

Abstract
Purpose
We investigated the population pharmacokinetics and exposure-response relationship of nilotinib in patients with newly diagnosed chronic myeloid leukemia (CML) in chronic phase.
Methods
Nilotinib was given at 300 mg or 400 mg twice daily. Serum concentration data (sparse and full pharmacokinetic profiles) were obtained from 542 patients over 12 months. A population pharmacokinetic analysis was performed using nonlinear mixed-effect modeling. Exposure-response relationships were explored graphically or using logistic regression models.
Results
Nilotinib concentrations were stable over 12 months. Patients in the 400 mg twice-daily arm had an 11.5% higher exposure than did those in the 300 mg twice-daily arm, and the relative bioavailability of nilotinib 400 mg twice daily was 0.84 times that of 300 mg twice daily. Patient demographics did not significantly affect nilotinib pharmacokinetics. The occurrence of all grade total bilirubin elevation was significantly higher in patients with higher nilotinib exposure, and a positive correlation was also observed between nilotinib exposure and QTcF change on electrocardiograms from baseline. There was no significant relationship between nilotinib exposure and major molecular response at 12 months.
Conclusions
There is a less than proportional dose-exposure relationship between nilotinib 300 mg and 400 mg twice-daily doses. Blood level testing is unlikely to play an important role in the general management of patients with newly diagnosed CML treated with nilotinib.
Keywords: Nilotinib, Population pharmacokinetics, Exposure-response relationship, Chronic myeloid leukemia
Introduction
Nilotinib (Tasigna®; Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA), a potent and selective BCR-ABL tyrosine kinase inhibitor (TKI), was recently approved in over 40 countries, including the European Union, the United States, and Japan, for the treatment of patients with newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia in chronic phase (CML-CP). The drug is also approved worldwide for patients with Ph+ CML-CP and accelerated phase (CML-AP) who are resistant to or intolerant of imatinib.
Nilotinib is approximately 30% absorbed and 98% bound to plasma proteins. It is metabolized primarily in the liver through hydroxylation and oxidation mediated by the cytochrome P4503A4 (CYP3A4) enzyme [1]. Although minor metabolites are formed in this process, unchanged nilotinib is the main circulating drug component in the serum, and it is primarily responsible for the drug's pharmacologic activity [1]. Nilotinib is 30-fold more potent than imatinib in cellular assays and maintains activity against most imatinib-resistant mutant forms of BCR-ABL [2–4].
After data from single-arm, open-label, phase 2 studies conducted by the Gruppo Italiano Malattie Ematologiche dell' Adulto (GIMEMA) [5] and the M. D. Anderson Cancer Center [6] suggested substantially faster and higher responses with nilotinib in the frontline CML-CP setting compared with historical imatinib data [7], the phase 3 randomized Evaluating Nilotinib Efficacy and Safety in Clinical Trials–Newly Diagnosed Patients (ENESTnd) study was initiated, comparing nilotinib doses at 300 mg and 400 mg twice daily with imatinib 400 mg once daily in patients with newly diagnosed Ph+ CML-CP. Nilotinib at both doses demonstrated significantly higher rates of major molecular response (MMR), complete cytogenetic response, and complete molecular response [8–10] and resulted in significantly lower rates of transformation to CML-AP or CML in blast crisis (CML-BC) compared with imatinib [11].
Similar to other TKIs, nilotinib exhibited a moderate to high interpatient variability in its pharmacokinetics (PK) in patients with imatinib-resistant or -intolerant CML [12]. Thus, identification of factors contributing to its PK variability and understanding of correlations between PK exposure and treatment outcomes may be important for nilotinib therapy. The population PK profile of nilotinib in patients with newly diagnosed CML has not been reported. In this first report we evaluated the population PK and exposure-response relationship of nilotinib in patients with newly diagnosed CML-CP in the ENESTnd study.
Materials and Methods
Study design
The present analyses were based on the 12-month data cutoff of ENESTnd (Clinical Trials.gov number: NCT00471497). The study, including this PK subanalysis, was conducted in accordance with the Declaration of Helsinki, and the protocol was reviewed by the ethics committee or institutional review board at each participating institution. All subjects were required to give written informed consent.
The design and methodology of the ENESTnd study have been reported previously [11]. Briefly, eligible patients were ≥18 years of age and diagnosed with Ph+ CML-CP within 6 months of study entry. Patients had not received treatment for CML, except for hydroxyurea or anagrelide. They were randomly assigned in a 1:1:1 ratio to receive nilotinib 300 mg twice daily, nilotinib 400 mg twice daily, or imatinib 400 mg once daily. Dose escalation of nilotinib or crossover between the two nilotinib arms was not allowed on the core study. Patients were instructed to take nilotinib at least 1 hour before or 2 hours after meal intake and to avoid grapefruit, star fruit, pomegranate, and Seville oranges or juices and products containing these fruits. Concomitant administration of medications that are known to be strong CYP3A4 inhibitors or inducers or have the potential to prolong the QTc interval were prohibited during nilotinib therapy. Patients who had QTcF >450 ms at baseline were excluded from the study.
The primary efficacy variable was the rate of MMR at 12 months after the start of study therapy. MMR was defined as a BCR-ABL transcript level ≤0.1%, measured by real-time quantitative reverse transcriptase polymerase chain reaction and expressed on the International Scale [8–10, 13]. Molecular assessments were performed on blood predose on day 1; at the end of months 1, 2, and 3; and every 3 months thereafter.
For comparison of nilotinib PK profiles between the 300 mg twice-daily and 400 mg twice-daily dosing regimen, full PK samples were obtained from a subset of patients (n=34) in both nilotinib arms at predose and 1, 2, 3, 5, 8, and 12 hours postdose on any day after day 8. Patients were required to take nilotinib for at least 3 consecutive days without dose interruption or dose modification prior to the full PK sample collection. In addition, sparse trough serum concentration (Cmin) and peak (3 ± 1 hour postdose) serum concentration (Cmax) PK samples were collected in the majority of patients at predose and 3 ± 1 hours postdose on day 1, day 8, and at the end of months 3 (day 84), 6 (day 168), 9 (day 252) and 12 (day 336).
Serum concentrations of nilotinib were measured using a validated liquid chromatography-tandem-mass spectrometry assay at the Novartis Institutes for Biomedical Research (East Hanover, NJ, USA) as previously described [14]. Using 100 μL of serum sample, the lower limit of quantification was determined to be 2.5 ng/mL. Within-study assay validation showed a precision of 2.1% to 5.6% and an accuracy of 98.8% to 101.0% for quality-control samples at a concentration range of 2.5 ng/mL to 5,000 ng/mL.
Pharmacokinetic data analysis
For the full PK data set, a standard noncompartmental analysis was performed using WinNonlin Pro (Version 5.0; Pharsight, Mountain View, CA, USA), to obtain the following parameters: Cmin, Cmax, area under the concentration-time curve from time 0 to the last sampling point over a dosing interval (AUC0–tlast), and apparent oral clearance (CL/F).
For the pooled PK data set consisting of full and sparse PK samples, a nonlinear mixed-effect modeling approach was used to evaluate the population PK of nilotinib. Patients who received nilotinib and had at least one evaluable PK measurement were included in the analysis. The nonlinear mixed-effect modeling was performed using NONMEM Version VI (double precision, level 2.0; Icon Development Solutions, Ellicott City, MD, USA), with the first-order conditional estimation method with interaction. S-Plus Version 8.1 (Insightful Corp, Seattle, WA, USA) was used for graphic analyses.
Standard model-building approaches were used during model development [15]. The structural PK model was a two-compartment model, with nilotinib absorption being described by a zero-order process and its elimination described by a first-order process. For an initial exploration of the potential covariates on nilotinib PK, steady-state nilotinib Cmin was plotted against the covariates. Steady-state Cmin was defined as the PK samples taken within ± 3 hours of the scheduled time (i.e., between 9 and 15 hours post prior dose) and after at least 3 days of nilotinib dosing without any dose interruption or modification. Formal covariate selection was performed by a stepwise forward selection procedure, based on the change in the minimum objective function. When two models were related as hierarchical, a difference in minimum objective function values of >6.63 was considered significant (at the 0.01 level, approximated by the χ2 distribution with 1 degree of freedom) for inclusion of the covariate into the model. Other factors, such as size of the estimated random effects, random errors, precision of parameter estimate, general interpretability, and clinical relevance, were also considered for selecting the covariates.
The following covariates were tested for their significance on relative bioavailability (F1), clearance (CL), or volume of distribution of central compartment (V1): dose, ethnicity, race, sex, age, body weight, and clinical laboratory parameters such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), hemoglobin, and total bilirubin levels.
Interindividual variabilities in the model parameters CL, V1, and volume of distribution of peripheral compartment (V2) were estimated using the exponential error model. The variability of nilotinib concentrations was modeled using a combined random-effects model that included both an additive and proportional error term.
Exposure-response analysis
For exposure-response analysis, the PK exposure measures were average daily AUC (AUC0–24) and Cmin at steady state. AUC was used because it presents the overall drug exposure in individual patients, and the efficacy and safety endpoints tested were long-term measures. Cmin was also included because it is a preferred exposure measure that is practically more feasible to obtain for therapeutic drug monitoring.
The model-predicted AUC0–24 of nilotinib in individual patients was calculated as follows:
where F1 and CL were patient-specific estimates of the F1 and CL obtained from the population PK analysis. Average dose was computed up to the time point of the efficacy measures, taking into account dose interruptions or reductions during the treatment period.
Model-predicted Cmin at steady state was calculated using the standard equation for a two-compartment model with zero-order absorption [16], taking into account the dosing information (dose interruptions or reductions) for individual patients.
The potential relationship between nilotinib exposure and safety variables was explored in the following two ways: (1) safety variables according to observed nilotinib Cmin values categorized into quartile (Q) groups (averaged Cmin values for individual patients were calculated based on the measurements obtained on day 8 and at the end of months 3, 6, 9, and 12 and then pooled among patients in both nilotinib arms to derive the quartile groups); (2) safety variables in relation to model-predicted AUC0–24 or model-predicted Cmin using a logistic regression model, where baseline safety laboratory values were included as the potential significant covariates. Safety variables included hemoglobin, absolute neutrophil count, platelet count, total bilirubin, ALT, AST, phosphate, lipase, and amylase. Additionally, an exploratory analysis using mixed-effects modeling was performed to assess the relationship between serum nilotinib concentration and QTcF change on electrocardiogram from baseline in patients who had time-matched nilotinib concentration (i.e., Cmin and Cmax) and QTcF data on days 8 and 84 after initiation of nilotinib treatment.
The potential relationship between nilotinib exposure and efficacy variables, such as MMR at 12 months and the BCR-ABL transcript ratio at 12 months, was explored by summarizing the efficacy measures according to observed nilotinib Cmin values categorized into quartile groups. A logistic regression analysis was also performed for MMR at 12 months in relation to model-predicted AUC0–24 or model-predicted Cmin.
Results
Patient demographics
Full PK profiles were obtained from 17 patients in each nilotinib arm. The final data set for the population PK analysis included a total of 4,936 samples from 542 nilotinib-treated patients (including the 34 patients who participated in a full PK assessment). The patient baseline demographics and disease characteristics were balanced between the two nilotinib arms (Table 1).
Table 1. Patient baseline demographics and disease characteristics.
| Demographic variable | Nilotinib 300 mg twice daily (n=275) | Nilotinib 400 mg twice daily (n=267) |
|---|---|---|
| Age (years) | ||
| Median (range) | 47 (18–85) | 46 (18 – 81) |
| Weight (kg) | ||
| Median (range) | 69.9 (41.4–162.0) | 73.0 (34.5 – 125.3) |
| Sex, n (%) | ||
| Male | 154 (56.0) | 168 (62.9) |
| Female | 121 (44.0) | 99 (37.1) |
| Race, n (%) | ||
| White | 166 (60.4) | 174 (65.2) |
| Black | 12 (4.4) | 11 (4.1) |
| Asian | 74 (26.9) | 64 (24.0) |
| Native American | 0 | 1 (0.4) |
| Other | 23 (8.4) | 17 (6.4) |
| Ethnicity, n (%) | ||
| Hispanic/Latino | 55 (20.0) | 41 (15.4) |
| Chinese | 12 (4.4) | 10 (3.7) |
| Indian (Indian subcontinent) | 4 (1.5) | 1 (0.4) |
| Japanese | 29 (10.5) | 22 (8.2) |
| Mixed ethnicity | 5 (1.8) | 6 (2.2) |
| Other | 167 (60.7) | 186 (69.7) |
| Missing | 3 (1.1) | 1 (0.4) |
Full PK profiles of nilotinib
There was overlap in the mean serum concentration-time profiles of nilotinib obtained at steady state in the 300 mg twice-daily and 400 mg twice-daily arms of the full PK cohort (Fig. 1). Geometric mean nilotinib AUC0–tlast values were 11,865 ng∙h/mL in patients in the 300 mg twice-daily arm and 13,656 ng∙h/mL in patients in the 400 mg twice-daily arm, representing an 11.5% higher exposure for patients in the 400 mg twice-daily arm (Table 2).
Fig. 1. Mean serum concentration-time profiles of nilotinib obtained at steady state in the full PK cohort.
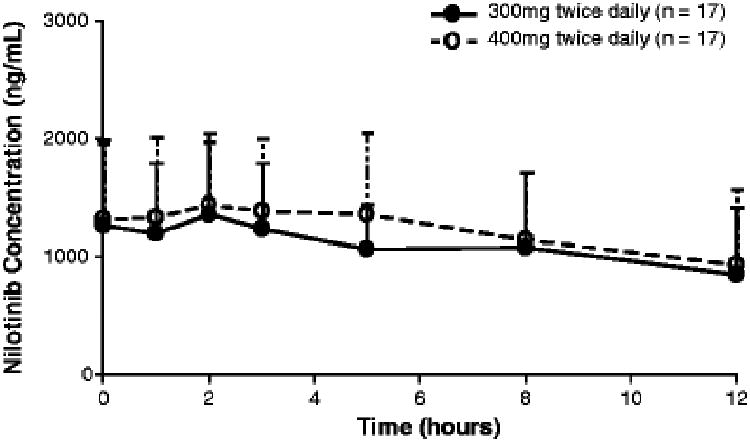
Table 2. Steady state PK parameters of nilotinib in the full PK cohort.
| Cmin (ng/mL) | Cmax (ng/mL) | AUC0–tlast (ng∙h/mL) | Tmaxa (h) | CL/F (L/h) | |
|---|---|---|---|---|---|
| Nilotinib 300 mg twice daily (n=16)b | |||||
| Geometric mean (CV%) | 1,123 (64.1) | 1,360 (58.6) | 11,865 (56.8) | 2.00 (0.00, 7.95) | 25.28 (56.8) |
| Nilotinib 400 mg twice daily (n=15)b | |||||
| Geometric mean (CV%) | 1,239 (51.9) | 1,595 (47.0) | 13,656 (51.3) | 2.00 (0.00, 7.95) | 29.3 (51.3) |
Tmax values are presented as median (range).
Full PK samples were collected from 17 patients in each arm, 3 of whom (1 in the 300 mg twice-daily arm and 2 in the 400 mg twice-daily arm) did not have sufficient data points for the calculation of full PK parameters and were excluded from the analyses shown in this table.
CV, coefficient of variation; Tmax, time to maximum concentration.
Population PK of nilotinib
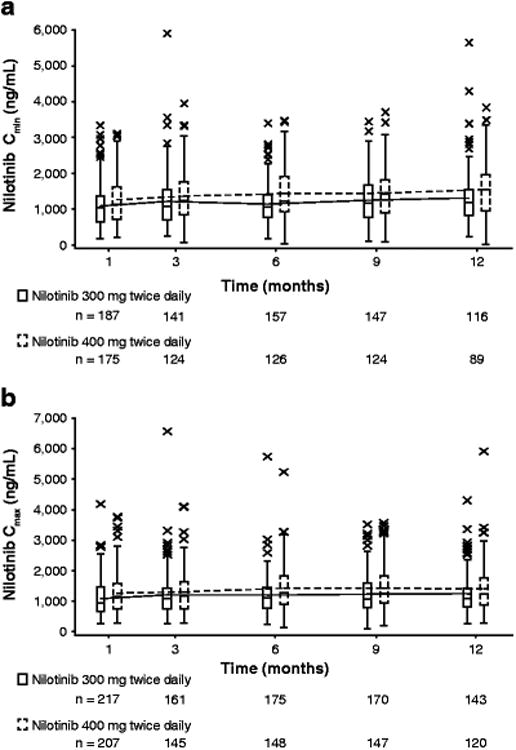
The steady-state nilotinib Cmin and Cmax values determined at the end of months 1, 3, 6, 9, and 12 are shown in Fig. 2. Nilotinib concentrations were found to remain relatively stable over the 12-month treatment course in patients in both nilotinib arms. The average nilotinib Cmin values were 1,158 and 1,340 ng/mL (Fig. 2a) and the average Cmax values were 1,172 and 1,346 ng/mL (Fig. 2b) in the 300 mg and 400 mg twice-daily arms, respectively.
Fig. 2. Nilotinib Cmin (a) and Cmax (b) over time in the 300 mg twice-daily and 400 mg twice-daily arms.
The goodness-of-fit plots for the final population PK model is shown in Figure 3.The parameter estimates from the final population PK model are summarized in Table 3. A less than proportional dose-exposure relationship is evident between the nilotinib 300 mg and 400 mg twice-daily doses. The F1 of 400 mg twice-daily nilotinib was found to be 0.843 times that of the 300 mg twice-daily dose. Thus, the ratio of population predicted exposure (AUC0–24) of nilotinib 400 mg to 300 mg twice daily was approximately 1.12.
Fig. 3.
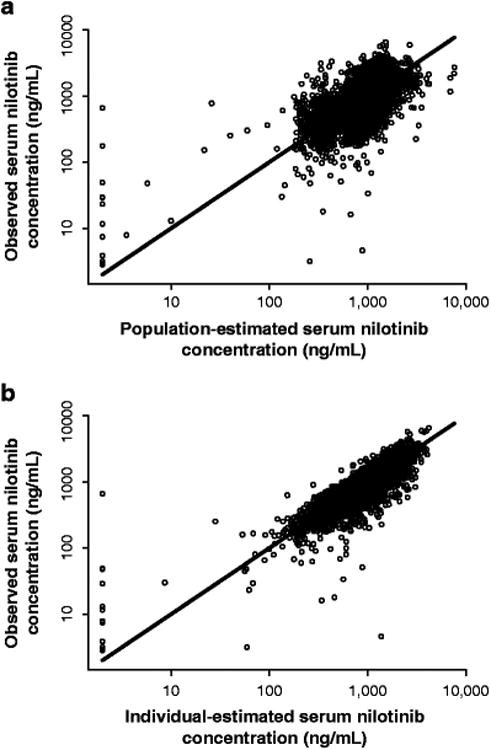
Goodness-of-fit plots for the population PK model: observed concentrations versus population (a) and individual (b) predictions. Values <1 (including observed values reported below LLOQ) are plotted at 1.0. The line of identity is plotted as a reference
Table 3. Parameter estimates (standard error) from the final population PK model.
| Parameter | Population mean | Interindividual variability, % (SE) |
|---|---|---|
| D1=θD1 (h) | 9.88 (0.112) | NE |
| Lag=θLag (h) | 0.67 h (fixed) | NE |
| F1=θF1·θDose I(Dose ≥ 400 mg) · θSexI(Sex = Male) | NE | |
| θf1 | 1 (fixed) | |
| θDose | 0.843 (0.0296) | |
| θSex | 0.903 (0.0326) | |
| CL=θCL·exp[θCL,TBIL(TBIL–0.5)]·exp[θCL,AST(AST–0.5)] | 37.6 (1.4) | |
| θCL (L/h) | 21.0 (0.662) | |
| θCL,TBIL | −0.256 (0.0214) | |
| θCL,AST | −0.0277 (0.0104) | |
| V1=θV1 (L) | 58.0 (23.6) | 79.2 (14.2) |
| V2=θV2 (L) | 181 (25.4) | 98.9 (19.7) |
| Q=θQ (L/h) | 66.5 (19.9) | NE |
| σ1 (%) | 29.8 (0.917) | – |
| σ2 (ng/mL) | 110 (17.4) | – |
SE, standard error; θDose, dose effect on F1; θSex, sex effect on F1; θCL TBIL, normalized total bilirubin level on CL; θCL,AST, normalized aspartate aminotransferase level on CL; σ1, proportional random error; σ2, additive random error; D1, zero-order input duration; Lag, lag time for absorption; F1, relative bioavailability; CL, clearance; NE, not estimated; Q, intercompartmental clearance; V1, volume of distribution of central compartment; V2, volume of distribution of peripheral compartment.
Interindividual variability (%) is reported as 100% × square root of the estimated variance. Its standard error is reported as 100% × [square root(estimated variance + estimate standard error) – square root(estimated variance)]. NONMEM reported estimated interindividual variances (standard errors) as 0.141 (0.0109), 0.627 (0.246), and 0.978 (0.428), respectively for CL, V1, and V2. The model also estimated a covariance (standard error) between CL and V1 as 0.163 (0.0581), which is a correlation of 0.548.
In male patients, nilotinib F1 was estimated to be 0.903 times that of female patients (Table 3), suggesting that male patients had an approximately 10% lower bioavailability or 10% lower systemic exposure than female patients at the same dose level. However, patient age and body weight were not found to be significant factors affecting nilotinib PK. There was no significant difference in nilotinib PK between Japanese and non-Japanese patients or across various racial groups, such as white, black, Asian, and other races (data not shown).
Among the clinical laboratory parameters evaluated, normalized total bilirubin and AST levels were identified as statistically significant covariates affecting nilotinib CL. While addition of the statistically significant covariates into the final model resulted in only a small decrease in the interindividual variability of CL (from 40.0% to 37.6%), an increase in total bilirubin from the upper limit of normal (ULN) to 1.5 × ULN, 2 × ULN, and 2.5 × ULN was predicted to result in a 12%, 23%, and 32% decrease in CL, respectively. In contrast, an increase in AST from ULN to 1.5 × ULN and 2 × ULN was predicted to result in only a 1% and 3% decrease in CL, respectively. The interpatient variability in V1 and V2 were estimated to be 79.2% and 98.9%, respectively (Table 3).
Exposure-safety relationship
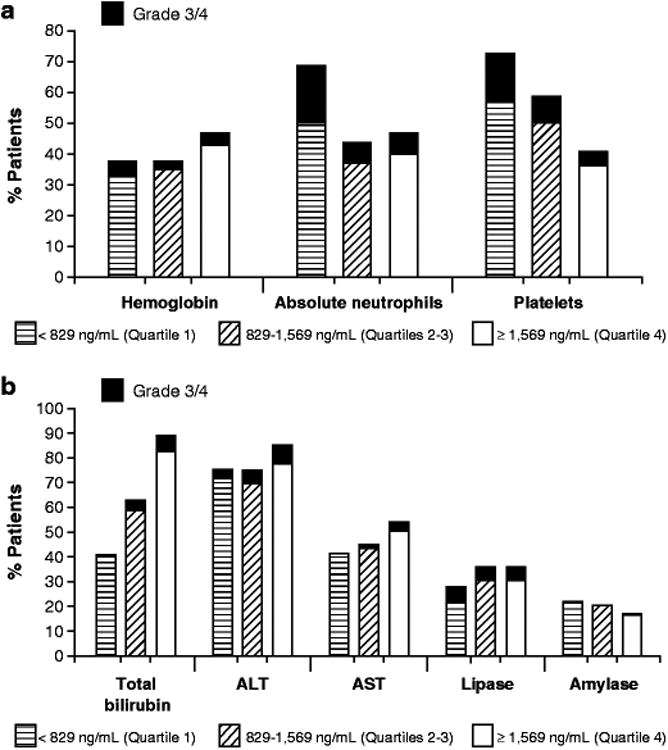
No apparent correlation was observed between nilotinib exposure and all grade abnormalities (i.e., newly occurring or worsening abnormalities from baseline) of hemoglobin, absolute neutrophil count, platelet count, ALT, AST, lipase, or amylase levels (Fig. 4a, 4b). However, the occurrence of all grade elevations in total bilirubin was higher in patients with higher nilotinib exposure (Fig 4b). For patients with nilotinib Cmin in Q1 (<829 ng/mL), Q2/3 (829–1,569 ng/mL), and Q4 (>1,569 ng/mL), all grade elevations in total bilirubin were 39.8%, 58.1%, and 82.3%, respectively. Logistic regression confirmed a significant positive correlation between nilotinib AUC0–24 and all grade elevations in total bilirubin, where baseline total bilirubin (BIL) level (median: 6.84 μmol/L; range: 2.42–37.64 μmol/L) was also a significant covariant affecting the incidence of all grade total bilirubin elevation. The logistic regression model was estimated as follows:
Fig. 4. All grade and grade 3/4 reductions in hemoglobin, absolute neutrophils, and platelets (a) and in blood chemistry elevations (b) according to nilotinib Cmin.
where p is the probability of occurrence of total bilirubin elevation, and logit(p) is the natural logarithm of p/(1 - p). Thus, with the median AUC0-24 of 25.9 μg∙h/mL and median baseline total bilirubin level of 6.84 μmol/L, the predicted incidences of all grade total bilirubin elevation were 29.4%, 46.5%, and 59.5% respectively, for the AUC0–24 values of 16, 24, and 32 μg∙h/mL (AUC value of 24 μg∙h/mL corresponds to the typical AUC in male patients who received nilotinib 300 mg twice daily).
Similarly, statistically significant positive correlations were observed between nilotinib AUC0–24 and the incidence of all grade toxicities related to hemoglobin reductions and lipase elevation, but the slope was relatively flat. For example, for AUC0–24 values of 16, 24, and 32 μg·h/mL, the predicted incidences of all grade abnormalities were 31.7%, 36.4%, and 39.9% for hemoglobin and 17.4%, 23.8%, and 29.3% for lipase, respectively. Higher baseline hemoglobin and lipase values were also associated with a higher incidence of hemoglobin and lipase abnormalities, respectively.
The incidences of grade 3/4 hematologic (Fig. 4a) and biochemical (Fig. 4b) abnormalities were generally low in all Cmin quartile groups. Although the occurrence of grade 3/4 elevations in total bilirubin appeared to be numerically higher in patients with higher nilotinib exposure (0.9%, 4.8%, and 7.1% in Cmin Q1, Q2–3, and Q4 groups, respectively), the relationship was not statistically significant based on logistic regression. Incidences of grade 3/4 reductions in platelet count and absolute neutrophil count were shown to be highest in the lowest Cmin quartile (Fig. 4a). This cannot be explained by any known drug effects and may be an artifact of the relatively small sample size in each quartile. There was no clear correlation between nilotinib Cmin and grade 3/4 abnormalities of other hematologic and biochemical parameters.
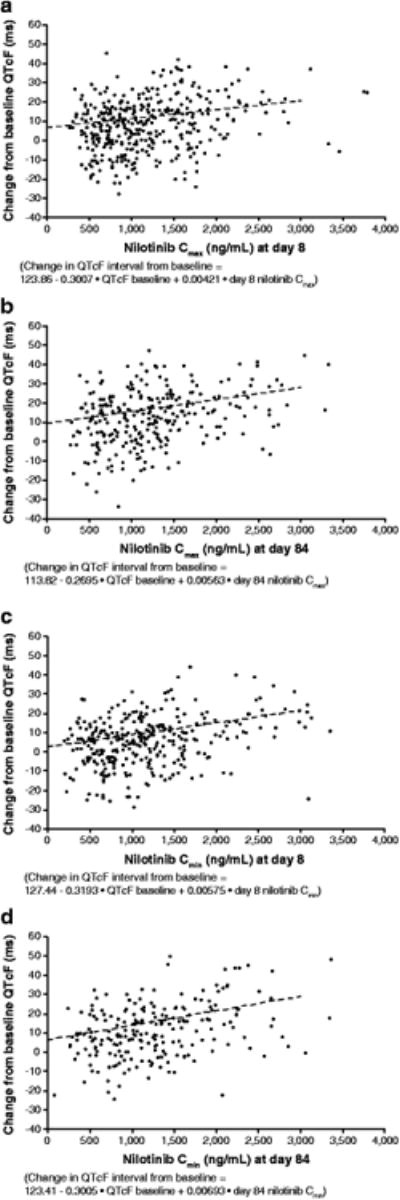
A positive correlation was noted between serum nilotinib concentrations (both Cmin and Cmax) and QTcF change on electrocardiogram from baseline for patients who had time-matched nilotinib concentration and QTcF data (Fig. 5). A-000 ng/mL increase in the serum nilotinib concentration was associated with a 4.2-ms (based on Cmax data obtained on day 8; Fig. 5a) to 6.9-ms (based on Cmin data obtained on day 84; Fig. 5d) increase in QTcF.
Fig. 5. QTcF change from baseline according to nilotinib Cmax at days 8 (a) and 84 (b) and according to nilotinib Cmin at days 8 (c) and 84 (d).
Exposure-efficacy relationship
Patients with higher nilotinib Cmin (Q2/3 or Q4) tended to have lower BCR-ABL ratios at 12 months than patients with lower Cmin (Q1) (Table 4). The relationship was weakly positive, but not statistically significant (P=0.097, Cochran-Mantel-Haenszel test for ordinal variables).
Table 4. BCR-ABL ratio at 12 months in relation to the observed nilotinib average Cmin.
| Nilotinib average Cmin over 12 months | ||||
|---|---|---|---|---|
|
| ||||
| Q1 <829 ng/mL (n=71a+42b) | Q2/3 829–1,569 ng/mL (n=123a+106b) | Q4 >1,569 ng/mL (n=46a+67b) | P valuec | |
| Cumulative BCR-ABL ratio at 12 months, n (%) | ||||
| ≤0.0032% | 1 (0.9) | 10 (4.4) | 8 (7.1) | 0.074 |
| ≤0.01% | 9 (8.0) | 23 (10.0) | 15 (13.3) | 0.460 |
| ≤0.1% | 45 (39.8) | 101 (44.1) | 52 (46.0) | 0.750 |
| ≤1% | 78 (69.0) | 186 (81.2) | 94 (83.2) | 0.372 |
| ≤10% | 93 (82.3) | 206 (90.0) | 100 (88.5) | 0.097 |
Average Cmin for individual patients was calculated based on the measurements obtained on day 8 and at the end of months 3, 6, 9, and 12 and then pooled among patients in both nilotinib arms to derive the quartiles.
Patients receiving nilotinib 300 mg twice daily.
Patients receiving nilotinib 400 mg twice daily.
Cochran-Mantel-Haenszel test for ordinal variables.
There was no apparent relationship between the observed nilotinib Cmin and MMR rate at 12 months (Table 5). Among evaluable patients, the MMR rate was 46.9% in patients with nilotinib Cmin <829 ng/mL (Q1, n=113), 48.6% in patients with Cmin 829 ng/mL to 1569 ng/mL (Q2/3, n=229), and 51.5% in patients with Cmin >1,569 ng/mL (Q4, n=113). No significant correlations between the model-predicted Cmin and MMR at 12 months or model-predicted AUC0–24 and MMR at 12 months were identified through logistic regression analysis.
Table 5. MMR in relation to the observed nilotinib average Cmin.
| Nilotinib average Cmin over 12 months | |||
|---|---|---|---|
|
| |||
| Q1 <829 ng/mL (n=71a+42b) | Q2/3 829–1,569 ng/mL (n=123a+106b) | Q4 >1,569 ng/mL (n=46a+67b) | |
| MMR at 12 months | |||
| n observedc | 96 | 208 | 101 |
| MMR, n (%) | 45 (46.9) | 101 (48.6) | 52 (51.5) |
| 95% CId | 36.6, 57.3 | 41.6, 55.6 | 41.3, 61.6 |
| MMR difference vs Q1 | |||
| (95% CIe) | NA | 1.7 (−10.4, 13.8) | 4.6 (−9.3, 18.6) |
| MMR difference vs Q2/3 | |||
| (95% CIe) | NA | NA | 2.9 (−9.0, 14.8) |
Average Cmin for individual patients was calculated based on the measurements obtained on day 8 and at the end of months 3, 6, 9, and 12 and then pooled among patients in both nilotinib arms to derive the quartiles.
Patients receiving nilotinib 300 mg twice daily.
Patients receiving nilotinib 400 mg twice daily.
Number of patients with observed data at 12 months. All patients with atypical transcripts at baseline or missing RQ-PCR evaluation at 12 months (whatever the reason) were excluded from this patient set. Percentages were computed based on evaluable patients.
Pearson-Clopper 95% 2-sided CI.
Asymptotic Wald 95% 2-sided CI.
CI, confidence interval.
Discussion
This is the first report evaluating the population PK profile and exposure-response relationship of nilotinib in patients with newly diagnosed CML-CP. The results suggested a less than proportional dose-exposure relationship for nilotinib between the 300 mg and 400 mg twice-daily doses, presumably due to the dose-limited absorption of nilotinib in humans. The occurrence of all grade total bilirubin elevation was found to be significantly higher in patients with higher nilotinib exposure, but no significant relationship was noted between nilotinib exposure and MMR at 12 months.
The final population PK model provided reasonable fitting to the observed nilotinib concentration data. All parameter estimates had reasonable precision, although the additive random error was high relative to the lower limit of quantification of the assay. This may be attributed to several factors such as assay error, errors in recording of dates and times of drug administration and PK sample collections, and intrasubject variability.
In a previous study in which nilotinib was administered as escalating doses from 50 mg to 1,200 mg daily to patients with imatinib-resistant or -intolerant CML, a general dose-exposure proportionality for nilotinib was observed over the dose range of 50 to 400 mg once daily, but there was no further appreciable increase in exposure with once-daily doses above 400 mg [17]. Because a 300-mg twice-daily dose of nilotinib was not tested previously, it was uncertain whether the plateau in the dose-exposure relationship of nilotinib started at 400 mg or somewhere between the 200 mg and 400 mg doses. By evaluating the PK profiles of nilotinib 300 mg twice-daily and 400 mg twice-daily, the present study confirmed a less than proportional dose-exposure relationship for nilotinib between these two doses. The less than proportional dose-exposure relationship remained after the gender effect was accounted for in the population PK model, suggesting that this finding is not likely attributable to the slightly higher percentage of male patients in the nilotinib 400 mg twice-daily arm.
The interpatient variability in nilotinib PK was moderate in the present study, and it appeared to be similar between the 300 mg and 400 mg twice-daily doses. Such variations are comparable with other TKIs, such as imatinib (49% CV in AUC with doses of 25 to 1,000 mg daily) [18] and dasatinib (56% CV in Cmax with the 100 mg once-daily dose) [19]. A number of factors may contribute to this variability, including interpatient variability in drug absorption or genetic polymorphisms that affect CYP3A4 activity [20–22].
Population PK analysis suggests that sex is a statistically significant covariate on nilotinib bioavailability. Male patients were found to have an approximately 10% lower bioavailability or 10% lower systemic exposure than female patients. The exact mechanisms for the lower nilotinib bioavailability in male patients are unknown, but might be attributed to the physiological differences in the gastrointestinal tract (GI) between males and females (i.e., shorter gastric emptying and GI transit time in males than in females and/or different bile acid composition) [23–25]. However, since the observed extent of the difference is relatively small, such a sex effect is unlikely to be clinically meaningful for nilotinib therapy. Other demographic variables, such as age, body weight, and racial group did not significantly affect nilotinib PK. Thus, the results suggest that patient demographics are not a clinically important factor contributing to interpatient variability in nilotinib PK and exposure.
Normalized AST and total bilirubin levels were found to be significant covariates affecting nilotinib CL. Based on the final population PK model, an increase in normalized AST from ULN to 1.5 × ULN and 2 × ULN was predicted to result in only a 1% and 3% decrease in CL, respectively, suggesting such a covariate effect is not clinically relevant. While the present analysis suggested that a substantial elevation in total bilirubin might be associated with a moderate increase in nilotinib exposure, the underlying mechanism for such an effect of total bilirubin on nilotinib CL is unclear. It may be a reflection of the relationship between nilotinib exposure and bilirubin level, since in the PK-safety analysis higher nilotinib exposure was shown to be significantly correlated with a greater incidence of all grade elevation in total bilirubin. As no parallel strong association between nilotinib CL and AST or ALT was observed, the association between nilotinib CL and total bilirubin is unlikely to be a result of altered hepatic function.
The correlation between nilotinib exposure and all grade total bilirubin elevation may be attributed to the potential inhibitory effect of nilotinib on the UGT1A1 gene enzyme activity since in a previous in vitro study nilotinib was shown to be a competitive inhibitor of UGT1A1, which encodes a key enzyme in bilirubin conjugation [26]. The UGT1A1 promoter repeat polymorphism is also reported to increase the risk of hyperbilirubinemia in patients receiving nilotinib [27]. Such an effect was not assessed in our study because information on the UGT1A1 genotype of individual patients was not available when the analysis was performed. Further evaluation is needed to investigate the relative impact of nilotinib concentration and UGT1A1 polymorphisms on the occurrence of total bilirubin elevation. Nevertheless, in clinical trials, bilirubin elevations were reversible and the observed hyperbilirubinemia was clinically manageable in patients receiving nilotinib therapy [11, 17]. Thus, the ultimate impact of an association between nilotinib exposure and bilirubin elevations on patient outcomes would likely be limited.
Like many TKIs, nilotinib has been associated with an increase in QTcF [1]. However, the incidence of cardiac-related adverse events was low in patients with newly diagnosed CML-CP [28]. In this study, electrocardiogram recording was performed at the time of “trough” and “peak” PK sample collection. Thus, a simple statistical analysis and presentation was used to correlate to the QTcF data with the time-matched Cmin and Cmax levels. The positive correlation between nilotinib concentration and QTcF change indicates that a patient with higher nilotinib blood levels is more likely to develop a greater QTcF prolongation. Thus, in the case of QT prolongation, nilotinib dosage may be reduced or temporarily withheld.
Results from PK-efficacy analyses show a weakly positive, but statistically insignificant, association between the degree of nilotinib exposure and BCR-ABL transcript reduction at 12 months. No clear association was noted between the degree of exposure and MMR at 12 months. Although leukemic cell concentrations of nilotinib were not determined in this study due to feasibility issues, a general correlation between nilotinib serum and leukemic cell concentrations would be expected. Previous studies have shown that the movement of nilotinib into CML cells is independent of the influx transporter OCT1 [29, 30]. Among the various efflux transporters (i.e., ABCB1, ABCG2) that are identified to be expressed in CML cells, none of them appear to play an important role in the cellular retention of nilotinib [30]. These observations, together with the findings that nilotinib exposure was only 13.4% higher in patients in the 400 mg twice-daily arm than in the 300 mg twice-daily arm (versus the 33% higher exposure expected according to dose proportionality), support the clinical results that both doses had similar efficacy.
The relationship between nilotinib exposure and clinical outcomes described in this report was not as obvious as that observed previously in patients treated with imatinib [31]. This may reflect the fact that covariates for response and safety become less clinically relevant with a more potent TKI such as nilotinib. In addition, the observed less prominent exposure-response relationship for nilotinib could have been attributed to the high inter- and intrapatient variability. While the present analysis, based on a well-controlled clinical trial, seems to suggest that blood level testing is unlikely to play an important role in the general management of patients with newly diagnosed Ph+ CML treated with nilotinib, additional studies are needed to further define the role of blood level testing in circumstances that may arise in the clinic, such as nonadherence to treatment or in cases of potential drug-drug interactions.
Acknowledgments
We thank Yen-Lin Chia, PhD, and William Sallas, PhD, for their contribution to the analyses described herewith. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. We thank Erinn Goldman, PhD for medical editorial assistance with this manuscript.
Footnotes
Conflicts of interest: Dr Larson received research support, honoraria, and consulting fees from Novartis; Dr Yin is an employee and stock holder of Novartis; Dr Hochhaus has received research support, honoraria, and consulting fees from Novartis and Bristol-Myers Squibb; Dr Saglio has received honoraria and consulting fees from Novartis and Bristol-Myers Squibb; Dr Clark has received research support, honoraria, and consulting fees from Novartis; Dr Nakamae has received honoraria from Novartis and Bristol-Myers Squibb; Dr Gallagher is a Novartis employee and stock holder; Dr. Eren Demirhan is a Novartis employee; Dr Hughes has received research support, honoraria, and consulting fees from Novartis and Bristol-Myers Squibb; Dr Kantarjian has received research support and consulting fees from Novartis; and Dr le Coutre has received research support and honoraria from Novartis and consulting fees from Novartis and Bristol-Myers Squibb.
Clinical Trials.gov number: NCT00471497
References
- 1.Tasigna [package insert] East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2010. [Google Scholar]
- 2.Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer. 2006;94(12):1765–1769. doi: 10.1038/sj.bjc.6603170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, Mestan J, Trappe J, Wartmann M, Fabbro D. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 2010;1804(3):445–453. doi: 10.1016/j.bbapap.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 4.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG, Griffin JD. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Rosti G, Palandri F, Castagnetti F, Breccia M, Levato L, Gugliotta G, Capucci A, Cedrone M, Fava C, Intermesoli T, Cambrin GR, Stagno F, Tiribelli M, Amabile M, Luatti S, Poerio A, Soverini S, Testoni N, Martinelli G, Alimena G, Pane F, Saglio G, Baccarani M GIMEMA CML Working Party. Nilotinib for the frontline treatment of Ph(+) chronic myeloid leukemia. Blood. 2009;114(24):4933–4938. doi: 10.1182/blood-2009-07-232595. [DOI] [PubMed] [Google Scholar]
- 6.Cortes JE, Jones D, O'Brien S, Jabbour E, Konopleva M, Ferrajoli A, Kadia T, Borthakur G, Stigliano D, Shan J, Kantarjian H. Nilotinib as front-line treatment for patients with chronic myeloid leukemia in early chronic phase. J Clin Oncol. 2010;28(3):392–397. doi: 10.1200/JCO.2009.25.4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, Cornelissen JJ, Fischer T, Hochhaus A, Hughes T, Lechner K, Nielsen JL, Rousselot P, Reiffers J, Saglio G, Shepherd J, Simonsson B, Gratwohl A, Goldman JM, Kantarjian H, Taylor K, Verhoef G, Bolton AE, Capdeville R, Druker BJ. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 8.Branford S, Fletcher L, Cross NC, Muller MC, Hochhaus A, Kim DW, Radich JP, Saglio G, Pane F, Kamel-Reid S, Wang YL, Press RD, Lynch K, Rudzki Z, Goldman JM, Hughes T. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood. 2008;112(8):3330–3338. doi: 10.1182/blood-2008-04-150680. [DOI] [PubMed] [Google Scholar]
- 9.Hughes T, Deininger M, Hochhaus A, Branford S, Radich J, Kaeda J, Baccarani M, Cortes J, Cross NC, Druker BJ, Gabert J, Grimwade D, Hehlmann R, Kamel-Reid S, Lipton JH, Longtine J, Martinelli G, Saglio G, Soverini S, Stock W, Goldman JM. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37. doi: 10.1182/blood-2006-01-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Branford S, Cross NC, Hochhaus A, Radich J, Saglio G, Kaeda J, Goldman J, Hughes T. Rationale for the recommendations for harmonizing current methodology for detecting BCR-ABL transcripts in patients with chronic myeloid leukaemia. Leukemia. 2006;20(11):1925–1930. doi: 10.1038/sj.leu.2404388. [DOI] [PubMed] [Google Scholar]
- 11.Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C, Pasquini R, Clark RE, Hochhaus A, Hughes TP, Gallagher N, Hoenekopp A, Dong M, Haque A, Larson RA, Kantarjian HM the ENESTnd Investigators. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka C, Yin OQ, Sethuraman V, Smith T, Wang X, Grouss K, Kantarjian H, Giles F, Ottmann OG, Galitz L, Schran H. Clinical pharmacokinetics of the BCR-ABL tyrosine kinase inhibitor nilotinib. Clin Pharmacol Ther. 2010;87(2):197–203. doi: 10.1038/clpt.2009.208. [DOI] [PubMed] [Google Scholar]
- 13.Muller MC, Cross NC, Erben P, Schenk T, Hanfstein B, Ernst T, Hehlmann R, Branford S, Saglio G, Hochhaus A. Harmonization of molecular monitoring of CML therapy in Europe. Leukemia. 2009;23(11):1957–1963. doi: 10.1038/leu.2009.168. [DOI] [PubMed] [Google Scholar]
- 14.Yin OQ, Gallagher N, Li A, Zhou W, Harrell R, Schran H. Effect of grapefruit juice on the pharmacokinetics of nilotinib in healthy participants. J Clin Pharmacol. 2010;50(2):188–194. doi: 10.1177/0091270009336137. [DOI] [PubMed] [Google Scholar]
- 15.Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic—pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20(5):511–528. doi: 10.1007/BF01061469. [DOI] [PubMed] [Google Scholar]
- 16.Rowland M, Tozer TN. Clinical pharmacokinetics: concepts and applications 1995 [Google Scholar]
- 17.Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, Hochhaus A, Griffin JD, Hoelzer D, Albitar M, Dugan M, Cortes J, Alland L, Ottmann OG. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 18.Peng B, Hayes M, Resta D, Racine-Poon A, Druker BJ, Talpaz M, Sawyers CL, Rosamilia M, Ford J, Lloyd P, Capdeville R. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–942. doi: 10.1200/JCO.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Hochhaus A, Kantarjian HM, Agrawal S, Roy A, Pfister M, Chen T, Bleickardt E, Nicaise C, Shah N. Dasatinib pharmacokinetics and exposure-response (E-R): relationship to safety and efficacy in patients (pts) with chronic myeloid leukemia (CML) J Clin Oncol. 2008;26(15):175s. abstract 3590. [Google Scholar]
- 20.Schmidli H, Peng B, Riviere GJ, Capdeville R, Hensley M, Gathmann I, Bolton AE, Racine-Poon A. Population pharmacokinetics of imatinib mesylate in patients with chronic-phase chronic myeloid leukaemia: results of a phase III study. Br J Clin Pharmacol. 2005;60(1):35–44. doi: 10.1111/j.1365-2125.2005.02372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wojnowski L. Genetics of the variable expression of CYP3A in humans. Ther Drug Monit. 2004;26(2):192–199. doi: 10.1097/00007691-200404000-00019. [DOI] [PubMed] [Google Scholar]
- 22.Picard S, Titier K, Etienne G, Teilhet E, Ducint D, Bernard MA, Lassalle R, Marit G, Reiffers J, Begaud B, Moore N, Molimard M, Mahon FX. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109(8):3496–3499. doi: 10.1182/blood-2006-07-036012. [DOI] [PubMed] [Google Scholar]
- 23.Datz FL, Christian PE, Moore J. Gender-related differences in gastric emptying. J Nucl Med. 1987;28(7):1204–1207. [PubMed] [Google Scholar]
- 24.Stephen AM, Wiggins HS, Englyst HN, Cole TJ, Wayman BJ, Cummings JH. The effect of age, sex and level of intake of dietary fibre from wheat on large-bowel function in thirty healthy subjects. Br J Nutr. 1986;56(2):349–361. doi: 10.1079/bjn19860116. [DOI] [PubMed] [Google Scholar]
- 25.Nicolas JM, Espie P, Molimard M. Gender and interindividual variability in pharmacokinetics. Drug Metab Rev. 2009;41(3):408–421. doi: 10.1080/10837450902891485. [DOI] [PubMed] [Google Scholar]
- 26.Fujita KI, Sugiyama M, Akiyama Y, Ando Y, Sasaki Y. The small-molecule tyrosine kinase inhibitor nilotinib is a potent noncompetitive inhibitor of the SN-38 glucuronidation by human UGT1A1. Cancer Chemother Pharmacol. 2011;67(1):237–241. doi: 10.1007/s00280-010-1445-3. [DOI] [PubMed] [Google Scholar]
- 27.Singer JB, Shou Y, Giles F, Kantarjian HM, Hsu Y, Robeva AS, Rae P, Weitzman A, Meyer JM, Dugan M, Ottmann OG. UGT1A1 promoter polymorphism increases risk of nilotinib-induced hyperbilirubinemia. Leukemia. 2007;21(11):2311–2315. doi: 10.1038/sj.leu.2404827. [DOI] [PubMed] [Google Scholar]
- 28.Larson RA, Hochhaus A, Saglio G, Rosti G, Lopez JL, Stenke L, Nakamae H, Goldberg SL, Wang MC, Gallagher NJ, Hoenekopp A, Ortmann CE, Hughes TP, Kantarjian HM. Cardiac safety profile of imatinib and nilotinib in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): results from ENESTnd. Blood. 2010;116(21):944–945. abstract 2291. [Google Scholar]
- 29.White DL, Saunders VA, Dang P, Engler J, Zannettino AC, Cambareri AC, Quinn SR, Manley PW, Hughes TP. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006;108(2):697–704. doi: 10.1182/blood-2005-11-4687. [DOI] [PubMed] [Google Scholar]
- 30.Davies A, Jordanides NE, Giannoudis A, Lucas CM, Hatziieremia S, Harris RJ, Jorgensen HG, Holyoake TL, Pirmohamed M, Clark RE, Mountford JC. Nilotinib concentration in cell lines and primary CD34(+) chronic myeloid leukemia cells is not mediated by active uptake or efflux by major drug transporters. Leukemia. 2009;23(11):1999–2006. doi: 10.1038/leu.2009.166. [DOI] [PubMed] [Google Scholar]
- 31.Larson RA, Druker BJ, Guilhot FA, O'Brien SG, Riviere GJ, Krahnke T, Gathmann I, Wang Y. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008;111(8):4022–4028. doi: 10.1182/blood-2007-10-116475. [DOI] [PubMed] [Google Scholar]