

Abstract
S100B protein is elevated in the brains of patients with early stages of Alzheimer's disease and Down's syndrome. S100A4 is correlated with the development of metastasis. Both proteins bind to p53 tumor suppressor. We found that both S100B and S100A4 bind to the tetramerization domain of p53 (residues 325-355) only when exposed in lower oligomerization states and so they disrupt the tetramerization of p53. In addition, S100B binds to the negative regulatory and nuclear localization domains, which results in a very tight binding to p53 protein sequences that exposed the tetramerization domain in their C terminus. Because the trafficking of p53 depends on its oligomerization state, we suggest that S100B and S100A4 could regulate the subcellular localization of p53. But, the differences in the way these proteins bind to p53 could result in S100B and S1004 having different effects on p53 function in cell-cycle control.
Keywords: affinity, binding, fluorescence anisotropy, S100A4, S100B
The tumor suppressor p53 protein is a homotetrameric transcription factor that regulates several cellular processes. In response to a subset of stresses, p53 prevents tumorigenic transformation through the induction of cell cycle arrest or apoptosis. Its crucial role is reflected in that >50% of human cancers contain mutations in this gene (1-4). The protein is divided into four main functional domains: the N-terminal transactivation domain (residues 15-29), the core domain, which contains the specific DNA binding activity (residues 102-292), the tetramerization domain (residues 325-355), and the negative regulatory domain (residues 367-393) (Fig. 1a) (5).
Fig. 1.

Domain organization of p53 protein. (a) Schematic representation of human p53 protein and its functional domains: the transactivation domain (residues 15-29), core DNA binding domain (residues 102-292), tetramerization domain (residues 325-355), and negative regulatory domain (residues 367-393). (b) Detail of the amino acid sequence of the C-terminal region of p53 (residues 293-393). The peptides used in this article are delimited by boxes [NLS (residues 305-322), N-tet (residues 325-339), NES (residues 340-351), and NRD (367-393)] or underlined in the sequence [TET (residues 325-355)].
In an unstressed cell, p53 tumor suppressor is maintained at low levels. When the cell is challenged by stress, however, p53 is activated through posttranslational modifications that increase its stability. The regulation of protein stability is one of the most effective mechanisms of controlling p53 function. Key to this process is MDM2, an E3 ligase that targets p53 for ubiquitination (2, 6). As p53 works essentially as a transcription factor, nuclear transport and retention are also crucial regulators of p53 activity. p53 is a nucleocytoplasmic shuttling protein that contains both nuclear localization sequences (NLSs) and nuclear export sequences (NESs); the latter are recognized by the nuclear transport factor CRM-1 (6-10). Tetramerization has also been found to be relevant in p53 activation (11). p53 binds to its DNA response elements most efficiently as a tetramer, and tetrameric p53 is most effective for transactivation (12-17). These three levels of control of p53 activity (stability, cellular localization, and tetramerization) are tightly cross-regulated. For instance, tetrameric p53 is less efficient at entering the nucleus than monomeric p53 (10), p53 tetramerization occludes a NES signal, thereby ensuring nuclear retention of the DNA-binding form of p53 (9), and nuclear import and export are essential for MDM2-mediated p53 degradation (18).
The S100 protein family is a highly conserved group of EF-hand calcium-binding proteins with molecular masses ranging from 10 to 12 kDa. More than 20 S100 proteins with seemingly distinct functions and tissue distributions have been identified to date (19). Members of this family form either homodimeric or heterodimeric complexes with one another (19-22). Although S100 genes are structurally and evolutionary related, the intracellular location and expression pattern of each individual S100 protein is distinct (23-25). Additionally, each member is under complex transcriptional regulation (26, 27). Overexpression of S100B in the brain of individuals with Down's syndrome and in the brain of patients with Alzheimer's disease and AIDS has led to the hypothesis that S100B plays a role in common neuro-pathologies associated with these diseases (28-31). On the other hand, interest has been focused on S100A4 because its expression is strongly associated with the stimulation of metastasis (32-36). However, the exact mechanism by which S100A4 takes part in the formation of metastasis is poorly understood.
S100B and S100A4 interact with p53. S100B binds to p53 in a calcium-dependent manner, found from affinity chromatography experiments, and protects p53 from aggregation (37). A peptide corresponding to the negative regulatory domain (residues 367-388 of p53) binds S100B, as based on the NMR chemical shifts in S100B (38). Plasmon resonance shows that S100B binds to a p53 fragment that comprises residues 319-393. Competition experiments reveal that the tetramerization domain (residues 327-352 of human p53) binds to S100B more tightly than does the negative regulatory domain (39). S100A4 was shown to bind p53 by using coimmunoprecipitation and far Western blot experiments (40) and plasmon resonance (41). The last 30 residues of p53 (residues 364-393 in human p53) are important for binding, as the level of GST pull-down with GST-p53Δ30 was much less efficient than with full-length p53 (40).
There is some controversy about the effect of S100B on p53 function. Although some reports support the notion that S100B contributes to nuclear accumulation and activation of p53 function (42, 43), others suggest S100B overexpression decreases p53 transcriptional activity (44). Similarly, S100A4 overexpression seems to interfere with p53 activity (40).
In this article, we compare the binding of human S100B and S100A4 with the C-terminal region of p53 (p53CT, residues 293-393) by fluorescence anisotropy and analytical ultracentrifugation (AUC). This region contains several functional domains: the NLS, the tetramerization domain, the NES (contained within the tetramerization domain), and the negative regulatory domain. Our results showed that S100B and S100A4 bind differently to peptides comprising these domains. Moreover, S100B binds 40 times more tightly than S100A4 to p53CT. Interestingly, the binding affinities for p53 constructs containing the tetramerization domain depend on p53 oligomerization status. S100 proteins bind preferentially to small oligomeric forms of p53. These results suggest that S100 proteins might play a role in the control of p53 function by influencing its oligomerization state, however, to a characteristic degree depending on which S100 protein is involved.
Materials and Methods
Plasmid Construction and Protein Purification. Plasmids pRSET-untagS100B, pRHislipoTEV-hS100A, and pRHislipoTEV-p53CT were constructed to express human S100B, human S100A4, and human p53 C-terminal domain (residues 293-393), respectively. Plasmid construction, protein expression, and protein purification are described in detail in Supporting Text, which is published as supporting information on the PNAS web site.
Fluorescein Labeling of Proteins. Proteins were labeled by using 5′ (and 6′) carboxyfluorescein succinimidyl ester (Molecular Probes) in potassium phosphate buffer, pH 6.4 and 30 mM NaCl. At this pH the acylation of α-amino groups from primary amines is favored over that of ε-groups (45). An approximate ratio of 1:1 fluorescein ester/protein was used. After incubation at room temperature for 1 h with stirring, the sample was applied to an analytical GF column Tricorn Superdex 75 10/300 GL (Amersham Pharmacia) to separate the protein from the nonincorporated fluorescein ester. The fluorescein-labeled protein was separated from the unlabeled one by reverse-phase HPLC (Waters 600 equipped with a 996 PDA detector) on an analytical reverse-phase C8 column (Vydac, Hesperia, CA) with a gradient from 25% to 35% buffer B [buffer A, 0.1% (vol/vol) trifluoroacetic acid (TFA) in water and buffer B, 5% (vol/vol) water, 0.1% (vol/vol) TFA in acetonitrile].
Peptide Synthesis. Peptides were synthesized and purified as described (46).
Fluorescence Anisotropy. Measurements were performed at 25°C by using a PerkinElmer LS-50b luminescence spectrofluorimeter equipped with a Hamilton microlab M dispenser. Reactions were carried out in buffer PIS (physiological ionic strength, 25 mM Tris, pH 7.5/10 mM CaCl2/99.2 mM NaCl/1 mM DTT) or PIS-Ca buffer (25 mM Tris, pH 7.5/129.2 mM NaCl/0.1 mM EDTA/1 mM DTT). Fluorescence anisotropy was measured with excitation at 480 nm and emission at 525 nm. The bandwidths were changed depending on the amount of the labeled molecule used. The labeled peptide or protein was placed in the cuvette in a volume of 1 ml, and the binding protein was placed in the dispenser (240 or 300 μl). Additions of 6 μl were titrated at 1-min intervals, the solution was stirred for 30 s, and the anisotropy was measured. Dissociation constants (Kd) were calculated by fitting the anisotropy and fluorescence titration curves (corrected for the dilution effect) by using kaleida-graph (Synergy Software, Reading, PA). The following equation was used for the single-site model (one titrant molecule binding to one labeled molecule):
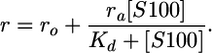 |
To fit data to a single-site plus linear drift model the following equation was used:
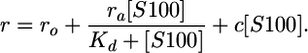 |
Finally, the following equation was used to fit data to a two-site model (two molecules of titrant binding to one labeled molecule):
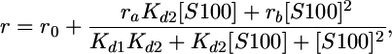 |
where, ro is the initial anisotropy, ra is the difference in anisotropy between unbound and bound to one titrant molecule, rb is the difference in anisotropy between unbound and bound to two titrant molecules, Kd is the dissociation constant (Kd1 is the tighter and Kd2 the weaker), [S100] is the concentration of the S100 protein in molar (M) units and always in terms of concentration of dimer, and c is the constant of the linear drift.
AUC. The equilibrium sedimentation experiments were performed on a Beckman Optima XL-I ultracentrifuge by using a Ti-60 rotor and six-sector cells at speeds of 30,000 and 40,000 rpm and at 25°C in a volume of 50 μl. Samples were considered to be at equilibrium as judged by comparing several scans at each speed. Runs were carried out in physiological ionic strength buffer. Fluorescein-labeled peptides were used at concentrations of 4 or 6 μM, and S100 proteins were used at concentrations ranging from 50 to 200 μM. Absorbance at 495 nm and interference data were recorded on reaching equilibrium (typically after 6-12 h). The absorbance data were used to calculate the concentrations of free peptide and peptide-protein complex, and interference data provided total concentration of the protein at the reference position. The dissociation constant was calculated as Kd = ([free peptide]/[complex])·[free protein]. Data were processed and analyzed with ultraspin software (www.mrc-cpe.cam.ac.uk).
Results
S100B and S100A4 Binding to Peptides Derived from the C-Terminal Region of p53 by Fluorescence Anisotropy. We synthesized a set of fluorescein-labeled peptides derived from p53 to compare S100B and S100A4 binding with the C-terminal region of p53 (Fig. 1b). The peptides used were: NLS (residues 305-322 of human p53), NES (residues 340-351), tetramerization domain peptide (TET) (residues 325-355), and negative regulatory domain peptide (NRD) (residues 367-393). As NES is the C-terminal part of the tetramerization domain, we also synthesized a peptide that represents only the N-terminal part (N-tet, residues 325-339). Fluorescence anisotropy experiments were carried out at physiological ionic strength.
S100B bound to the NLS, N-tet, NES, TET, and NRD peptides, although binding of S100B to NLS was too weak to be quantified. For all other combinations, the Kd of binding was calculated by using the mean of two independent experiments (Table 1). Binding experiments carried out in a buffer without calcium confirmed that binding is calcium-dependent (data not shown).
Table 1. Binding affinity of S100B and S100A4 for p53CT and p53CT-derived peptides by using fluorescence anisotropy.
|
Kd, μM
|
||
|---|---|---|
| Peptide (residues) | S100B | S100A4 |
| NLS (305-322) | n.q. | n.d. |
| N-tet (325-339) | 172 ± 4 | 150 ± 4 |
| NES (340-351) | 302 ± 7 | 135 ± 3 |
| TET (325-355) | 112 ± 7 | 17 ± 1 |
| NRD (367-393) | 102 ± 3 | n.q. |
| p53CT (293-393) | 0.25 ± 0.05 | 10 ± 2* |
| 16 ± 2 | 50 ± 1 | |
Experiments were carried out by using peptide at a concentration of 0.5 μM, with the exception of TET and p53CT, which were used at 0.1 μM. Values were obtained by fitting the binding data to a single-site model, with the exception of TET data, which fit to a single-site model plus a linear drift, and p53CT data, which fit to a two-site model. n.q., not quantifiable although detectable; n.d., not detectable.
Kd values for S100A4 binding to p53CT were obtained by fitting the change in total fluorescence.
A similar set of experiments was carried out with S100A4 protein. S100A4 clearly binds to the N-tet, NES, and TET peptides. No binding of S100A4 to NLS was detected, and although weak binding of S100A4 to NRD was detected, it was not possible to quantify it. The Kd for S100A4 binding to N-tet, NES, and TET was calculated as for S100B (Table 1).
These results show that S100B and S100A4 have different patterns of binding to peptides derived from the C-terminal region of p53. Apart from the obvious differences in binding to NLS and NRD peptides, there is an interesting quantitative difference in the way these two proteins bind to the TET and peptides derived from it. The binding affinities to N-tet are very similar. However, S100A4 binds more than two times tighter than S100B to the NES peptide and seven times tighter to the TET (Table 1).
AUC Analysis of S100B and S100A4 Binding to p53-Derived Peptides. To confirm the results obtained by fluorescence anisotropy and to attempt to measure the Kd of the weak complexes, we carried out AUC experiments. This type of AUC experiment is based on the different sedimentation behavior of small peptides compared with peptide-protein complexes. The TET is tetrameric at the concentrations used for these AUC experiments (4-6 μM) (47), which precludes its use. As a control, we used a monomeric mutant of TET with a leucine-to-proline mutation in residue 344 of p53, TET-L344P (48).
Results obtained by AUC are shown in Table 2. Kds obtained for binding to N-tet and NES peptides and for S100B binding to NRD are in very good agreement with those determined by fluorescence anisotropy. Additionally, we were able to quantify the binding affinity of S100B to NLS peptide. The Kd obtained is 570 μM. This finding confirms that S100B binds to NLS but not as tightly as to the other peptides of the C-terminal region of p53. A potential complex between S100A4 and NLS was possibly detected, although the signal was very close to the detection limit (Kd ≈1, 100 μM) and may simply represent background. We were also able to measure the binding affinity of S100A4 to NRD. S100A4 binds eight times more weakly than S100B to the NRD, with a Kd of 700 μM. The values obtained for TET-L344P were in good agreement with the results obtained for this peptide in fluorescence anisotropy experiments (see below).
Table 2. S100B and S100A4 binding to p53-derived peptides studied by AUC.
| S100B
|
S100A4
|
|||
|---|---|---|---|---|
| Peptide (residues) | Kd, μM | n | Kd, μM | n |
| NLS (305-322) | 570 ± 50 | 9 | 1,140 ± 130 | 4 |
| N-tet (325-339) | 180 ± 20 | 5 | 130 ± 20 | 4 |
| NES (340-351) | 260 ± 20 | 5 | 85 ± 7 | 5 |
| TET-L344P (325-355) | 77 ± 11 | 6 | 44 ± 7 | 5 |
| NRD (367-393) | 82 ± 6 | 5 | 700 ± 50 | 6 |
Kds were calculated as the mean of the total experiments carried out with that peptide-protein combination, and the standard error of the mean was calculated by using prism software (GraphPad, San Diego). n is the number of experiments for each combination.
Binding of S100B and S100A4 to the C-Terminal Region of p53. We then compared the binding of S100B and S100A4 to a recombinant protein consisting of the C-terminal region of p53 (p53CT, residues 293-393) by fluorescence anisotropy. All of the peptides studied above are contained within p53CT. S100B and S100A4 were titrated into fluorescein-labeled p53CT. Binding was detected in both cases but we were able to detect a full binding curve only for S100B, which indicates that S100B binds more tightly than S100A4 to p53CT (Fig. 2a). Data for S100B fit to a two-site model where two dimers of S100B bind sequentially to one molecule of p53CT. Kds for binding were obtained as the mean of two independent experiments (Table 1).
Fig. 2.

Binding of S100B and S100A4 to p53CT (residues 293-393 of p53). (a) Comparison of the binding of S100B and S100A4 to 0.1 μM fluorescein-labeled p53CT by using fluorescence anisotropy. S100B binding curve fits to a two-site model (continuous line). Concentration of dimeric protein is in molar units (M). (b) Binding of S100A4 to fluorescein-labeled p53CT analyzed by fluorescence. The continuous line shows the fit to a two-site model. Concentration of dimeric S100A4 is in molar units (M).
At the high concentrations of S100A4, which would be required to observe a full binding curve, S100A4 induces a large change in total fluorescence. Such large changes in fluorescence affect anisotropy measurements and, under these conditions, anisotropy cannot be used to quantify binding. In these cases, the change in total fluorescence can be used instead to obtain the Kd of binding. The fluorescence data generated upon S100A4 binding fit to a two-site model (Fig. 2b). Kds for binding were obtained as the mean of two independent experiments (Table 1). S100B binds 40 times more tightly than S100A4 to p53CT. The binding affinity of p53 to S100A4 had been measured previously with an optical biosensor (41) and determined to be much tighter than we determined for S100A4 binding to p53CT. This difference is unlikely to be caused by binding sites for S100A4 in p53 regions other than p53CT (40). Immobilized S100A4 might not be properly folded (41), somehow exposing novel binding sites for p53 that are not accessible in solution.
Effects of p53 Oligomerization on Binding of S100B. The concentration of the TET had a profound effect on binding of S100B and S100A4. In the fluorescence anisotropy experiments described above, Kds were measured by adding S100 proteins to 0.1 μM of the TET (Table 1). At 0.5 μM peptide, we were not able to increase the concentration of S100 protein to obtain a full binding curve, especially with S100B. The dissociation constant of the tetramerization domain is ≈2 μM (47). Accordingly, the range of concentrations of the TET used for the fluorescence anisotropy experiments is such that we can alter significantly its state of oligomerization.
To check the effects of oligomerization, we repeated the experiments by using TET-L344P, which is a monomeric mutant (48); TET-L344A and TET-L348A, both of which are dimeric (49); and WT TET. Fluorescence anisotropy experiments were done with S100B and fluorescein-labeled TET-L344P, TET-L344A, TET-L348A, and TET. The TET and TET mutants were used at four different concentrations (0.1, 0.5, 2, and 5 μM). Interestingly, binding of S100B to monomeric TET-L344P is not affected by the concentration of peptide used in the experiment, and approximately the same binding curves are obtained in each case (Fig. 3a). Data fit to a single-site model and the Kd values were obtained as the mean of two independent experiments (Table 3).
Fig. 3.

Effect of p53 oligomerization on S100B binding. Binding of S100B to different concentrations of fluorescein-labeled TET-L344P (a), TET-L344A (b), TET-L348A (c), and TET (d) peptides by using fluorescence anisotropy is shown. Concentration of S100B dimer is expressed in molar units (M).
Table 3. Dissociation constants of TET and TET mutants from S100B.
|
Kd, μM
|
||||
|---|---|---|---|---|
| Peptide concentration, μM | TET-L344P | TET-L344A | TET-L348A | TET |
| 0.1 | 50 ± 1.4 | 49 ± 1.2 | 40 ± 0.9 | 103 ± 5.5 |
| 0.5 | 51 ± 1.5 | 55 ± 1.4 | 49 ± 1.1 | >400 |
| 2 | 56 ± 1.8 | 57 ± 1.7 | 59 ± 1.4 | >400 |
| 5 | 58 ± 1.5 | 94 ± 3.6 | 94 ± 2.9 | >400 |
Data are the means of two independent experiments.
TET-L344A and TET-L348A behaved similarly (Fig. 3 b and c). The binding curves are similar, in both cases, for the lower peptide concentrations (0.1, 0.5, and 2 μM) but the curves obtained for the highest concentration of peptide (5 μM) clearly indicate weaker binding. This finding is probably representative of the presence of a certain population of tetrameric peptide at that concentration, as these mutants are capable of tetramerization at very high concentrations (unpublished data). The magnitude of the Kd confirmed the qualitative result (Table 3).
For TET the effect of oligomerization in binding to S100B was clearly evident. Only when the lowest concentration of peptide was used was a clear binding curve obtained (Fig. 3d). At higher peptide concentrations binding was detected but was in the linear range of binding and a full-binding curve was not obtained. Clearly the higher the TET concentration, the lower the affinity, resulting in almost undetectable binding at 5 μM peptide. The effects of the tetramerization of p53 on the binding affinity to S100B were also evident when fluorescein-labeled p53CT was used (data not shown).
Apart from the moderate effect that TET mutations have on the binding of S100B, these results show that S100B binds to the monomers/dimers of the TET and that tetramerization prevents S100B binding. We cannot distinguish whether S100B binds to both monomers and dimers of p53 or only monomers.
The effect of p53 oligomerization on the binding of S100B could also explain why when using plasmon resonance Delphin et al. (39) found binding to p53 C-terminal region to be 10 times tighter than we determined in solution by using fluorescence anisotropy. Immobilization of proteins in BIAcore experiments can affect their conformation, and so immobilization of p53 might impair its oligomerization and, therefore, result in a tighter binding to S100B.
Effects of p53 Oligomerization on Binding of S100A4. A similar set of experiments was carried out with S100A4, TET-L344P, and TET. As with S100B, binding of S100A4 to monomeric TET-L344P was not affected by the concentration of peptide used in the experiment (Fig. 4a and Table 4). However, there was an evident effect of concentration on the binding affinity of S100A4 for TET (Fig. 4b and Table 4). The higher the concentration of peptide, the weaker was the binding to S100A4. These results show that S100A4, as S100B, binds to the lower oligomerization states of the TET and tetramerization prevents S100A4 binding.
Fig. 4.

Effect of p53 oligomerization on S100A4 binding. Binding of S100A4 to different concentrations of fluorescein-labeled TET-L344P (a) and TET (b) by using fluorescence anisotropy is shown. Concentration of S100A4 dimer is expressed in molar units (M).
Table 4. Dissociation constants of TET and TET-L344P from S100A4.
|
Kd, μM
|
||
|---|---|---|
| Peptide concentration, μM | TET-L344P | TET |
| 0.1 | 36 ± 1.8 | 17 ± 0.7 |
| 0.5 | 32 ± 1.5 | >100 |
| 2 | 29 ± 0.9 | >100 |
| 5 | 39 ± 1.3 | >100 |
Data are the means of two independent experiments.
Discussion
Binding of S100B and S100A4 to p53: Differences and Similarities. p53 has a key role in the control of the cell cycle under different stress situations. Given its important role, p53 is subjected to tight regulation, and protein-protein interactions are thought to be relevant in the control of p53 function (1, 5). S100B and S100A4, proteins of the S100 calcium binding family, interact with p53 (37, 40, 41). The interactions of p53 with S100 family members are particularly intriguing because they link p53 biology to calcium-mediated signal transduction pathways (50).
In this article, we show that both S100B and S100A4 bind preferentially to the tetramerization domain of p53 when exposed in lower oligomerization states and accordingly disrupt the tetramerization of p53. Our results may explain the apparent controversy about the effect of S100B overexpression on p53 activity. Although S100B has a cooperative effect in the activation of p53 activity (42, 43), very high levels of S100B could prevent p53 tetramerization and consequently inhibit its activity (44).
Despite the high conservation among S100 proteins, we found significant differences in the binding of S100B and S100A4 to p53. Whereas S100B binds to different peptides within the p53 C-terminal region, in the same affinity range, S100A4 binds well only to TET and peptides contained within it. This result is in contrast with those of Grigorian et al. (40) who determined, based on immunoprecipitation and far Western blot analysis, that the deletion of the last 30 residues of p53 significantly reduces the binding of S100A4. We found that S100B binds 40 times more tightly than S100A4 to p53CT. This finding reflects the synergistic effect of the multiple binding sites that this p53 region has for S100B.
Relevance of S100-p53 Interactions for p53 Subcellular Localization and Function. p53 is a transcription factor, which means it has to accumulate in the nucleus. Although p53 is able to enter the nucleus as a tetramer there are indications that the p53 monomer can enter the nucleus more efficiently (10). Several studies have suggested that other proteins may be involved in the regulation of p53 subcellular localization (10). S100B enhances the nuclear accumulation of p53 (43). In this article we have shown that S100B and S100A4 disrupt the tetramerization equilibrium of p53. The presence of S100 proteins could increase the cytoplasmic population of p53 monomers and consequently the efficiency of p53 nuclear transportation. It would be interesting to assess whether other proteins of the S100 family share this feature.
A very interesting level of control seems to have evolved to ensure the sequestration of active p53 within the nucleus. Stommel et al. (9) proposed a model in which regulated p53 tetramerization occludes its NES. Binding of S100B and S100A4 could also mask NES and help p53 to stay in the nucleus. However, S100B and S100A4 could diverge in the way they affect p53 nuclear accumulation. CRM-1 export factor binds to its target sequences with a Kd of ≈0.5 μM (51, 52). As S100B binds tighter to p53CT than S100A4, CRM-1 could more easily compete off S100A4 from S100-p53 complexes.
Although nuclear import is essential for p53 activation, it is also needed for its degradation. Nuclear import and nuclear export have been shown to be essential for MDM2-mediated degradation of p53, and modifications in the C terminus, in addition to ubiquitination, are acquired in the nucleus and are crucial for cytoplasmic degradation of p53 (18). An increased shuttling of p53 into the nucleus could also be a way to promote its degradation. S100B binding to the C-terminal negative regulatory domain could protect p53 from acquiring the modifications that target it for degradation. S100A4 is more unlikely to play this role as we showed it binds very weakly to this domain of p53. It is tempting to speculate that high levels of S100A4 could promote p53 import to the nucleus and consequently increase the targeting of p53 for degradation and that this could be the basis of the causative effect of S100A4 in metastasis.
We have shown that S100B and S100A4 have a common role in the regulation of p53 tetramerization. However, the differences we found between the binding of S100B and S100A4 to p53 could represent a divergence in the way these two S100 proteins have evolved to assist p53 in the control of the cell cycle.
Supplementary Material
Acknowledgments
We thank Caroline Blair for TEV protease purification, Dr. Assaf Friedler and Karoly Von Gloss for peptide synthesis, and Dr. Roger Barraclough and Prof. Philip S. Rudland for helpful discussions. M.R.F.-F. was supported by a European Molecular Biology Organization Long-Term Fellowship. This work was supported by the Medical Research Centre and Cancer Research UK.
Author contributions: M.R.F.-F. and A.R.F. designed research; M.R.F.-F. performed research; D.B.V. contributed new reagents/analytic tools; M.R.F.-F., D.B.V., and A.R.F. analyzed data; and M.R.F.-F. wrote the paper.
Abbreviations: NLS, nuclear localization sequence; NES, nuclear export sequence; TET, tetramerization domain peptide; NRD, negative regulatory domain peptide; AUC, analytical ultracentrifugation.
References
- 1.Levine, A. J. (1997) Cell 88, 323-331. [DOI] [PubMed] [Google Scholar]
- 2.Vousden, K. H. (2000) Cell 103, 691-694. [DOI] [PubMed] [Google Scholar]
- 3.Vousden, K. H. (2002) Biochim. Biophys. Acta 1602, 47-59. [DOI] [PubMed] [Google Scholar]
- 4.Vousden, K. H. & Lu, X. (2002) Nat. Rev. Cancer 2, 594-604. [DOI] [PubMed] [Google Scholar]
- 5.Ko, L. J. & Prives, C. (1996) Genes Dev. 10, 1054-1072. [DOI] [PubMed] [Google Scholar]
- 6.Vousden, K. H. & Woude, G. F. (2000) Nat. Cell Biol. 2, E178-E180. [DOI] [PubMed] [Google Scholar]
- 7.Zhang, Y. & Xiong, Y. (2001) Science 292, 1910-1915. [DOI] [PubMed] [Google Scholar]
- 8.Shaulsky, G., Goldfinger, N., Ben-Ze'ev, A. & Rotter, V. (1990) Mol. Cell. Biol. 10, 6565-6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stommel, J. M., Marchenko, N. D., Jimenez, G. S., Moll, U. M., Hope, T. J. & Wahl, G. M. (1999) EMBO J. 18, 1660-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang, S. H. & Clarke, M. F. (2001) Eur. J. Biochem. 268, 2779-2783. [DOI] [PubMed] [Google Scholar]
- 11.Chene, P. (2001) Oncogene 20, 2611-2617. [DOI] [PubMed] [Google Scholar]
- 12.Halazonetis, T. D. & Kandil, A. N. (1993) EMBO J. 12, 5057-5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hainaut, P., Hall, A. & Milner, J. (1994) Oncogene 9, 299-303. [PubMed] [Google Scholar]
- 14.Pietenpol, J. A., Tokino, T., Thiagalingam, S., el-Deiry, W. S., Kinzler, K. W. & Vogelstein, B. (1994) Proc. Natl. Acad. Sci. USA 91, 1998-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarunina, M., Grimaldi, M., Ruaro, E., Pavlenko, M., Schneider, C. & Jenkins, J. R. (1996) Oncogene 13, 589-598. [PubMed] [Google Scholar]
- 16.McLure, K. G. & Lee, P. W. (1998) EMBO J. 17, 3342-3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinberg, R. L., Veprintsev, D. B. & Fersht, A. R. (2004) J. Mol. Biol. 341, 1145-1159. [DOI] [PubMed] [Google Scholar]
- 18.O'Keefe, K., Li, H. & Zhang, Y. (2003) Mol. Cell. Biol. 23, 6396-6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schafer, B. W. & Heizmann, C. W. (1996) Trends Biochem. Sci. 21, 134-140. [DOI] [PubMed] [Google Scholar]
- 20.Donato, R. (1991) Cell Calcium 12, 713-726. [DOI] [PubMed] [Google Scholar]
- 21.Donato, R. (1999) Biochim. Biophys. Acta 1450, 191-231. [DOI] [PubMed] [Google Scholar]
- 22.Zimmer, D. B., Cornwall, E. H., Landar, A. & Song, W. (1995) Brain Res. Bull 37, 417-429. [DOI] [PubMed] [Google Scholar]
- 23.El Naaman, C., Grum-Schwensen, B., Mansouri, A., Grigorian, M., Santoni-Rugiu, E., Hansen, T., Kriajevska, M., Schafer, B. W., Heizmann, C. W., Lukanidin, E. & Ambartsumian, N. (2004) Oncogene 23, 3670-3680. [DOI] [PubMed] [Google Scholar]
- 24.Mandinova, A., Atar, D., Schafer, B. W., Spiess, M., Aebi, U. & Heizmann, C. W. (1998) J. Cell Sci. 111, 2043-2054. [DOI] [PubMed] [Google Scholar]
- 25.Hsieh, H. L., Schafer, B. W., Cox, J. A. & Heizmann, C. W. (2002) J. Cell Sci. 115, 3149-3158. [DOI] [PubMed] [Google Scholar]
- 26.Cohn, M. A., Hjelmso, I., Wu, L. C., Guldberg, P., Lukanidin, E. M. & Tulchinsky, E. M. (2001) Nucleic Acids Res. 29, 3335-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen, D. S. & Barraclough, R. (1996) Biochem. Soc. Trans. 24, 352S. [DOI] [PubMed] [Google Scholar]
- 28.Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., White, C. L., 3rd & Araoz, C. (1989) Proc. Natl. Acad. Sci. USA 86, 7611-7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marshak, D. R., Pesce, S. A., Stanley, L. C. & Griffin, W. S. (1992) Neurobiol. Aging 13, 1-7. [DOI] [PubMed] [Google Scholar]
- 30.Sheng, J. G., Mrak, R. E. & Griffin, W. S. (1994) J. Neurosci. Res. 39, 398-404. [DOI] [PubMed] [Google Scholar]
- 31.Stanley, L. C., Mrak, R. E., Woody, R. C., Perrot, L. J., Zhang, S., Marshak, D. R., Nelson, S. J. & Griffin, W. S. (1994) J. Neuropathol. Exp. Neurol. 53, 231-238. [DOI] [PubMed] [Google Scholar]
- 32.Ebralidze, A., Tulchinsky, E., Grigorian, M., Afanasyeva, A., Senin, V., Revazova, E. & Lukanidin, E. (1989) Genes Dev. 3, 1086-1093. [DOI] [PubMed] [Google Scholar]
- 33.Lloyd, B. H., Platt-Higgins, A., Rudland, P. S. & Barraclough, R. (1998) Oncogene 17, 465-473. [DOI] [PubMed] [Google Scholar]
- 34.Davies, M. P., Rudland, P. S., Robertson, L., Parry, E. W., Jolicoeur, P. & Barraclough, R. (1996) Oncogene 13, 1631-1637. [PubMed] [Google Scholar]
- 35.Davies, B. R., Davies, M. P., Gibbs, F. E., Barraclough, R. & Rudland, P. S. (1993) Oncogene 8, 999-1008. [PubMed] [Google Scholar]
- 36.Ambartsumian, N. S., Grigorian, M. S., Larsen, I. F., Karlstrom, O., Sidenius, N., Rygaard, J., Georgiev, G. & Lukanidin, E. (1996) Oncogene 13, 1621-1630. [PubMed] [Google Scholar]
- 37.Baudier, J., Delphin, C., Grunwald, D., Khochbin, S. & Lawrence, J. J. (1992) Proc. Natl. Acad. Sci. USA 89, 11627-11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rustandi, R. R., Drohat, A. C., Baldisseri, D. M., Wilder, P. T. & Weber, D. J. (1998) Biochemistry 37, 1951-1960. [DOI] [PubMed] [Google Scholar]
- 39.Delphin, C., Ronjat, M., Deloulme, J. C., Garin, G., Debussche, L., Higashimoto, Y., Sakaguchi, K. & Baudier, J. (1999) J. Biol. Chem. 274, 10539-10544. [DOI] [PubMed] [Google Scholar]
- 40.Grigorian, M., Andresen, S., Tulchinsky, E., Kriajevska, M., Carlberg, C., Kruse, C., Cohn, M., Ambartsumian, N., Christensen, A., Selivanova, G. & Lukanidin, E. (2001) J. Biol. Chem. 276, 22699-22708. [DOI] [PubMed] [Google Scholar]
- 41.Chen, H., Fernig, D. G., Rudland, P. S., Sparks, A., Wilkinson, M. C. & Barraclough, R. (2001) Biochem. Biophys. Res. Commun. 286, 1212-1217. [DOI] [PubMed] [Google Scholar]
- 42.Scotto, C., Deloulme, J. C., Rousseau, D., Chambaz, E. & Baudier, J. (1998) Mol. Cell. Biol. 18, 4272-4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scotto, C., Delphin, C., Deloulme, J. C. & Baudier, J. (1999) Mol. Cell. Biol. 19, 7168-7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin, J., Blake, M., Tang, C., Zimmer, D., Rustandi, R. R., Weber, D. J. & Carrier, F. (2001) J. Biol. Chem. 276, 35037-35041. [DOI] [PubMed] [Google Scholar]
- 45.Gaudriault, G. & Vincent, J. P. (1992) Peptides 13, 1187-1192. [DOI] [PubMed] [Google Scholar]
- 46.Friedler, A., Hansson, L. O., Veprintsev, D. B., Freund, S. M., Rippin, T. M., Nikolova, P. V., Proctor, M. R., Rudiger, S. & Fersht, A. R. (2002) Proc. Natl. Acad. Sci. USA 99, 937-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakamoto, H., Lewis, M. S., Kodama, H., Appella, E. & Sakaguchi, K. (1994) Proc. Natl. Acad. Sci. USA 91, 8974-8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lomax, M. E., Barnes, D. M., Hupp, T. R., Picksley, S. M. & Camplejohn, R. S. (1998) Oncogene 17, 643-649. [DOI] [PubMed] [Google Scholar]
- 49.Mateu, M. G. & Fersht, A. R. (1998) EMBO J. 17, 2748-2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikura, M. & Yap, K. L. (2000) Nat. Struct. Biol. 7, 525-527. [DOI] [PubMed] [Google Scholar]
- 51.Askjaer, P., Bachi, A., Wilm, M., Bischoff, F. R., Weeks, D. L., Ogniewski, V., Ohno, M., Niehrs, C., Kjems, J., Mattaj, I. W. & Fornerod, M. (1999) Mol. Cell. Biol. 19, 6276-6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paraskeva, E., Izaurralde, E., Bischoff, F. R., Huber, J., Kutay, U., Hartmann, E., Luhrmann, R. & Gorlich, D. (1999) J. Cell Biol. 145, 255-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.