambia Government/University College London Medical School working group on mother-child transmission of HIV. Aids. 2000;14:441–8. doi: 10.1097/00002030-200003100-00019. [DOI] [PubMed] [Google Scholar]
- 35.Marlink R, Kanki P, Thior I, Travers K, Eisen G, Siby T, et al. Reduced rate of disease development after HIV-2 infection as compared to HIV-1. Science. 1994;265:1587–90. doi: 10.1126/science.7915856. [DOI] [PubMed] [Google Scholar]
- 36.de Silva TI, Cotten M, Rowland-Jones SL. HIV-2: the forgotten AIDS virus. Trends in microbiology. 2008;16:588–95. doi: 10.1016/j.tim.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Andersson S, Norrgren H, da Silva Z, Biague A, Bamba S, Kwok S, et al. Plasma viral load in HIV-1 and HIV-2 singly and dually infected individuals in Guinea-Bissau, West Africa: significantly lower plasma virus set point in HIV-2 infection than in HIV-1 infection. Archives of internal medicine. 2000;160:3286–93. doi: 10.1001/archinte.160.21.3286. [DOI] [PubMed] [Google Scholar]
- 38.Popper SJ, Sarr AD, Travers KU, Gueye-Ndiaye A, Mboup S, Essex ME, et al. Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. The Journal of infectious diseases. 1999;180:1116–21. doi: 10.1086/315010. [DOI] [PubMed] [Google Scholar]
- 39.Gottlieb GS, Hawes SE, Agne HD, Stern JE, Critchlow CW, Kiviat NB, et al. Lower levels of HIV RNA in semen in HIV-2 compared with HIV-1 infection: implications for differences in transmission. Aids. 2006;20:895–900. doi: 10.1097/01.aids.0000218554.59531.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rawson JM, Landman SR, Reilly CS, Mansky LM. HIV-1 and HIV-2 exhibit similar mutation frequencies and spectra in the absence of G-to-A hypermutation. Retrovirology. 2015;12:60. doi: 10.1186/s12977-015-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:9530–5. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lundberg DS, Yourstone S, Mieczkowski P, Jones CD, Dangl JL. Practical innovations for high-throughput amplicon sequencing. Nature methods. 2013;10:999–1002. doi: 10.1038/nmeth.2634. [DOI] [PubMed] [Google Scholar]
- 44.Jabara CB, Jones CD, Roach J, Anderson JA, Swanstrom R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:20166–71. doi: 10.1073/pnas.1110064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holtz CM, Mansky LM. Variation of HIV-1 mutation spectra among cell types. Journal of virology. 2013;87:5296–9. doi: 10.1128/JVI.03576-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mansky LM. Forward mutation rate of human immunodeficiency virus type 1 in a T lymphoid cell line. AIDS research and human retroviruses. 1996;12:307–14. doi: 10.1089/aid.1996.12.307. [DOI] [PubMed] [Google Scholar]
- 47.Mansky LM. The mutation rate of human immunodeficiency virus type 1 is influenced by the vpr gene. Virology. 1996;222:391–400. doi: 10.1006/viro.1996.0436. [DOI] [PubMed] [Google Scholar]
- 48.Mansky LM, Preveral S, Selig L, Benarous R, Benichou S. The interaction of vpr with uracil DNA glycosylase modulates the human immunodeficiency virus type 1 In vivo mutation rate. Journal of virology. 2000;74:7039–47. doi: 10.1128/jvi.74.15.7039-7047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harari A, Ooms M, Mulder LC, Simon V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. Journal of virology. 2009;83:295–303. doi: 10.1128/JVI.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, et al. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–9. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 51.Langlois MA, Beale RC, Conticello SG, Neuberger MS. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic acids research. 2005;33:1913–23. doi: 10.1093/nar/gki343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Current biology : CB. 2004;14:1385–91. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 53.Knoepfel SA, Salisch NC, Huelsmann PM, Rauch P, Walter H, Metzner KJ. Comparison of G-to-A mutation frequencies induced by APOBEC3 proteins in H9 cells and peripheral blood mononuclear cells in the context of impaired processivities of drug-resistant human immunodeficiency virus type 1 reverse transcriptase variants. Journal of virology. 2008;82:6536–45. doi: 10.1128/JVI.00554-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–84. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 55.Polson AG, Bass BL. Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J. 1994;13:5701–11. doi: 10.1002/j.1460-2075.1994.tb06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic acids research. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minoche AE, Dohm JC, Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome biology. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y, et al. Sequence- specific error profile of Illumina sequencers. Nucleic acids research. 2011;39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R, et al. Characterizing and measuring bias in sequence data. Genome biology. 2013;14:R51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schirmer M, Ijaz UZ, D’Amore R, Hall N, Sloan WT, Quince C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic acids research. 2015;43:e37. doi: 10.1093/nar/gku1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van den Hoecke S, Verhelst J, Vuylsteke M, Saelens X. Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing. BMC genomics. 2015;16:79. doi: 10.1186/s12864-015-1284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou S, Jones C, Mieczkowski P, Swanstrom R. Primer ID Validates Template Sampling Depth and Greatly Reduces the Error Rate of Next-Generation Sequencing of HIV-1 Genomic RNA Populations. Journal of virology. 2015;89:8540–55. doi: 10.1128/JVI.00522-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14508–13. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Acevedo A, Brodsky L, Andino R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature. 2014;505:686–90. doi: 10.1038/nature12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lou DI, Hussmann JA, McBee RM, Acevedo A, Andino R, Press WH, et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:19872–7. doi: 10.1073/pnas.1319590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gregory MT, Bertout JA, Ericson NG, Taylor SD, Mukherjee R, Robins HS, et al. Targeted single molecule mutation detection with massively parallel sequencing. Nucleic acids research. 2016;44:e22. doi: 10.1093/nar/gkv915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alvarez M, Matamoros T, Menendez-Arias L. Increased thermostability and fidelity of DNA synthesis of wild-type and mutant HIV-1 group O reverse transcriptases. Journal of molecular biology. 2009;392:872–84. doi: 10.1016/j.jmb.2009.07.081. [DOI] [PubMed] [Google Scholar]
- 69.Matamoros T, Kim B, Menendez-Arias L. Mechanistic insights into the role of Val75 of HIV- 1 reverse transcriptase in misinsertion and mispair extension fidelity of DNA synthesis. Journal of molecular biology. 2008;375:1234–48. doi: 10.1016/j.jmb.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 70.Beard WA, Stahl SJ, Kim HR, Bebenek K, Kumar A, Strub MP, et al. Structure/function studies of human immunodeficiency virus type 1 reverse transcriptase. Alanine scanning mutagenesis of an alpha-helix in the thumb subdomain. The Journal of biological chemistry. 1994;269:28091–7. [PubMed] [Google Scholar]
- 71.Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded- RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15:5376–88. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang JH, Luo X, Nie Y, Su Y, Zhao Q, Kabir K, et al. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology. 2003;109:15–23. doi: 10.1046/j.1365-2567.2003.01598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coffey LL, Beeharry Y, Borderia AV, Blanc H, Vignuzzi M. Arbovirus high fidelity variant loses fitness in mosquitoes and mice. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16038–43. doi: 10.1073/pnas.1111650108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meng T, Kwang J. Attenuation of human enterovirus 71 high-replication-fidelity variants in AG129 mice. Journal of virology. 2014;88:5803–15. doi: 10.1128/JVI.00289-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Van Slyke GA, Arnold JJ, Lugo AJ, Griesemer SB, Moustafa IM, Kramer LD, et al. Sequence-Specific Fidelity Alterations Associated with West Nile Virus Attenuation in Mosquitoes. PLoS pathogens. 2015;11:e1005009. doi: 10.1371/journal.ppat.1005009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zeng J, Wang H, Xie X, Li C, Zhou G, Yang D, et al. Ribavirin-resistant variants of foot-and- mouth disease virus: the effect of restricted quasispecies diversity on viral virulence. Journal of virology. 2014;88:4008–20. doi: 10.1128/JVI.03594-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rawson JM, Heineman RH, Beach LB, Martin JL, Schnettler EK, Dapp MJ, et al. 5,6- Dihydro-5-aza-2’-deoxycytidine potentiates the anti-HIV-1 activity of ribonucleotide reductase inhibitors. Bioorganic & medicinal chemistry. 2013;21:7222–8. doi: 10.1016/j.bmc.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beach LB, Rawson JM, Kim B, Patterson SE, Mansky LM. Novel inhibitors of human immunodeficiency virus type 2 infectivity. The Journal of general virology. 2014;95:2778–83. doi: 10.1099/vir.0.069864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao F, Vidal N, Li Y, Trask SA, Chen Y, Kostrikis LG, et al. Evidence of two distinct subsubtypes within the HIV-1 subtype A radiation. AIDS research and human retroviruses. 2001;17:675–88. doi: 10.1089/088922201750236951. [DOI] [PubMed] [Google Scholar]
- 80.Li Y, Hui H, Burgess CJ, Price RW, Sharp PM, Hahn BH, et al. Complete nucleotide sequence, genome organization, and biological properties of human immunodeficiency virus type 1 in vivo: evidence for limited defectiveness and complementation. Journal of virology. 1992;66:6587–600. doi: 10.1128/jvi.66.11.6587-6600.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Y, Kappes JC, Conway JA, Price RW, Shaw GM, Hahn BH. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. Journal of virology. 1991;65:3973–85. doi: 10.1128/jvi.65.8.3973-3985.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hahn BH, Shaw GM, Arya SK, Popovic M, Gallo RC, Wong-Staal F. Molecular cloning and characterization of the HTLV-III virus associated with AIDS. Nature. 1984;312:166–9. doi: 10.1038/312166a0. [DOI] [PubMed] [Google Scholar]
- 83.Gao F, Robertson DL, Carruthers CD, Morrison SG, Jian B, Chen Y, et al. A comprehensive panel of near-full-length clones and reference sequences for non-subtype B isolates of human immunodeficiency virus type 1. Journal of virology. 1998;72:5680–98. doi: 10.1128/jvi.72.7.5680-5698.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ndung’u T, Renjifo B, Essex M. Construction and analysis of an infectious human Immunodeficiency virus type 1 subtype C molecular clone. Journal of virology. 2001;75:4964–72. doi: 10.1128/JVI.75.11.4964-4972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodenburg CM, Li Y, Trask SA, Chen Y, Decker J, Robertson DL, et al. Near full-length clones and reference sequences for subtype C isolates of HIV type 1 from three different continents. AIDS research and human retroviruses. 2001;17:161–8. doi: 10.1089/08892220150217247. [DOI] [PubMed] [Google Scholar]
- 86.Kong LI, Lee SW, Kappes JC, Parkin JS, Decker D, Hoxie JA, et al. West African HIV-2- related human retrovirus with attenuated cytopathicity. Science. 1988;240:1525–9. doi: 10.1126/science.3375832. [DOI] [PubMed] [Google Scholar]
- 87.Kumar P, Hui HX, Kappes JC, Haggarty BS, Hoxie JA, Arya SK, et al. Molecular characterization of an attenuated human immunodeficiency virus type 2 isolate. Journal of virology. 1990;64:890–901. doi: 10.1128/jvi.64.2.890-901.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kuhnel H, von Briesen H, Dietrich U, Adamski M, Mix D, Biesert L, et al. Molecular cloning of two west African human immunodeficiency virus type 2 isolates that replicate well in macrophages: a Gambian isolate, from a patient with neurologic acquired immunodeficiency syndrome, and a highly divergent Ghanian isolate. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:2383–7. doi: 10.1073/pnas.86.7.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.von Briesen H, Andreesen R, Rubsamen-Waigmann H. Systematic classification of HIV biological subtypes on lymphocytes and monocytes/macrophages. Virology. 1990;178:597–602. doi: 10.1016/0042-6822(90)90361-t. [DOI] [PubMed] [Google Scholar]
- 90.Schulz TF, Whitby D, Hoad JG, Corrah T, Whittle H, Weiss RA. Biological and molecular variability of human immunodeficiency virus type 2 isolates from The Gambia. Journal of virology. 1990;64:5177–82. doi: 10.1128/jvi.64.10.5177-5182.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gervaix A, West D, Leoni LM, Richman DD, Wong-Staal F, Corbeil J. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:4653–8. doi: 10.1073/pnas.94.9.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hattori N, Michaels F, Fargnoli K, Marcon L, Gallo RC, Franchini G. The human immunodeficiency virus type 2 vpr gene is essential for productive infection of human macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:8080–4. doi: 10.1073/pnas.87.20.8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shibata R, Miura T, Hayami M, Ogawa K, Sakai H, Kiyomasu T, et al. Mutational analysis of the human immunodeficiency virus type 2 (HIV-2) genome in relation to HIV-1 and simian immunodeficiency virus SIV (AGM) Journal of virology. 1990;64:742–7. doi: 10.1128/jvi.64.2.742-747.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abada P, Noble B, Cannon PM. Functional domains within the human immunodeficiency virus type 2 envelope protein required to enhance virus production. Journal of virology. 2005;79:3627–38. doi: 10.1128/JVI.79.6.3627-3638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vodicka MA, Goh WC, Wu LI, Rogel ME, Bartz SR, Schweickart VL, et al. Indicator cell lines for detection of primary strains of human and simian immunodeficiency viruses. Virology. 1997;233:193–8. doi: 10.1006/viro.1997.8606. [DOI] [PubMed] [Google Scholar]
- 96.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–2. [Google Scholar]
- 97.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–3. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–81. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wolfinger R, Oconnell M. Generalized Linear Mixed Models – a Pseudo-Likelihood Approach. J Stat Comput Sim. 1993;48:233–43. [Google Scholar]
- 102.Venables WN, Ripley BD. Modern Applied Statistics with S. Fourth. New York: Springer; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.