

Abstract
Equilibrium and kinetic hydrogen exchange experiments show that cytochrome c is composed of five foldon units that continually unfold and refold even under native conditions. Folding proceeds by the stepwise assembly of the foldon units rather than one amino acid at a time. The folding pathway is determined by a sequential stabilization process; previously formed foldons guide and stabilize subsequent foldons to progressively build the native protein. Four other proteins have been found to show similar behavior. These results support stepwise protein folding pathways through discrete intermediates.
Keywords: cytochrome c, hydrogen exchange, stability labeling
To understand how proteins fold to their native state, it will be necessary to define the structure of the intermediate forms that they pass through, as for any other biochemical mechanism. This effort has proven to be exceptionally difficult. Partially folded kinetic intermediates live for <1 s, and they cannot be isolated and studied by the usual structural methods. Spectroscopic observations widely used to detect fast kinetic folding events provide very little structural detail and can be misleading in respect to structure formation. Theoretical methods are not yet able to simulate the folding of sizeable proteins.
These problems can be avoided by studying proteins under native conditions. Under these conditions proteins predominantly occupy their lowest free energy state (1) but they must repeatedly unfold and refold, thermodynamically cycling through all possible higher-energy forms. The high-energy forms generally exist only at minuscule levels and therefore are invisible to most methods, which perceive only the overwhelmingly populated native state. These high-energy forms can be detected by hydrogen exchange (HX) methods because the predominant native structure makes no contribution to the HX rates that one measures. Hydrogens that are protected in the native protein can exchange with solvent only when their protecting H bonds are transiently broken as the protein searches through its higher-energy forms.
HX measurements provide amino acid-resolved information that can in favorable cases define the structure of partially folded intermediate forms, their equilibrium and kinetic parameters, and their interconversions. Results obtained for cytochrome c (Cyt c) show that it is composed of five subglobally cooperative unfolding/refolding units, called foldons, color-coded in Fig. 1. This article integrates our research and prior information for Cyt c and other proteins. All of these results consistently show that proteins at equilibrium are composed of foldon building blocks and that they fold kinetically by the stepwise assembly of their foldon units, guided by the same factors that determine the native state.
Fig. 1.

Cyt c color-coded to indicate its component foldon units.
Materials and Methods
WT equine Cyt c (type VI from Sigma) was further purified as required. The pseudo-WT (pWT) protein and its mutants were expressed in Escherichia coli and purified as described (2). The pWT protein has no N-terminal acetylation; also, two peripheral histidine residues that can misligate to the heme iron and complicate kinetic folding (H26N and H33N) have been replaced (2). D2O (99.9%) was from Isotec, and other chemicals were from Sigma and Fisher. Experimental details are given for each experiment in the text and figure legends. For more detailed description see ref. 3.
Results
Native-State HX (NHX). In an experiment known as NHX, H-to-D exchange was measured for a large number of amide hydrogens in Cyt c in denaturant concentrations far below the melting transition. Fig. 2 shows NHX results for WT equine Cyt c, a recombinant pWT variant, and two slightly destabilized pWT mutants.
Fig. 2.

Equilibrium NHX results color-coded as in Fig. 1. Data are for WT Cyt c (12), recombinant pWT Cyt c, and two pWT variants [pDr 7.0 and 30°C (WT) or 20°C (pWT)]. The amide NHs shown in gray have residual protection in the unfolded forms.
Eq. 1 was used to translate measured HX rates (kex) for each hydrogen into the free energy of the governing H bond opening reaction (ΔGHX).
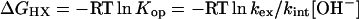 |
[1] |
Eq. 1 is pertinent for bimolecular exchange (EX2) conditions where HX rate is determined by the fraction of time that the protecting H bond is transiently severed, given by Kop. The EX2 condition holds when reformation of the protecting H bonded structure is faster than the chemical exchange rate for the unprotected hydrogen (kcl > kch = kint[OH-]). Amide HX is catalyzed only by OH- ion above ≈pH 4; kint is the intrinsic second-order rate constant for each amide (4, 5). In the monomolecular exchange (EX1) limit, where structural reclosing is slower than the chemical exchange rate, every opening reaction allows exchange, and the measured HX rate is equal to the opening rate (kcl < kch; kex = kop).
Fig. 2 shows that the amide hydrogens of Cyt c exchange over a large range of rates. Neighboring hydrogens often exchange at very different rates and with no dependence on denaturant concentration, indicating that the exchange of most hydrogens is determined by small local structural fluctuations that break one protecting H bond at a time (6) (although see ref. 7).
Some hydrogens show a large dependence on denaturant (large dΔGHX/d[denaturant] = m). They exchange by way of sizeable unfolding reactions that transiently expose considerable new surface to the denaturing solvent. Increasing denaturant selectively promotes these large unfoldings, and they ultimately come to dominate the exchange of all of the hydrogens that they expose. This phenomenon is seen as a merging of sets of hydrogens into given HX isotherms.
The hydrogens that merge into each isotherm are exposed to exchange by a common unfolding. Each isotherm represents a set of neighboring amino acids, and thus identifies one or two continuous segments that concertedly unfold and refold (foldons). The HX parameters of each isotherm reveal the free energy of each unfolded state relative to the native state (ΔGHX; Eq. 1) and the amount of protein surface that each concerted unfolding exposes [related to m (8)].
The Foldon Substructure of Cyt c. In WT Cyt c (Fig. 2), all of the H bonded hydrogens in the N-terminal and C-terminal α-helices are exposed in a common unfolding reaction that represents the transient global unfolding (9). These results, when extrapolated to higher denaturant, quantitatively predict the measurable global melting transition (10). In pWT Cyt c (Fig. 2) amides in the C-terminal helix show the same behavior as for WT Cyt c and register about the same global unfolding free energy.
Most residues in the N-terminal helix of WT Cyt c join the global unfolding of the C-terminal helix. However, the amide hydrogens of Cys-14, Ala-15, and His-18, at the heme attachment site, retain some protection in the globally unfolded state, apparently because of the rigid heme structure. Their exchange continues to be dominated by local fluctuations from the predominantly populated native state. In pWT Cyt c, the entire N-terminal helix retains some stability in the transiently unfolded state, ≈1.5 kcal/mol, for reasons that are unclear.
An unfolding reaction at lower free energy and with less surface exposure exposes all of the amides in the 60's helix and apparently also an Ω-loop (green helix and loop in Fig. 1). Thus these structural elements act as a concerted subglobal folding/unfolding unit.
Similar results for a few hydrogens placed in the neck of the yellow Ω-loop and the red Ω-loop suggest still lower lying concerted unfolding units. The reality of the red loop foldon is confirmed by a kinetic version of the NHX experiment, described below. Other HX experiments done as a function of decreasing pH show that the yellow Ω-loop segment nested within the two yellow neck segments (Fig. 1) acts as a separate lowest free energy foldon (11).
In summary, the equilibrium NHX experiment uses destabilants to promote and make visible otherwise hidden unfolding reactions. The results identify and characterize five separate foldon units that account for the entire Cyt c structure (Fig. 1). The same foldons are seen in pWT Cyt c that does not have the histidine residues known to cause misfolding-dependent, three-state kinetic folding in WT Cyt c.
The Pathway Option. The five Cyt c unfolding steps array in a ladder of increasing free energy and increasing surface exposure in the order of nested yellow, red, yellow, green, and blue. These relationships suggest that the different foldons might unfold in a pathway sequence (12), as in Eq. 2, where each foldon unit is represented by its color coding (Fig. 1) in either its folded (uppercase) or unfolded (lowercase) form.
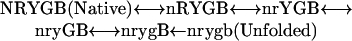 |
[2] |
If so, then the reverse sequence must depict the major Cyt c refolding pathway because these observations are made under equilibrium native conditions. Each unfolding reaction must be matched by an equal and opposite refolding reaction, or else equilibrium would not be maintained.
The Unfolding Sequence by Stability Labeling. To test the pathway model, a series of stability labeling experiments were performed. If unfolding is sequential as in Eq. 2, then each higher energy unfolded state contains all of the lower-lying unfoldings. In this case, if the stability of any given foldon is changed, then the unfolding of the altered foldon and all higher-lying partially unfolded forms (PUFs) that contain it should be equally affected, whereas lower-lying PUFs should be unaffected.
In reduced Cyt c, the heme iron binds its Met-80-S ligand more strongly by 3.2 kcal/mol than in oxidized Cyt c [independently measured (13)]. The red loop is stabilized by this same amount (Fig. 3A), as might be expected, because Met-80 is in the red loop foldon and the red unfolding breaks the S-Fe ligation (13). The higherlying PUFs are equally stabilized, as expected for a sequential unfolding model but not for independent foldon unfolding or nonfoldon models. Interestingly, the final global unfolding is stabilized by an additional 2 kcal/mol. This finding suggests that the last unfolding step finally exposes the buried heme iron (14), which carries a destabilizing charge in oxidized Cyt c but is neutral in the reduced form.
Fig. 3.

Stability labeling results showing the change in the free energy level of the Cyt c unfolded states caused by selective perturbations. Compared are: reduced and oxidized WT Cyt c (A); the effect of a Glu-62-Gly mutation in pWT Cyt c (B); and the effect of a Glu-66 -Ala mutation in pWT Cyt c (C). The free energy levels compare values for the isotherm marker protons at zero denaturant. The nested-yellow unit is omitted in these comparisons; its protons exchange too rapidly to measure under the conditions used.
Fig. 3B compares results for pWT Cyt c and a Glu-62-Gly variant (Fig. 2). The surface Gly-62 mutation does not perturb the native structure but it does remove a stabilizing ion pair interaction in the yellow neck (Glu-62 to Lys-60). The mutation equally destabilizes the yellow unit and the higher-lying PUFs but not the lower-lying red foldon, as expected for sequential foldon unfolding (15).
Figs. 2 and 3C compare results for pWT Cyt c and a Glu-66-Ala variant. This surface mutation does not perturb the native structure but it does remove a stabilizing salt link (green helix to red loop). The red loop unfolding and all higher-lying PUFs are equally destabilized as expected for a sequential foldon unfolding model.
In summary, these stability labeling results favor a sequential unfolding model and therefore an equal and opposite refolding sequence, as in Eq. 2.
Kinetic Folding Intermediate by HX Pulse Labeling. The experiments just described measure equilibrium properties of the unfolding ensemble at native conditions. Further experiments were done to study the kinetic sequence starting from either the unfolded protein or the folded protein.
WT Cyt c folds kinetically in a three-state way at neutral pH. The intermediate that accumulates can be characterized by a recently reconfigured kinetic HX pulse labeling experiment (3, 16). Cyt c initially unfolded and deuterated in concentrated denaturant (D2O) was diluted into refolding conditions in a stopped-flow apparatus. The protein starts to fold but then encounters a large barrier. Conditions were arranged to maximize transient intermediate accumulation (85% of the population in the intermediate state). The intermediate was H labeled by a brief high pH pulse (50 ms) in a second mixing to speed D-to-H exchange. During the pulse, each exchangeable deuteron is replaced by a solvent hydrogen at a rate that depends on the equilibrium and kinetic parameters of the protecting structure. A third mix lowers the pH and produces the more stable heme-reduced protein. These conditions halt further exchange and preserve the H labeling imposed during the pulse for subsequent analysis by 2D NMR.
Fig. 4 shows the H labeling obtained in the trapped intermediate as a function of the pulse pH. At pH <8.5, exchange is in the EX2 mode and labeling increases with pH, dependent on the stability of protecting structure at each amide. At higher pH, exchange enters the EX1 region. H labeling of the different amides becomes independent of pH and plateaus to a level that is determined by the HX pulse time and the opening rate of each protecting H bond within the intermediate. These results reveal: (i) the structure of the trapped intermediate in terms of its H bonding pattern at 45 amino acid residues; (ii) the stability against opening of each H bond (EX2 region) in the transient intermediate state; and (iii) the kinetics for the loss and reformation of each protecting H bond (EX1 region).
Fig. 4.

HX pulse-labeling results. Unfolded deuterated WT Cyt c (pDr 7.5, 4.2 M GdmCl, 10°C) was refolded by denaturant dilution (pH 6, 10°C, 0.23 M GdmCl, H2O). A kinetic intermediate, blocked by a histidine to heme misligation barrier, was H labeled in a 50-ms, high-pH pulse. The refolded native protein was analyzed by 2D NMR. The black dashed curves indicate the labeling expected for each amide in the absence of HX protection. The colored curves fit the labeling actually obtained within the intermediate alone (EX2 and EX1) after correction for extraneous effects (3, 16).
All of the amides in the segments that form the native N-terminal and C-terminal helices are well protected. The two helices are fully formed, and they dock at their native interface (3, 16). All other regions are unprotected except for minor protection at a few native-like β-turns (e.g., His-33 and Thr-78, see also Figs. 2 and 5) and some scattered amides, which are probably buried. This early kinetic intermediate is structurally indistinguishable from the initial intermediate indicated by equilibrium NHX (Fig. 2). The equilibrium stability measured in these two different experiments agree to within 1.2 kcal/mol, with much of the difference probably caused by the different conditions used (10°C, high pH vs. 30°C, pD 7).
Fig. 5.

Kinetic NHX results. Predeuterated native WT Cyt c was exposed to a 75-ms H labeling pulse at high pH and 20°C, then quenched to slow HX conditions and analyzed by 2D NMR (20). The labeling of the foldon markers is shown in bold. The black curve depends on the rate for formation of a stable high pH intermediate (measured by CD and fluorescence) and shows the EX1 labeling that would be obtained during the 75-ms pulse caused by unfolding to this state. Comparison of the EX1 labeling of the markers for the different foldons at given pH values shows that the kinetic order for unfolding of the different foldon units is first red, then yellow, then green, and finally blue.
The entire protein population folds through this intermediate (16). The barrier that leads to the intermediate in three-state folding is identical to the barrier that rate-limits folding under two-state conditions (fluorescence folding experiments). They have the same thermodynamic (ΔG‡, ΔH‡, ΔS‡) and structural (m‡) parameters (17). Because the rate-limiting two-state barrier is clearly on the main folding pathway, the intermediate that it leads to must also be on-pathway.
In the three-state case, folding is known to be blocked after N/C bihelix formation by an error-repair barrier caused by misligation to the exposed heme iron of a peripheral histidine. Either His-26 or His-33 ligands to the heme Fe at the Met-80 position (Fig. 1). Both histidines are in the green loop. The misligation holds the green loop out of place and so, according to Eq. 2, should block the next pathway step, green unit formation, as is seen.
In summary, kinetic pulse labeling results correspond rather precisely with equilibrium NHX and Eq. 2.
The Unfolding Sequence by Kinetic NHX. Another experiment, called kinetic NHX, can in favorable cases probe the kinetic order in which the different foldons unfold. Two conditions are necessary. The HX measured must be controlled by the unfolding reactions to be characterized and the HX rate must be in the EX1 mode, controlled by the rate of foldon unfolding (kex = kop).
To obtain EX1 behavior, high pH is required. At high pH the exchange rate becomes too fast to measure directly. A pulse technique was used (18, 19). Predeuterated native WT Cyt c was exposed to a brief D-to-H exchange-labeling pulse at high pH, then quenched into slow HX conditions where the H labeling obtained during the pulse could be measured at leisure by 2D NMR.
Fig. 5 shows the color-coded H labeling imposed at the measurable amides in each of the foldon units when the native protein was exposed to a 75-ms labeling pulse at the pH values indicated (20). The black curve depends on the pH-driven rate for formation of a stable high pH intermediate (measured by CD and fluorescence), called state V (21). This curve shows the EX1 labeling that would be obtained during the 75-ms pulse caused by unfolding to state V, which is evidently equivalent to nrygB in Eq. 2 (see below).
All six measurable hydrogens in the red loop (Fig. 5) show the same value for Kop in the EX2 region, and they plateau to the same exposure rate (kop) in the high pH EX1 region (between pH 10.5 and 11.5). The spread observed mostly reflects the different kint values in Eq. 1. This result confirms the impression given in Fig. 2 that the red loop unfolds as a concerted foldon unit. Also, the red unit unfolding rate, given by the EX1 plateau level, is equal to the molecular unfolding rate in this same pH range (measured in the high denaturant unfolding rollover region and extrapolated to zero denaturant). These results indicate that the red unfolding step is limited by the same early barrier that limits global unfolding at high denaturant (20), i.e., it is early and on the main unfolding pathway. The secondary upsweep at higher pH (pH ≈12) is caused by the additional labeling that occurs when Cyt c is driven to begin unfolding to the stable high-pH state V intermediate (Fig. 5, black curve).
The exchange of most remaining amide hydrogens is controlled by fast local fluctuations (dotted curves in Fig. 5). As the pulse pH increases, these sites become increasingly labeled in an EX2 manner consistent with their rates at low pH. They do not become limited by an EX1 reaction. To determine foldon behavior, we focus on the marker protons (thick curves in Fig. 5) whose exchange is controlled by foldon unfolding.
The yellow markers are seen to approach an EX1 labeling plateau that is lower than for the red unit. This result indicates an unfolding rate for the yellow neck foldon that is slightly slower than for the red unit. Again here the secondary upsweep in labeling at higher pH, conforming with the black curve in Fig. 5, shows that the yellow unit also unfolds as part of the high pH transition to state V. The green and blue marker protons remain unlabeled. Below pH 11.5, they unfold much less than once in 75 ms.
Still higher pH drives Cyt c to unfold to the high pH state V intermediate, at the black curve rate. The green foldon unfolds and is labeled at precisely this rate. These results [plus fluorescence and CD (20)] show that state V has all but the blue foldon unfolded. Still higher pH finally drives even the blue foldon to unfold on the 75-ms time scale.
The unfolding rates measured in these experiments are obviously accelerated by the high pH used but they specify the kinetic order of foldon unfolding. The unfolding order is first red (independently shown to be early and on the main unfolding pathway), then yellow, then green, and finally blue (independently shown to be first on the main folding pathway), all just as predicted by the pathway sequence in Eq. 2.
Discussion
Foldons and Folding: Other Proteins, Perturbants, and Methods. The variety of experimental results just described consistently show that Cyt c is composed of concerted foldon units that unfold and refold in a stepwise sequential pathway, as in Eq. 2. Is this behavior peculiar to Cyt c, perhaps as an artifact of the bound heme group, or caused by some peculiarity of guanidinium (Gdm)-Cl, or is HX itself somehow misleading? Apparently not. Similar experiments find similar results for other nonheme proteins, other perturbants (pressure, temperature, pH), and other non-HX methodologies, as summarized in Fig. 6.
Fig. 6.

Foldon behavior in other proteins. Published NHX results for apo Cyt b562 (22) and ribonuclease H (27) and sulfhydril reactivity results for triosephosphate isomerase (TIM) (29) are summarized. [Reproduced with permission from ref. 22 (Copyright 2002, American Chemical Society; ref. 27 (Copyright 1996, Nature Publishing Group, www.nature.com); and ref. 29 (Copyright 2002, Elsevier Science Ltd.).]
NHX results for apoCyt b562 (Fig. 6) reveal three component foldon units that fold in the order helix 2 plus helix 3 plus part of helix 4, then helix 4, and finally helix 1 (22-24). The same foldons were found in experiments done as a function of high pressure (24), urea (23), and GdmCl (22). Destabilizing mutations caused discrete forms to populate at equilibrium where they could be solved by high-resolution NMR (25, 26). Excellent agreement with the NHX results (Fig. 6) was found. These studies show that native-like secondary structure tends to be conserved in the non-native intermediates, whereas tertiary interactions are malleable (26).
NHX results for ribonuclease H (Fig. 6) capture three foldon units (27). Multiple probing experiments including HX pulse labeling, quenched molten globule HX, and fragment synthesis/stabilization support their foldon nature and show that they construct a kinetic folding pathway (28).
Fig. 6 also shows some results for dimeric triosephophate isomerase, the prototypical (β/α)8 triosephosphate isomerase barrel protein (the most common enzyme fold). In this case, global and subglobal unfolding reactions were measured as a function of increasing denaturant but the experiment does not depend on HX. Rather the chemical reactivity of 47 buried Cys-SH groups was measured. The results demonstrate three concerted foldon units, each involving two or three entire helices plus other unspecified strands and loops. Experiments analogous to stability labeling (above) show that these foldons unfold in a sequential stepwise pathway manner (29).
In the OspA protein of Borrelia, equilibrium (EX2) and kinetic (EX1) NHX experiments and related mutational studies found six foldons and show that at least some of them are involved in a kinetic folding pathway (30, 31).
In a number of other proteins, the NHX experiment measured the global unfolding but did not successfully find subglobal unfolding behavior. For HX experiments to detect subglobal foldons, some rather demanding requirements must be met (3). An unusually large dynamic range in ΔG (8 kcal/mol or more) and m is necessary. The separation between isotherms must be large relative to the spread within each one, which is not negligible (Figs. 2 and 6). To be found by equilibrium NHX, a partially unfolded state must be more stable than any other state that exposes the same hydrogens. To be found by kinetic NHX, a partially unfolded state must be separated from more unfolded states by a kinetic barrier that is larger than any prior unfolding barrier. In both modes, measured HX must be dominated by concerted unfolding reactions in competition with ubiquitous local fluctuation pathways.
These severe restrictions militate against foldon detection. Yet foldons have now been detected in five proteins. This finding suggests that foldons occur in many more proteins than have so far been detected.
The Folding Pathway by Sequential Stabilization. It seems likely that protein molecules composed of separately cooperative foldon building blocks should tend to fold in a stepwise manner, one foldon unit at a time. We find that to be the case.
What determines the order of foldon formation? Fig. 7 places the different foldon units within Cyt c and shows the contacts that connect them in the native structure. Experiment shows that the N/C bihelix forms first. Why should this be? HX results indicate that this unit represents the most stable part of the native protein. Also, the literature shows that proteins tend to dock their N- and C-terminal elements first, apparently for evolutionary reasons (32).
Fig. 7.

Graphical contact map for the different foldons in Cyt c and the residue contacts that connect them in the native protein (39).
Subsequent folding apparently proceeds in the native context. In the native protein, the N/C bihelical unit creates a docking surface that interacts only with the green helix and loop (Fig. 7). It can only guide and stabilize the green foldon. In turn, the green foldon must be in place to support further native-like structure formation. Indeed, experiment shows that the green unit does fold next. The yellow neck strands grow out of the ends of the two green units (Fig. 1). They can form only after both green units are in place, and they must precede the nested-yellow loop. Again, we find that to be the case. Finally, all of these preformed structures are necessary to guide and stabilize the formation of the least-stable red and nested-yellow Ω-loops.
These consistencies suggest that the folding pathway builds the native structure by progressively assembling its foldon units in a “sequential stabilization” sequence determined by the way the various foldons interact in the native protein. The folding sequence that emerges can be usefully represented by a conventional pathway energy profile with explicit intermediates and barriers (Eq. 2).
The Hidden Multistate Nature of Protein Molecules. Equilibrium and kinetic experimentation over the last 50 years has established the view that monomeric proteins are highly cooperative, all or none, two-state structures. This view is not consistent with the various observations of multistate foldon behavior just described. A number of factors have conspired to hide the multistate nature of protein molecules.
Partially folded intermediates are generally not seen in equilibrium melting experiments because, in the transition zone where melting is measured, stability is close to zero. When any unit is lost, the entire protein unfolds. Further, a simple derivation shows that the fractional population of any intermediate species reaches a maximum in the melting transition zone where it is maximally hidden. Under more stabilizing conditions, PUFs can retain significant stability and may be detected by NHX methods.
Partially folded intermediates are not seen in two-state kinetic folding where, by definition, stable intermediates can form only after the rate-limiting step. In fact, the rate-limiting step in two-state folding does occur as an initial event (17, 33). Thus kinetic intermediates are hidden. Equilibrium NHX is independent of kinetic barriers and therefore can detect intermediates that are otherwise kinetically hidden. An early folding barrier aids the kinetic NHX experiment, which requires that the unfolding of the foldon to be detected must be followed by an even larger barrier (20).
Folding intermediates do accumulate in three-state kinetic folding and can be detected by many spectroscopic methods. However, these methods do not provide the structural information necessary to illuminate the significance of the kinetic events that they detect. The functional significance of populated kinetic intermediates has often been attributed to spurious features of the folding landscape (34). The detailed structural information provided by HX and related results now shows that these forms can represent increasingly native-like structures on the main folding pathway.
Intermediates are not seen in most theoretical simulations. They will be seen only when the models used incorporate subglobally cooperative foldon units.
In summary, results now available correct a long-standing inference drawn from previous studies. Proteins can act thermodynamically and kinetically as multistate structures, determined by their component foldon units. This behavior, although fundamental, escapes most methodologies under the experimental conditions normally used. Not surprisingly, foldon behavior has functional significance (see also refs. 11 and 35).
Conclusions
A quantity of evidence shows that some proteins are composed of separately cooperative foldon building blocks. Foldons tend to be coincident with secondary structural elements. Secondary structures are intrinsically cooperative units (11, 36). They retain this property when built into globular proteins. The same secondary structures are ubiquitous. Thus foldon behavior seems likely to be general. The successful demonstration of foldon substructure in five different proteins, despite their hidden nature, underscores this conclusion.
It seems likely that protein folding should proceed by the stepwise assembly of their separately cooperative units, as we find. The folding steps follow each other in a conventional pathway that progressively builds the native structure. The order of steps tracks the way that the foldon units interact in the native protein, pointing to a sequential stabilization process. A corollary is that folding pathways might be linear or branched or parallel, as determined by the arrangement of the foldon units in the native protein.
In the same way, any model that pictures the many amino acids as individual folding units must lead to a very large ensemble of alternative parallel pathways, as simulations show (34, 37, 38). However, experimental results now find a small number of discrete partially native forms in the folding landscape rather than a nearly undifferentiated continuum. All other partially folded forms must, of course, exist, but apparently only at higher free energy.
It is noteworthy that the guidance of structure formation by sequential stabilization appears widely. Examples include fragment complementation, domain swapping, the structuring of natively unfolded proteins when they find their target molecules, and the recruitment of polypeptides and proteins into preexisting amyloid structures.
Acknowledgments
This work was supported by National Institutes of Health Grant GM31847.
Abbreviations: Cyt c, cytochrome c; HX, hydrogen exchange; NHX, native-state HX; EX1, monomolecular exchange; EX2, bimolecular exchange; PUF, partially unfolded form; pWT, pseudo-WT; Gdm, guanidinium.
References
- 1.Anfinsen, C. B. (1973) Science 181, 223-230. [DOI] [PubMed] [Google Scholar]
- 2.Rumbley, J. N., Hoang, L. & Englander, S. W. (2002) Biochemistry 41, 13894-13901. [DOI] [PubMed] [Google Scholar]
- 3.Krishna, M. M. G., Hoang, L., Lin, Y. & Englander, S. W. (2004) Methods 34, 51-64. [DOI] [PubMed] [Google Scholar]
- 4.Bai, Y., Milne, J. S., Mayne, L. & Englander, S. W. (1993) Proteins 17, 75-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connelly, G. P., Bai, Y., Jeng, M. F. & Englander, S. W. (1993) Proteins 17, 87-92. [DOI] [PubMed] [Google Scholar]
- 6.Maity, H., Lim, W. K., Rumbley, J. N. & Englander, S. W. (2003) Protein Sci. 12, 153-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wooll, J. O., Wrabl, J. O. & Hilser, V. J. (2000) J. Mol. Biol. 301, 247-256. [DOI] [PubMed] [Google Scholar]
- 8.Myers, J. K., Pace, C. N. & Scholtz, J. M. (1995) Protein Sci. 4, 2138-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bai, Y., Milne, J. S., Mayne, L. & Englander, S. W. (1994) Proteins 20, 4-14. [DOI] [PubMed] [Google Scholar]
- 10.Mayne, L. & Englander, S. W. (2000) Protein Sci. 9, 1873-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishna, M. M. G., Lin, Y., Rumbley, J. N. & Englander, S. W. (2003) J. Mol. Biol. 331, 29-36. [DOI] [PubMed] [Google Scholar]
- 12.Bai, Y., Sosnick, T. R., Mayne, L. & Englander, S. W. (1995) Science 269, 192-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu, Y., Mayne, L. & Englander, S. W. (1998) Nat. Struct. Biol. 5, 774-778. [DOI] [PubMed] [Google Scholar]
- 14.Cohen, D. S. & Pielak, G. J. (1995) J. Am. Chem. Soc. 117, 1675-1677. [Google Scholar]
- 15.Maity, H., Maity, M. & Englander, S. W. (2004) J. Mol. Biol. 343, 223-233. [DOI] [PubMed] [Google Scholar]
- 16.Krishna, M. M. G., Lin, Y., Mayne, L. & Englander, S. W. (2003) J. Mol. Biol. 334, 501-513. [DOI] [PubMed] [Google Scholar]
- 17.Sosnick, T. R., Mayne, L. & Englander, S. W. (1996) Proteins 24, 413-426. [DOI] [PubMed] [Google Scholar]
- 18.Milne, J. S., Mayne, L., Roder, H., Wand, A. J. & Englander, S. W. (1998) Protein Sci. 7, 739-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sivaraman, T. & Robertson, A. D. (2001) Methods Mol. Biol. 168, 193-214. [DOI] [PubMed] [Google Scholar]
- 20.Hoang, L., Bédard, S., Krishna, M. M. G., Lin, Y. & Englander, S. W. (2002) Proc. Natl. Acad. Sci. USA 99, 12173-12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boeri, E., Ehrenberg, A., Paul, K. G. & Theorell, H. (1953) Biochim. Biophys. Acta 1, 273-282. [DOI] [PubMed] [Google Scholar]
- 22.Chu, R., Pei, W., Takei, J. & Bai, Y. (2002) Biochemistry 41, 7998-8003. [DOI] [PubMed] [Google Scholar]
- 23.Fuentes, E. J. & Wand, A. J. (1998) Biochemistry 37, 3687-3698. [DOI] [PubMed] [Google Scholar]
- 24.Fuentes, E. J. & Wand, A. J. (1998) Biochemistry 37, 9877-9883. [DOI] [PubMed] [Google Scholar]
- 25.Feng, H., Takei, J., Lipsitz, R., Tjandra, N. & Bai, Y. (2003) Biochemistry 42, 12461-12465. [DOI] [PubMed] [Google Scholar]
- 26.Feng, H., Zhou, Z. & Bai, Y. (2005) Proc. Natl. Acad. Sci. USA, in press.
- 27.Chamberlain, A. K., Handel, T. M. & Marqusee, S. (1996) Nat. Struct. Biol. 3, 782-787. [DOI] [PubMed] [Google Scholar]
- 28.Chamberlain, A. K. & Marqusee, S. (2000) Adv. Protein Chem. 53, 283-328. [DOI] [PubMed] [Google Scholar]
- 29.Silverman, J. A. & Harbury, P. B. (2002) J. Mol. Biol. 324, 1031-1040. [DOI] [PubMed] [Google Scholar]
- 30.Yan, S., Kennedy, S. & Koide, S. (2002) J. Mol. Biol. 323, 363-375. [DOI] [PubMed] [Google Scholar]
- 31.Yan, S., Gawlak, G., Smith, J., Silver, L., Koide, A. & Koide, S. (2004) J. Mol. Biol. 338, 811-825. [DOI] [PubMed] [Google Scholar]
- 32.Krishna, M. M. G. & Englander, S. W. (2005) Proc. Natl. Acad. Sci. USA 102, 1053-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Plaxco, K. W., Simons, K. T. & Baker, D. (1998) J. Mol. Biol. 277, 985-994. [DOI] [PubMed] [Google Scholar]
- 34.Onuchic, J. N. & Wolynes, P. G. (2004) Curr. Opin. Struct. Biol. 14, 70-75. [DOI] [PubMed] [Google Scholar]
- 35.Hoang, L., Maity, H., Krishna, M. M. G., Lin, Y. & Englander, S. W. (2003) J. Mol. Biol. 331, 37-43. [DOI] [PubMed] [Google Scholar]
- 36.Zimm, G. H. & Bragg, J. K. (1959) J. Chem. Phys. 31, 526-535. [Google Scholar]
- 37.Bryngelson, J. D., Onuchic, J. N., Socci, N. D. & Wolynes, P. G. (1995) Proteins 21, 167-195. [DOI] [PubMed] [Google Scholar]
- 38.Dill, K. A. & Chan, H. S. (1997) Nat. Struct. Biol. 4, 10-19. [DOI] [PubMed] [Google Scholar]
- 39.Rumbley, J., Hoang, L., Mayne, L. C. & Englander, S. W. (2001) Proc. Natl. Acad. Sci. USA 98, 105-112. [DOI] [PMC free article] [PubMed] [Google Scholar]