

Abstract
Engineered T cells are currently in clinical trials to treat patients with cancer, solid organ transplants, and autoimmune diseases. However, the field is still in its infancy. The design, and manufacturing, of T cell therapies is not standardized and is performed mostly in academic settings by competing groups. Reliable methods to define dose and pharmacokinetics of T cell therapies need to be developed. As of mid-2016, there are no US Food and Drug Administration (FDA)–approved T cell therapeutics on the market, and FDA regulations are only slowly adapting to the new technologies. Further development of engineered T cell therapies requires advances in immunology, synthetic biology, manufacturing processes, and government regulation. In this review, we outline some of these challenges and discuss the contributions that pathologists can make to this emerging field.
Keywords: T cells, synthetic biology, cellular therapy, immunotherapy, cellular engineering
INTRODUCTION
T lymphocytes have diverse physiological functions, including fighting infections, preventing damaging inflammation, and controlling or eliminating tumors. However, T cells sometimes fail in these roles, leading to chronic infection, autoimmune disease, and cancer. The goal of T cell engineering is to augment the natural functions of T cells through ex vivo manipulation, including special culture conditions, genetic manipulation, and synthetic biology approaches. However, making heterogeneous bulk populations of T cells into therapies presents several immunological, regulatory, and engineering challenges. These challenges and recent advances are presented below.
In particular, we discuss current clinical applications of T cell therapies, government regulation of the development of new T cell therapies, sources of cells for T cell therapies, genetic engineering technologies, recent advances in synthetic biology of T cells, and safety mechanisms. Currently, the dominant therapeutic paradigm is fire and forget, whereby a large population of therapeutic T cells is manufactured (often with gene modification) and then infused into a patient. Toxicities are usually treated with broad immunosuppressive agents. New engineering approaches to enhance T cell functionality and to incorporate control modules to enhance safety and efficacy of T cell therapeutics are critical to advance the field.
As the engineering and manufacturing of T cell therapeutics become more complex, physicians trained in pathology and transfusion medicine will oversee the collection and manufacturing of these cellular products and assist clinical teams with the selection and administration of appropriate T cell therapies (Figure 1a). For existing T cell therapies, pathologists will be asked to answer important questions about cell trafficking, site of action, and effects on diseased tissues. Pathologists will be at the forefront of understanding how these new T cell therapies function in vivo. Thus, an understanding of the many aspects of T cell engineering is becoming increasingly important for the practicing pathologist.
Figure 1.
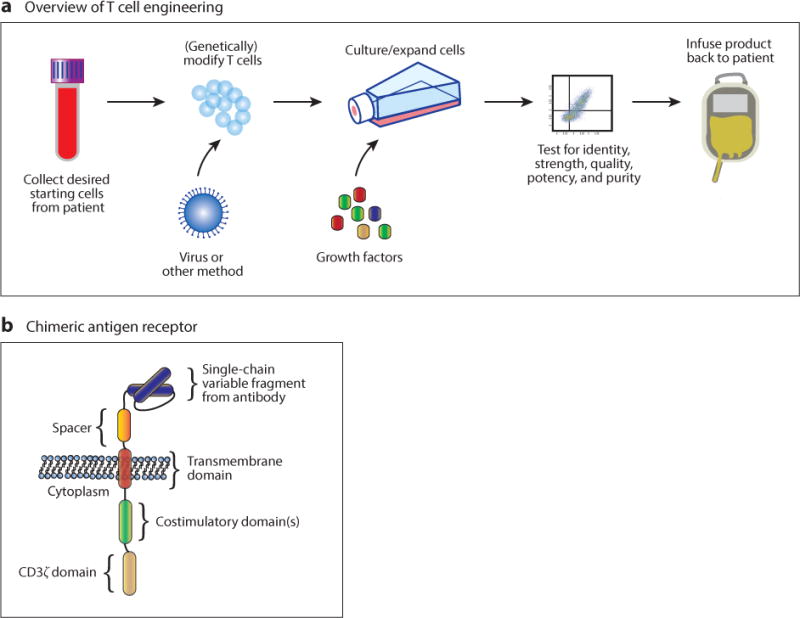
Overview of T cell engineering and a chimeric antigen receptor (CAR) structure. (a) T cell engineering currently involves harvest of T cells from a patient or allogeneic donor, modification of the T cells through genetic engineering via viral infection or other specialized culture conditions, expansion, testing, and reinfusion (1). (b) Diagram of a third-generation CAR. Currently, it is difficult to predict the function of a particular CAR component when combined with others, so each CAR must be optimized individually. This lack of modularity is an impediment to the rapid production of CARs with unique desired properties (1, 2).
CURRENT THERAPEUTIC APPLICATIONS OF ENGINEERED T CELLS
In the past decade, there has been a rapid increase in the number of engineered T cell therapies entering clinical trials. Here, we briefly review the major types of engineered T cell therapies currently used in human trials, with a focus on anticancer T cells. Although there have been some striking successes, the field of engineered T cell therapies is still hampered by the artisanal approach of many different laboratories working without common standards or preclinical models. The massive investments of pharmaceutical and biotechnology companies in engineered T cell technologies (1) could potentially bring a level of standardization to the process of new therapy design and development.
Unmanipulated allogeneic T cells have long been used to induce graft versus cancer effect in patients with hematopoietic malignancies who have received hematopoietic stem cell transplants (2, 3). However, more sophisticated approaches to inducing antitumor effects of T cells were first described by Rosenberg and colleagues (4–6) at the National Cancer Institute. This group harvested autologous tumor-infiltrating lymphocytes (TILs) from 20 patients with metastatic melanoma and adoptively transferred the TILs with IL-2. Eleven of the 20 patients showed objective responses (4).1 Many modifications to this protocol, including to the culture conditions of the autologous T cells, have been made over the years. Some patients treated with TILs have experienced complete remission of their tumors (5). However, successes with melanoma may not be easily generalizable because these tumors are considered particularly immunogenic. It has proven more difficult to expand TILs from other solid tumors (6).
Instead of relying on the specificities of endogenous T cells, a genetic engineering approach has also been used to treat cancer patients. T cell receptors recognizing tumor antigens (such as MART-1 and gp100) have been cloned from TILs. The receptors are then genetically inserted into autologous T cells, which are infused into patients with metastatic melanoma and other cancers (7–10). Although many patients in these studies showed cancer regression, some patients also experienced destruction of normal melanocytes. Another drawback of this approach is that the patients must express the human leukocyte antigen (HLA) molecule on which the exogenous T cell receptor (TCR) is restricted, in the above cases HLA-A∗0201 (an example of major histocompatibility complex restriction).
One method to avoid the challenges of major histocompatibility complex restriction of T cell receptors involves the chimeric antigen receptor (CAR) (Figure 1b). Polyclonal autologous T cells expressing CARs have been used to successfully treat patients with B cell–derived tumors refractory to standard therapies (11–16). CARs contain an antibody-derived single-chain variable fragment (scFv) with intracellular domains derived from T cell receptor–associated signaling molecules (such as CD3ς) and stimulatory receptors (such as CD28 and 4-1BB). Many different CAR configurations and combinations of domains have been tested in mouse models and clinical trials. The details of how CARs are constructed have been reviewed in detail elsewhere (17–19). The overall picture from initial work is that CAR T cell design and testing involve a process of trial and error because the precise effects of certain design decisions on T cell functions are only just starting to be elucidated. For example, the affinity of the antigen-binding portion of the CAR affects anticancer functions in difficult to predict ways. Some reports show that selecting an scFv with decreased affinities for antigen promotes antitumor effects and spares normal tissue that might express low levels of a tumor antigen (20, 21). In another example, CAR T cell function depends on the length of the spacer domain between the scFv and the T cell membrane. Mutations preventing binding of the CAR to endogenous Fc receptors are also critical in some cases (22). Intracellularly, there are many variables that affect CAR function. CD28-derived costimulatory domains promote an effector memory phenotype and enhance glycolysis in T cells. Conversely, 4-1BB intracellular domains promote CD8+ central memory phenotype and mitochondrial biogenesis (23). These examples show that the design principles allowing modular construction of CAR T cells with predictable function from well-characterized parts still need to be elucidated.
Although many groups have focused on cancer and infectious diseases (24) as targets for engineered T cell therapies, inflammatory and autoimmune diseases are also potential targets for engineered cellular therapeutics. Regulatory T cells (Tregs), a subset of CD4+ T cells, enforce tolerance to a variety of self and nonself antigens. Treg deficiencies may contribute to the pathogenesis of human autoimmune and autoinflammatory diseases (25). Ex vivo expanded Tregs are currently in clinical trials for the treatment of type 1 diabetes and to prevent organ rejection after solid organ transplants. Initial results from a phase 1 trial of polyclonal Treg infusion in patients with type 1 diabetes show that the cells are safe and persist for up to a year after infusion (26). In organ transplant patients, populations of Tregs expanded and enriched for alloreactive clones using the donor’s allogeneic B cells have the potential to be more effective than polyclonal Tregs at preventing organ rejection and establishing long-term graft tolerance in organ recipients (27, 28).
THE US FOOD AND DRUG ADMINISTRATION AND THE CHALLENGE OF T CELL THERAPEUTICS
As more clinical trials of T cell therapies are developed, pathologists with training in transfusion medicine and clinical trials will play key roles to ensure compliance with evolving federal regulations. T cell therapies are very different from traditional small molecules and biologics. In particular, it is difficult to establish the precise identity of T cell products, to determine dose, and to quantify pharmacokinetic and pharmacodynamic properties. Therefore, although the regulatory regime for T cell therapies has similarities to traditional drugs, there are many areas of policy under active development.
In the United States, regulatory oversight of gene therapy and cell therapy is performed by the Center for Biologics Evaluation and Research, which is part of the US Food and Drug Administration (FDA) (29). Despite the promise of and excitement surrounding T cell therapies, as of November 2016, there are no FDA-licensed engineered T cell therapeutics. Such licensure would indicate that the agency is satisfied that a new therapy is safe and effective and would be required before marketing a genetically engineered cell therapy.
There is an inherent tension between the desire for new therapies to be made available quickly to patients and the need to ensure safety and efficacy. Indeed, a recent analysis showed that the amount and quality of clinical trial evidence required for FDA approval of drugs and biologics is quite variable. This finding is likely due to the varying levels of risk that are acceptable to the FDA given the seriousness of the disease target, the availability of alternative therapies, and the documented safety risks of a given therapy (30). For engineered T cell therapies, the pathway to FDA approval is subject to the same variables. Importantly, there is not a well-defined path for engineered T cell approval, and the FDA guidance documents explicitly acknowledge that the novelty of this therapeutic area means that there is flexibility in the pathway to approval. Two anti-CD19 CAR T cell therapeutics (from Juno Therapeutics and Kite Pharma) have received “breakthrough designations” to speed their pathways to licensure. As the FDA gains experience regulating engineered cellular therapies, it is likely that the pathway to licensure will become more routine and perhaps accelerated in some cases.
Statutes, Regulations, and Guidances for Cellular Therapies
The regulation of engineered T cells is controlled by three sets of government documents, listed here according to increasing level of detail: statutes, which are laws passed by Congress; regulations (principally Title 21 of the Code of Federal Regulations), which are written rules that implement the statutes; and guidances, which are documents that reflect the FDA’s recommendations of how to comply with specific statutes; and regulations. The FDA regularly releases guidances to keep up with advances in technology and answer questions not directly addressed by statutes or regulations (29).
Engineered T cell products are considered to be human cells, tissues, and cellular and tissue-based products (HCT/Ps) for the purposes of FDA regulation. How an HCT/P is regulated depends upon the manufacturing process of the product. In particular, the FDA has a risk-based regulatory framework for HCT/Ps. Products that are minimally manipulated, intended for homologous use, and meet additional criteria for being low risk are regulated only by Section 361 of the Public Health Service Act of 1944 and 21 CFR 1271. These laws and regulations are primarily focused on preventing transmission of infection through rules about donor eligibility and current good tissue practice (cGTP) manufacturing. These products do not require clinical data for marketing. Examples of such products include connective tissue, such as skin or bone, and reproductive cells and tissues, such as semen and oocytes (29).
Regulation of Engineered T Cell Products
Engineered T cell products, owing to the requirement for significant ex vivo manipulation, are regulated by both Section 361 and Section 351 of the Public Health Service Act of 1944. Therefore, engineered T cell products require extensive testing in the context of an investigational new drug (IND) application. In addition to the measures designed to prevent the spread of infectious diseases, engineered T cell products require premarketing evidence of safety and efficacy (31). Knowledge of the requirements for regulatory approval of T cell therapies is important for investigators and pathologists who oversee cellular therapy facilities. There are unique challenges to meeting FDA requirements that may not be anticipated when manufacturing processes are initially being designed. In particular, donor eligibility rules and qualification processes for all equipment, ingredients, reagents, and procedures used in the manufacturing process can present difficult challenges during the development of novel T cell therapies. These considerations and others are addressed in depth by the manufacturing regulations discussed in the next section.
Current Good Manufacturing Practice and Current Good Tissue Practice: What Are They?
Current good manufacturing practice (cGMP) is a set of regulations outlined in 21 CFR Parts 210 and 211 concerning the personnel, physical manufacturing facility, equipment, processes, packaging, distribution, and record keeping that must go into the manufacturing of biologics and drugs. Therefore, cGMP is a mandated program to ensure the drug meets the identity, strength, quality, purity, and potency that it is represented to possess. cGMP requires quality control and assurance programs for the manufacturing process. Therefore, cGMP is about more than just a particular GMP production facility (32). The role of cGMP for investigational T cell therapies is complex because the principles of cGMP applied to large commercial production runs are not appropriate for small-scale cell therapy preparations used in Phase I clinical trials. Therefore, the FDA issued a guidance in 2008 clarifying that cGMP manufacturing during phase 1 production requires (a) effective written procedures; (b) equipment, facility, and manufacturing controls; and (c) accurate and consistent record keeping. However, this document acknowledges that some products, such as gene therapy products and cellular therapies, include novel manufacturing methods and product assays. These products present difficulties in retaining samples due to limited shelf life and small sample sizes (33). Therefore, phase I cell therapy products can be exempted from full cGMP regulations, but full compliance with cGMP regulations is required by the time of licensure (34).
There are also separate regulations that apply to HCT/Ps called cGTP, which are found in 21 CFR 1271. These rules overlap partially with cGMP rules and compliance with cGMP ensures compliance with many cGTP rules. However, cGTP rules include specific regulations focusing on donor eligibility requirements, prevention of the spread of communicable disease, packaging and shipping requirements, and other areas that are not directly addressed by cGMP. In particular, for allogeneic donors, the FDA requires screening for infection with human immunodeficiency virus (HIV)-1/2, hepatitis B virus (HBV), hepatitis C virus (HCV), and Treponema pallidum. For T cell products, additional testing for human T lymphotropic virus type 1 and 2 and cytomegalovirus are also required (32). The required testing for T. pallidum is an example of the extremely conservative approach taken by the FDA in many matters of donor testing. The last documented case of transfusion-transmitted syphilis occurred in the United States in 1966, yet syphilis testing is still required of allogeneic donors (35, 36). For autologous donors, testing is recommended for HIV-1/2, HBV, and HCV, mostly for the purposes of ensuring the safety of the workers involved in product manufacturing (32). Given the many technical and medical issues that can arise during the determination of donor eligibility, such as how to deal with false-positive test results, the involvement of a pathologist or other physician with experience in this area is very important for engineered T cell manufacturing facilities.
In summary, adherence to cGMP and cGTP is required for FDA licensing of engineered T cell products. Owing to the complexity of complying with FDA regulations and the desire to maintain consistent standards, cell therapy laboratories may choose to become accredited by the Foundation for the Accreditation of Cellular Therapy (FACT) or AABB (formerly the American Association of Blood Banks). Although historically, they have different areas of focus in hematopoietic stem cell transplantation (FACT) or blood banking (AABB), these voluntary accrediting organizations have developed standards and provide support to cellular manufacturing facilities. Accreditation by one of these organizations should be considered by facilities engaged in therapeutic T cell manufacturing, particularly for the eventual purpose of ensuring reimbursement from health insurance companies and government agencies.
The Investigational New Drug Application
The first step in the clinical development of a new engineered T cell therapeutic occurs when a sponsor submits an IND application to the FDA (21 CFR 312). The IND application must include data on a product’s pharmacology and toxicity. For engineered T cells, these data can be difficult to obtain because cells do not have traditional pharmacological parameters, such as an elimination half-life or a standard dose measurement. Therefore, proof-of-concept studies in animal models are important for establishing a reasonable approach to using engineered T cells in phase I clinical trials. Specific safety concerns for engineered T cell products that must be addressed are tumor formation and immunological rejection (34).
For initial clinical trials, the FDA also requires investigators to identify testing that allows for verification of product safety and effectiveness, which can be very challenging with complex cellular therapy products (37). Purity and sterility testing is required at all stages of development and generally includes cell counts; viability; and the absence of aerobic and anaerobic bacteria, fungus, and endotoxins. Testing for potency, which is required for licensure, can be difficult for some cell therapy products because they have complex or incompletely understood functions. Therefore, the FDA allows for progressive potency assay implementation during product development (34, 38). Importantly for pathologists, although the federal Clinical Laboratory Improvement Amendments of 1988 (CLIA) regulations apply to laboratories carrying out some tests for product safety testing (such as testing for many infectious agents), purity and potency testing is exempted from CLIA under most conditions (39).
In summary, T cell therapies are highly regulated in the United States. A familiarity with the regulations is important for medical directors of cellular therapy manufacturing facilities and investigators who are seeking to translate new cellular therapies into clinical trials. The technology driving the development of engineered T cell therapies is moving much faster than the federal regulators in charge of overseeing it. Therefore, pathologists who are involved with T cell therapy trials are likely to encounter unique regulatory challenges that require close collaboration with the FDA.
GENETIC ENGINEERING TECHNOLOGIES
Many T cell therapies require genetic engineering. Examples include the ablation of endogenous genes, replacement of endogenous genes with modified versions, or addition of new synthetic genes. In some cases, multiple genetic modifications, such as endogenous TCR inactivation followed by transgene insertion, are required for effective therapeutic T cell production (40). In this section, we review methods for making site-specific changes in the T cell genome. We also discuss approaches to inserting transgenes into T cell genomes. Many others have reviewed the underlying biochemistry of these technologies (41–44), therefore the focus of this section is on the successful use of these approaches to genetically modify human T cells.
There are several approaches to making site-specific genetic modification of mammalian cells. All rely on nucleases that cut DNA in a specific location, activating either the nonhomologous end-joining (NHEJ) DNA repair pathway or the homologous recombination (HR) repair pathway. NHEJ often results in insertions or deletions at the site of the double-strand break. Therefore, if a double-strand break is targeted to the coding region of a gene, a frameshift mutation or a nonsense mutation is likely to be introduced into the targeted gene (40, 45). HR is an alternative DNA repair pathway where a cell uses a homologous stretch of DNA to repair either a single- or double-strand break. Single-strand breaks in particular have much less risk of being repaired with indels than double-strand breaks (46). Specific gene modification has been described using targeted nucleases that induce DNA strand breaks. These breaks are repaired by HR with exogenous DNA templates that serve to introduce specific mutations of interest. Such approaches have led to several reported successes in site-specific gene modification in human T cells (47–49).
Several different classes of site-specific nucleases have been used for gene editing in T cells. These include zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and Cas9 nucleases (Table 1). Each method has advantages and disadvantages, which are discussed in more detail in the section titled Site-Specific Nucleases.
Table 1.
A comparison of gene editing technologies used in T cell engineering
| DNA-binding protein | Binding unit | Binding-site restrictions | Nuclease | Successful use in primary T cells? | Commercially available? |
|---|---|---|---|---|---|
| ZFN | Each finger binds 3 bp of DNA | Two different adjacent sites separated by a spacer, guanine-rich; binding is context dependent, and limited specificities are available | FokI dimer | Yes | Yes |
| TALEN | Each TALE repeat binds 1 bp of DNA | Two different adjacent sites separated by a spacer, T at the 5′ end of target sequences | FokI dimer | Yes | Yes |
| MegaTAL | Combination of TALE repeats and meganuclease | Meganuclease target site must be present | Meganuclease | Yes | No |
| Cas9 | 20 bp of a RNA-DNA hybrid | 3 bp of PAM immediately following the target site | Cas9 | Yes | Yes |
Abbreviations: Cas9, clustered regularly interspaced short palindromic repeats associated protein 9; megaTAL, a synthetic protein that is a combination of TALE DNA binding domains and a meganuclease; PAM, protospacer-adjacent motif; TALE, transcription activator-like effector; TALEN, transcription activator-like effector nuclease; ZFN, zinc finger nuclease.
Site-Specific Nucleases
Zinc finger DNA-binding domains recognize specific 3-bp DNA sequences. For site-specific DNA cutting, multiple site-specific zinc finger domains are assembled into two separate modules that bind 9–18 bp upstream or downstream of a target site. Each of the two zinc finger modules are fused to endonuclease domains of the FokI restriction enzyme designed to require heterodimerization to cut (50). When the two zinc finger modules bind to the two adjacent target sites, the FokI domains are brought together, forming a functional enzyme that initiates a double-strand break in the short spacer between the two binding sites (42). However, ZFNs can be difficult to design due to context-dependent binding of DNA, toxicity, off-target effects, and limitations on the sequences that can be targeted (51). ZFNs have been used to knock out chemokine receptors CXCR4 or CCR5 in human CD4+ T cells to make the cells resistant to HIV infection (44). They have also been used successfully to edit specific gene targets in T cells and insert larger gene cassettes using a viral vector (49).
TALENs are similar to ZFNs because both use modular DNA-binding domains coupled to FokI nucleases. Cutting at a specific site likewise requires two TALENs to bind upstream and downstream of a target site to allow FokI dimerization (52). TALEN repeats have the advantage of recognizing single base pairs rather than sets of 3 bp, allowing for much easier design of sequence-specific proteins (53, 54). However, TALENs are difficult to clone and deliver with lentivirus because the transcription activator-like effector (TALE) repeats have a propensity to recombine (55). Furthermore, each TALE repeat contains 33–35 amino acids, which makes the size of TALENs capable of site-specific recognition quite large. TALENs have been used to knock out specific genes in human T cells (56–58). A variation on the use of the TALE repeats involves linking them to a meganuclease, which has both a sequence-specific DNA-binding function and nuclease activity. Using this approach, a chimeric megaTAL was able to target and disrupt the TCR-α locus in primary human T cells through targeting an 11-bp sequence with a TALE array and a nearby 22-bp sequence with a meganuclease (59). This approach avoids the necessity of making two flanking proteins. However, engineering meganucleases to bind to specific DNA sequences is complex, limiting the usefulness of this technology in T cell engineering (60).
Cas9-based approaches to genetic engineering use the bacterial Cas9 nuclease and an RNA guide that is complementary to the sequence being targeted for cleavage. The guide RNA is designed with a 20-bp region of homology to the target, followed by the 80 nucleotides required for binding to Cas9. Targeting specific sequences in the human genome is restricted by the protospacer-adjacent motif site, a short 3-bp sequence adjacent to the target region that is recognized by Cas9. This system has the advantage of its easy production of the guide RNAs that give Cas9 specificity; however, early reports have shown significant off-target cleavage (61), which may require additional optimization of the Cas9 protein (62). In T cells, Cas9-based systems have been used to successfully ablate CXCR4 (47, 63), TCR-α (40), and the HIV genome (64), and to edit specific genes via HR (47). Importantly, off-target editing was not reported for Jurkat T cells modified with Cas9 (40). Additional characterization of the off-target effects of each of the systems discussed above is necessary to determine which combines the highest efficiency and specificity.
Viral Transgenesis
Viruses are a common delivery vehicle for transgenes used in T cell engineering (65). Retroviruses, particularly replication-incompetent gammaretroviruses and lentiviruses (based on HIV), have been used extensively in T cell engineering (65). Initial experiences with gammaretroviral vectors for gene therapy raised important safety concerns. For example, patients with X-linked severe combined immunodeficiency due to deficiency of the common gamma chain cytokine receptor had their hematopoietic stem cells treated with a gammaretrovirus encoding the missing gene. However, several patients in the initial cohort developed T cell leukemia and were found to have viral integration sites near oncogenes (66). Gammaretrovirus-mediated gene transfer into mature T cells has a much better safety record, perhaps due to the low propensity of mature T cells for uncontrolled proliferation. In one study that followed patients who received anti-HIV CAR T cells, there was no evidence of enrichment for certain integration sites in more than 500 patient-years of follow-up (24). Gammaretroviral vectors and lentiviral vectors have a payload capacity of approximately 8–10 kb. Certain lentiviral vectors have the advantage of infecting nondividing T cells (67, 68). These retroviruses integrate preferentially into transcriptionally active regions (lentivirus) or transcriptional start sites (gammaretrovirus), which may increase the risk of oncogenesis in genetically modified cells (69).
Transposon-Based Transgenesis
Transposons, such as Sleeping Beauty and piggyBac, provide nonviral approaches to permanent transgenesis in T cells. The SB transposon system requires delivery into cells of a plasmid with the desired payload flanked by domains with both inverted and direct DNA repeats and a second plasmid expressing the transposase (70). In T cells, delivery of these two plasmids has been accomplished with electroporation. This approach has been used successfully to manufacture anti-CD19 CAR T cells for clinical trials (71). A direct comparison of lentiviral and SB transgenesis in primary human T cells showed that the efficiency of expression of a transgenic TCR was slightly lower in cells electroporated with SB plasmids; however, the integration sites of SB also were less likely to be inside of transcribed genes than lentivirus integration sites (72). The efficiency of transposition decreases as the size of the payload increases with maximum payload sizes in the range of 10–20 kb, depending on the target cell type (73).
The similar piggyBac transposon system has also been used to genetically modify primary human T cells (74); however, this transposon has a nonrandom integration pattern with a preference for integration into transcribed genes (75). Transposons have several advantages over viruses in the production of transgenic T cells, including the relatively lower cost of producing GMP-grade DNA and the potentially higher payload capacity.
RNA-Based Expression
Unlike the viral and transposon-based approaches discussed above, electroporation of primary T cells with mRNA does not permanently alter the cell’s genome. RNA also appears to have lower toxicity than DNA when electroporated into cells. RNA has been used to express CARs in primary human T cells (76). RNA degrades rapidly, and protein expression from electroporated mRNA decreases after several days. RNA electroporation may be a useful technique when the function of an engineered T cell is needed for only a short period of time or when repeated dosing of a patient is feasible. The safety advantages of RNA electroporation for therapeutic T cell manufacturing are discussed in more detail in the section titled Transient Transfection with RNA.
CHALLENGES OF ALLOGENEIC T CELL THERAPIES
As discussed above, many successful anticancer T cell therapies, including TIL and CAR therapy, have used autologous T cells as the source material. However, an off-the-shelf allogeneic engineered T cell product would be very beneficial for patients. First, it would allow scaling up of manufacturing and immediate availability for patient care. Second, it would benefit patients who are lymphopenic or who cannot tolerate or wait for autologous cell collection and autologous cell manufacturing. Below in this section, we review past experience with allogeneic T cell therapies and current engineering approaches to make such therapies available to a wider patient population.
Immunology of Allogeneic T Cells
There are both immunological and technical challenges to the development of allogeneic T cell therapeutics. Human T cells express both HLA class I (A, B, and C) and HLA class II (DP, DQ, and DR) molecules (77). Due to the many polymorphisms in HLA genes in human populations, it is rare to find allogeneic donors with HLA genotypes similar enough to prevent either graft-versus-host disease (GVHD) or host rejection of the graft. Even if major histocompatibility antigens can be matched, therapeutic T cell rejection or GVHD could be mediated by polymorphic minor antigens. In hematopoietic stem cell transplantation, high-resolution matching between donor and recipient at HLA-A, HLA-B, HLA-C, and HLA-DRB1 is routinely performed. Currently, the probability of finding an allogeneic donor with an 8/8 match in the United States for patients of European ancestry is 75% and is lower for all other ethnic groups (78). Thus, the diversity of human HLA is a major barrier to the development of routine matching of donors to recipients for allogeneic T cell therapies.
Even if HLA antigens can be perfectly matched, there is a risk that other cell surface antigens on allogeneic T cells might be targeted by antibodies produced by the recipient. Such antibody formation is common (2–3% in antibody-negative recipients) after allogeneic red blood cell transfusion. This clinical experience is relevant because red blood cells do not express surface HLA molecules, but allogeneic red blood cells in most cases have potentially immunogenic single-amino acid polymorphisms in membrane proteins (79). To put the challenge of using allogeneic T cells into perspective, even autologous cells that have been modified with nonhuman sequences such as CARs or murine T cell receptors can elicit rejection from immunocompetent recipients with solid tumors or lymphoma (80, 81). Therefore, immune-mediated rejection is a major challenge for engineered T cell therapies.
Because of positive selection in the thymus, T cell receptors have a baseline ability to bind to HLA molecules. Perhaps as a result of this developmental process, T cells have high levels of reactivity against allogeneic HLA, estimated to be about 1–10% of T cells (82). Therefore, GVHD can develop if immunosuppressed patients are infused with allogeneic T cells expressing T cell receptors that may be alloreactive. Importantly, partial matching of T cell HLA alleles (such as T cells from family members) can lead to GVHD even in immunocompetent patients. Such cases have been described if the donor T cells are homozygous for HLA alleles for which the recipient is heterozygous (83). In addition, some immunocompetent patient populations, such as cardiac surgery patients, appear to have a higher risk for transfusion-associated GVHD (84). Therefore, there is a clinical risk of both rejection and GVHD in immunocompetent recipients of allogeneic T cells.
However, some immunocompetent patients establish long-term, stable chimerism after infusion of allogeneic blood products. Data from blood transfusion recipients undergoing elective surgery show that allogeneic T cells are rapidly eliminated after transfusion. In one study, 25 patients received at least one unit of nonleukoreduced red blood cells during elective surgery. Subsequent analysis showed that allogeneic T lymphocytes from the transfused unit undergo rapid elimination (99.9%), followed by expansion of a subpopulation of the donor lymphocytes between 2 and 7 days after transfusion (likely due to alloreactive clones). There is complete clearance of the donor T cells by 14 days (85). These data contrast with data from obstetric patients and trauma patients who show much higher levels of long-term engraftment with allogeneic fetal or donor lymphocytes, respectively. In trauma patients, long-term engraftment of donor lymphocytes has been documented years after the transfusion (86–88). Therefore, long-term chimerism with allogeneic T cells in the absence of GVHD or rejection is possible, but the mechanism by which such chimerism can be established is unknown. Tolerizing regimens based on an understanding of this biology could be clinically useful for the introduction of allogeneic T cell therapeutics. An alternative approach to protecting allogeneic T cells from elimination involves engineering resistance to alemtuzumab or chemotherapeutic agents in the cells (56, 89). However, this blunt-force approach is unlikely to lead to long-term tolerance of the allogeneic cells.
Preventing Rejection and Graft-Versus-Host Disease After Allogeneic T Cell Infusion
Allogeneic lymphocyte therapy (called donor lymphocyte infusion) is already well established as a treatment for patients who have received an allogeneic stem cell transplant for a hematological malignancy and who have residual disease. In such cases, the risk of rejection of the allogeneic T cells is lower given the successful engraftment of the donor’s hematopoietic system and tolerization to donor HLA. By contrast, most of these patients (approximately 70%) develop GVHD, which is correlated with lower risk of relapse of their malignancy (3, 90). Such donor-derived lymphocytes can also be modified to express a CAR specific for a tumor antigen (91) or selected and expanded to treat specific viral infections (92–94).
The use of third-party allogeneic cells enriched for viral reactivity may decrease the risk of GVHD because virus-specific TCRs may have less alloreactivity. One clinical trial used third-party Epstein–Barr virus reactive allogeneic T cells that were partially matched for the recipient’s HLA-A, -B, and -DR alleles. These cells were infused to treat patients with Epstein–Barr virus– mediated lymphoproliferative disease after stem cell or solid organ transplant. The investigators reported that 64% of patients responded to this therapy at 5 weeks, and there were no reported cases of GVHD (95). Another clinical trial using third-party antiviral T cells in post-hematopoietic stem cell transplant patients showed high rates of response at 6 weeks (66–78%, depending on the virus) with 2 of 50 enrolled patients experiencing de novo GVHD (96). Therefore, the virus-specific T cells may have less alloreactive potential, particularly in patients who are chronically immunosuppressed after solid organ or hematopoietic stem cell transplant.
Another approach to generate large populations of therapeutic T cells with known TCR specificities and/or CAR expression involves making induced pluripotent stem cells from mature T cells and then redifferentiating these induced pluripotent stem cells into T cells. These approaches could allow the production of large numbers of functional, genetically modified T cells with defined HLA types and TCR specificity (97–99).
Genetic Engineering to Make a Universal T Cell
Genetic engineering approaches may enable the production of off-the-shelf allogeneic T cell therapy products with minimal risk of rejection or GVHD. In particular, genetic ablation of the TCR and HLA molecules has been explored.
In one approach used by multiple groups, ZFNs were used to disrupt the genes expressing HLA-A and/or the α- and β-chains of the TCR (48, 100, 101). One group used electroporation of mRNA encoding ZFNs targeting conserved regions of the TCR α- or β-chains. Deletion efficiency of the TCR in activated primary human T cells was in the range of 15–40%, depending on which TCR chain was targeted. No off-target effects of the ZFNs were detected. This approach was also used to delete TCR in CD19 CAR-expressing cells, which were shown to retain cytotoxic activity against CD19-expressing target cells similar to the parental CAR T cell line (101). The same group subsequently showed efficient deletion of HLA-A2 (up to 57% deletion) in primary T cells from an HLA-A2 homozygous donor (100). Deletion of HLA-A is a useful technical accomplishment, but deletion of this single HLA locus alone is insufficient to prevent rejection given expression of other HLA molecules in T cells. However, loss of multiple HLA proteins would likely cause T cells to become susceptible to natural killer (NK) cell-mediated killing. Theoretically, these cells could be protected from NK cell killing in vivo by enforcing higher expression of nonclassical HLA molecules (102, 103). In vivo models for this complex engineering challenge need to be further developed.
Another research group infected previously activated primary human T cells with integrase-defective lentiviral vectors expressing ZFNs specific for two regions in the TCR-β gene. The efficiency of this approach was lower than the mRNA electroporation approach, with a loss of CD3 expression in approximately 7% of cells. Importantly, these CD3-negative T cells could be expanded with low-dose IL-7 and IL-15 in the absence of additional TCR signals. These cells were then transduced with a tumor antigen-specific TCR and shown to have antitumor activity in a mouse model (48).
TALENs have also been used to successfully ablate TCR-α and -β in primary human T cells. In this approach, electroporation of TALENs targeting the TCR-α or -β genes in primary human T cells led to 60% and 40% TCR knockout, respectively (57). However, the use of TALENs targeting two loci simultaneously was shown to induce translocations between the targeted chromosomes (89). Translocations or other types of chromosomal abnormalities are likely to be a feature of any genetic engineering approach that induces simultaneous double-strand breaks in multiple genomic locations.
A more recent publication compared three different approaches to TCR ablation in primary T cells: TALENs, Cas9, and megaTAL nucleases (a hybrid of a meganuclease with additional TAL repeat regions for added specificity) (40). This report showed low rates of successful TCR ablation using TALENs, but rates of approximately 75% using the megaTAL system and 85% with Cas9. No off-target editing events were detected with TALEN or Cas9; however, megaTAL editing caused off-target editing events in many genes, particularly in the KAT2B locus.
The successful use of Cas9 for knock in of specific sequences into primary human T cells could potentially allow the same technology to be used both for specific gene ablation and for addition of modified sequences (47). One important example is the production and clinical application of allogeneic TCR-ablated anti-CD19 CAR T cells, which promises to permit the development of universal allogeneic T cells for use in patients for whom autologous or stem cell donor-derived T cells are not available (104).
WHAT IS SYNTHETIC BIOLOGY?
The recent successes of engineered T cell therapies have paralleled the emergence of the field of synthetic biology. Synthetic biology differs from genetic engineering in that it aspires to use the approaches of classical engineering disciplines. Synthetic biology seeks to make cellular engineering into a systematic process rather than the nonstandardized trial and error process that often characterizes much of genetic engineering. Synthetic biology principles include designing to meet quantitative performance specifications set in advance, separating design from fabrication, using modular biological parts, and using standardized cell populations to allow for consistent results (105). Applying these principles to develop synthetic systems in mammalian cells is still an ongoing challenge because of the complexity of the systems and the difficulty of predicting the function of novel constructs (106, 107). Systems developed for one cell type or organism may not work in another environment. Therefore, some workers have proposed directed evolution as an alternative to a top-down engineering approach (107). Nonetheless, the philosophy of synthetic biology is having a major impact on the engineering of therapeutic T cells, as these sorts of viewpoints are focused on meeting the challenge of systematically altering T cell function. One example of the challenges of rational design is the stepwise optimization of CARs. Over the past 25 years, all components (including the ectodomain, hinge region, signaling, and costimulatory domains) of these receptors were optimized with painstaking trial and error work (108). In the selected examples in the sections titled Logic Gates, Small-Molecule Controls, and Docking Systems below, we discuss specific examples of T cell engineering that employ methods of synthetic biology and that could serve as building blocks for future advances (Figure 2).
Figure 2.
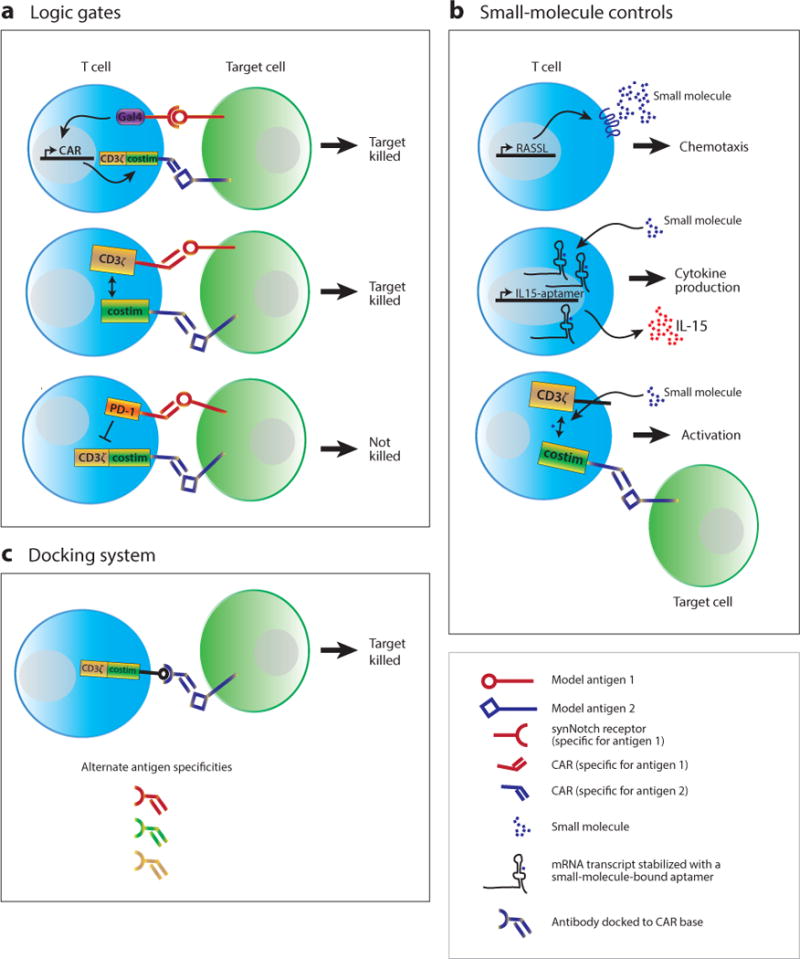
Diagram of control modules for therapeutic T cells. (a) Logic gates control T cell activation when two antigens are encountered at the same time. Examples include synthetic notch (synNotch) receptors to one antigen that drive chimeric antigen receptor (CAR) expression for a second antigen, CAR T cells in which signal 1 (CD3ς) and signal 2 (costimulation) are triggered by different CARs, and inhibitory CARs where the presence of an antigen blocks activation. (b) Small molecules have been used to control T cell function. Examples include orthogonal chemotaxis using RASSLs (receptors activated solely by a synthetic ligand), RNA aptamers that require small-molecule binding to stabilize specific RNA transcripts (such as IL-15), and small-molecule gated split CARs where both the small molecule and the target antigen are required for T cell activation (also an example of a logic gate). (c) Docking systems allow a single CAR with a common binding domain to use several different adapters with different antigen specificities. The adapters have defined pharmacokinetics, whereas the modified T cells do not. Abbreviations: CD3ς, a component of the T cell receptor; costim, costimulation; Gal4, a yeast transcription factor; PD-1, an inhibitory receptor expressed on T cells.
Logic Gates
CARs combine the binding affinity of an antibody with the signaling domains of the ς-chain of the TCR complex and costimulatory receptors, such as CD28 and 41BB. A challenge of current CAR therapies is that cells are limited to sensing a single antigen. One approach to engineering dual-antigen sensing is to construct an AND gate using a two independent CARs, one with the intracellular TCR-signaling domains and one with intracellular costimulatory domains, in the same cell. In theory, binding of both CARs at the same time to the same (or nearby) target cells should be required for activation and effector function (Figure 2a). However, such systems are in fact quite complex and difficult to tune appropriately.
One group that employed this approach found that extensive optimization was required to find receptor-antigen combinations that gave acceptable gating activity. This group initially used a strong anti-CD19 CAR with a CD3ς intracellular domain and a second prostate-specific membrane antigen CAR with CD28 and 41BB intracellular domains. Although they found synergistic effects when T cells expressed both CARs and were exposed to target cells with both antigens, the anti-CD19 CAR was able to activate T cells independently of the costimulatory signal from the second CAR. Only by weakening the CAR-antigen interaction in the CAR with a CD3ς intracellular domain could AND gate activity be observed (109). Another group, using a similar two-CAR system, showed a lack of AND gate activity in cytolytic activity assays but increased CAR T cell expansion (110). In this system, T cells expressed two CARs: a CAR specific for the tumor antigen ErbB2 with a CD3ς intracellular domain and a second CAR specific for tumor antigen MUC1 with a CD28 intracellular domain.
Another approach is the inhibitory CAR where the presence of a second antigen activates a CAR with an inhibitory intracellular domain for PD-1 or CTLA-4. These inhibitory CARs could inhibit classical CAR-induced effector functions against target cells. However, the expression levels of the target antigens and of the inhibitory CARs were critical factors in tuning the magnitude of the effect (111). Thus, all of these dual-antigen systems described here require optimization to generate the desired AND gate behavior.
A more recent report using a synthetic Notch (synNotch) receptor shows a potentially more robust AND gate with a high level of modularity in all its components. In this system, the synthetic receptor consists of an extracellular single-chain antibody connected to a portion of the human Notch receptor. Upon antigen binding to the synNotch receptor, the intracellular tail of the protein is cleaved, releasing a transcription factor that drives expression of a CAR with specificity for a second tumor antigen (112). In vivo, T cells expressing both a synNotch receptor and a synNotch-induced CAR specific for two different target antigens show strong specificity for tumors expressing both target antigens. There is minimal off-target killing of tumors expressing only one of the two antigens, even in the same animal (113). Owing to the modularity of the synNotch system, both binding specificity and transcriptional targets can be easily modified. Thus, the synNotch receptor is likely to be useful for synthetic biology projects in T cells beyond the control of CAR expression.
In summary, creating AND gates to control T cell activation is difficult, and careful tuning of each component is often necessary to get the desired behavior. Therefore, in the absence of rigorous quantitative models for the construction of appropriately tuned dual-CAR systems, repeated trial and error seems to be necessary. New technologies such as synNotch receptors may help create more robust AND gates; however, the robustness of this tool in other contexts requires further investigation.
Small-Molecule Controls
Various synthetic biology approaches to gain small-molecule control of T cell behavior have shown promise (Figure 2b). For example, engineered G protein–coupled receptors (GPCRs) that respond only to synthetic small-molecule ligands have been developed for each of the classes of GPCR (114). In a mouse model, primary mouse T cells expressing a synthetic GPCR responsive to the small-molecule ligand clozapine-N-oxide were able to specifically localize to implanted clozapine-N-oxide–releasing microspheres (115).
Another small-molecule control mechanism uses RNA aptamers in the 3′ untranslated region of genes. When bound to a small-molecule ligand, the RNA transcript is stabilized, and the gene product is translated. In the absence of ligand, the RNA degrades (116). Human T cells expressing a theophylline-binding aptamer to control IL-15 expression showed greater proliferation in the presence of theophylline (117).
Direct control of CAR function with small molecules has been described by splitting the CAR into two parts that can be brought together by a small-molecule dimerizer such as rapamycin (Figure 2b). Dimerization of the two components of the split CAR, each of which contains either FKBP or FRB domains, brings TCR-signaling domains into the cluster of receptors formed when a CAR-expressing T cell encounters a target cell. Both the target antigen and the small molecule must be present for CAR function (118).
Docking Systems
Currently, CAR T cell antigen specificities are hardwired by the scFv incorporated into the receptor. An alternative method of controlling CAR T cell activity and specificity is to make a universal CAR T cell whose CAR receptor can be docked with an antibody or antibody fragment that gives the cell different specificity (Figure 2c). This approach has been described by multiple groups using a variety of docking interactions, such as avidin-biotin, antibody-small molecule, and antibody-peptide binding pairs (119–123). However, these studies also showed the precise orientation of the antigen-binding components of these receptors affects CAR activity. Thus, significant optimization has to be performed to develop new specificities for such universal CAR T cells.
SAFETY OF ENGINEERED T CELL THERAPEUTICS
Engineered T cell therapeutics, like all medical treatments, have the potential to harm a patient. In many cases, these toxicities are difficult to predict for a given therapy or patient. However, multiple clinical trials with anticancer T cells have shown that even low-level expression of a tumor antigen on normal tissue can provoke on-target toxicity in healthy tissue.
For example, autologous CAR T cells recognizing ERBB2 (based on the drug trastuzumab) caused acute respiratory distress syndrome, diffuse alveolar damage, severe hypotension, and death in one patient with metastatic colon cancer. The authors speculate that low levels of the ERBB2 antigen on the lung epithelium triggered this response (124). This outcome was surprising given that the parent drug trastuzumab has a very good safety profile. Similar on-target toxicities in cell types with low-level antigen expression have been described in patients treated with anti-MAGE-A3 TCR-engineered T cells who developed necrotizing leukoencephalopathy (125) or cardiogenic shock (126). These two cases are instructive because they illustrate two possible mechanisms of toxicity. Low levels of MAGE-A expression in some neurons likely contributed to the neurological toxicities. By contrast, reactivity against peptides derived from the completely unrelated cardiomyocyte protein titin was likely responsible for heart failure in the second report. Thus, predictions of which tissues are likely to be targeted by engineered T cells with specificity for tumor antigens are difficult or impossible.
The gastrointestinal tract is also a site of on-target toxicity in anticancer-engineered T cell trials. Autologous CAR-expressing T cells targeting carboxy-anhydrase-IX (CAIX) were used to treat 12 patients with CAIX-expressing renal cell carcinoma. Two of the patients developed cholangitis, presumably due to low-level CAIX expression on the bile duct epithelium (127). Autologous T cells engineered to express a murine TCR targeting carcinoembryonic antigen were used to treat three patients with metastatic colorectal cancer. All three patients developed diarrhea and severe inflammatory colitis, which prompted the halting of the clinical trial (128). Retroviral DNA from the TCR-expressing vector was found in biopsies from throughout the patients’ gastrointestinal tract.
The experience with CAR T cells targeting CD19 has shown that these cells can cause cytokine release syndrome and macrophage activation syndrome due to the inflammation caused by a vigorous anticancer response. Cytokine release syndrome in the context of CAR T cell therapy is defined by fever for three consecutive days, massive increase in inflammatory cytokine levels, and hypoxia or neurological dysfunction. However, this acute toxicity appears to be reversible with immunosuppressive therapies, including anticytokine biologics (11, 129, 130). However, many patients treated with anti-CD19 T cells develop B cell aplasia and require infusions of immunoglobulin to maintain adequate plasma immunoglobulin levels.
Several different approaches have been employed to address the risks posed by on-target and off-target toxicities of engineered T cell therapies; see Reference 131 for an excellent comprehensive review. Many T cell therapies, in particular TILs and CAR T cells, have been used in patients with advanced cancer who have few other therapeutic options and a poor chance of long-term survival. Thus, there is a higher tolerance for risk in the products used to treat these patients. If T cell therapies are to be used routinely for conditions that are rarely lethal, safety becomes a much more important consideration.
Inducible Dimerization Kill Switches
There are many ways to increase the safety of engineered T cells. One of the most promising systems is based on a transgene that produces a truncated form of caspase-9 linked to a dimerizer domain. Inducible dimerization of this construct leads to activation of caspase-9, which is a central mediator of the mitochondrial death pathway. In vitro data show that more than 90% of T cells stably expressing this inducible caspase 9 (iCasp9) construct could be killed with the small-molecule dimerizer AP20187 (132). These results have been reproduced in a clinical trial with patients who were treated with retrovirally transduced iCasp9-expressing donor lymphocyte infusions after allogeneic stem cell transplant. Patients who developed GVHD were treated with the dimerizer drug AP1903, which led to a greater than 90% decrease in peripheral blood transgenic T cells within 30 min. All four patients had complete remission of GVHD (133). However, not all transgenic T cells were killed by the treatment, raising the possibility that a subset of alloreactive T cells that silence iCasp9 expression could rebound to cause GVHD or another pathology. There is some evidence that activated T cells are more efficiently deleted than resting T cells (134). Further follow-up showed that iCasp9 was not immunogenic in 10 patients followed for up to two years; however, more data are required to determine if treatment with AP1903 leads to increased risk of relapse, despite the persistence of some donor T cells after treatment (135).
An alternative homodimerization safety switch is based on small-molecule-induced dimerization of the intracellular signaling domain of Fas, which induces apoptosis via an alternative caspase-8-dependent pathway. This approach has been used to induce apoptosis in mouse thymocytes using a small molecule made by fusing two molecules of the drug calcineurin (136, 137) or in primary human T cells with the FKBP/AP1903 dimerizer system described above (138).
Herpes Simplex Virus Thymidine Kinase Kill Switch
One of the pioneering safety mechanisms for T cell therapy is the herpes simplex virus thymidine kinase (HSV-TK) gene. This gene, when introduced into donor lymphocytes with a retrovirus, makes the human cells susceptible to killing by the small-molecule drug ganciclovir. In the initial report, the successfully transduced cells were positively selected using a truncated version of the nerve growth factor receptor that was coexpressed with the viral thymidine kinase (139). In the initial investigation and in subsequent clinical trials using similar HSV-TK systems, patients who received modified donor T cells after allogeneic stem cell transplants and subsequently developed GVHD were successfully treated with ganciclovir (123). However, an anti-HSV-TK immune response has been described in some patients, which in some cases led to immune-mediated elimination of the genetically modified allogeneic T cells. Further work with HSV-TK showed that safety switches that make use of nonhuman proteins are likely to be immunogenic and cause the rejection of engineered T cells in all but the most profoundly immunosuppressed patients (140, 141). Patients who receive T cells transduced with HSV-TK are also prevented from receiving ganciclovir for cytomegalovirus infection.
Cell Surface Markers for Targeted Elimination
Another engineered safety mechanism is the insertion of a cell surface marker for which FDA-approved antibody drugs are available. For example, T cells transduced with a synthetic protein consisting of a CD8-α stalk with two rituximab (anti-CD20) mimotopes can be depleted with rituximab. Importantly, the anti-CD20 antibody of ofatumumab does not bind to the synthetic epitope, allowing for specific targeting of nontransduced CD20-positive cells (142). Similar approaches have been described with full-length CD20 (143), c-myc epitope tagging of TCRs (144), and with truncated versions of the human epidermal growth factor receptor. This truncated epidermal growth factor receptor lacks both ligand-binding and intracellular signaling domains but retains the epitope for cetuximab binding (145). These synthetic proteins can be used to both mark and select engineered T cells and as a safety mechanism to deplete cells in vivo. In vivo depletion occurs by both complement-mediated lysis of opsonized cells and antibody-mediated cell-dependent cytotoxicity. However, experimental results supporting these approaches have so far been confined to mouse models.
Transient Transfection with RNA
One particularly elegant way to limit the potential toxicity of an engineered T cell therapy is to electroporate autologous T cells with mRNA that transiently reprograms the cell to have a desired therapeutic effect. The mRNA is easily quantifiable and has a short half-life, so the pharmacokinetics of the modified T cells is more easily defined. Even the most effective kill switches described above do not delete all genetically modified cells, raising the possibility that engineered T cells may be impossible to completely eliminate in the event of severe toxicity.
In vitro assays with primary human T cells show that transgene expression from electroporated mRNA lasts for several days (146). This mRNA electroporation approach has been used in a clinical trial of CAR T cells targeting mesothelin because this tumor antigen is expressed on many normal tissues, leading to the possibility of on-target toxicities. The initial report of results shows that the mRNA persists for a few days, but there is clear antitumor effect with minimal on-target toxicity (76).
Designing T Cells for Safety
Permanently genetically modified T cells do not have well-defined pharmacokinetics and can persist for years after infusion. Therefore, robust nonimmunogenic safety mechanisms need to be developed if such therapies are going to be used in immunocompetent patients with chronic medical conditions. Some potential future safety measures include controlling the location of transgene integration (47); designing inhibitory receptors to prevent on-target, off-tumor toxicity (111); building in strict dependence on small-molecule ligands for survival or activity (117, 147); or making gene deletions leading to auxotrophic T cells. Importantly, turning “on” a T cell’s desired function with a small molecule is likely to be a more effective mechanism to prevent toxicities (such as cytokine release syndrome) than “off” switches, which do not completely eliminate a therapeutic T cell population. However, only an effective kill switch can stop T cell cancers that could arise from engineered cell populations. Although no instances of T cell cancer caused by engineered T cell therapies have been reported, such cases will eventually arise. Therefore, instead of being an afterthought, safety mechanisms will increasingly be an integral part of the design of T cell therapies in the future.
CONCLUSION
T cell therapies have the potential to become a third pillar of biomedicine (148). Just as biologic drugs have become a central part of the therapeutic arsenal in the past few decades, T cell therapies are poised to become therapeutic options for a variety of medical conditions. As discussed above, allogeneic and autologous T cells have already been extensively used in the treatment of hematopoietic malignancies. With the rapid development of genetic engineering tools and synthetic biology approaches, the pace of development of T cell–based therapies is likely to increase. Developing T cell therapies poses unique challenges, including a highly regulated manufacturing process and limited data on long-term safety. A familiarity with the techniques and challenges used to develop T cell therapies is critical for scientists, pathologists, and clinicians who will be working in the development and deployment of these drugs.
SUMMARY POINTS.
T cell therapies derived from tumor-infiltrating T cells and genetically engineered autologous T cells have already shown impressive results in clinical trials for melanoma and hematopoietic malignancies.
Government regulation of engineered T cell therapies are evolving due to the complex manufacturing of T cell products and the difficulties in defining the pharmacokinetic and pharmacodynamic properties of the cells.
Engineered T cells have the potential to be powerful therapeutic devices beyond cancer, but synthetic biology tools to control their behavior require further development.
Allogeneic donor-derived therapeutic T cells would make T cell therapies more widely available, but there are significant immunological barriers.
More robust safety mechanisms for engineered T cells need to be developed if T cells are to be used routinely for patients with chronic diseases.
Pathologists and transfusion medicine–trained physicians will increasingly be called upon to manage T cell product manufacturing and to investigate the mechanisms of action of these new drugs.
Acknowledgments
We thank Florinna Dekovic and Giulia Giunti for helpful suggestions for the FDA/regulatory section of the manuscript. We thank Crystal Ghosh and Verna Huang for assistance in finding references.
Glossary
- Allogeneic
cells or tissue containing major histocompatibility complex antigens different from those of an intended transplant recipient
- Autologous
cells or tissues derived from a patient for the patient’s own use
- Human leukocyte antigens (HLAs)
highly polymorphic cell surface antigens (also called the major histocompatibility complexes) that serve as a major immunological barrier to cell and tissue transplantation
- Major histocompatibility complex (MHC) restriction
developmentally imposed selection of T cell receptors for reactivity with self-MHC molecules in the thymus
- Chimeric antigen receptor (CAR)
a synthetic protein combining the antigen recognition domain of an antibody with T cell and costimulatory receptor intracellular signaling domains
- Single-chain variable fragment (scFv)
protein derived from the antigen-binding domain of an antibody used to give antigen specificity to synthetic antigen receptors
- Regulatory T cell (Treg)
a subset of CD4+ T cells that have anti-inflammatory properties
- US Food and Drug Administration (FDA)
the federal agency that regulates cellular therapies via the Center for Biologics Evaluation and Research
- Human cells, tissues, and cellular and tissue-based products (HCT/Ps)
the designation used by the FDA for all engineered T cell products
- Clinical Laboratory Improvement Amendments (CLIA)
US rules that govern laboratory testing on human specimens for clinical purposes
- Zinc finger nucleases (ZFNs)
synthetic DNA-cutting proteins consisting of DNA sequence-specific zinc finger–binding domains linked to the FokI nuclease
- Transcription activator-like effector nucleases (TALENs)
synthetic DNA-cutting proteins consisting of modular sequence-specific DNA-binding domains derived from Xanthomonas bacteria linked to the FokI nuclease
- Cas9 nucleases
RNA-guided DNA nucleases derived from bacteria; part of the CRISPR system of adaptive immunity
- Graft-versus-host disease (GVHD)
a group of inflammatory pathologies caused by allogeneic T cells transfused into a patient
- AND gate
a logic gate in which two inputs must both be present to generate an output
- Synthetic notch (synNotch) receptor
a synthetic protein consisting of a truncated Notch receptor with an extracellular scFv and intracellular transcription factor
Footnotes
Identical twins and syngeneic mice also can provide functionally autologous (major histocompatibility complex antigen identical) cells and tissues.
DISCLOSURE STATEMENT
J.H.E. and J.A.B. are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review. W.A.L. is the founder of Cell Design Labs and is a member of its scientific advisory board.
LITERATURE CITED
- 1.Barrett DM, Grupp SA, June CH. Chimeric antigen receptor- and TCR-modified T cells enter Main Street and Wall Street. J Immunol. 2015;195:755–61. doi: 10.4049/jimmunol.1500751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Depil S, Deconinck E, Milpied N, Sutton L, Witz F, et al. Donor lymphocyte infusion to treat relapse after allogeneic bone marrow transplantation for myelodysplastic syndrome. Bone Marrow Transplant. 2004;33:531–34. doi: 10.1038/sj.bmt.1704381. [DOI] [PubMed] [Google Scholar]
- 3.Kolb HJ, Schmid C, Barrett AJ, Schendel DJ. Graft-versus-leukemia reactions in allogeneic chimeras. Blood. 2004;103:767–76. doi: 10.1182/blood-2003-02-0342. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319:1676–80. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 5.Feldman SA, Assadipour Y, Kriley I, Goff SL, Rosenberg SA. Adoptive cell therapy—tumor-infiltrating lymphocytes, T-cell receptors, and chimeric antigen receptors. Semin Oncol. 2015;42:626–39. doi: 10.1053/j.seminoncol.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yannelli JR, Hyatt C, McConnell S, Hines K, Jacknin L, et al. Growth of tumor-infiltrating lymphocytes from human solid cancers: summary of a 5-year experience. Int J Cancer. 1996;65:413–21. doi: 10.1002/(SICI)1097-0215(19960208)65:4<413::AID-IJC3>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–29. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21:914–21. doi: 10.1038/nm.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter DL, Hwang WT, Frey V, Lacey SF, Shaw PA, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aac5415. 303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–23. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. 2015;373:1040–47. doi: 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Srivastava S, Riddell SR. Engineering CAR-T cells: design concepts. Trends Immunol. 2015;36:494–502. doi: 10.1016/j.it.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–47. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Jiang S, Fang C, Yang S, Olalere D, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3:125–35. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–90. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 24.Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–90. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 26.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7:315ra189. doi: 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant. 2013;13:3010–20. doi: 10.1111/ajt.12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang Q, Bluestone JA. Regulatory T-cell therapy in transplantation: moving to the clinic. Cold Spring Harb Perspect Med. 2013;3:a015552. doi: 10.1101/cshperspect.a015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey AM, Arcidiacono J, Benton KA, Taraporewala Z, Winitsky S. United States Food and Drug Administration regulation of gene and cell therapies. Adv Exp Med Biol. 2015;871:1–29. doi: 10.1007/978-3-319-18618-4_1. [DOI] [PubMed] [Google Scholar]
- 30.Downing NS, Aminawung JA, Shah ND, Krumholz HM, Ross JS. Clinical trial evidence supporting FDA approval of novel therapeutic agents, 2005–2012. JAMA. 2014;311:368–77. doi: 10.1001/jama.2013.282034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harvath L. A brief history of US FDA regulation of human cells and tissues. In: Areman EM, Loper K, editors. Cellular Therapy: Principles, Methods, and Regulations. Bethesda, MD: AABB; 2009. pp. 2–12. [Google Scholar]
- 32.Kennett S, Porter C, Bloom E, Wonnacott K. The FDA perspective on the manufacturing, production, and processing of regulated cellular therapies. In: Areman EM, Loper K, editors. Cellular Therapy: Principles, Methods, and Regulations. Bethesda, MD; AABB: 2009. pp. 18–25. [Google Scholar]
- 33.US Dep. Health Hum. Serv., Food Drug Adm. Guidance for Industry: CGMP for Phase 1 Investigational Drugs. Rockville, MD: FDA; 2008. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070273.pdf. [Google Scholar]
- 34.Au P, Hursh DA, Lim A, Moos MC, Jr, Oh SS, et al. FDA oversight of cell therapy clinical trials. Sci Transl Med. 2012;4:149fs31. doi: 10.1126/scitranslmed.3004131. [DOI] [PubMed] [Google Scholar]
- 35.Chambers RW, Foley HT, Schmidt PJ. Transmission of syphilis by fresh blood components. Transfusion. 1969;9:32–34. doi: 10.1111/j.1537-2995.1969.tb04909.x. [DOI] [PubMed] [Google Scholar]
- 36.Orton S. Syphilis and blood donors: what we know, what we do not know, and what we need to know. Transfus Med Rev. 2001;15:282–91. doi: 10.1053/tmrv.2001.26956. [DOI] [PubMed] [Google Scholar]
- 37.Gee AP. Product release assays. Cytotherapy. 1999;1:485–91. doi: 10.1080/0032472031000141309. [DOI] [PubMed] [Google Scholar]
- 38.US Dep. Health Hum. Serv., Food Drug Adm. Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products. Rockville, MD: FDA; 2011. http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM243392.pdf. [Google Scholar]
- 39.US Dep. Health Hum. Serv., Cent. Medicare & Medicaid Serv. Clinical Laboratory Improvement Amendments of 1988 (CLIA)—CLIA Applicability for Laboratory Testing Associated with Blood, Cells/Tissue, and Organs. Baltimore, MD: CMS; 2011. https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/Downloads/SCLetter11_08.pdf. [Google Scholar]
- 40.Osborn MJ, Webber BR, Knipping F, Lonetree CL, Tennis N, et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol Ther. 2016;24:570–81. doi: 10.1038/mt.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaj T, Gersbach CA, Barbas CF., III ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–34. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 43.Osborn MJ, Tolar J. Cellular engineering and disease modeling with gene-editing nucleases. In: Cathomen T, Hirsch M, Porteus M, editors. Genome Editing: The Next Step in Gene Therapy. New York: Springer; 2016. pp. 223–58. [Google Scholar]
- 44.Yuan J, Wang J, Crain K, Fearns C, Kim KA, et al. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4+ T cell resistance and enrichment. Mol Ther. 2012;20:849–59. doi: 10.1038/mt.2011.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. PNAS. 2008;105:5809–14. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis L, Maizels N. DNA nicks promote efficient and safe targeted gene correction. PLOS ONE. 2011;6:e23981. doi: 10.1371/journal.pone.0023981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. PNAS. 2015;112:10437–42. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–15. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, DeClercq JJ, Hayward SB, Li PW, Shivak DA, et al. Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 2016;44:e30. doi: 10.1093/nar/gkv1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25:786–93. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 51.Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, et al. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther. 2008;16:352–58. doi: 10.1038/sj.mt.6300357. [DOI] [PubMed] [Google Scholar]
- 52.Ousterout DG, Gersbach CA. The development of TALE nucleases for biotechnology. Methods Mol Biol. 2016;1338:27–42. doi: 10.1007/978-1-4939-2932-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 54.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–12. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 55.Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63. doi: 10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valton J, Guyot V, Marechal A, Filhol JM, Juillerat A, et al. A multidrug-resistant engineered CAR T cell for allogeneic combination immunotherapy. Mol Ther. 2015;23:1507–18. doi: 10.1038/mt.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berdien B, Mock U, Atanackovic D, Fehse B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014;21:539–48. doi: 10.1038/gt.2014.26. [DOI] [PubMed] [Google Scholar]
- 58.Menger L, Gouble A, Marzolini MA, Pachnio A, Bergerhoff K, et al. TALEN-mediated genetic inactivation of the glucocorticoid receptor in cytomegalovirus-specific T cells. Blood. 2015;126:2781–89. doi: 10.1182/blood-2015-08-664755. [DOI] [PubMed] [Google Scholar]
- 59.Boissel S, Jarjour J, Astrakhan A, Adey A, Gouble A, et al. MegaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 2014;42:2591–601. doi: 10.1093/nar/gkt1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006;34:e149. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–26. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hou P, Chen S, Wang S, Yu X, Chen Y, et al. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci Rep. 2015;5:15577. doi: 10.1038/srep15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaminski R, Chen Y, Fischer T, Tedaldi E, Napoli A, et al. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci Rep. 2016;6:22555. doi: 10.1038/srep22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suerth JD, Schambach A, Baum C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr Opin Immunol. 2012;24:598–608. doi: 10.1016/j.coi.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 66.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Investig. 2008;118:3132–42. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geng X, Doitsh G, Yang Z, Galloway L, Greene WC. Efficient delivery of lentiviral vectors into resting human CD4 T cells. Gene Ther. 2014;21:444–49. doi: 10.1038/gt.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frecha C, Levy C, Costa C, Negre D, Amirache F, et al. Measles virus glycoprotein-pseudotyped lentiviral vector-mediated gene transfer into quiescent lymphocytes requires binding to both SLAM and CD46 entry receptors. J Virol. 2011;85:5975–85. doi: 10.1128/JVI.00324-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Modlich U, Navarro S, Zychlinski D, Maetzig T, Knoess S, et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol Ther. 2009;17:1919–28. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh H, Huls H, Kebriaei P, Cooper LJ. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev. 2014;257:181–90. doi: 10.1111/imr.12137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh H, Figliola MJ, Dawson MJ, Olivares S, Zhang L, et al. Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PLOS ONE. 2013;8:e64138. doi: 10.1371/journal.pone.0064138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Field AC, Vink C, Gabriel R, Al-Subki R, Schmidt M, et al. Comparison of lentiviral and Sleeping Beauty mediated αβ T cell receptor gene transfer. PLOS ONE. 2013;8:e68201. doi: 10.1371/journal.pone.0068201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Turchiano G, Latella MC, Gogol-Doring A, Cattoglio C, Mavilio F, et al. Genomic analysis of Sleeping Beauty transposon integration in human somatic cells. PLOS ONE. 2014;9:e112712. doi: 10.1371/journal.pone.0112712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Manuri PV, Wilson MH, Maiti SN, Mi T, Singh H, et al. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum Gene Ther. 2010;21:427–37. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Galvan DL, Nakazawa Y, Kaja A, Kettlun C, Cooper LJ, et al. Genome-wide mapping of PiggyBac transposon integrations in primary human T cells. J Immunother. 2009;32:837–44. doi: 10.1097/CJI.0b013e3181b2914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–20. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Holling TM, van der Stoep N, Quinten E, van den Elsen PJ. Activated human T cells accomplish MHC class II expression through T cell-specific occupation of class II transactivator promoter III. J Immunol. 2002;168:763–70. doi: 10.4049/jimmunol.168.2.763. [DOI] [PubMed] [Google Scholar]
- 78.Gragert L, Eapen M, Williams E, Freeman J, Spellman S, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. 2014;371:339–48. doi: 10.1056/NEJMsa1311707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heddle NM, Soutar RL, O’Hoski PL, Singer J, McBride JA, et al. A prospective study to determine the frequency and clinical significance of alloimmunization post-transfusion. Br J Haematol. 1995;91:1000–5. doi: 10.1111/j.1365-2141.1995.tb05425.x. [DOI] [PubMed] [Google Scholar]
- 80.Davis JL, Theoret MR, Zheng Z, Lamers CH, Rosenberg SA, Morgan RA. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res. 2010;16:5852–61. doi: 10.1158/1078-0432.CCR-10-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heeger PS. T-cell allorecognition and transplant rejection: a summary and update. Am J Transplant. 2003;3:525–33. doi: 10.1034/j.1600-6143.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- 83.Wagner FF, Flegel WA. Transfusion-associated graft-versus-host disease: risk due to homozygous HLA haplotypes. Transfusion. 1995;35:284–91. doi: 10.1046/j.1537-2995.1995.35495216075.x. [DOI] [PubMed] [Google Scholar]
- 84.Takahashi K, Juji T, Miyamoto M, Uchida S, Akaza T, et al. Analysis of risk factors for post-transfusion graft-versus-host disease in Japan. Japanese Red Cross PT-GVHD Study Group. Lancet. 1994;343:700–2. doi: 10.1016/s0140-6736(94)91580-6. [DOI] [PubMed] [Google Scholar]
- 85.Lee TH, Paglieroni T, Ohto H, Holland PV, Busch MP. Survival of donor leukocyte subpopulations in immunocompetent transfusion recipients: frequent long-term microchimerism in severe trauma patients. Blood. 1999;93:3127–39. [PubMed] [Google Scholar]
- 86.Reed W, Lee TH, Norris PJ, Utter GH, Busch MP. Transfusion-associated microchimerism: a new complication of blood transfusions in severely injured patients. Semin Hematol. 2007;44:24–31. doi: 10.1053/j.seminhematol.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 87.Utter GH, Owings JT, Lee TH, Paglieroni TG, Reed WF, et al. Blood transfusion is associated with donor leukocyte microchimerism in trauma patients. J Trauma. 2004;57:702–7. doi: 10.1097/01.ta.0000140666.15972.37. discussion 7–8. [DOI] [PubMed] [Google Scholar]
- 88.Lee TH, Paglieroni T, Utter GH, Chafets D, Gosselin RC, et al. High-level long-term white blood cell microchimerism after transfusion of leukoreduced blood components to patients resuscitated after severe traumatic injury. Transfusion. 2005;45:1280–90. doi: 10.1111/j.1537-2995.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 89.Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–64. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 90.Scarisbrick JJ, Dignan FL, Tulpule S, Gupta ED, Kolade S, et al. A multicentre UK study of GVHD following DLI: Rates of GVHD are high but mortality from GVHD is infrequent. Bone Marrow Transplant. 2015;50:62–67. doi: 10.1038/bmt.2014.227. [DOI] [PubMed] [Google Scholar]
- 91.Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–73. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gerdemann U, Katari UL, Papadopoulou A, Keirnan JM, Craddock JA, et al. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol Ther. 2013;21:2113–21. doi: 10.1038/mt.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Papadopoulou A, Gerdemann U, Katari UL, Tzannou I, Liu H, et al. Activity of broad-spectrum T cells as treatment for AdV, EBV, CMV, BKV, and HHV6 infections after HSCT. Sci Transl Med. 2014;6:242ra83. doi: 10.1126/scitranslmed.3008825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330:1185–91. doi: 10.1056/NEJM199404283301703. [DOI] [PubMed] [Google Scholar]
- 95.Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. 2007;110:1123–31. doi: 10.1182/blood-2006-12-063008. [DOI] [PubMed] [Google Scholar]
- 96.Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–23. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31:928–33. doi: 10.1038/nbt.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 99.Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–26. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 100.Torikai H, Reik A, Soldner F, Warren EH, Yuen C, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122:1341–49. doi: 10.1182/blood-2013-03-478255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Riteau B, Menier C, Khalil-Daher I, Martinozzi S, Pla M, et al. HLA-G1 co-expression boosts the HLA class I-mediated NK lysis inhibition. Int Immunol. 2001;13:193–201. doi: 10.1093/intimm/13.2.193. [DOI] [PubMed] [Google Scholar]
- 103.Rouas-Freiss N, Marchal RE, Kirszenbaum M, Dausset J, Carosella ED. The α1 domain of HLA-G1 and HLA-G2 inhibits cytotoxicity induced by natural killer cells: Is HLA-G the public ligand for natural killer cell inhibitory receptors? PNAS. 1997;94:5249–54. doi: 10.1073/pnas.94.10.5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qasim W, Amrolia PJ, Samarasinghe S, Zhan H, Stafford S, et al. First clinical application of Talen engineered universal CAR19 T cells in B-ALL. Blood. 2015;126:2046. [Google Scholar]
- 105.Way JC, Collins JJ, Keasling JD, Silver PA. Integrating biological redesign: where synthetic biology came from and where it needs to go. Cell. 2014;157:151–61. doi: 10.1016/j.cell.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 106.Kwok R. Five hard truths for synthetic biology. Nature. 2010;463:288–90. doi: 10.1038/463288a. [DOI] [PubMed] [Google Scholar]
- 107.Silver PA, Way JC, Arnold FH, Meyerowitz JT. Synthetic biology: engineering explored. Nature. 2014;509:166–67. doi: 10.1038/509166a. [DOI] [PubMed] [Google Scholar]
- 108.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–26. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32:1059–70. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 111.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016;164:780–91. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164:770–79. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, et al. Engineering GPCR signaling pathways with RASSLs. Nat Methods. 2008;5:673–78. doi: 10.1038/nmeth.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Park JS, Rhau B, Hermann A, McNally KA, Zhou C, et al. Synthetic control of mammalian-cell motility by engineering chemotaxis to an orthogonal bioinert chemical signal. PNAS. 2014;111:5896–901. doi: 10.1073/pnas.1402087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Townshend B, Kennedy AB, Xiang JS, Smolke CD. High-throughput cellular RNA device engineering. Nat Methods. 2015;12:989–94. doi: 10.1038/nmeth.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen YY, Jensen MC, Smolke CD. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. PNAS. 2010;107:8531–36. doi: 10.1073/pnas.1001721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. PNAS. 2016;113:E450–58. doi: 10.1073/pnas.1524193113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. PNAS. 2016;113:E459–68. doi: 10.1073/pnas.1524155113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, et al. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18:6436–45. doi: 10.1158/1078-0432.CCR-12-1449. [DOI] [PubMed] [Google Scholar]
- 122.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72:1844–52. doi: 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Weissinger EM, Borchers S, Silvani A, Provasi E, Radrizzani M, et al. Long term follow up of patients after allogeneic stem cell transplantation and transfusion of HSV-TK transduced T-cells. Front Pharmacol. 2015;6:76. doi: 10.3389/fphar.2015.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–51. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–71. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–12. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–26. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Davila ML, Riviere I, Wang X, Bartido S, Park J, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–20. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gross G, Eshhar Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol. 2016;56:59–83. doi: 10.1146/annurev-pharmtox-010814-124844. [DOI] [PubMed] [Google Scholar]
- 132.Tey SK, Dotti G, Rooney CM, Heslop HE, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2007;13:913–24. doi: 10.1016/j.bbmt.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–83. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Budde LE, Berger C, Lin Y, Wang J, Lin X, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLOS ONE. 2013;8:e82742. doi: 10.1371/journal.pone.0082742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhou X, Di Stasi A, Tey SK, Krance RA, Martinez C, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood. 2014;123:3895–905. doi: 10.1182/blood-2014-01-551671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Spencer DM, Belshaw PJ, Chen L, Ho SN, Randazzo F, et al. Functional analysis of Fas signaling in vivo using synthetic inducers of dimerization. Curr Biol. 1996;6:839–47. doi: 10.1016/s0960-9822(02)00607-3. [DOI] [PubMed] [Google Scholar]
- 137.Belshaw PJ, Spencer DM, Crabtree GR, Schreiber SL. Controlling programmed cell death with a cyclophilin-cyclosporin-based chemical inducer of dimerization. Chem Biol. 1996;3:731–38. doi: 10.1016/s1074-5521(96)90249-5. [DOI] [PubMed] [Google Scholar]
- 138.Thomis DC, Marktel S, Bonini C, Traversari C, Gilman M, et al. A Fas-based suicide switch in human T cells for the treatment of graft-versus-host disease. Blood. 2001;97:1249–57. doi: 10.1182/blood.v97.5.1249. [DOI] [PubMed] [Google Scholar]
- 139.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–24. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 140.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mercier-Letondal P, Deschamps M, Sauce D, Certoux JM, Milpied N, et al. Early immune response against retrovirally transduced herpes simplex virus thymidine kinase-expressing gene-modified T cells coinfused with a T cell-depleted marrow graft: an altered immune response? Hum Gene Ther. 2008;19:937–50. doi: 10.1089/hum.2007.156. [DOI] [PubMed] [Google Scholar]
- 142.Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 2014;124:1277–87. doi: 10.1182/blood-2014-01-545020. [DOI] [PubMed] [Google Scholar]
- 143.Griffioen M, van Egmond EH, Kester MG, Willemze R, Falkenburg JH, Heemskerk MH. Retro-viral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica. 2009;94:1316–20. doi: 10.3324/haematol.2008.001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kieback E, Charo J, Sommermeyer D, Blankenstein T, Uckert W. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. PNAS. 2008;105:623–28. doi: 10.1073/pnas.0710198105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–63. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–59. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science. 350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Sci Transl Med. 2013;5:179ps7. doi: 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]