

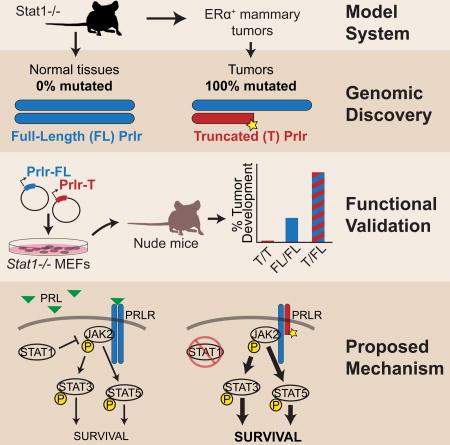
Summary
Estrogen receptor alpha-positive luminal tumors are the most frequent subtype of breast cancer. Stat1−/− mice develop mammary tumors that closely recapitulate the biological characteristics of this cancer subtype. To identify transforming events that contribute to tumorigenesis, we performed whole genome sequencing of Stat1−/− primary mammary tumors and matched normal tissues. This investigation identified somatic truncating mutations affecting the prolactin receptor (Prlr) in all tumor and no normal samples. Targeted sequencing confirmed the presence of these mutations in precancerous lesions, indicating this is an early event in tumorigenesis. Functional evaluation of these heterozygous mutations in Stat1−/− mouse embryonic fibroblasts showed that co-expression of truncated and wild type Prlr led to aberrant Stat3 and Stat5 activation downstream of the receptor, cellular transformation in vitro and tumor formation in vivo. In conclusion, truncating mutations of Prlr promote tumor growth in a model of human ERα+ breast cancer and warrant further investigation.
Keywords: Breast cancer, estrogen-receptor-positive, luminal, STAT1, mouse model, whole genome sequencing, PRLR
Graphical abstract
Introduction
The transcription factor STAT1 functions as a tumor suppressor in mammary gland epithelial cells (Chan et al., 2014; Chan et al., 2012; Schneckenleithner et al., 2011). Selective loss of STAT1 expression in breast cancer cells is associated with a significant percentage of human estrogen receptor alpha-positive (ERα+) luminal breast tumors (Chan et al., 2012). The lack of Stat1 expression in mice pre-disposes these animals to mammary adenocarcinoma development (Chan et al., 2012; Schneckenleithner et al., 2011). We demonstrated that spontaneous mammary tumors that develop in Stat1−/− female mice progressed in a manner similar to ERα+ progesterone receptor-positive invasive ductal carcinoma in humans (Chan et al., 2012). These tumors also displayed transcript expression profiles that clustered more closely with human ERα+ luminal breast cancers than other murine mammary tumor models and thus recapitulated the molecular characteristics of the luminal breast tumor subtype (Chan et al., 2012; Pfefferle et al., 2013).
To further identify the biological consequences of STAT1 loss in ERα+ luminal breast cancer, we set out to uncover genomic event(s) that fully transform the phenotype of mammary gland epithelial cells into cancer cells in the Stat1−/− mouse model. We performed whole genome sequencing of 14 primary Stat1−/− mammary tumors, 5 Stat1−/− ovarian hormone-independent tumors, as well as 3 Stat1−/− tumor-derived cell lines for a total of 22 independent Stat1−/− tumors (Figure 1). We compared genomic variations in tumor samples with those in control samples that consisted of 10 wild type, 5 tumor-free Stat1−/− mammary glands and 15 Stat1−/− tails. Our analysis revealed relatively few copy number variation (CNV) events in primary Stat1−/− mammary tumors, but a point mutation rate consistent with that observed in human breast cancers. A number of key genes reported in human cohorts were also mutated in the Stat1−/− mammary tumors including Trp53, Brca1, Mll3 and the Arid family. Strikingly, we identified a truncating mutation hotspot within the prolactin receptor (Prlr) with mutations affecting 100% of the Stat1−/− mammary tumor samples and 0% of control samples examined. Co-expression of full-length and truncated Prlr in immortalized Stat1−/− mouse embryonic fibroblasts (MEFs) led to activation of the downstream oncogenic substrates Stat3 and Stat5, transformation of MEFs in vitro, and tumor formation in mice.
Figure 1. Sample summary.

Samples from wild type 129/Sv (+/+) and Stat1 knockout (−/−) were used for whole genome sequencing in discovery and subsequent extension for targeted sequencing of Prlr. Mice with matched normal tail DNA used for analysis are indicated with a red tail. Ductal carcinoma in situ (DCIS) samples were prepared from formalin-fixed, paraffin embedded (FFPE) samples. See also Table S7 and Figure S2.
Results
Summary of somatic alterations
In a discovery set of 22 Stat1−/− tumor samples, using whole genome sequencing (WGS), we detected over 10,112 single nucleotide variants (SNVs) and 3,331 insertions and deletions (indels) within or near coding regions of known genes. Filtering and manual review reduced the set to 1,770 SNVs and 88 indels (Table S1). The 1,858 mutations occurred in 1,649 genes with 139 recurrently mutated genes (occurring in two or more samples) across all 22 tumors (Table 1 and Table S2). Mutational significance analysis revealed 16 significantly mutated genes (FDR<0.05, likelihood ratio test (LRT) method; Table S3) (Dees et al., 2012). The most recurrently and significantly mutated gene was the prolactin receptor (Prlr), found in 17 out of 22 samples. These Prlr mutations will be discussed in greater detail below. Beyond the Prlr gene, mutations were observed in many of the same key genes and pathways reported previously for human breast and ovarian cancer, including Trp53, DNA repair genes (Brca1, Rad50, Rfc2, Poln, and Polr2a), chromatin modifiers (Arid1a and Arid1b), transcription factors (Zfp335, Zfp523, Zfp119a, Zfp119b), and kinases and phosphatases (Ptprb, Pik3r2, Pik3cd, Mapk7 and Src)(Banerji et al., 2012; Cancer Genome Atlas Network, 2012; Ellis et al., 2012; Jones et al., 2010; Stephens et al., 2012; Wiegand et al., 2010). Other mutations of note were in Ip6k2, which encodes a protein that affects the growth suppressive and apoptotic activities of interferon-beta in ovarian cancers (Morrison et al., 2001); Tiam1, which is a t-lymphoma invasion and metastasis-inducing protein; and Esrrg, which encodes the estrogen-related receptor gamma protein. No significant differences were observed in mutation frequencies between ovarian dependent and independent tumors or cell lines although we are admittedly underpowered to detect such differences.
Table 1.
Recurrently mutated genes with mutations in more than two tumors from WGS. See also Table S1, Table S2, and Table S3.
| Gene Common Name | Mutations (n) | Samples (n) | HGNC symbol |
|---|---|---|---|
| Prlr* | 17& | 17& | PRLR |
| Olfr1062* | 5 | 5 | N/A |
| Mll3 | 4 | 4 | MLL3 |
| 4932431P20Rik* | 4 | 4 | WDR87 |
| Gm10750 | 4 | 4 | N/A |
| Gatsl3 | 3 | 3 | GATSL3 |
| Gnl1* | 3 | 3 | GNL1 |
| Esrrg* | 3 | 3 | ESRRG |
| Galnt5* | 3 | 3 | GALNT5 |
| BC006779 | 3 | 3 | N/A |
| Rbbp6 | 3 | 3 | RBBP6 |
| Slc39a12* | 3 | 3 | SLC39A12 |
| Zfp335 | 3 | 3 | ZNF335 |
| Gm6369 | 3 | 3 | N/A |
| Fbxl7 | 4 | 3 | FBXL7 |
| Krt15* | 3 | 3 | KRT15 |
| Tgoln1* | 3 | 3 | TGOLN2 |
| Gm16372 | 3 | 3 | N/A |
| Trp53* | 3 | 3 | TP53 |
| Taar7e* | 3 | 3 | N/A |
| 4930503E14Rik* | 3 | 3 | N/A |
| ENSMUSG00000077055 | 3 | 3 | N/A |
| Tmem181b-ps | 3 | 3 | TMEM181 |
| Gm10601 | 3 | 3 | N/A |
| A230087F16Rik | 3 | 3 | N/A |
| Gm11867 | 3 | 3 | N/A |
| Gm16957 | 3 | 3 | N/A |
| Vmn2r90 | 3 | 3 | N/A |
‘Mutations’ = total number of mutations identified
‘Samples’ = number of samples with at least one mutation.
Multiple mutations within the same gene per sample are possible.
Prlr mutations were called in 17/22 tumor samples during initial calling from WGS data. Manual review and subsequent validation assays confirmed mutations in 22/22 tumor samples.
Significantly mutated genes (Table S3)
The numbers of mouse whole genomes sequenced in our study (n=22) limit the direct comparison of mutation frequencies. Despite this, we compared our cohort to mutations frequently observed in human luminal breast cancers by identifying genes mutated at >5% frequency in the human TCGA luminal A and B cohort (n=699) (Cancer Genome Atlas Network, 2012). We observed Trp53 and Mll3 mutations at frequencies comparable to the human dataset with 14% vs 16% and 18% vs 8%, respectively (Figure S1). The lack of Pik3ca and Map3k1 mutations is perhaps expected in the context of Prlr truncation (see below) given that their activity is downstream of Prlr and therefore activating mutations in these genes may not be required for tumor formation. Gata3 mutations were also not observed in our dataset although we previously showed upregulation of Gata3 in Stat1−/− tumors, consistent with ERα+ human breast cancer (Chan et al., 2012).
Summary of copy number variation results
As a positive control, in each tumor we verified that the Stat1 exon 3–5 deletion was detectable by read-depth based CNV analysis using CopyCat. Only moderate additional copy number changes were observed in tumor samples (Table S9). Virtually no CNV events were observed in wild type or Stat1−/− tumor-free mammary glands (Table S9).
Recurrent truncating mutations of Prlr
As described above, the most recurrently mutated gene in Stat1−/− mammary tumors was that of prolactin receptor (Prlr) (Table 1). Prlr mutations were not observed in any matched normal tails (0/17), wild type mammary glands (0/10), or tumor-free Stat1−/− mammary glands (0/5). Our alignment and variant calling pipelines and further manual inspection of WGS data for the Prlr region revealed Prlr mutations in a total of 21/22 tumors (Table 2 and Appendix 1). Only the TAC246 tumor sample had no evidence of a Prlr mutation in the initial discovery WGS dataset. All discovery tumor samples were further Sanger sequenced for the Prlr region of interest to validate the observed indels (Table S4 and Appendix 2). Sequence traces consistent with the WGS mutations were confirmed for 19 of 21 tumors. Traces for two samples were ambiguous. However, detection of indels from Sanger traces is difficult and this, in our experience, represents a very high indel validation rate. MiSeq sequencing of a formalin-fixed paraffin-embedded (FFPE) sample from the TAC246 tumor identified a Prlr mutation in this sample that was missed in the original discovery set by WGS (giving a sensitivity of 95.5% for the ~30× WGS approach of detecting Prlr mutations). An additional second mutation was detected by MiSeq data of an FFPE sample from the TAC247 tumor that was also missed by WGS. As a result, 100% of the original discovery samples were found to contain at least one Prlr mutation (Figure S2). Extension sequencing, by the Sanger method, was performed on an additional 10 tumors and 35 non-tumor samples from 10 additional mice (Appendix 3). Non-tumor samples included 10 normal tails, 8 uteri, 7 ovaries, 8 livers, and 2 mammary glands. Prlr mutations were observed in all additional tumors and none of the non-tumor samples (Figure S2).
Table 2.
Summary of Prlr mutations in discovery and extension sets. See also Table S4, Table S5, Table S10, Appendix 1, Appendix 2, Appendix 3, and Appendix 4.
| Start | Stop | Variant | Sample | Variant Effect | AA change |
Set | WGS | PCR Sanger |
PCR MiSeq |
|---|---|---|---|---|---|---|---|---|---|
| 10258182 | 10258182 | G/0 | B3R15 L. tho. | frame_shift_del | p.G330fs | Discovery | Y | Y | N/A |
| 10258180 | 10258180 | C/0 | B3R1R2L1 R. tho. | frame_shift_del | p.P329fs | Discovery | Y | Y | N/A |
| 10258151 | 10258151 | G/0 | OVX3L2 R. tho.* | frame_shift_del | p.L320fs | Discovery | Y | Y | N/A |
| 10258151 | 10258151 | G/0 | OVX6R2 L. cerv.* | frame_shift_del | p.L320fs | Discovery | Y | Y | N/A |
| 10258147 | 10258147 | G/0 | SSM1 | frame_shift_del | p.E318fs | Discovery | Y | Y | N/A |
| 10258139 | 10258140 | 0/TGAGGACGAGC | SSM2 | frame_shift_ins | p.E319fs | Discovery | Y | Y | N/A |
| 10258195 | 10258195 | A/0 | SSM3 | frame_shift_del | p.K334fs | Discovery | Y | Y | N/A |
| 10258180 | 10258180 | C/0 | TAC171 R. tho.* | frame_shift_del | p.P329fs | Discovery | Y | Y | N/A |
| 10258147 | 10258147 | G/0 | TAC183 R. tho.* | frame_shift_del | p.E318fs | Discovery | Y | Y | N/A |
| 10258147 | 10258148 | 0/GATGGCT | TAC186 L. cerv.* | frame_shift_ins | p.E318fs | Discovery | Y | Y | N/A |
| 10258178 | 10258186 | ATCCGGGTC/0 | TAC246 R. ing. | in_frame_del | p.Y328* | Discovery | N | N | Y |
| 10258147 | 10258147 | G/0 | TAC247 L. cerv. | frame_shift_del | p.E318fs | Discovery | N | N | Y |
| 10258153 | 10258168 | CTAATGCCATCCCATT/0 | TAC247 L. cerv. | frame_shift_del | p.L320fs | Discovery | Y | A | Y |
| 10258182 | 10258182 | G/0 | TAC266 L. tho. | frame_shift_del | p.G330fs | Discovery | Y | Y | N/A |
| 10258151 | 10258154 | GGCT/0 | TAC268 L. tho. | frame_shift_del | p.R319fs | Discovery | Y | Y | N/A |
| 10258147 | 10258147 | G/0 | TAC269 L. cerv. | frame_shift_del | p.E318fs | Discovery | Y | Y | N/A |
| 10258180 | 10258180 | C/0 | TAC270 L. cerv. | frame_shift_del | p.P329fs | Discovery | Y | Y | N/A |
| 10258147 | 10258148 | GA/0 | TAC270 L. ing. | frame_shift_del | p.E318fs | Discovery | Y | Y | N/A |
| 10258179 | 10258179 | T/A | TAC271 R. tho. | nonsense | p.Y328* | Discovery | Y | A | N/A |
| 10258146 | 10258168 | CGAGCGGCTAATGCCATCCCATT/0 | TAC272 L. tho. | frame_shift_del | p.E318fs | Discovery | Y | Y | N/A |
| 10258195 | 10258195 | A/0 | TAC273 L. tho. | frame_shift_del | p.K334fs | Discovery | Y | Y | N/A |
| 10258146 | 10258149 | CGAG/0 | TAC273 R. ing. | frame_shift_del | p.E318fs | Discovery | Y | Y | N/A |
| 10258147 | 10258147 | G/T | TAC274 R. ing. | nonsense | p.E318* | Discovery | Y | Y | N/A |
| 10258184 | 10258184 | G/0 | OVX13R1R2 L. ing.* | frame_shift_del | p.G330fs | Extension | N/A | Y | N/A |
| 10258181 | 10258181 | C/0 | TAC297 L. tho. | frame_shift_del | p.P329fs | Extension | N/A | Y | N/A |
| 10258148 | 10258149 | AG/0 | TAC298 L. tho. | frame_shift_del | p.E318fs | Extension | N/A | Y | N/A |
| 10258147 | 10258147 | G/T | TAC299 R. tho. | nonsense | p.E318* | Extension | N/A | Y | N/A |
| 10258181 | 10258181 | C/0 | TAC300 L. tho. | frame_shift_del | p.P329fs | Extension | N/A | Y | N/A |
| 10258194 | 10258194 | T/0 | TAC300 R. ing. | frame_shift_del | p.K334fs | Extension | N/A | Y | N/A |
| 10258151 | 10258169 | GGCTAATGCCATCCCATTC/0 | TAC301 L. tho. | frame_shift_del | p.R319fs | Extension | N/A | Y | N/A |
| 10258142 | 10258149 | AGGACGAG/0 | TAC302 R. cerv. | frame_shift_del | p.E316fs | Extension | N/A | Y | N/A |
| 10258184 | 10258184 | G/0 | TAC311 L. cerv. | frame_shift_del | p.G330fs | Extension | N/A | Y | N/A |
| 10258147 | 10258147 | G/T | TAC311 L. ing. | nonsense | p.E318* | Extension | N/A | Y | N/A |
| 10258222 | 10258223 | 0/A | TAC299 L. tho. | frame_shift_ins | p.D343fs | DCIS | N/A | N/A | Y |
| 10258151 | 10258173 | GGCTAATGCCATCCCATTCCAAA/- | TAC312 L. tho. | frame_shift_del | p.L320fs | DCIS | N/A | N/A | Y |
| 10258177 | 10258177 | T/- | TAC314 L. tho. | frame_shift_del | p.Y328fs | DCIS | N/A | N/A | Y |
| 10258151 | 10258173 | GGCTAATGCCATCCCATTCCAAA/- | TAC319 L. ing. | frame_shift_del | p.L320fs | DCIS | N/A | N/A | Y |
| 10258147 | 10258147 | G/- | TAC319 R. ing. | frame_shift_del | p.E318fs | DCIS | N/A | N/A | Y |
| 10258154 | 10258191 | TAATGCCATCCCATTCCAAAGAGTATCCGGGTCAAGGT/- | TAC322 L. tho. | frame_shift_del | p.L320fs | DCIS | N/A | N/A | Y |
| 10258147 | 10258147 | G/- | TAC323 L. tho. | frame_shift_del | p.E318fs | DCIS | N/A | N/A | Y |
indicates ovarian-hormone independent tumor.
Abbreviations: WGS – whole genome sequencing; Y, Yes – mutation observed; N, No – mutation not observed; N/A – mutation data not available for technology; A – mutation calling ambiguous.
Note: a mutation for ‘TAC246 R. ing.’ was not observed in WGS or PCR Sanger assay but was observed by PCR MiSeq method.
In order to determine whether Prlr mutations are an early tumor-initiating event, ductal carcinoma in situ (DCIS) components were identified from FFPE blocks of additional Stat1−/− mammary glands. DCIS DNA samples were amplified for the mutated Prlr region and sequenced by MiSeq (Appendix 4). In total, 7 of 9 (77.8%) DCIS samples showed evidence of truncating Prlr mutations. Immunohistochemical analysis also indicated that activated Stat3 and Stat5 were present in a majority of the atypical cells in DCIS (Figure S3) consistent with our previous results for pStat3/5 in primary Stat1−/− tumors (Chan et al., 2014). These results suggest that mutations in the Prlr allele and activation of the Prlr pathway are an early event during tumorigenesis of Stat1−/− mammary epithelial cells.
The final set of 40 Prlr mutations observed included 32 frame-shift-deletions, 3 frame-shift-insertions, 4 non-sense SNVs and 1 in-frame deletion introducing a stop-gain (Table 2, Table S5, and Figure 2). All mutations were located within an 85 base pair window (chr15:10258139-10258223 of the mouse reference genome build mm9). These mutations are predicted to produce a truncated Prlr protein, only 317 to 349 amino acids (aa) in length compared to the 608 aa full-length wild type Prlr (Figure S4). The truncated forms result in loss of most, but not all, of the Prlr cytoplasmic tail. They share the first 285 aa with the known ‘S1b’ short-form with 32 to 64 additional amino acids and total lengths ranging between the ‘S1c’ and ‘S1a’ forms (Bole-Feysot et al., 1998; Pujianto et al., 2010). Examination of WGS data showed that nearly all Prlr mutations, except for the SSM1 tumor cell line, appeared to be heterozygous. Although definitive determination of zygosity from Sanger and MiSeq data was challenging, especially for FFPE samples, we did not find any additional samples in the extension set that were obviously homozygous for the truncation mutation. To identify germline variants that may produce a similar effect, we searched the Sanger Mouse Sequencing Project (version 5)(Keane et al., 2011; Yalcin et al., 2011) and identified a single missense variant (rs46169444) and no indels within this hotspot region.
Figure 2. Mutational hotspot analysis of Prlr.

The diagrams depict the full-length 608-amino acid residue coding region of Prlr (ENSMUST00000124470; Ensembl v67) that is encoded by 9,900 base pairs. Panel A depicts mutations identified in the original discovery set of tumors by whole genome sequencing and panel B depicts those identified in the extension/validation set (including DCIS samples) by Sanger/MiSeq sequencing. A total of 32 frame-shift deletions, 3 frame-shift insertions, 4 nonsense SNVs and 1 in-frame deletion introducing a stop codon were identified in Prlr that cluster in 2 hotspots around residues 318 and 330. See also Figure S4, Table S5, Table S10, Appendix 1, Appendix 2, Appendix 3, and Appendix 4.
Functional significance of the truncated form of Prlr protein
Since all but one of the observed Prlr mutations in primary Stat1−/− mammary tumors occurred in one of two alleles and all of the primary tumors examined so far displayed constitutive Prlr pathway activation, we hypothesized that heterodimers of full-length (FL) and truncated (T) Prlr may be the cause of constitutive Prlr activation and thus the tumorigenic phenotype of the Stat1−/− mammary epithelial cells. Endogenous expression of the FL and T Prlr isoforms was verified in Stat1−/− mammary tumor cell lines (SSM1, SSM2 and SSM3) harboring these mutations by immunoprecipitation and Western blotting (Figure S5A). In contrast to the SSM2 and SSM3 tumor cell lines, which are heterozygous for the mutation, the SSM1 tumor cell line was homozygous (Table 2 and Figure S5A) and failed to display constitutive Prlr-Jak2-Stat3/5 signaling (Chan et al., 2014). To directly examine the activity of the two Prlr isoforms, FL or T Prlr (aa residues 1 to 317) were expressed either alone or together in non-transformed Stat1−/− murine embryonic fibroblasts (MEFs). Expression of the Prlr isoforms was confirmed by flow cytometry using a Prlr-specific monoclonal antibody (Figure S5B) (Chan et al., 2014). In the absence of exogenous Prl stimulation, phosphorylation of Stat3 and Stat5 was detected in cells expressing both FL and T Prlr, but not in cells expressing either FL Prlr homodimers or T Prlr homodimers alone (Figure 3A). Therefore, co-expression of FL and T Prlr led to phosphorylation and activation of Stat3 and Stat5 in the absence of exogenous Prl stimulation.
Figure 3. Co-expression of full-length and truncated Prlr promote Stat3 and Stat5 activation, cellular transformation in vitro and tumor formation in vivo.

(A) Stat1−/− MEFs expressing full-length (FL), truncated (T) Prlr or both (FL/T) were stained for phosphorylated Stat3 (pStat3) or phosphorylated Stat5 (pStat5) (y-axis). Rabbit (Rb) IgG was used as an isotype control. MEFs also expressed mJak.IRES.GFP (x-axis) to mediate signaling downstream of the Prlr proteins. (B) Stat1−/− MEFs expressing the indicated Prlr constructs were analyzed by anchorage-independent soft agar assay. The number of colonies was counted after single cells had been cultured for 3 weeks. (C) Stat1−/− MEFs alone (−), Stat1−/− MEFs transduced with vector alone (Jak2) or vector expressing full-length (FL/FL-Jak2), truncated (T/T-Jak2) or both (FL/T-Jak2) Prlr were implanted into nude mice. Stat1−/− MEFs expressing Kras were used as positive control. Tumor growth was monitored over time. The percentages of animals that developed palpable tumors in each experimental group were plotted. * p<0.002, ** p<0.0001. See also Figure S3, Figure S5.
To further determine the biological significance of FL and T Prlr heterodimers, Stat1−/− MEFs expressing both FL and T Prlr were analyzed for their ability to grow in an anchorage-independent manner. Cells expressing FL and T Prlr developed significantly more colonies than control MEFs (p = 0.0013), or those expressing FL alone (p = 0.0013) or T alone (p = 0.0015) when plated in soft agar (Figure 3B and Figure S5C). Stat1−/− MEFs expressing both FL and T Prlr also developed significantly more tumors in nude mice than MEFs expressing vector alone (p = 1.041E-5), FL Prlr alone (p = 0.0017) or T Prlr alone (p = 7.648E-6) (Figure 3C). Tumor formation also occurred more quickly in mice that received FL/T expressing MEFs than FL alone (p=0.03), T alone (p=0.0001), vector alone (p=0.0005), or MEFs alone (p=0.0001; Figure S5D). FL/T expressing MEFs formed tumors at a frequency similar to the Kras expressing positive control (p = 1.0), although at a significantly slower rate (p<0.0001). Taken together, these results indicate that FL/T Prlr heterodimers promote activation of oncogenic Stat3 and Stat5, anchorage-independent growth and transformation of non-transformed Stat1−/− MEFs.
PRLR mutations and isoform usage in human breast cancers
To assess the prevalence of PRLR mutations in human breast cancers, we examined human breast cancer exome sequence data from 991 patients made publicly available through the TCGA data portal. Using the published MAF file, 4 mutations in PRLR, including 2 SNVs and 2 indels, were identified (Cancer Genome Atlas Network, 2012). One of these mutations, an indel (L360fs), causes a truncating mutation in the human PRLR exon 10 (ENST00000382002, Ensembl v70_37), analogous to that observed in the mouse Stat1−/− mammary tumors. Manual review of alignment data for this exon identified 4 additional truncating indels at E313fs (2/47 reads), L315fs (2/44), L360fs (35/42) and K460fs (3/100) from samples TCGA-B6-A0X7, TCGA-A2-A04R, TCGA-AC-A3EH and TCGA-AR-A5QQ, respectively. L360fs, E313fs and L315fs were found in luminal subtype breast cancers whereas K460fs was in a basal breast cancer. The Exome Aggregation Consortium (ExAC) reports only 4 individuals with rare (allele frequency < 0.00001) germline truncating mutations in PRLR at A597fs, N568fs, S27*, and W180* (Lek et al., 2015).
There are currently 8 to 10 reported complete protein-coding transcript isoforms for human PRLR according to Ensembl (ENSG00000113494, release 79), UCSC (PRLR, GRCh37/hg19) and UniProt (P16471, Entry version 175) that can be broadly grouped as long, intermediate and short PRLR isoforms (Bole-Feysot et al., 1998) (Figure S6 and Table S6). We investigated the possibility of an increase in the expression of truncated (T) (i.e., short) PRLR relative to full-length (FL) (i.e., long) PRLR in human ERα+ luminal breast cancer. We hypothesize that a skewing towards more T PRLR expression could be functionally equivalent to PRLR truncation. The expression ratio of FL to T PRLR was calculated using the TCGA human breast RNA-seq datasets based on counts for isoform-specific junctions (see Methods). We also analyzed the STAT1 expression status of human luminal and basal/Her2 breast cancers and stratified each tumor subtype into STAT1-low and STAT1-high. In a previous report, we demonstrated that STAT1 was specifically down-regulated in tumor cells but not in stromal cells in ERα+ luminal breast cancers (Chan et al., 2012). Although the human breast tumor TCGA RNA-seq datasets were generated from whole tumors, we observed an overall significant reduction in STAT1 expression level in the luminal breast cancer subtype compared to basal and Her2 subtypes (p = 2.484E-06), reflecting the selective downregulation of STAT1 in these tumor cells and consistent with our previous report (Chan et al., 2012). We then analyzed the FL/T Prlr ratio in STAT1-low and STAT1-high tumors among each subtype. We observed a significant increase in T PRLR expression relative to FL PRLR expression (i.e., lower FL/T ratio) in STAT1-low samples compared to STAT1-high samples among luminal subtype tumors (p=0.0077; Figure 4). No significant difference in FL/T ratio was observed among basal/Her2 breast tumors (p=0.723). These data indicate that there may be a preferential usage of the truncated PRLR isoform in tumor cells with reduced STAT1 expression among ERα+ luminal breast cancers.
Figure 4. PRLR isoform usage versus STAT1 expression in human TCGA breast cancer RNA-seq data.

STAT1 expression levels from TCGA RNA-seq data were binned into tertiles (low, medium, high) (mid panels) and separated into luminal or basal/Her2 breast cancer subtypes. The ratios of full-length (FL) to truncated (T) PRLR isoform expression (FL/T ratio values) calculated in terms of junction per million (JPM) were compared using a Wilcoxon rank sum test with continuity correction between low and high STAT1 expressing groups (top panels). For reference, read counts of FL and T are plotted (bottom panels). See also Figure S6 and Table S6.
Discussion
In this study, we identified recurrent gene mutations that were associated with the tumorigenic landscape of ERα+ Stat1−/− luminal mammary gland tumors. Several of these genes have also been reported as significantly mutated in human breast cancers, underscoring the biological significance of these mutations in the pathogenesis of this disease. Our study also revealed a potential mechanism whereby ERα+ luminal breast cancer initiates and progresses. Loss of Stat1 expression in mammary cells favors acquisition of mutations in an 85 base pair hotspot of exon 10 of the Prlr gene (ENSMUST00000124470), resulting in a truncation of the cytoplasmic tail of the prolactin receptor (Prlr). Concurrent expression of full-length (FL) and truncated (T) Prlr in the absence of Stat1 promotes phosphorylation and activation of the oncogenic Stat3 and Stat5, anchorage-independent growth of mouse embryonic fibroblasts, and tumor formation in nude mice.
PRLR is a transmembrane homodimeric receptor with an extracellular region that binds prolactin (PRL). It functions as a cytokine receptor, and activates second messenger cascades including the JAK2-STAT3/5, JAK-RUSH, RAS-RAF-MAPK, and PI3K pathways (Aksamitiene et al., 2011; Helmer et al., 2010; Rui et al., 1994). Over 75% of human ERα+ breast cancers display persistent PRLR-JAK2-STAT3/5 signaling (Chan et al., 2014). Activation of PRLR-JAK2-STAT3/5 signaling has been implicated in the up-regulation of steroid hormone receptor expression and malignant progression of breast cancer (Chan et al., 2014; Fiorillo et al., 2013). There is also support for an association between PRLR allelic variations and breast cancer risk (Bogorad et al., 2008; Lee et al., 2007; Mong et al., 2011; Vaclavicek et al., 2006). In lobular neoplasia, amplification of PRLR may also be important for pathogenesis and progression (Tran-Thanh et al., 2011). Additionally, mouse mammary cancer models support a role for Prlr signaling in tumor progression. Elevated production of Prl ligand, driven by the Neu promoter, causes development of carcinomas in mice resembling human luminal breast carcinomas (Arendt et al., 2011). We have also shown that loss of Stat1 expression results in unopposed Prlr signaling, promotes expansion of mammary luminal progenitor cells, leads to development of ductal carcinoma in situ (DCIS) and finally to invasive mammary carcinomas (Chan et al., 2014). Lack of Prlr signaling has the opposite effect whereby Prlr-deficiency delays tumor onset in the C3(1)SV40T model of mammary cancer (Oakes et al., 2007). Similarly, pharmacological inhibition of Jak2 (BMS-911543) not only abrogates mammary tumor formation but also causes regression of established Stat1−/− mammary tumors, demonstrating that constitutive activation of the Prlr-Jak2-Stat3/5 pathway promotes tumor progression and maintenance (Chan et al., 2014). Therefore, there is strong evidence supporting the involvement of Prlr signaling in the pathogenesis of ERα+ breast cancer.
The mechanism by which the PRLR pathway is activated during the development of ERα+ breast cancer is, however, less clear. Although elevated serum PRL levels have been associated with increased risk of developing ERα+ breast cancer (Tworoger et al., 2015), it has been difficult to definitively show a causal relationship between PRLR pathway activation and breast cancer progression in those individuals who exhibited high PRL levels prior to diagnosis. In a recent study by Tworoger and colleagues, half of the patients with ERα+ breast cancer did not show high plasma PRL levels when blood samples were collected <10 years prior to diagnosis (Tworoger et al., 2013), suggesting that the association of Prlr signaling and development of ERα+ breast cancer may be more complex than simply elevated PRL production. In our previous report, we showed that excess production of Prl ligand was not the cause of constitutive activation of Prlr-Jak2-Stat3/5 signaling in Stat1−/− mammary tumors (Chan et al., 2014). In this study, we sought to identify a mechanism whereby constitutive oncogenic Prlr signaling was established and maintained in the absence of aberrant over-production of Prl. We showed that heterodimers consisting of full-length and truncated Prlr activate Jak2-Stat3/5 in the absence of exogenous Prl stimulation. Prlr truncation and Stat3/5 activation were also observed in DCIS in Stat1−/− mammary glands, indicating that persistent Prlr signaling mediated by Prlr mutations was an early tumorigenic event. These results suggest that FL/T PRLR heterodimers could contribute to ERα+ breast cancer development in patients with normal PRL levels due to the intrinsic ability of the heterodimers to signal without ligand stimulation. Since this observation was made in mice and cell lines without Stat1 expression, it is likely that the ability of the heterodimers to confer a tumorigenic effect would be correlated with the loss of the negative regulator that normally controls Prlr signaling. We previously showed that approximately half of ERα+ luminal breast cancers displayed selective down-regulation of STAT1 expression in tumor cells (Chan et al., 2012). Given that the STAT1-SOCS1 pathway negatively regulates PRLR signaling (Chan et al., 2014), it would be interesting to determine whether breast cancer cells in patients with normal PRL levels have down-modulated STAT1 and/or SOCS1 expression. Our study also indicated that truncated Prlr homodimers alone failed to transform Stat1−/− MEFs or promote tumor formation in mice (Figure 3). This is consistent with the finding that truncated Prlr lacks the Stat5 binding sites at 496 and 597 residues (Figure 2 and Figure S4) and that Stat5 activation is necessary for transformation. Full-length homodimers also failed to transform Stat1−/− MEFs in vitro and showed significantly reduced oncogenic activity in vivo. Therefore, it seems that coexpression of FL and T Prlr is required to collaborate with STAT1 loss and promote tumor progression.
One might expect that Prl-mediated tumor induction in the case of the NRL-PRL transgenic model (Arendt et al., 2011) and constitutive Prlr pathway activation in the case of Stat1−/− tumors (Chan et al., 2014) would lead to similar pathological outcomes. However, the tumors developed in Stat1−/− mice were mechanistically different from the tumors developed in NRL-PRL transgenic mice reported by Arendt et al. (Arendt et al., 2011; Chan et al., 2014). Arendt et al. found that Prl-induced mouse mammary carcinomas were heterogeneous with respect to histology, ER/PR expression, and signaling cascades (e.g., Stat5 signaling) and were insensitive to ER-mediated signaling. Our model is less heterogeneous, independent of over-production of Prl ligand and sensitive to ER-mediated signaling. In addition, although Jak2 is required for initiation of the NRL-PRL tumors, it is not essential for tumor maintenance. In contrast, Stat1−/− tumors require Jak2 activation for both initiation and progression. While the tumor-initiating cell population in the Stat1−/− tumors is the luminal progenitor subtype (Chan et al., 2014), it is not clear from which specific cell compartment the NRL-PRL tumors are derived. Given the distinct mode of tumorigenesis, the potential difference in targeted tumor-initiating cell populations and the presence of Stat1 in the NRL-PRL model, it is likely that these differences might explain the different endocrine sensitivity and biological outcome of the two models.
We observed four PRLR truncating mutations in human breast cancer TCGA datasets. To our knowledge only two exome sequencing studies of DCIS have been published with 11 and 9 cases, respectively, and neither report any PRLR mutations (Banerji et al., 2012; Kim et al., 2015). It is possible that PRLR truncation may occur as other types of genomic alterations such as larger scale deletions, gene fusions, or translocations that are not easily detected from TCGA exome or RNA-seq data. More detailed structural analyses of breast cancer genomes will be required to further explore this possibility. Changes in the expression of naturally occurring alternative transcript isoforms may also provide a mechanism of PRLR deregulation. The human S1a and S1b truncated PRLR isoforms are generated by alternative splicing. Their roles in the development of ERα+ breast cancer remain to be clarified. Dufau and colleagues reported an increase in mRNA expression of the full-length PRLR over the short S1a and S1b PRLR isoforms in human breast cancers (Meng et al., 2004). Our analyses compared all of the known short isoforms (S1a, S1b, Δ4 S1b, ΔS4–Δ7/11, Δ7/11, ΔS1) to the full-length isoform and observed a significant increase in expression of the short isoforms over full-length Prlr in STAT1-low luminal but not basal breast cancer. Since tumor subtypes of the datasets used in the Dufau study were not classified, it was difficult to directly compare their results to our current study.
The biological significance of the Prlr short isoforms has been controversial. Some studies indicate that the Prlr short isoforms act as dominant negatives and block signaling from full-length Prlr (Hu et al., 2001). However, expression of the Prlr short isoform alone is sufficient to rescue the mammary gland differentiation defect in Prlr+/− mice (Binart et al., 2003), indicating that Prlr short isoform is not a dominant negative, and FL Prlr and Prlr short isoform heterodimers can indeed transduce signals. None of these past studies examined the ability of the FL Prlr and Prlr short isoform heterodimers to transform normal cells. We demonstrate the ability of these heterodimers to transform mouse embryonic fibroblasts and promote tumor formation in nude mice. This model also recapitulated the pStat3/5 activation we observed in primary tumors and DCIS samples. However, these experiments were performed using MEFs rather than the primary cell of origin and future studies should assess the tumorigenic property of Prlr heterodimers in Stat1−/− mammary epithelial cells (MECs) with endogenous levels of Jak2. We speculate that the truncated Prlr short form is able to prolong signaling in the absence of Prl ligand because of its increased half-life on the cell surface. Phosphorylation of Ser349 on Prlr recruits the b-TrCP ubiquitin-protein ligase (Li et al., 2006). This interaction is important for ubiquitin-dependent degradation of the Prlr to terminate signaling. The truncated Prlr described in our current study lacks this critical Ser residue, suggesting that it may be insensitive in b-TrCP-mediated degradation. Consistent with this hypothesis, phosphorylation on Ser349 is diminished in human breast cancer cell lines, leading to an increase in Prlr expression levels (Li et al., 2006). In addition, although FL Prlr and the truncated S1b isoform have similar binding affinity to growth hormone, the level of specific binding by S1b is significantly higher than that of FL Prlr on COS-1 or HEK293 cells transfected with either isoform (Hu et al., 2001; Trott et al., 2003). These results suggest that the cell surface expression of S1b Prlr is elevated compared to that of FL Prlr. Therefore, it is conceivable that increased FL/T Prlr heterodimer expression on the cell surface mediates receptor and Jak2 clustering such that autophosphorylation and activation is possible without Prl ligand engagement.
Anti-tumor agents targeting the PRLR pathway are being investigated for patients with ERα+ breast cancer. For example, LFA102, an anti-PRLR antibody, blocks PRLR pathway activation by either inhibiting PRLR dimerization or locking the PRLR dimer in an inactive conformation without affecting PRL ligand binding (Damiano et al., 2013). Since truncated PRLR expression is preferentially increased in ERα+ breast cancer and FL/T PRLR heterodimers display constitutive activation as shown in our current study, it would be of interest to examine whether LFA102 is able to block heterodimerization of full-length and truncated PRLR in future studies. Unfortunately, LFA102 failed to show antitumor efficacy in a recent Phase 1 clinical trial for metastatic breast cancer (Agarwal et al., 2016). Direct inhibition of Jak2 using small molecule inhibitors will also be worthy of investigation in breast cancers with Prlr activation. Future studies should also aim to clarify the biological outcome of signaling crosstalk between the FL/T Prlr heterodimers and the estrogen receptor pathway since combination therapy targeting both pathways may be beneficial. In summary, our findings provide a mechanism whereby ERα+ luminal breast cancer is initiated and maintained, and pose hypotheses of translational and clinical significance in the treatment of this most common human breast cancer subtype.
Experimental Procedures
Mice
Stat1−/− mammary gland adenocarcinomas have been previously characterized in our laboratory (Chan et al., 2014; Chan et al., 2012; Shankaran et al., 2001). Wild type (WT) 129S6/SvEv and Stat1tm1Rds/tm1Rds (Stat1−/−) mice were purchased from Taconic Farms (Germantown, NY). Stat1−/− mammary tumors of approximately 10 mm in diameter were harvested from 10–18 month old retired breeders. Tumor-free Stat1−/− mice about 8-months of age were used as tumor-free controls. To obtain ovarian hormone-independent mammary tumors, ovaries were surgically removed from primary tumor-bearing mice as previously reported (Chan et al., 2014). If tumors did not respond to estrogen-deprivation and grew progressively, tumors were harvested. Tails from both tumor-free and tumor-bearing Stat1−/− mice were also harvested as normal controls. All animal experiments were carried out according to the guidelines of the American Association for Laboratory Animal Science under a protocol approved by the Animal Studies Committees and performed in AAALAC-accredited specific pathogen-free facilities at Washington University School of Medicine in St. Louis.
Cell cultures
The SSM1, SSM2 and SSM3 Stat1−/− mammary tumor cell lines were cultured as previously described (Chan et al., 2012).
Sample Acquisition
Genomic DNAs were purified from tumor-free mammary glands, whole tumors or tails using QIAGEN DNeasy Blood & Tissue kit according to manufacturer's instructions (QIAGEN, Germantown, MD). Fifty-two samples were whole genome sequenced for discovery purposes and an additional 54 samples were sequenced using the Sanger protocol for targeted validation and extension of the Prlr findings (Figure 1 and Figure S2). The discovery set included 14 primary Stat1−/− mammary tumors, 5 OH-independent tumors and the 3 cell lines (SSM1, SSM2 and SSM3) for a total of 22 tumors from 20 individual mice (2 mice had two tumors each). An additional 10 WT and 5 tumor-free Stat1−/− mammary glands from 13 additional mice were also sequenced as controls (two mice had both tumor-bearing and tumor-free mammary glands). From the Stat1−/− mice, 15 tails were sequenced as matched normal samples for 12 primary tumors, 2 OH-independent tumors and 5 tumor-free mammary glands. For 2 primary tumors, 3 OH-independent tumors, 3 cell lines and 10 wild type mammary glands without matched tails, a pooled sample of 2 normal tails was used as reference for somatic variant calling (See Table S7 for extensive details of all samples).
Whole Genome Sequencing
The yield and integrity of native genomic DNA was verified by a PicoGreen assay to determine mass (Invitrogen, Carlsbad, CA). Small insert dual indexed Illumina paired end libraries were constructed with the KAPA LTP sample prep kits according to the manufacturer's recommendations (KAPA Biosystems, Woburn, MA) with a few exceptions (Supplemental Experimental Procedures). Each genome was loaded on a HiSeq2000 version 3 flow cell according to the manufacturer's recommendations (Illumina, San Diego, CA). 2 × 101 bp read pairs were generated for each sample, yielding an average of 37.1× sequence coverage for the tumor genomes and 27.2× sequence coverage for the normal genomes (Table S8). The reference-aligned whole genome sequence data and sample details for 52 tumor and non-tumor mouse tissues have been submitted to NCBI SRA study SRP061941, BioProject PRJNA248457.
Reference alignment and somatic variant detection
The Genome Modeling System, an integrated analysis information management system, was used for preliminary analysis of sequence data as previously described (Supplemental Experimental Procedures) (Griffith et al., 2015). Alignment was performed against the mouse reference genome (mm9) and variants annotated with our custom annotator against Ensembl (version 67). Further filtering of SNVs and indels was performed to (1) exclude random, MT and Y contig events; (2) exclude germline events defined as greater than 5% variant allele frequency (VAF) in the normal sample; (3) exclude events with greater than 500 reads at the site and (4) exclude variants from the Sanger Mouse Genomes Project (v2), (5) exclude variants that appear in 2 or more of the 10 WGS data sets from mammary tissue samples obtained from wild type mice. These represent likely systematic artifacts of our alignment and calling pipelines. Further manual review of all SNVs, indels, and CNVs was performed in the Integrative Genome Viewer (IGV) to eliminate false positives arising from likely read mapping artifacts. Finally, analysis was performed on all reviewed and somatic variants (Table S1) to identify the recurrent (Table S2) and significantly mutated genes (Table S3) for SNVs and indels. Significantly mutated genes were determined using the Mutational Significance in Cancer (MuSiC) pipeline (version 0.4), including non-coding mutations in the background mutation rate calculation (Dees et al., 2012). Reviewed CNV events are also summarized in Table S9. Genomic visualizations (Figure S1A) were created with the GenVisR Bioconductor package (Skidmore et al., 2016).
Validation and Extension Sequencing of Prlr by Sanger and MiSeq
Based on the region in which truncating Prlr mutations were observed in the discovery set (chr15:10258139-10258195; mm9) two sets of primers were designed to encompass this region with approximately 50 or 100 bp additional flanking sequence on each side, respectively (Table S10 and Supplemental Experimental Procedures). Primers were tailed (p1k / m13 reverse) and ordered from Integrated DNA Technologies (IDT, Coralville, IA). Amplification was performed in a Bio-Rad thermocycler (Bio-Rad Laboratories, Inc., Hercules, CA). A Lonza flash gel was run to confirm product. Sequencing reactions were performed and loaded on a 3730 DNA analyzer (Life Technologies). Bases were called from sequence trace files using phred and then assembled against a reference scaffold of the amplicon sequence using phrap (Ewing and Green, 1998; Ewing et al., 1998). Sequence variants were identified by manual review of assemblies and sequence traces in Consed (Gordon et al., 1998). We performed Sanger sequencing as described above on the original 22 tumor samples to validate Prlr variants that were called from WGS data and to extend the Prlr findings to 10 additional tumors and 35 non-tumor samples (Figure 1 and Figure S2). For two tumors in the original discovery set, additional FFPE samples were obtained, and sequenced on a MiSeq Illumina instrument using products of the same PCR protocol described above. Finally we sequenced an additional 9 FFPE samples by MiSeq, to evaluate the presence of Prlr mutations in DCIS tissues.
Review of PRLR sequence data from TCGA human breast cancers
The mutated region of mouse Prlr (chr15:10258139-10258195; mm9) and the flanking 50 base pairs on each side were aligned to the human reference genome using BLAST to identify the homologous region of PRLR in the human genome (chr5:35066045-35066101; hg19). This region in human PRLR was then extended to include the entirety of the affected “long” exon of PRLR (Figure S6) as well as the upstream-most exon-intron boundary and exon to identify a target region of interest (chr5:35065191-35068387; hg19) for manual review in human sequence data using IGV. Manual review focused on identifying truncating mutations or deletions of the long exon. For a mutation to be considered credible at least a 3% tumor VAF was required. A total of 991 tumor/normal pairs of exome sequence data were investigated from the breast TCGA project including 501 luminal A, 198 luminal B, 171 basal, 77 Her2, 31 normal-like and 13 of unknown subtype (Cancer Genome Atlas Network, 2012).
Exome analysis of TCGA human breast cancers
Genes with somatic mutations for mouse data (Table S1, n=22) and human luminal breast cancers from TCGA (n=699) were compared (Figure S1). Within the comparison, mutation types were restricted to nonsynonymous coding and RNA mutations to ensure results were directly comparable. Using biomaRt, H.sapiens and M.musculus ensembl IDs were annotated with orthologs from the other species. Genes without an ortholog with a one to one mapping, were excluded from the analysis. Instances in which a sample had > 1 mutation in the same gene/sample were treated as having a single mutation.
RNA-seq analysis of TCGA human breast cancers
Breast cancer RNA-seq level 3 data corresponding to 10/10/2013 from the firehose pipeline (gdac.broadinstitute.org) were obtained from the TCGA data portal. A total of 775 breast tumor samples were represented with 376 luminal A, 181 luminal B, 131 basal, 65 Her2 and 22 normal-like subtype. Read counts per kilobase per million (RPKM) values were calculated as: Number of mapped reads/(transcript length in bp/1,000)/(total reads/1,000,000) and used to define STAT1 and PRLR gene expression levels. Tumor samples (excluding normal-like) were divided into tertiles to define low, intermediate, and high STAT1 expression groups (Figure 4). Junction fragments per million (JPM) values were defined as the raw counts for each junction divided by the sum of all junctions multiplied by one million. PRLR full-length (FL) and Truncated (T or short) isoform expression were estimated from the JPM values of FL and T isoform-specific junctions as follows (Figure S6):
The ratio of PRLR isoform expression was defined as the log2 value of FL expression divided by T expression. As illustrated in Figure S6, the short human Prlr isoforms are generated by alternative splicing and will be defined as ‘Truncated’ for clarification purposes. It should also be noted that human FL and intermediate isoform expression were grouped as "full-length" since it is unachievable to extract FL expression alone using junction data from TCGA.
Immunoprecipitation and western blotting analysis
SSM1, SSM2 and SSM3 cells were lysed using complete RIPA buffer and Prlr was immunoprecipitated using anti-mPrlr (clone 5A12) as previously described (Chan et al., 2014). Membrane was blotted with biotinylated anti-mPrlr and streptavidin-anti-hamster-IR800, and scanned using the Li-Cor Odyssey detection system (Lincoln, NE).
Immunohistochemistry
Immunohistochemical analyses of Stat3 and Stat5 on Stat1−/− DCIS lesions were performed as described in a previous study (Chan et al., 2014).
Expression of full-length and truncated Prlr
Full-length (FL) or truncated (T) Prlr were cloned into pLVX-Het-1 or pLVX-Het-2, respectively (Clontech, Mountain View, CA). The shortest predicted variant of mutated Prlr (residues 1 to 317) was used as the T Prlr isoform, as shown in Figure S4. Stat1−/− murine embryonic fibroblasts (MEFs) were transduced with lentivirus expressing either FL or T Prlr alone, or both FL and T Prlr together. In cells expressing both FL and T Prlr, FL Prlr was transduced first. Prlr-positive cells were sorted by flow cytometry using the anti-Prlr Ab clone 5A12 and sorted cells were subsequently transduced with T Prlr. Since MEFs did not express sufficient Jak2 to mediate signaling, Stat1−/− MEFs were also transduced with mouse Jak2.IRES.GFP and sorted for GFP-positive cells.
Flow cytometry
Cell surface expression of Prlr in Stat1−/− MEFs was confirmed using a biotinylated monoclonal antibody against murine Prlr (clone 5A12) (Chan et al., 2014) and streptavidin-PE (SA-PE, eBioscience, San Diego, CA). To examine basal activation of Stat3 and Stat5, MEFs were serum-deprived in 0.05% FBS for 16 hours before analysis. Cells were fixed and permeabilized according to manufacturer’s protocol (BD Biosciences, San Jose, CA). Tyrosine phosphorylation of Stat3 and Stat5a/5b was detected using antibodies specific for the phosphorylated forms of each Stat (Cell Signaling, Danvers, MA). Monoclonal rabbit IgG was used as isotype control for gating.
Soft agar assay
Single cell suspensions of 20,000 or 50,000 Stat1−/− MEFs expressing FL, truncated Prlr or both were mixed in 0.3% noble agar in DMEM and plated on top of 0.6% noble agar. Each condition was plated in triplicate in p60 dishes. The number of colonies in each dish was counted after 3 weeks using a Scan 100 colony counter.
Analysis of tumorigenicity in nude mice
Female NCr nude mice (Taconic) were implanted with 1 ×106 of immortalized Stat1−/− MEFs expressing Jak2 alone, FL Prlr/Jak2, T Prlr/Jak2, FL/T Prlr/Jak2 or Kras in 100 µl vehicle. Tumor diameter was measured twice weekly. Animals were censored when progressively growing palpable tumors of at least 2 mm were detected. On Day 73, all remaining mice were sacrificed and evaluated for evidence of tumors prior to considering animals to be tumor-free.
Statistical analyses
Wilcox rank sum test with continuity correction was performed to test for an association between PRLR isoform ratio and STAT expression group using R 3.1.0. Unpaired t test was used to determine the statistical significance between control and experimental groups in the soft agar assay. All tests are two-sided and a p-value ≤ 0.05 was considered significant. Fisher’s exact test was used to compare tumor formation in each group of nude mice implanted with Stat1−/− MEFs to the FL/T expressing group.
Supplementary Material
Acknowledgments
We thank the Production group for sequence data production; Analysis Pipeline group for developing the automated sequence analysis pipelines; the LIMS group for developing tools and software to manage samples and sequencing; and the Systems group for providing the IT infrastructure and HPC solutions required for sequencing and analysis. We would also like to thank Nathan Dees and Scott Smith for their help with adapting analysis pipelines for the mouse genome during the early phase of this project. O.L.G. was supported by the National Cancer Institute (NIH NCI K22 CA188163). M.G. was supported by the National Human Genome Research Institute (NIH NHGRI K99 HG007940). This work was funded by grants to R.K.W. from the National Human Genome Research Institute (NIH NHGRI U54 HG003079) and to R.D.S from the National Cancer Institute (NIH NCI R01 CA043059) and Bristol-Myers Squibb.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
O.L.G. led sequencing experiments and data analysis. S.R.C. led laboratory experiments. O.L.G, S.R.C., M.G., K.K., Z.L.S., J.H., H.S., L.T., K.L.K., and W.V., performed data analysis and prepared figures and tables. S.R.C., J.A.A, C.A., D. R., A.P.M., H.S., D.R., M.B., and R.S.F. developed laboratory methods and performed laboratory experiments. O.L.G., S.R.C., M.G., R.S.F., R.K.W., R.D.S. and E.R.M. developed the project concept and experimental design. C.A.M. and D.E.L. provided informatics support. O.L.G. and S.R.C. wrote the manuscript with input from M.G., K.K., Z.L.S., R.S.F., R.D.S., and E.R.M.
Accession Numbers
The reference-aligned whole genome sequence data and sample details for 52 tumor and non-tumor mouse tissues have been submitted to NCBI SRA study SRP061941, BioProject PRJNA248457.
References
- 1.Agarwal N, Machiels JP, Suarez C, Lewis N, Higgins M, Wisinski K, Awada A, Maur M, Stein M, Hwang A, et al. Phase I Study of the Prolactin Receptor Antagonist LFA102 in Metastatic Breast and Castration-Resistant Prostate Cancer. Oncologist. 2016;21:535–536. doi: 10.1634/theoncologist.2015-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aksamitiene E, Achanta S, Kolch W, Kholodenko BN, Hoek JB, Kiyatkin A. Prolactin-stimulated activation of ERK1/2 mitogen-activated protein kinases is controlled by PI3-kinase/Rac/PAK signaling pathway in breast cancer cells. Cellular signalling. 2011;23:1794–1805. doi: 10.1016/j.cellsig.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arendt LM, Rugowski DE, Grafwallner-Huseth TA, Garcia-Barchino MJ, Rui H, Schuler LA. Prolactin-induced mouse mammary carcinomas model estrogen resistant luminal breast cancer. Breast cancer research : BCR. 2011;13:R11. doi: 10.1186/bcr2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binart N, Imbert-Bollore P, Baran N, Viglietta C, Kelly PA. A short form of the prolactin (PRL) receptor is able to rescue mammopoiesis in heterozygous PRL receptor mice. Molecular endocrinology. 2003;17:1066–1074. doi: 10.1210/me.2002-0181. [DOI] [PubMed] [Google Scholar]
- 6.Bogorad RL, Courtillot C, Mestayer C, Bernichtein S, Harutyunyan L, Jomain JB, Bachelot A, Kuttenn F, Kelly PA, Goffin V, et al. Identification of a gain-of-function mutation of the prolactin receptor in women with benign breast tumors. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14533–14538. doi: 10.1073/pnas.0800685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocrine reviews. 1998;19:225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan SR, Rickert CG, Vermi W, Sheehan KC, Arthur C, Allen JA, White JM, Archambault J, Lonardi S, McDevitt TM, et al. Dysregulated STAT1-SOCS1 control of JAK2 promotes mammary luminal progenitor cell survival and drives ERalpha(+) tumorigenesis. Cell Death Differ. 2014;21:234–246. doi: 10.1038/cdd.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, Lonardi S, Arthur C, Young LJ, Levy DE, et al. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast cancer research : BCR. 2012;14:R16. doi: 10.1186/bcr3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damiano JS, Rendahl KG, Karim C, Embry MG, Ghoddusi M, Holash J, Fanidi A, Abrams TJ, Abraham JA. Neutralization of prolactin receptor function by monoclonal antibody LFA102, a novel potential therapeutic for the treatment of breast cancer. Molecular cancer therapeutics. 2013;12:295–305. doi: 10.1158/1535-7163.MCT-12-0886. [DOI] [PubMed] [Google Scholar]
- 12.Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC, Mooney TB, Callaway MB, Dooling D, Mardis ER, et al. MuSiC: identifying mutational significance in cancer genomes. Genome research. 2012;22:1589–1598. doi: 10.1101/gr.134635.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome research. 1998;8:186–194. [PubMed] [Google Scholar]
- 15.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome research. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 16.Fiorillo AA, Medler TR, Feeney YB, Wetz SM, Tommerdahl KL, Clevenger CV. The prolactin receptor transactivation domain is associated with steroid hormone receptor expression and malignant progression of breast cancer. The American journal of pathology. 2013;182:217–233. doi: 10.1016/j.ajpath.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome research. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 18.Griffith M, Griffith OL, Smith SM, Ramu A, Callaway MB, Brummett AM, Kiwala MJ, Coffman AC, Regier AA, Oberkfell BJ, et al. Genome Modeling System: A Knowledge Management Platform for Genomics. PLoS Computational Biology. 2015;11:e1004274. doi: 10.1371/journal.pcbi.1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helmer RA, Panchoo M, Dertien JS, Bhakta SM, Hewetson A, Chilton BS. Prolactin-induced Jak2 phosphorylation of RUSH: a key element in Jak/RUSH signaling. Molecular and cellular endocrinology. 2010;325:143–149. doi: 10.1016/j.mce.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu ZZ, Meng J, Dufau ML. Isolation and characterization of two novel forms of the human prolactin receptor generated by alternative splicing of a newly identified exon 11. The Journal of biological chemistry. 2001;276:41086–41094. doi: 10.1074/jbc.M102109200. [DOI] [PubMed] [Google Scholar]
- 21.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Jr, Vogelstein B, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, Chung YJ, Lee SH. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015;6:7597–7607. doi: 10.18632/oncotarget.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SA, Haiman CA, Burtt NP, Pooler LC, Cheng I, Kolonel LN, Pike MC, Altshuler D, Hirschhorn JN, Henderson BE, Stram DO. A comprehensive analysis of common genetic variation in prolactin (PRL) and PRL receptor (PRLR) genes in relation to plasma prolactin levels and breast cancer risk: the multiethnic cohort. BMC medical genetics. 2007;8:72. doi: 10.1186/1471-2350-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O'Donnell-Luria A, Ware J, Hill A, Cummings B, et al. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv. 2015 doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Clevenger CV, Minkovsky N, Kumar KG, Raghunath PN, Tomaszewski JE, Spiegelman VS, Fuchs SY. Stabilization of prolactin receptor in breast cancer cells. Oncogene. 2006;25:1896–1902. doi: 10.1038/sj.onc.1209214. [DOI] [PubMed] [Google Scholar]
- 27.Meng J, Tsai-Morris CH, Dufau ML. Human prolactin receptor variants in breast cancer: low ratio of short forms to the long-form human prolactin receptor associated with mammary carcinoma. Cancer research. 2004;64:5677–5682. doi: 10.1158/0008-5472.CAN-04-1019. [DOI] [PubMed] [Google Scholar]
- 28.Mong FY, Kuo YL, Liu CW, Liu WS, Chang LC. Association of gene polymorphisms in prolactin and its receptor with breast cancer risk in Taiwanese women. Molecular biology reports. 2011;38:4629–4636. doi: 10.1007/s11033-010-0596-y. [DOI] [PubMed] [Google Scholar]
- 29.Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. The Journal of biological chemistry. 2001;276:24965–24970. doi: 10.1074/jbc.M101161200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oakes SR, Robertson FG, Kench JG, Gardiner-Garden M, Wand MP, Green JE, Ormandy CJ. Loss of mammary epithelial prolactin receptor delays tumor formation by reducing cell proliferation in low-grade preinvasive lesions. Oncogene. 2007;26:543–553. doi: 10.1038/sj.onc.1209838. [DOI] [PubMed] [Google Scholar]
- 31.Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, Torres-Arzayus MI, Brown M, Egan SE, Wahl GM, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome biology. 2013;14:R125. doi: 10.1186/gb-2013-14-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pujianto DA, Curry BJ, Aitken RJ. Prolactin exerts a prosurvival effect on human spermatozoa via mechanisms that involve the stimulation of Akt phosphorylation and suppression of caspase activation and capacitation. Endocrinology. 2010;151:1269–1279. doi: 10.1210/en.2009-0964. [DOI] [PubMed] [Google Scholar]
- 33.Rui H, Kirken RA, Farrar WL. Activation of receptor-associated tyrosine kinase JAK2 by prolactin. The Journal of biological chemistry. 1994;269:5364–5368. [PubMed] [Google Scholar]
- 34.Schneckenleithner C, Bago-Horvath Z, Dolznig H, Neugebauer N, Kollmann K, Kolbe T, Decker T, Kerjaschki D, Wagner KU, Muller M, et al. Putting the brakes on mammary tumorigenesis: loss of STAT1 predisposes to intraepithelial neoplasias. Oncotarget. 2011;2:1043–1054. doi: 10.18632/oncotarget.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 36.Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, Griffith M. GenVisR: Genomic Visualizations in R. Bioinformatics. 2016 doi: 10.1093/bioinformatics/btw325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tran-Thanh D, Arneson NC, Pintilie M, Deliallisi A, Warren KS, Bane A, Done SJ. Amplification of the prolactin receptor gene in mammary lobular neoplasia. Breast cancer research and treatment. 2011;128:31–40. doi: 10.1007/s10549-010-1025-6. [DOI] [PubMed] [Google Scholar]
- 39.Trott JF, Hovey RC, Koduri S, Vonderhaar BK. Alternative splicing to exon 11 of human prolactin receptor gene results in multiple isoforms including a secreted prolactin-binding protein. Journal of molecular endocrinology. 2003;30:31–47. doi: 10.1677/jme.0.0300031. [DOI] [PubMed] [Google Scholar]
- 40.Tworoger SS, Eliassen AH, Zhang X, Qian J, Sluss PM, Rosner BA, Hankinson SE. A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development. Cancer research. 2013;73:4810–4819. doi: 10.1158/0008-5472.CAN-13-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tworoger SS, Rice MS, Rosner BA, Feeney YB, Clevenger CV, Hankinson SE. Bioactive prolactin levels and risk of breast cancer: a nested case-control study. Cancer Epidemiol Biomarkers Prev. 2015;24:73–80. doi: 10.1158/1055-9965.EPI-14-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaclavicek A, Hemminki K, Bartram CR, Wagner K, Wappenschmidt B, Meindl A, Schmutzler RK, Klaes R, Untch M, Burwinkel B, Forsti A. Association of prolactin and its receptor gene regions with familial breast cancer. The Journal of clinical endocrinology and metabolism. 2006;91:1513–1519. doi: 10.1210/jc.2005-1899. [DOI] [PubMed] [Google Scholar]
- 43.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. The New England journal of medicine. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yalcin B, Wong K, Agam A, Goodson M, Keane TM, Gan X, Nellaker C, Goodstadt L, Nicod J, Bhomra A, et al. Sequence-based characterization of structural variation in the mouse genome. Nature. 2011;477:326–329. doi: 10.1038/nature10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.