

Abstract
Understanding regulation of α-synuclein has long been a central focus for Parkinson’s disease (PD) researchers. Accumulation of this protein in the Lewy body or neurites, mutations in the coding region of the gene and strong association of α-synuclein encoding gene multiplication (duplication/triplication) with familial form of PD have indicated the importance of this molecule in pathogenesis of the disease. Several years of research identified many potential faulty pathways associated with accumulation of α-synuclein inside dopaminergic neurons and its transmission to neighboring ones. Concurrently, an appreciable body of research is growing to understand the epigenetic and genetic deregulation of α-synuclein that might contribute to the disease pathology. Completion of the ENCODE (Encyclopedia of DNA Elements) project and recent advancement made in the epigenetic and trans factor mediated regulation of each gene, has tremendously accelerated the need to carefully understand the epigenetic structure of the gene (SNCA) encoding α-synuclein protein in order to decipher the regulation and contribution of α-synuclein to the pathogenesis of PD. We have also analyzed the detailed epigenetic structure of this gene with knowledge from ENCODE database, which may open new avenues in α-synuclein research. Interestingly, we have found that the gene contains several transcriptionally activate histone modifications and associated potential transcription factor binding sites in the non-coding areas that strongly suggest alternative regulatory pathways. Altogether this review will provide interesting insight of α-synuclein gene regulation from epigenetic, genetic and post-transcriptional perspectives and their potential implication in the PD pathogenesis.
1. Introduction
Parkinson’s disease (PD) is a late-onset neurodegenerative disease that affects a significant portion of elderly populations worldwide (Beitz, 2014). Recent epidemiological data suggests this disease is increasing every year, with 50,000 to 60,000 new cases in the USA alone in addition to the 1 million patients already suffering (National Parkinson’s Disease Foundation; http://www.pdf.org/en/parkinson_statistics) (Beitz, 2014). The clinical features of the disease include cardinal motor dysfunctions, such as postural instability, resting tremor, bradykinesia, and rigidity (Beitz, 2014; Sherer et al., 2012; Thomas and Beal, 2011). In conjunction, several non-motor symptoms are also associated with the disease: anosmia, sleep disturbances, anxiety or depression, loss of bladder control, constipation, etc. (Beitz, 2014; Sherer et al., 2012; Thomas and Beal, 2011). PD cases are generally classified under two categories based on its origin. The first type contains a known or inherited familial genetic abnormalities, called familial PD (autosomal dominant or recessive). This accounts for only 10% or less of the total cases. The second type is called sporadic PD cases without any known familial history and consists of around 90% or more of the cases,(Gasser, 2009). Generally, degeneration of around 70% dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) of the midbrain region takes place before the clinical symptoms sets in (Heisters, 2011; Morrish et al., 1998; Postuma et al., 2010). Recently Kordower et al. in a report studied 28 post-mortem brain samples ranging from 1–27 years post diagnosis of PD and showed that 50% to 90% of tyrosine hydroxylase(TH)-positive DA neuronal death in the SNpc occurs within relatively early years after the disease diagnosis and negligible loss (<20%) thereafter. Interestingly, although a variable degree of loss (33%–80%) of nigral melanin-containing neurons was observed across the disease duration, there was higher number of melanin containing neurons remained as compared to TH-positive neurons at any point analysis (Kordower et al., 2013).
One of the most prominent pathophysiological hallmarks of this disease is the presence of intracytoplasmic inclusion bodies called Lewy bodies, or Lewy neurites (Spillantini et al., 1997). These inclusion bodies are present in the neurons containing α-synuclein (α-SYN) aggregates (Spillantini et al., 1997). The dynamic process of α-SYN aggregation through the formation of different intermediate species mainly contributes to α-synucleinopathy in PD (Stefanis, 2012). The contribution of α-SYN in the disease pathogenesis is further highlighted by the discovery of the genetic components in PD, which first came into the picture through genetic abnormalities in the α-SYN coding gene called SNCA, when familial mutations and gene multiplications were discovered in that particular locus (Farrer et al., 2004; Ibanez et al., 2004; Kruger et al., 1998; Polymeropoulos et al., 1997; Singleton et al., 2003; Zarranz et al., 2004). Later on, numerous other genetic or environmental factors—such as imbalances in genetic dosage, post-translational modifications, exposure to toxins and pesticides/herbicides, oxidative stress and more recently, epigenetic deregulation—have been implicated in dysregulation of α-SYN in PD (Basu et al., 2015; Cristovao et al., 2012; Guhathakurta et al., 2017; Mouradian, 2012; Tanner et al., 2011; Venda et al., 2010). These factors have been either implicated in increased α-SYN protein expression or in the generation of misfolded intermediates of the protein, which ultimately contribute to α-synucleinopathy in PD. Altogether, it can be said that environmental factors and age-associated changes—along with some common genetic variants identified through candidate gene approach or whole genome association studies—play important roles in the pathogenesis of the sporadic form of the disease (Gasser et al., 2011).
A lot of effort has been made over the years to understand what factor(s) instigate α-SYN protein aggregation and how this event leads to neurodegeneration. However, significant progress has not been made in understanding the epigenetic or transcriptional regulation of α-SYN through considering its detailed genetic structure. Several single nucleotide polymorphisms (SNP) in the regulatory element of SNCA have been identified as genetic risk factors for PD, but their contribution to the aberrant regulation of α-SYN has only been investigated by using in vitro systems where implications from epigenetic underpinnings were largely overlooked (Basu et al., 2017). With the completion of the ENCODE project (Encyclopedia of DNA Elements), we can now understand the complex gene regulation pattern of eac h gene by considering all cis and trans elements associated with DNA (Consortium, 2012). In this review, the present understanding of genetic and epigenetic regulation of SNCA will be discussed and it will also highlight the importance of the enormous amount of ENCODE data in SNCA regulation. This will open a new avenue for research related to the regulation of α-SYN expression in dopaminergic neurons, helping researchers understand the behavior of this molecule under severe pathological conditions such as PD.
2. Regulation of α-synuclein expression and Parkinson’s disease
α-SYN is a synaptic protein consisting of 140 amino acids (Maroteaux et al., 1988). The gene coding for this protein resides on chromosome 4 in humans and spans around 114 kb region in the genome (Touchman et al., 2001). α-SYN belongs to a family of synuclein proteins where two other members, such as β and γ synuclein, are also present and the genes coding for them are well-conserved across the species (George, 2002). Expression of α-SYN can be regulated at various stages of its development like any other gene expression system. Interestingly, deregulation of this gene has long been associated with PD (Stefanis, 2012; Venda et al., 2010).
The first piece of evidence pointing to the fact that α-SYN gene deregulation is directly associated with PD came from familial PD cases where coding region mutation resulted in the 53rd amino acid substitution from alanine to Threonine in Italian and Greek families (Polymeropoulos et al., 1997). Subsequently, two other mutations at the 30th (Alanine to Proline) and the 46th (Glutamine to Lysine) were discovered in families of Greek and Spanish descent, which led to an autosomal dominant pattern of the disease inheritance along with early-onset (Kruger et al., 1998; Zarranz et al., 2004). These coding mutations altered amino acids in the N–terminal domain of the protein, which has a role in this protein′s aggregation (Gallegos et al., 2015; Recchia et al., 2004). Involvement of α-SYN gene deregulation in pathogenesis of PD was further strengthened by the findings of SNCA locus multiplication. Both triplication and duplication of SNCA were strongly associated with severity and early onset in familial PD (Chartier-Harlin et al., 2004; Singleton et al., 2003). Gene multiplication was also shown to increase the level of α-SYN, which can be correlated with the higher expression (Miller et al., 2004). Moreover, contribution of this gene in the pathogenesis of PD in a dose-dependent manner became even clearer when it was shown that gene triplication produces a more severe form of the disease than SNCA duplication (Fuchs et al., 2008). This evidence from familial PD cases clearly indicates that any faulty regulatory mechanism, which can lead to a higher amount of α-SYN production in the brain, can be strongly associated with PD pathogenesis.
Based on this, it is conceivable that epigenetic or transcriptional deregulation of this gene may lead to a deranged expression of α-SYN, which can ultimately produce α-SYN pathology as observed in PD and other neurodegenerative disorders. Based on this strong evidence from familial PD cases, several studies have been conducted on idiopathic PD cases investigating the impact of genetic and epigenetic factors that may regulate the α-SYN expression by compromising transcription and post-transcriptional RNA processing under this pathologic condition. Several SNPs identified throughout SNCA were found to interfere with the normal transcriptional regulation of the gene and the binding of micro RNA (miRNA). Some of the SNPs were found to be strongly associated with PD in spite of their functional role not being fully understood (Junn et al., 2009; Kabaria et al., 2015; Maraganore et al., 2006; Rhinn et al., 2012; Simon-Sanchez et al., 2009).
Apart from SNPs, some transcription factors, gene-upstream regulatory elements, and epigenetic factors were all shown to contribute towards the enhanced expression of α-SYN (Chiba-Falek and Nussbaum, 2001; Jowaed et al., 2010; Scherzer et al., 2008). Interestingly, studies have shown that the specifically SNpc of sporadic post-mortem PD brains have considerably higher α-SYN transcript as compared to the control brains (Chiba-Falek et al., 2006). Moreover, by using UV-assisted laser capture microdissection, Grundemann et al. confirmed that significantly higher levels of α-SYN mRNA were present in dopaminergic cells of PD brains at a single cell level (Grundemann et al., 2008). Ageing also plays a significant role in deregulated expression of α-SYN. In a study by Chu and Kordower, it was shown that α-SYN levels were significantly higher in the SN regions of aged control samples as compared to the younger ones and this increased levels of the protein correlated significantly with decreased levels of TH immunoreactivity in the DA neurons (Chu and Kordower, 2007). Therefore, it can then be said that these genetic and epigenetic regulatory elements have profound roles in controlling α-SYN expression in the neurons. Consequently, understanding the role of different epigenetic and transcription factors in the regulation of α-SYN in PD is extremely important to completely decipher the role of this molecule in pathophysiology of the disease.
2.1 Epigenetic regulation
Epigenetic regulation is considered the regulation system that can control a gene expression without modifying its DNA sequence in a heritable fashion (Egger et al., 2004). This regulating mechanism can be modified by its cellular environment, which in turn directly influences the gene expression. Ever since the concept of epigenetics was made by Conrad Waddington in the 1940s, the description of epigenetics has been under constant re-construction, not only due to expansive discoveries of regulating elements which can control gene expression independent of cis-elements, but also because the understanding of complex gene regulation pattern keeps increasing (Jaenisch and Bird, 2003; Waddington, 2012). Studying the regulation of gene expression by epigenetic elements is important since epigenetic architecture of a gene determines its transcriptional activity based on the cellular environment. With increasing knowledge of genomic complexity and thanks to the completion of ENCODE, we now have a better understanding of how these epigenetic elements regulate the gene expression (Consortium, 2012).
Epigenetic regulation of gene expression can be divided into three main categories: regulation by (i) DNA methylation; (ii) histone post-translational modifications; and (iii) non-coding RNAs, small RNA interferences (Egger et al., 2004). In the following section, we will discuss the effect of DNA methylation and histone modifications on α-SYN regulation in PD. Involvement of microRNA and the 3′-UTR in regulating α-SYN expression will be discussed in a separate section.
2.1.1 Contribution of DNA methylation in regulation of α-synuclein expression
DNA methylation at the 5th carbon of cytosine (5mC) base is the first epigenetic modification that was considered as the heritable change that can repress the gene expression (Cheng and Blumenthal, 2011; McGhee and Ginder, 1979; Suzuki and Bird, 2008). Recent studies dealing with synaptic plasticity, activity-induced brain functions, learning, and memory proved that DNA methylation marks can be changed in adult neural tissues based on environmental cues (Guo et al., 2011; Lubin et al., 2008; Ma et al., 2009; Martinowich et al., 2003; Miller and Sweatt, 2007). Usually DNA methylation happens to the cytosines located adjacent to guanine residues, known as CpG methylation (Bird, 1980). And when such CpGs reside in a DNA segment with a higher C+G ratio than expected, they are called CpG islands or CGIs (Deaton and Bird, 2011; Illingworth and Bird, 2009). Mostly these islands are present in the gene-promoters and tend to be unmethylated (Deaton and Bird, 2011). DNA methylation-mediated transcriptional repression of tumor suppressing genes has long been known in cancer epigenetics (Baylin, 2005). However, the role of DNA methylation in regulating neurodegenerative diseases like PD has not been explored in detail. Only a handful of reports are available except for the gene encoding for α-SYN (Coupland et al., 2014; Lu et al., 2013).
Control of α-SYN expression in PD is relatively well explored, as far as the transcriptional or epigenetic regulation of SNCA is concerned. Interestingly, the human α-SYN gene, SNCA, harbors one large CpG island of 591 bp at its 5′ end (Fig. 1). This CpG island covers the transcription start sites and the associated regulatory region of the gene. As shown in Figure 1A, these CpG residues are densely distributed within the CGI and also extend into the intron1 region of the gene. Moreover, the observed CpG distribution in the upstream exon 2 area containing intron1 and promoter (+1 to −2648 bp relative to ATG start codon) is significantly higher than what is expected from normal distribution of the DNA bases (Fig. 1). However, the similar area of the gene in both mice and rats was not significantly enriched in CpGs as compared to that of a human, meaning these animals lack a CpG island within this area (Fig. 1B). Thus, it can be envisaged that the gene regulation by epigenetic factors might differ between the human and rodent α-SYN gene. This entire regulatory area in human SNCA harbors binding sites for several transcription factors, some of which also contain CpG sequence (Jowaed et al., 2010). Therefore, it can be seen that methylation of those CpGs can impede binding of several transcription factors, limiting the transcriptional activity of the gene. However, less methylation or hypomethylation of CpG dinucleotides in the regulatory region of the gene may result in overproduction of the transcript and thus create more protein. This is particularly important because it has been seen that the α-SYN protein level is directly correlated with the gene dosage, i.e. higher expression (Fuchs et al., 2008). As it is known that hypomethylation of a gene is positively associated with its higher expression, it is of immense importance to investigate whether SNCA expression is at all regulated by its DNA methylation of regulatory region and, if so, whether hypomethylation is associated with the disease. Aberrant methylation of CpGs is associated with a wide range of diseases (Jowaed et al., 2010).
Fig. 1. CpG distribution in the SNCA 5′ regulatory region.
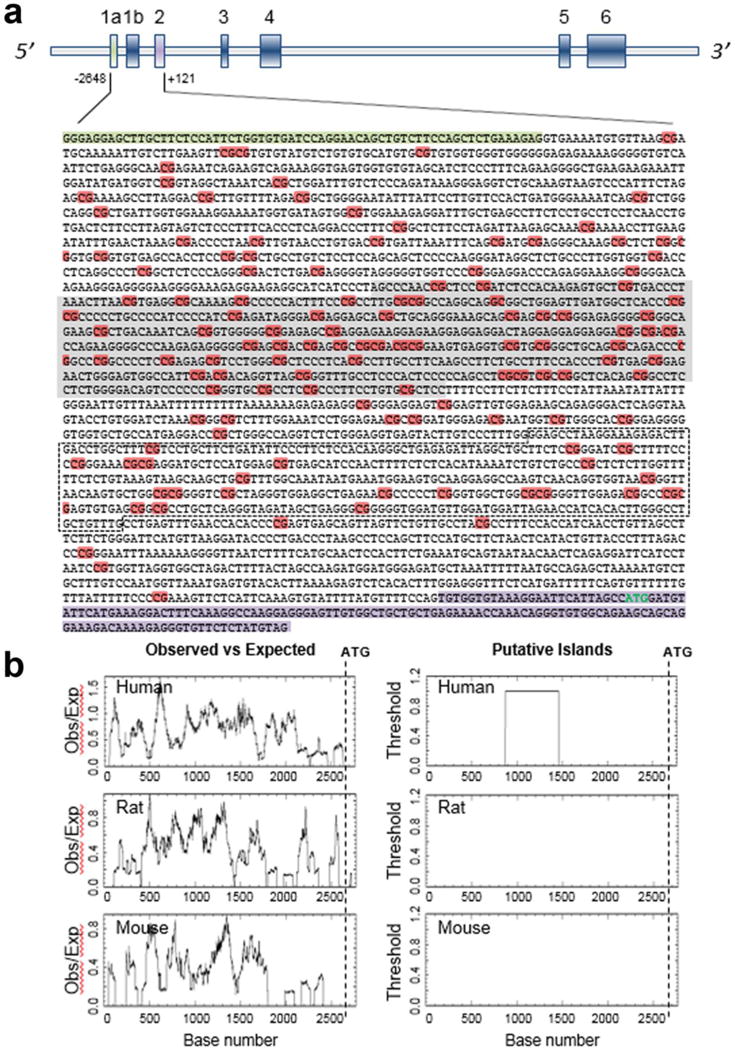
Human SNCA gene has 6 exons with two alternative non-coding exon 1s. The translation start codon is located in exon 2. The vertical bars represent each exon. The sequence from +121 to −2,648 with respect to ATG (+1) (marked in green), encompassing both non-coding exons, intron1 and exon 2, shows very high density of CG bases as highlighted in orange. The presence of the CpG island was predicted using the CpGPLOT program (http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/) with default parameter; the predicted island of 591 base pairs is highlighted in grey. The black dashed marked box represents the region in intron1 whose CpG methylation status has been analyzed by most of the studies (A). Comparative CpG density between human, mouse and rat α-SYN gene (−2,648 to +1) was done using the same program as mentioned above. Analysis revealed a significant difference in CpG base distribution between human and other species when the same region of the gene was tested. Only human showed the presence of a CpG island >200 bp, and the ratio between the observed CpG distribution over expected region ranged mostly in between 1.0 to 1.5. The ratio went down to 0.8 when the other two species were analyzed for CpG distribution for the same region (B). The cut-off for the ratio was set at 0.6.
These important scientific facts have led to investigations on the methylation status of SNCA regulatory regions. Jowaed et al. first studied the significance of the methylation status of these CGIs in the regulation of SNCA in PD cases (Jowaed et al., 2010). Interestingly, the group found that it was not the methylation status of the promoter CGI; rather it was the methylation structure of intron1 CGI that controls the gene expression. At the same time, they also found that the mean methylation level of this area is 10 fold lower in post-mortem human brains as opposed to that of SK-N-SH cells, a human neuroblastoma cell line. Most importantly, they noted significant hypomethylation of SNCA intron1 CpG islands in different regions of brain tissue belonging to PD patients as compared to the controls (Jowaed et al., 2010). Almost the same time, Matsumoto et al. also demonstrated that methylation status of the intron1 CGI of SNCA can regulate this gene’s expression with hypomethylation status being more actively transcribed (Matsumoto et al., 2010). They also showed that HEK-293 cells, an embryonic kidney cell line, express more α-SYN as compared to the known dopaminergic cell line called SH-SY5Y. HEK-293 cells showed a decrease of methylation with a correspondingly increased doses of dopamine, which correlated well with increased protein expression. However, they did not compare the methylation pattern of the same region between HEK-293 cells and SH-SY5Y, which expressed considerably low α-SYN protein at the basal level. Nor did the authors explain how such a high level of DNA methylation in the intron1 region of HEK-293 cells could support a comparatively higher basal levels of α-SYN production (Matsumoto et al., 2010). Importantly, they found significant hypomethylation of said region in the SNpc of PD patients as opposed to controls (0% in patients vs. 100% in controls) (Matsumoto et al., 2010). At this juncture, it is interesting to compare the results of Matsumoto et al. with Wang et al. who also investigated methylation patterns of similar regions in the SNCA intron1 (Matsumoto et al., 2010; Wang et al., 2013). The former study found a relatively higher basal level of α-SYN protein in HEK-293 cells where CGI of intron1 was methylated around 94%. However, they found a relatively lower level of α-SYN in case of SH-SY5Y cells (Matsumoto et al., 2010). The study by Wang et al. reported that the region of SNCA has only 49% methylation in SH-SY5Y cells (Wang et al., 2013). Therefore, it can be said that HEK-293 cells express more α-SYN protein as compared to SH-SY5Y cells, despite having significantly higher methylation at the regulatory region. This might indicate that distinct regulation mechanisms exist in these two different cells.
Another report published by Desplats et al. showed a 2-fold decrease in global DNA methylation in the cortex of human post-mortem PD and DLB (Diffuse Lewy Body) samples with significant hypomethylation of the SNCA intron1 region (Desplats et al., 2011). Interestingly, the authors found that the level of maintenance methyltransferase DNMT1 was significantly decreased in the cortex of both groups of patients, which might have resulted in the global hypomethylation in patient groups (Desplats et al., 2011). The next report which came in 2011 described the status of SNCA intron1 methylation in post-mortem Lewy Body Disease brains using the Next Generation Sequencing (NGS) platform (de Boni et al., 2011). The investigators had detailed information about the patients including Braak staging information for the disease. The overall methylation levels in these two CGIs in different brain regions were far lower, irrespective of sample cohort, and varied from 0.6% (promoter CGI) to 2.8% (in case of intron 1 CGI). Thus, the methylation levels did not vary between cases and controls. However, when comparison of the tissue-specific methylation between different LBD cases with the controls were made, they found relatively significant hypermethylation in the SNCA intron1 in putamen tissues belong to LBD limbic and LBD diffuse neocortical stages with no change in other brain areas. As for CGI in the SNCA promoter region, authors found a non-significant hypomethylation (de Boni et al., 2011). This apparent difference between their findings and the two previous studies can be attributed to the limited availability of fresh frozen brain tissues in the previous studies, as well as different methodologies. In a continuation of their previous finding, a recent report was published in 2015 that demonstrated that SNCA intron1 methylation in brain tissue is slightly increased in aged populations as compared to fetal populations (de Boni et al., 2015). However, no significant change was observed between adult samples of 19–60 years of age with that of seniors more than 60 years old. Most importantly, the authors showed a significant variation of methylation pattern in SNCA intron1 between neuronal and non-neuronal cells of brain tissue (de Boni et al., 2015). At the same time, no sex bias was observed in methylation pattern (de Boni et al., 2015). Likewise, a recent report by our group investigating post-mortem PD and control brain samples found no association of SNCA-intron1 methylation and PD. Nor did we find any significant correlation between α-synuclein protein levels and intron1 methylation in PD (Guhathakurta et al., 2017).
The next group of reports discussed the role of intron1 methylation in regulation of SNCA in leucocytes (peripheral blood mononuclear cell; PBMC) of PD patients. One report by Tan et al. found significant hypomethylation in SNCA intron1 in the patients (5.90%) as compared to healthy controls (7.69%) (Tan et al., 2014). Interestingly, the methylation level was even lower in early-onset PD (EOPD) than in late-onset PD (Tan et al., 2014). Another report by Ai et al. also demonstrated hypomethylation of intron1 CGI in PD patients (Ai et al., 2014). Pihlstrom et al. reported the same significant hypomethylation in SNCA intron1 in PBMC of PD patients as compared to controls (Pihlstrom et al., 2014). This significance was not observed, however, when they analyzed the cortex samples from 12 post-mortem PD patients and an equal number of controls. This discrepancy between two different sets of tissues and individuals was due to a relatively lower number of brain tissue samples (Pihlstrom et al., 2014). Along with this, they found a strong association (p = 5.9×10−5 in leucocyte and p=0.023 in brain tissue) of “G allele” of an SNP within the promoter irrespective of the tissue analyzed, which may serve as a Quantitative Trait Locus (QTL) for the disease (Pihlstrom et al., 2014).
Reports by Richter et al. and Song et al., though, failed to observe any difference in methylation in intron1 of SNCA from peripheral blood leucocytes of PD patients (Richter et al., 2012; Song et al., 2014). This difference in studies might arise partly because different tissues might have different methylation patterns; also, different or additional cytosine residues in the intron1 region might have been considered (apart from those 23 CpGs as shown in Fig. 1). It is worth noting that the patient groups studied in each report might vary in their medication histories as well (Song et al., 2014). Especially since the treatment with L-DOPA (L-3,4-dihydroxyphenylalanine) in PD patients can alter the pool of S-adenosyl methionine (SAM) or S-adenosyl homocysteine (SAH), which can directly affect cytosine methylation in the genome (Obeid et al., 2009; Song et al., 2014).
Recently, two more reports examined the status of SNCA intron1 methylation from PBMC in patients with PD or DLB (Funahashi et al., 2017; Wei et al., 2016). In the first report, the authors examined a Chinese Han population of 91 PD patients and demonstrated a significant hypomethylation in the intron1 but with no difference in α-SYN mRNA expression. They also investigated four PD-implicated SNPs identified in GWAS studies in their sample cohort and observed that rs3756063 is correlated with hypomethylation in the patients (Wei et al., 2016). In another study conducted among Japanese DLB patients (n=20), the authors observed a significant decrease in overall methylation of SNCA intron1 and site-specific hypomethylation. Just as in the previous group, they also failed to observe any increase in α-SYN mRNA levels in PBMC, which could be related to the hypomethylation in the patients (Funahashi et al., 2017). Although the hypomethylation of SNCA intron1 in the pathogenesis of PD remains controversial with both positive and negative outcomes, the DNA methylation of this region in regulation of α-SYN in PD pathogenesis provides some valuable insights into the complex deregulation of SNCA. It is also important to add since epigenetic mark like the DNA methylation status of a gene is considered as heritable and stable, effort has been made to consider using SNCA or some other PD-implicated gene’s methylation status in the peripheral tissues as a biomarker for PD (Jakubowski and Labrie, 2017). Although the hypothesis looks appealing but we must also acknowledge that DNA methylation pattern may vary across different tissues or cell types and that this association of SNCA intron1 methylation pattern with PD is not unanimous. Indeed, several groups found no significant correlation with this gene’s methylation and PD (de Boni et al., 2011; Guhathakurta et al., 2017; Richter et al., 2012; Song et al., 2014). However, contradictory findings might have arisen due to the different ethnicities in the samples of each study, an admixture of different cell types in the samples, limitation of sample size, etc. A recent study by Stoger et al. reported an elevated level of 5-hydroxymethylcytosine in the cerebella of PD samples as opposed to the controls without any significant difference in the global methylation pattern (Stöger et al., 2017). Moreover, in a disease like PD where selective neurodegeneration happens in the SN region of brain, the oxidative stress and cellular environments for that matter are very different as compared to other cell types in the body. It might be possible that this typical cellular environment affect the epigenome of DA neurons altogether differently unlike any other tissues in pathologic conditions of PD. Therefore it is debatable whether peripheral epigenetic marker like DNA methylation can be used as biomarker for detection of PD. Findings related to differential methylation pattern of the same region across different cell types also became evident when we explored ENCODE data for methylation analysis (Fig. 2A). The Hela-S3 cell shows significantly higher methylation, whereas K562 (chronic myelogenous cells), GM12878 (lymphoblastoid cells), and H1-hESC (human embryonic stem cells) showed sparse methylation pattern.
Fig. 2. Differential epigenetic landscape of SNCA promoter in different cell lines.
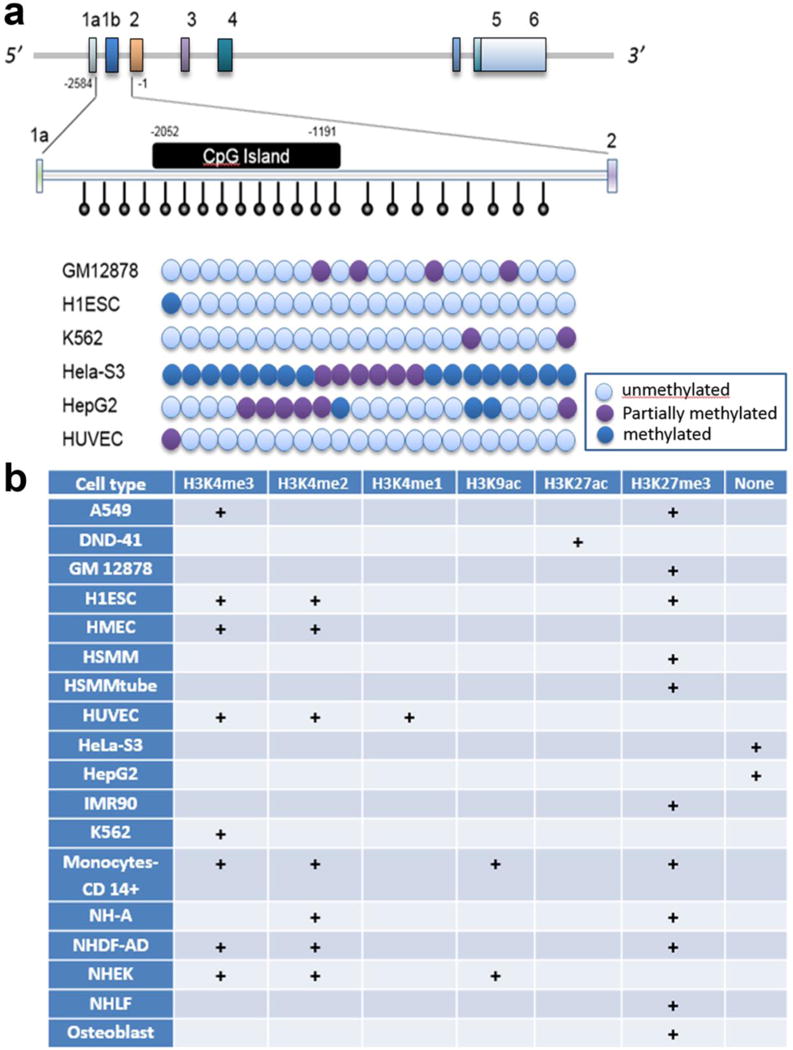
SNCA promoter and upstream region was analyzed for methylation status of its CpG rich area and occupancy of different transcription related histone marks. The entire analyses is based on the data from ENCODE using UCSC genome browser. ENCODE data showed the presence of a CpG island of around 861 bp with the starting base slightly upstream when compared to that of our analysis using CpGPLOT program as shown in Figure 1 (−2052 to −1191 bp in case of ENCODE analysis Vs −1777 to −1187 bp using CpGPLOT program; all positions are designated considering ATG as +1) (A). Twenty-two CpG sites distributed over the SNCA promoter/intron1 area were analyzed using 450K methylation bead array in the ENCODE database. The relative position of those 22 CpG sites within exon 1a to exon 2 is shown by downward vertical bars with round head. The methylation statuses of all 22 CpGs for 6 different cell lines are summarized in the figure: The blue ball represents unmethylated cytosines, pink represents methylated cytosines, and purple shows partially methylated cytosines. The ENCODE database showed that GM12878, H1ESC, HUVEC and K562 remains majorly unmethylated for respective CpGs, HeLa-S3 exhibits a heavily methylated pattern, and HepG2 cells demonstrates partial methylation across the 22 CpG sites. Different histone PTMs associated with transcriptional activity were also assessed from ENCODE database for similar areas of SNCA across different cell lines, as demonstrated by the matrix representation (B). H3K4me1/2/3, H3K9ac, H3K27ac represent active state of chromatin where as H3K27me3 and absence of any histone PTMs (designated as “None”) are associated with inactive state of chromatin. Each cell type shows possession of one or more different types of histone PTMs in similar or slightly different areas in the promoter, and their presence is designated by +. A549, human adenocarcinomic alveolar basal epithelial cell line; DND-41, human T cell leukemic cell line; GM12878, human lymphoblastoid cell line; H1ESC, human embryonic cell line; HMEC, human mammary epithelial cell line; HSMC, human skeletal muscle and myoblast; HSMMtube, skeletal muscle myotubes differentiated from HSMM cell line; HUVEC, human umbilical vein endothelial cells; HeLa-S3, clonal derivative from HeLa cells; HepG2, human liver hepatocellular cells; IMR90, human fetal lung cell line; K562, chronic myelogenous leukemia cell line; NH-A, normal human astrocyte cells; NHDF-AD, normal human dermal fibroblast cells; NHEK, normal human epidermal keratinocytes; NHLF, normal human fibroblast cells.
This evidence indicates that apart from DNA methylation-mediated epigenetic regulation, other epigenetic and genetic mechanisms are also working together towards regulating this gene expression in different types of cells. And this varied status of DNA methylation in different cells of origin might correlate with the gene expression pattern of respective cell type. Thus, we must strictly adhere to the neuronal cells if we are to investigate neuronal SNCA expression. More comprehensive research is necessary to understand this complex epigenetic regulation of the gene and to shed light on synucleinopathy as observed in PD.
2.1.2 Role of Histone post-translational modifications in regulation of α-SYN
Epigenetic regulation mediated by post-translational modification (PTM) of histone tails is one of the most important ways to regulate gene expression (Consortium, 2004; Koch et al., 2007). The amino acid residues of four different histone tails of an octamer undergo several types of PTMs. For example, lysine residues can undergo acetylation, biotinylation, methylation, ubiquitination, SUMOylation; arginine residues can have methylation, citrullination; glutamic acid residues can undergo ADP-ribosylate; serine/threonine residues can undergo phosphorylation (Latham and Dent, 2007). All these modifications have a different impact on genome, e.g. histone acetylation mainly causes de-condensation of chromatin structure and allows transcriptional activation to have a similar type of polarity with the phosphate backbone of DNA (Harrison and Dexter, 2013). Histone H3-lysine 4 trimethylation (H3K4me3) is also considered to be a transcriptionally favorable mark for any gene expression (Consortium, 2012). Despite being such a critical factor for gene expression regulation, it is still almost unexplored in the field of SNCA expression regulation by histone PTMs. However, it has been shown that neurodegeneration is generally associated with global histone deacetylation or hypoacetylation (Harrison and Dexter, 2013). Interestingly, Kontopoulos et al. showed that in the dopaminergic neurons of Drosophila, α-SYN present in the nucleus and prevented histone acetylation, leading to neurotoxicity (Kontopoulos et al., 2006). Recently another study showing overexpression of α-SYN in flies as well as in neuronal cells such as SH-SY5Y can induce H3K9me1 and H3K9me2 levels which in turn modulated H3K9me1/2 methyltransferase (EHMT2)-target gene expression and ultimately resulted in deregulated synaptic activity (Sugeno et al., 2016). In 2013, a report was published which described how α-SYN accumulation in neuronal cell lines leads to the reduction of PKCδ by down regulating p300 protein, acetyltransferase activity, reducing p65 acetylation, and by blocking NF-κB. This pathway was described as a neuroprotective mechanism (Jin et al., 2011). With that, it can be understood that α-synuclein can regulate neurodegeneration epigenetically but the role remains controversial. Liu et al. showed that the SN region of rats, when injected with MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), had significant increases in α-SYN, proteinase-activated receptor 2 (PAR2), and phosphorylation of p65 subunit of NF-κB (Liu et al., 2014). The activation of NF-κB then increased the histone H3 acetylation at the promoter of Snca gene, which led to increased transcription (Liu et al., 2014). The histone acetylation-mediated Snca activation was stopped by down regulating PAR2. It is well known that over-production of α-SYN protein in the neurons contributes to synucleinopathy in PD and other neurodegenerative disorders (Grundemann et al., 2008; Miller et al., 2004). Therefore, it will be important to know how epigenetic factors could lead to deregulated transcription that facilitates an over-production of α-SYN.
Since epigenetic tuning is one of the major players for gene regulations, further investigation is needed to understand how histone modifications of SNCA directly regulate the availability of transcription factors that can get access to the gene.
After the successful completion of the ENCODE project, it is clear that each gene can be regulated in many ways by the presence of different histone PTMs in the regulatory regions of a gene (Consortium, 2012). SNCA is one such gene whose high resolution epigenetic architecture is now available thanks to the completion of ENCODE. If we explore the ENCODE data related to histone modifications of SNCA, an array of complex and variant histone marks on the gene are readily available. It is interesting that different histone marks vary widely in different cell types in the same region of the SNCA. As shown in Figure 2B, the promoter area of SNCA is marked with the transcriptional repressive histone mark called H3K27me3 (histone H3 lysine 27 trimethylation) in IMR90, osteoblast, GM12878, NHLF, HSMM, and HSMM tube cells. Some other cell types—such as HMEC, HUVEC, K562, or NHEK—harbor the transcriptional permissive or initiation marks H3K4me3/2/1 and/or H3K9ac on the SNCA promoter region. This indicates that the gene is transcriptionally active in these cell types. On the other hand, cell types such as A549, H1ESC, CD14+ monocytes, and NHDF-AD pose both the repressive and permissive marks together, encompassing similar areas of SNCA regulatory regions and demonstrating a possibility for bivalent promoter which may either be poised for transcription or remain inactive. At the same time, Hela-S3 or HepG2 cells were not found to be enriched with any type of transcription-related histone marks at the promoter area of the gene, possibly reflecting a non-transcriptional SNCA in those cell types (Fig. 2B). Knowing the epigenetic architecture of a gene’s promoter is particularly important to understand the status of that gene’s transcription in different cell types of the body.
Based on this information, one can predict how a gene becomes transcriptionally active or inactive under disease or pathological conditions. As mentioned above, several studies have been carried out comparing promoter/intron1-methylation structure of SNCA in PD subjects and ethnically matched controls, which were mired controversy. However, comprehensive knowledge of histone structure and the methylation status of SNCA in the PD patients would likely lead to a fair understanding of the cause for the gene’s deregulated expression. By using the NIH roadmap epigenomics project website (http://www.roadmapepigenomics.org), one can access the extensive epigenetic data for any gene from various types of tissues belonging to adult or fetal samples. Likewise, the detail epigenetic information including enrichment of histone marks and DNA methylation data for SNCA from human mid brain SN tissues is readily accessible from this database. In order to understand the complete fidelity of a eukaryotic gene transcription from its initiation to the production of mature transcript, knowing the recruitment of RNA polymerase II (Pol II) and its phosphorylation state of heptapeptide repeat of the c-terminal domain (CTD) is equally important (Brookes and Pombo, 2009). For example, in a case of transcription of active genes, recruitment of Pol II with phosphorylated 5th serine residue within its CTD (S5P) in the transcription start site (TSS) region along with the enrichment of S2P modified Pol II in the coding regions is required (Brookes and Pombo, 2009). This S2P modified Pol II in turn recruits H3K36me3 (histone H3 lysine 36 trimethylation)-specific histone methyl transferase in the gene body to ensure elongation and the generation of a mature transcript (Brookes and Pombo, 2009). On the other hand, absence of Pol II modified at S2P in the gene body marks paused or poised state of transcription in any gene (Brookes and Pombo, 2009). Similarly, it was shown in cases of bivalent promoters marked by both H3K4me3 and H3K27me3 marks at the promoter, the gene is enriched in Pol II S5P while no or low S2P modifications were found as these genes yield in immature transcripts (Brookes and Pombo, 2009; Voigt et al., 2013). As discussed above, because different cell types show the presence of different histone marks at the promoter of SNCA, the transcriptional efficiency could be regulated differently depending on epigenetic marks and the presence of Pol II modifications. It would be encouraging to understand the epigenetic status of the SNCA promoter in the dopaminergic neurons that shows the deregulated expression of α-SYN in PD. We also explored the detailed histone structure of this gene extending from the promoter to the 3′UTR using the UCSC genome browser for ENCODE (https://genome.ucsc.edu/ENCODE/). In addition to finding the presence of peculiar epigenetic marks at the gene’s promoter region in different cell types, as mentioned above, we also noticed significant enriched modifications of H3K27ac (histone H3 lysine 27 acetylation) and H3K4me1 (histone H3 lysine 4 monomehylation) towards the 3′ side of the gene (chromosome 4: 90,657,342–90,771,539; hg19), specifically in the intron4 region (Fig. 3). SNCA intron4 is the longest intron of the gene which spans around 93 kb region of the genome. Significantly, two distinct overlapping peaks of these two histone marks (H3Kme1 and H3K27ac) are present (Fig. 3). Both of these peaks in intron4 are partially overlapped by relatively weaker signals of H3K4me3. H3Kme1 occupancy is usually observed downstream of promoters, especially at the enhancers (Heintzman et al., 2007). Similarly, it has been seen that H3K27ac signals are often associated with active regulatory elements such as enhancers, whereas H3K4me3 is typically enriched at active promoters (Ernst et al., 2011; Heintzman et al., 2007). Co-enrichment of both H3K4me1 and H3K27ac in the genomic regions, interestingly, has been used as a tool to identify the presence of potential enhancer elements (Ernst et al., 2011; Feng et al., 2015; Heintzman et al., 2009; Heintzman et al., 2007; Malik et al., 2014). Therefore, it can be understood from the enrichment of these two histone modifications that SNCA harbors enhancer elements in the intron4 region (Fig. 3). Out of 7 different cell lines studied in the ENCODE for this purpose, HUVEC and NHEK cell lines alone showed the co-presence of these two histone marks at intron4. Cells K562, GM12878, and H1ESC showed no presence of modification. The HSMM and NHLF cell lines, on the other hand, did show the presence of only the distal peak of H3K4me1/H3K27ac towards the 3′ end of the gene. To further confirm the presence of enhancer elements in the intron4, we looked for p300 binding. It has been shown that the binding of this transactivator protein at the genomic regions along with H3K27ac and H3K4me1 also marks enhancer sites (Feng et al., 2015; Heintzman et al., 2009). As expected, the presence of p300 at those two genomic areas of SNCA intron4 showed strong signals of H3K4me1 and H3K27ac. DNaseI hypersensitive regions were also noticed in those areas, fortifying this enhancer prediction. We also observed binding of Pol IIA to that region along with several transcription factors like GATA-2, AP-2c, c-FOS, and c-JUN in a cell-specific manner in in-silico analysis. However, weak occupancy of Pol IIA at some enhancer regions as compared to its enrichment at the 5′ promoter sites has also been documented genome-wide (Heintzman et al., 2007). It is worth noting that enhancer elements are usually located far from the gene promoters in upstream or downstream locations and (mostly in the intergenic or intronic areas) control their target gene (Blackwood and Kadonaga, 1998; Heintzman et al., 2007). However, the possibility that these enhancers serve as part of the transcriptional machinery for the other downstream or upstream neighboring genes of SNCA cannot be ruled out. Therefore, it is important to investigate the regulation of SNCA by these enhancer elements under pathologic conditions like PD. In a recent study conducted by Soldner et al., it was shown that SNCA expression in neuronal cells is actually affected by the distal enhancer elements housed in the intron 4 of the gene (Soldner et al., 2016). Moreover, this elegantly designed study using CRISPR/cas9-based genome editing technique also demonstrated that the G allele of the PD associated SNP rs356168, which is located exactly within the intron 4 distal enhancer region (Fig. 3), can suppress binding of brain specific transcription factors, EMX2 and NKX6-1, at the enhancer site. These factors usually favor suppression of the enhancer activity upon binding to the DNA. The hindrance in the binding of these transcription factors due to risk associated G allele of rs356168 resulted in the generation of an active enhancer element which in turn promoted enhanced transcription of the gene (Soldner et al., 2016). This path breaking report not only pointed out the importance of non-coding variants in regulation of SNCA, it also highlighted how in-depth analysis of the gene’s histone modifications is important in identifying new regulatory elements for their expression.
Figure 3. SNCA gene regulation depends on its non-coding region.
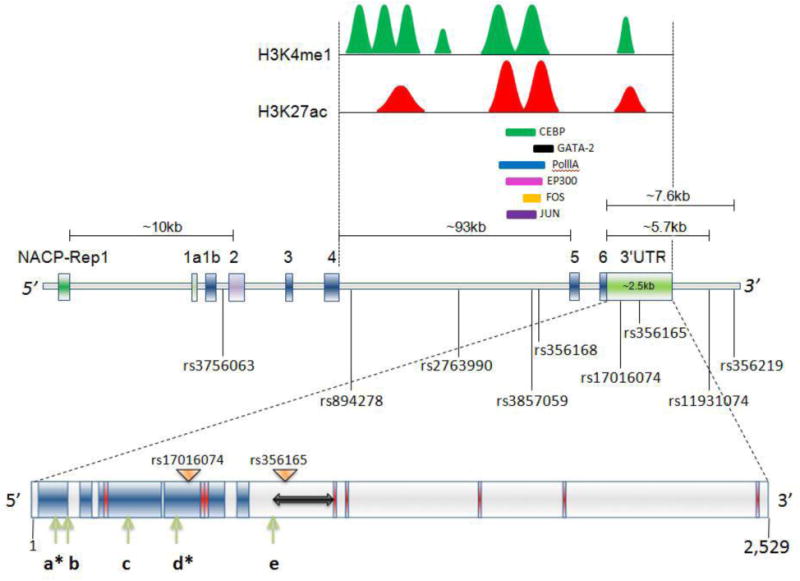
SNCA gene is around 114 kb long which encompasses 6 exons and one very long intron4/5 of around 93 kb. A polymorphic repeat-based enhancer element is present at around 10 kb upstream of ATG known as NACP-Rep1. The intron 4/5 area harbors two distinct regions where transcriptionally active marks H3K27ac (red peaks) and H3K4me1 (green peaks) signals are present. Several transcription factors along with polymerase IIA (Pol IIA) bind to those areas cell type specifically, some of which are shown in the figure. The entire gene harbors thousands of SNPs; of which some significantly demonstrate a repeated association with PD are shown in the figure with their respective IDs and relative position. Two SNPs which were significantly associated with the disease (rs11931074, rs356219) are located around 5.7 and 7.6 kb downstream of the stop codon respectively. The 3′-UTR region of SNCA is approximately 2,529 nucleotides and harbors several motifs and microRNA (miR) binding sites. Features of the long 3′-UTR of SNCA and its regulatory elements are shown: regions in red depict alternative poly (A) signals; arrows labeled a through e are indicative of micro-RNA binding sites (miR-7, miR-34b, miR-34b, miR-153 and miR-34C respectively). Asterisks illustrate miR-7 and miR-153 binding sites that result in significant repression of SNCA. The double-sided arrow represents the LINE region of the SNCA-3′-UTR and dark-blue areas represent the conserved regions of 3′-UTR across the species. Locations of the two SNPs in the UTR that are implicated in PD are shown by the triangles labeled with their respective IDs. The 3′-UTR motifs and conserved sequences are depicted according to Sotiriou et al. (2009). The binding sites of all the microRNAs shown here are adopted from references as discussed in the text (Doxakis, 2010; Kim et al., 2013; Choi et al., 2014; Kabaria et al., 2015). The locations of the H3K27ac/H3K4me1 peaks are adopted from ENCODE. The peak heights, distribution and area under the peaks are not in exact scale.
2.2 SNCA upstream promoter repeat length polymorphism and its association with PD
A promoter and its upstream enhancer-like regulatory regions are very important for controlling gene expression, as the proper coordination between these cis elements can regulate gene expression. Certain epigenetic or genetic changes can block or interfere with the transcriptional regulation of a gene (Jaenisch and Bird, 2003). Therefore, it is always important to know in detail how one gene expression is regulated by different genetic or epigenetic factors. Likewise, the regulatory region of SNCA is also functionally dissected and several important regulatory regions were found which can control its transcription (Chiba-Falek and Nussbaum, 2001). One such area is this gene’s intron1 region which has been shown to possess the capacity to regulate gene expression (Chiba-Falek and Nussbaum, 2001; Jowaed et al., 2010; Matsumoto et al., 2010). As discussed earlier, the CpG methylation within this area remains critical in the regulation of this gene in different cell lines and post-mortem human tissues. Another region also shown to play a key role in SNCA regulation is a polymorphic dinucleotide repeat region located around 10 kb upstream of the translational start site of the gene (Touchman et al., 2001; Xia et al., 1996). This polymorphic locus is called NACP-Rep1 (D4S3481). It is composed of different numbers of dinucleotide repeats and is basically designated as (TC)x (TT)1 (TC)y (TA)z (CA)w, where letters x to z designate variable number of repeats (8 to 13) of corresponding dinucleotides (Chiba-Falek and Nussbaum, 2001; Chiba-Falek et al., 2003; Touchman et al., 2001; Xia et al., 1996). Based on the repeat numbers, five alleles were identified that vary in size by multiples of dinucleotides in different populations (Chiba-Falek and Nussbaum, 2001; Chiba-Falek et al., 2003; Farrer et al., 2001; Mellick et al., 2005). Based on the convention set by Xia et al., these alleles are designated as −1, 0, +1, +2, and +3 and they mainly differ by the number of repeat variations in CA dinucleotides (Chiba-Falek et al., 2003; Xia et al., 1996). However, later reports confirmed that this allelic variation can also arise from differences in other dinucleotide repeats present in the NACP-Rep1 locus such as “TC” or “TA” (Farrer et al., 2001). This polymorphic locus was later described as essentially tri-allelic with 259 base pair (bp) being the short allele, 261 bp being the intermediate, and 263 bp as the long allele (Lill et al., 2012; Maraganore et al., 2006). Interestingly, Chiba-Falek and Nussbaum found that this gene contained significantly different promoter activity and alleles of the NACP-Rep1 locus by using in vitro promoter assay in SH-SY5Y cells (Chiba-Falek and Nussbaum, 2001). The promoter constructs with +1 or +3 allele with higher (11 or 13) CA dinucleotide repeats showed 2.5 to 3 folds higher reporter activity as compared to the 0 allele (10 CA repeats) (Chiba-Falek and Nussbaum, 2001). This observation of repeat number-based increases in promoter activity in the neuronal cell lines is very important since the same effect may also be expected for endogenous transcription of SNCA in PD patients. Individuals with higher repeat alleles will have more α-SYN transcript as compared to people with lower repeat alleles, which may lead to the PD phenotype. As discussed earlier, SNCA gene dosage-based increases (duplication or triplication) in α-SYN protein and the severity of PD is well known in the familial form of PD (Chartier-Harlin et al., 2004; Farrer et al., 2004; Ibanez et al., 2004; Singleton et al., 2003). It is also shown that higher α-SYN transcription is associated with idiopathic PD (Chiba-Falek et al., 2006; Grundemann et al., 2008).
Supporting this idea, several association studies were conducted on different ethnic populations across the globe, revealing positive associations with either +1 or +3 alleles of NACP-Rep1 locus and PD (Farrer et al., 2001; Kruger et al., 1999; Maraganore et al., 2006; Mellick et al., 2005; Mizuta et al., 2002; Tan et al., 2003). Along with the disease association, one study conducted on a Saskatchewan population found a positive association between higher repeat alleles of NACP-Rep1 locus and early-onset PD (Rajput et al., 2009). Interestingly, a study carried out on an Australian population showed that the 0 repeat allele of this locus suggests less transcription of the gene and therefore plays a protective role in the PD population (Mellick et al., 2005). Moreover, another study found significantly less α-SYN transcripts in blood of the PD patients who had protective alleles as compared to other patients with different genotypes (Fuchs et al., 2008). But they did not find such an effect in brain tissue samples (Fuchs et al., 2008). However, Linnertz et al. used a larger cohort of control brain tissue samples and found the lower repeat allele or the protective allele is significantly correlated with lower expression of α-SYN in the regions of the brain that are known to be affected by PD (Linnertz et al., 2009). On the contrary, some studies failed to find any significant correlation between NACP-Rep1 polymorphism and PD in different populations (Izumi et al., 2001; Khan et al., 2001; Parsian et al., 1998; Soldner et al., 2016; Spadafora et al., 2003). It is important to note that a large multipoint study conducted by Maraganore et al. on different NACP-Rep1 association studies and freshly recruited subjects revealed a positive correlation between the higher repeat 263 bp allele and PD, whereas the lower repeat allele 259 bp was found when PD was spared (Maraganore et al., 2006). The reason behind this upstream promoter dinucleotide repeat length-based regulation of gene expression is not very clear, but Chiba-Falek et al. showed that Poly (ADP Ribose) Polymerase-1, PARP-1, might play a repressive role by binding nonspecifically to the NACP-Rep1 area in regulating α-SYN transcription (Chiba-Falek et al., 2005). Chemical-based inhibition of PARP-1 enzymatic activity led to increased promoter activity in full-length NACP-Rep1 containing reporter construct but the PARP-1 inhibition led to decreased promoter activity in a construct lacking an NACP-Rep1 region (Chiba-Falek et al., 2005). This indicates secondary binding sites of PARP-1 in the SNCA upstream promoter/enhancer area or a cooperative function of the molecule with other transcription factors in α-SYN regulation. The regulation of SNCA transcription through NACP-Rep1 is quite intriguing given the complexity of the structure of the cis element. Luciferase assay with an NACP-Rep1 region demonstrated a significant negative effect on transcription, but longer repeat alleles of the locus or construct including the immediate upstream or downstream region of NACP-Rep1 locus showed an additive increase in transcription (Chiba-Falek and Nussbaum, 2001). Moreover, the increase in promoter/enhancer activity based on increments in dinucleotide numbers was also found to be non-linear since 11 repeat alleles could transcribe more than 12 or 13 repeat alleles (Chiba-Falek and Nussbaum, 2001). It is necessary to inspect if this increase in dinucleotide repeats can create binding sites for more transcription factors or if it can change the conformation of the cis-trans binding interface of that region, which may hamper transcriptional efficiency of SNCA.
2.3 Regulation of α-SYN transcription by trans factors
Regulation of transcription by trans factors is considered one of the most important molecular phenomena in terms of gene expression. However, transcription factor binding-mediated regulation of SNCA is rather unappreciated. Very few studies are available that detail this process in the SNCA gene regulation. In 2005, a study was published which identified the CCAAT/enhancer-binding protein β (C/EBPβ) as the transcriptional regulator for α-SYN in the neuroblastoma cell line called SH-SY5Y (Gomez-Santos et al., 2005). It was already known that the SNCA promoter region encompasses binding sites for this family of transcription factors (Vaughan et al., 1998). The authors showed that C/EBPβ induces α-SYN under dose-dependent dopamine stress in SH-SY5Y cells (Gomez-Santos et al., 2005). Next significant study came out in 2008 and it showed that the GATA family of transcription factors can regulate SNCA transcription (Scherzer et al., 2008). In their detailed study, the authors identified 10 GATA binding motifs present throughout the regulatory region of mouse Snca. However, GATA-1 binds specifically to Snca intron1 region but nowhere else on the promoter (Scherzer et al., 2008). Binding of GATA-1 caused a significantly higher expression of Snca in the erythroid precursor cell line. However, this transcription factor does not express itself in brain tissues. It was found that instead of GATA-1, another family member of GATA, i.e. GATA-2, is highly expressed in human SN, superior frontal cortex regions, and also in the dopaminergic neuronal cell line named SH-SY5Y. Concurrently, GATA-2 occupies the same binding regions as GATA-1 in the Snca regulatory region. As expected, knocking down of GATA-2 reduced α-SYN expression significantly in the dopaminergic cell line which indicates that GATA-2 might play a positive role in human transcriptional regulation of SNCA in the dopaminergic neurons (Scherzer et al., 2008). Its interaction with SNCA intron1 in post-mortem PD brain samples, or a possible change in its expression with disease pathology—was not studied. A study by Clough et al. demonstrated a regulation of Snca gene expression by two transcription factors in the PC12 cell line, which are structurally zinc finger proteins (Clough et al., 2009). Among the two identified transcription factors in the study, ZSCAN 21 was found to act as a transcriptional activator by binding to the intron1 region. On the other hand, the other transcription factor, called ZNF219—which binds to the 5′ upstream region relative to intron1—showed a complex mode of function by activating or repressing Snca (Clough et al., 2009). It was shown that ZNF219 acts as a transcriptional activator in the promoter luciferase construct lacking the intron1 region, whereas the same factor acts in the opposite fashion in the presence of intron1 (Clough et al., 2009). Also, this transcription factor failed to activate transcription in conditions where either all three or only one of the binding sites of ZNF219 were present in the luciferase construct. But this effect was reversed when only two binding sites were present, making the pattern of regulation yet to be fully interpreted.
Recently, Brenner et al. studied the three transcription factors C/EBPβ, GATA-2, and ZSCAN21 in PD patients and post-mortem brain samples (Brenner et al., 2015). They showed that ZSCAN21 binds to intron1 of SNCA as expected, but also that GATA-2 specifically binds to the intron2 region while C/EBPβ doesn’t demonstrate a significant binding at any of its four putative binding sites in the SNCA promoter in any different brain regions studied (Brenner et al., 2015). The expression of ZSCAN21 in different brain regions was found to be very low as compared to two other transcription factors. The binding sites for ZSCAN21 and GATA-2 were also screened for any PD-related SNPs, but no significant variant that can alter the binding of these factors to SNCA in PD conditions were found. At the same time, it is important to mention that the binding sites of GATA-2 on the SNCA regulatory region can be regulated by cytosine methylation which has been shown to vary between PD and age-matched control subjects (Jowaed et al., 2010). These apparent discrepancies in binding and expression might have arisen due to the study of different cellular systems. However, these consecutive studies proved again the importance of the intronic region in regulating gene expression (Brenner et al., 2015; Clough et al., 2009; Scherzer et al., 2008). Recently, a new study demonstrated p53 mediated transcriptional activation of α-SYN (Duplan et al., 2016). They demonstrated a strong binding of p53 to the SNCA promoter and its activity was modulated expression of the protein. α-SYN transcript level was induced by p53 overexpression and as expected, the level lowered by knocking down the p53 level (Duplan et al., 2016). All these studies pointed out the importance of cis-trans regulation of α-SYN in the pathogenesis of PD. Comprehensive studies investigating complex interactions between epigenetic and transcription factors in the pathology of PD would help to understand its complex biology.
2.4 Post-transcriptional regulation of α-SYN expression by micro RNAs and alternative splicing
In addition to the several layers of regulations described above—such as epigenetic mechanisms, transcriptional regulations, and promoter polymorphism-mediated regulations, SNCA expression is fine-tuned by post-transcriptional regulatory mechanisms as well. Generally, the 3′-UTR (3′-untranslated region) of a gene transcript plays a major role in generating different lengths of mRNA transcripts tissue-specifically by using alternative polyadenylation signals (Licatalosi and Darnell, 2010). Usually gene harbors cis sequences for several micro RNA (miRNA) binding sites and RNA binding proteins (Licatalosi and Darnell, 2010; Wang and Yi, 2014). Consequently, naturally occurring SNPs within those recognition sequences can alter the binding of miRNAs to the target mRNAs, which can ultimately jeopardize the stability and expression of the RNAs. Recently, careful reconsideration of the global RNA sequencing data by Miura et al. revealed neuron-specific bias in long 3′-UTR-containing transcripts. This indicates a complex post-transcriptional regulation of gene expression in the brain (Miura et al., 2013). α-SYN mRNA has a long 3′-UTR region of around 2529 nucleotide (nt) which is significantly longer than its coding sequence (Fig. 3) (Rhinn et al., 2012; Sotiriou et al., 2009). Bioinformatics analysis of the 3′-UTR of α-SYN sequence using Poly A signal Miner (http://dnafsminer.bic.nus.edu.sg/PolyA.html) revealed the presence of 8 alternative Poly A signals (Fig. 3) as found by others as well (Sotiriou et al., 2009). By using these alternative Poly A signals, an array of short (290–560 nt) to moderate (1070 nt), to long (~2520 nt) 3′-UTR containing α-SYN mRNAs are generated in different tissues (Rhinn et al., 2012; Sotiriou et al., 2009). It is important to note that most of the tissues harbor α-SYN transcripts containing smaller 3′-UTR (574 nt) (95%) and that part of the 3′-UTR is evolutionarily conserved across the species (Sotiriou et al., 2009). Recently, it has been shown that the existence of α-SYN transcripts contain longer 3′-UTR (≥560 to 2520 nt) in the cerebral cortex of PD patients as compared to age-matched controls (Rhinn et al., 2012). Moreover, it has also been found that the α-SYN transcripts containing long 3′-UTR tend to localize away from the nerve terminals and more towards the mitochondria, indicating possible mitochondria-mediated selective regulation for this transcript as well (Rhinn et al., 2012). Discovery of these short and long 3′-UTR-length α-SYN transcripts in different tissues and PD brain samples respectively, paves the way to identify the common or disease-specific miRNA or SNP-mediated regulation of α-SYN.
The most explored mechanism for post-transcriptional gene regulation is microRNA-mediated gene repression. The miRNAs are the small non-coding RNA molecules that are being transcribed from non-protein coding sequences of a gene (Doxakis, 2013). These RNA molecules are relatively small (around 17 to 24 nucleotide long) and exert their repressive function by binding to the complementary sequence on the 3′-UTR of target mRNA sequence (Doxakis, 2010). So far, 1,881 miRNAs from human are reported in the database (http://www.mirbase.org) and each of these has more than one target mRNA. miRNA-mediated gene regulation has long been implicated in PD and the presence of several different miRNAs has been shown in different tissues of PD patients (Cardo et al., 2013; Kim et al., 2007; Ma et al., 2013; Maciotta et al., 2013; Margis et al., 2011). Among different genes which are associated with PD, SNCA remains one such gene whose post-transcriptional regulation is mediated by miRNA (Doxakis, 2010; Junn et al., 2009; Ma et al., 2013; Maciotta et al., 2013). Two microRNAs which received maximum attention regarding modulation of α-SYN expression are miR-7 and miR-153 (Doxakis, 2010; Junn et al., 2009). Both of these miRNAs and their common target gene SNCA are expressed at high levels in neurons of different regions of the midbrain (Doxakis, 2010; Junn et al., 2009; Mouradian, 2012). The binding sites for these two miRNAs on the 3′-UTR of α-SYN mRNA are only 335 bases apart and both of these sites are evolutionarily conserved among vertebrate species (Mouradian, 2012). Interestingly, it was shown that miR-7 and miR-153 together repressed SNCA expression by 47% in a co-transfection experiment in HEK-293 cells and that a point mutation on the binding region of these miRNAs significantly reduced this repression (Doxakis, 2010). Similarly, Junn et al. showed that inhibiting miR-7 binding by chemically modified anti-miR mRNA inhibitor increased the α-SYN mRNA level in SH-SY5Y cells (Junn et al., 2009). Interestingly, in a recent study done by Kim et al. who sequenced the miR-7 and miR-153 binding sites on SNCA among 262 PD patients, it was found that a single patient harbored a C to A SNP (designated as SNCA 3′-UTR 464 C>A) within the miR-153 binding site (Kim et al., 2013). The SNP was predicted to change the binding kinetics of miR-153 to α-SYN mRNA and compensate for the repressive effect of this microRNA on α-SYN expression. The patient who harbored this mutation developed PD at an early age without any familial history of the disease (Kim et al., 2013). However, any direct correlation between this rare variant and the disease pathogenesis couldn’t be made (Kim et al., 2013). More studies need to be done on the implications of these two miRNA (miRNA-7 and miRNA-153) mediated α-SYN expressions regarding PD pathogenesis. Since these two miRNA binding sites fall within the part of a conserved 3′-UTR sequence of SNCA, it would be quite interesting to investigate the miR-7 and miR-153 binding profiles in PD patients. Apart from these two miRNA sites, two more miRNAs, miR-34b and miR-34c binding sites are present in the longer transcripts of SNCA that have also been shown to modulate α-SYN expression by direct interaction with the gene’s 3′UTR (Kabaria et al., 2015; Rhinn et al., 2012). Moreover, these two miRNAs were found to have been downregulated in PD conditions (Minones-Moyano et al., 2011).
In a well-organized study by Rhinn et al., it was demonstrated that single nucleotide changes made by two SNPs, rs356165 and rs78991202, in the binding sites of miR-34b could reverse the effect of this miRNA on α-SYN gene expression under dopamine treatment in SH-SY5Y cells (Rhinn et al., 2012). It is noteworthy to mention that SNP rs356165 has been consistently shown to be associated with PD in independent studies and in large scale meta-analyses alike (Cardo et al., 2012; Nalls et al., 2014). Interestingly, the study by Sotiriou et al. discovered a minor presence (5%) of α-SYN transcript containing a longer 3′-UTR (more than 574 nt long) in different human tissues (Sotiriou et al., 2009). Therefore, they concluded that this SNP (rs356165)—which is located within the longer 3′-UTR of α-SYN transcript—only reflects a very low percentage of association between it and PD (Sotiriou et al., 2009). Thus, it is understood that SNCA expression can be downregulated by miRNA binding under different experimental models and that these miRNAs are enriched in the brain (Doxakis, 2010; Junn et al., 2009; Kabaria et al., 2015). It would be interesting to sequence those particular miRNA binding sites of SNCA-3′-UTR in PD patients who have higher transcripts of α-SYN. This might help in discovering functional SNPs/novel mutations that might interfere with the binding of miRNAs, which could result in higher α-SYN expression.
Another important molecular event which influences RNA processing and its expression is alternative splicing (Keren et al., 2010; Mazin et al., 2013; Xu et al., 2002). This splicing mechanism has gained a great deal of interest as it has been shown to generate a variety of proteins from a single pre-mature transcript (Wang et al., 2015). Significant amounts of mammalian genes undergo alternative splicing, which ultimately generates a huge array of proteins that outnumbers the total number of genes that are present in the body (International Human Genome Sequencing, 2004; Mazin et al., 2013; McLean et al., 2012; Wang et al., 2015; Xu et al., 2002). α-SYN gene is no exception, as it also undergoes alternative splicing (Beyer and Ariza, 2013; McLean et al., 2012). The fascinating part of this is that some differentially spliced α-SYN transcripts are shown to be associated with oxidative stress, PD, and other neurodegenerative diseases (Beyer et al., 2004; Kalivendi et al., 2010; McLean et al., 2012). As shown in Figure 1, SNCA contains mainly 6 exons with some alternative non-coding exons in the 5′-UTR. Four different alternatively spliced mRNAs are generated as follows: (i) SNCA-140 where all six exons are present; (ii) SNCA-126 where exon3 is missing; (iii) SNCA-112 where exon5 is spliced out; and (iv) SNCA-98 where both exon3 and exon5 are missing (Beyer et al., 2008; Campion et al., 1995; Ueda et al., 1994). The importance of these alternatively spliced transcripts of SNCA could be huge, as these partially-truncated transcripts generate a range of α-SYN protein specie which are prone to aggregate depending on which part of the protein domain is missing due to an alternative splicing event (Beyer and Ariza, 2013). A study by Beyer et al. showed an increase in SNCA-112 isoform in the frontal cortex of patients with Dementia with Lewy body disease (Beyer et al., 2004). The study also showed that SNCA-98 is a brain-specific isoform of α-SYN as opposed to the other three alternatively spliced transcripts (Beyer et al., 2008). Moreover, this transcript level was significantly higher in the frontal cortices of PD post-mortem brains and Lewy body disease (Beyer et al., 2008). A later study, however, found significantly higher expressions of SNCA-140 and SNCA-126 in the SN region of PD brains as compared to matched controls (McLean et al., 2012). At the same time, they failed to identify any over-representation of SNCA-98 in the frontal cortex of PD patients but they found this transcript to be significantly higher in the SN region as opposed to the cortex of the PD samples (McLean et al., 2012). Another important study conducted by Kalivendi et al. showed that SNCA-112 transcript becomes increasingly prevalent in the SN region of MPTP mice models after 7 and 14 days of treatment (Kalivendi et al., 2010). The same study also demonstrates that SNCA-112 isoform got dose-dependently upregulated in the SH-SY5Y cell line following treatment with oxidative stress-inducing chemicals such as MPP+, rotenone, and 6-hydroxydopamine (6-OHDA) (Kalivendi et al., 2010). These investigations all indicate that alternative splicing of SNCA is a tissue-specific phenomenon and is strongly correlated with oxidative stress and neurodegenerative diseases. It will be of interesting to investigate this further in order to see the underlying molecular mechanism and learn how oxidative stress or pathogenic conditions like PD in the dopaminergic neurons can aggravate production of specific transcript of SNCA, which ultimately cause the degenerative changes.
2.5 Regulation of SNCA transcription by single nucleotide polymorphisms (SNPs) and its impact on PD
The role of SNPs in the pathogenesis of PD has been shown to be unequivocal; these natural genetic variants play an important role as a disease marker if they are overrepresented in concerned disease populations (Satake et al., 2009; Simon-Sanchez et al., 2009). Several genes have been shown to be associated with PD based on their functional characteristics which were found to be compromised in PD patients. This genetic malfunctioning could also be due to the presence of fewer single nucleotide mutations in coding regions of the gene, as seen in the case of α-SYN and its association with familial forms of PD (Appel-Cresswell et al., 2013; Kruger et al., 1998; Lesage et al., 2013; Pasanen et al., 2014; Polymeropoulos et al., 1997; Proukakis et al., 2013; Zarranz et al., 2004). However, in cases of idiopathic PD—which are significantly more prevalent than familial forms—there are multiple SNPs that have been studied irrespective of their position or functionality on the gene. These SNPs were compared between PD patients and healthy individuals of a homogenous population to discover if any allele of these variants was significantly overrepresented in the patients. If we look in the NCBI database for SNPs on SNCA, around 32,334 SNPs can be found that are located at different positions of the gene. Of this enormous number of SNPs, not all are associated with (or predicted to be associated with) PD. However, a significant number of them were found to have an association with PD in different populations (Nalls et al., 2014). To date, several genome-wide association studies (GWAS) have been performed with PD patient populations, which revealed significant associations of some SNPs located towards the 3′ end of SNCA with PD (Fung et al., 2006; International Parkinson Disease Genomics et al., 2011; Satake et al., 2009; Simon-Sanchez et al., 2009). A recently performed systematic meta-analysis summed up the significance of the SNPs found to be associated with PD in different studies (pdgene.org) (Nalls et al., 2014). However, the role of most of these associated SNPs in the SNCA regulation is yet to be explored. Two significant linkage disequilibrium (LD) blocks (one encompassing intron4 to downstream of the 3′-UTR area and another one at the promoter encompassing region at the 5′ side of gene) were found to have been significantly associated with PD (Mueller et al., 2005). SNPs located within this 3′ LD block area were shown to play a role in gene processing (Mueller et al., 2005). Linnertz et al. showed that protective genotypic combinations of the two SNPs within this 3′ LD block, namely rs356219 and rs356165, were strongly correlated with lower mRNA levels of SNCA in the temporal cortex and midbrain region that encompassed the SN (Linnertz et al., 2009). Interestingly, it was also shown that SNPs in the non-protein coding region of the gene, such as the intronic region (rs2736990) or the 3′-UTR (rs356165), even SNPs located further downstream of the 3′-UTR (rs356219; ~7.65 kb down of stop codon) are strongly associated with enrichment of one splicing variant of SNCA called SNCA-112 in the frontal cortex (Beyer, 2006; McCarthy et al., 2011). Likewise, it was shown using a luciferase construct containing the minor allele of SNP (rs17016074)—which is located between the two frequently used Poly A sites in the 3′-UTR—that there was an upregulated expression of α-SYN in SH-SY5Y cells (Sotiriou et al., 2009). It can be interpreted that people genotyped for minor allele of rs17016074 might express more α-SYN. Interestingly, the study by Rhinn et al. showed that the risk allele of another intron4 variant (rs356168) is strongly associated with the longer 3′-UTR containing mRNA of SNCA in human cortical tissues (Rhinn et al., 2012). The authors also found this intronic variant to be in tight LD with a 3′-UTR SNP rs356165 located 892 bp after the stop codon. At the same time, another study demonstrated functional significance of an SNP (rs3756063) located within the 5′ LD block of the gene, showing the strong association between the G allele of the SNP and the hypomethylation in PD patients (Pihlstrom et al., 2014).
It was also predicted that this SNP might alter the binding of GATA-1 on the intron1 of SNCA due to its vicinity to the GATA-1 binding site; however, no experiment has been performed to prove this concept. It is conceivable that SNPs located within the promoter/regulatory region of the gene may alter the epigenetic environments that could interfere with transcription factor binding. But SNPs located within the middle or end of large introns of SNCA—like intron4 (rs2736990, rs3857059, rs894278) and also further downstream of the 3′-UTR of the gene (rs11931074, rs356219)—have consistently been found to be associated with idiopathic PD (International Parkinson Disease Genomics et al., 2011; Satake et al., 2009; Simon-Sanchez et al., 2009). It was predicted that these SNPs might play a role in RNA stability, miRNA binding, or even in alternative splicing (Satake et al., 2009; Simon-Sanchez et al., 2009). However, exact functionality of these SNPs in the SNCA regulation in PD has yet to be determined. It is will be fascinating to learn how these intronic or intergenic SNPs contribute to expression of SNCA, which can lead to PD. To our surprise, we found that the two neighboring SNPs, rs356168 and rs3857059 (only 807 bp apart), which were significantly associated with PD, are positioned within the putative enhancer region of intron4 (Fig. 3). It is quite possible that the polymorphic alleles of these two SNPs might alter the binding of important trans factors at that area which in turn can influence the overall transcriptional activity of the gene under pathologic conditions such as PD. As mentioned earlier, a recent study by Soldner et al. indeed found that the SNP rs356168 can alter enhancer activity of this gene (Soldner et al., 2016). This again indicates the importance of studying the functional aspects of intronic polymorphic variants in relation to transcriptional activity of the gene. Additionally, we also observed that two 3′-UTR variants—such as rs17016074 (located within the core 3′-UTR region of 600 bp) and rs356165 (located in the longer 3′-UTR region)—are also located within the H3K27ac/H3K4me1 signal spanning area of the 3′-UTR of SNCA. It is quite possible that the risk allelic combinations of these SNPs which were found to be strongly associated with PD might contribute towards SNCA transcription by modulating regulatory factor binding to this secondary transcriptionally active area of the gene.
3. Conclusion
Regulation of α-SYN has long been a major focus for PD researchers. Since the discovery of the genetic component of familial PD—that is, SNCA multiplication, the presence of genetic mutations in familial PD, and also the presence of α-SYN protein in the Lewy bodies—a constant effort has been made to decipher the abnormal regulation of this gene in pathological conditions (Kruger et al., 1998; Lesage et al., 2013; Pasanen et al., 2014; Polymeropoulos et al., 1997; Proukakis et al., 2013; Singleton et al., 2003; Spillantini et al., 1997; Zarranz et al., 2004). However, relatively little research has been made towards the epigenetic or transcriptional or post-transcriptional regulation of α-SYN as compared to post-translational modifications of this protein to understand its aggregation pattern. In this review, we have discussed the potential importance of histone post-translational modifications in the regulation of SNCA, which has never been discussed in the literature in detail.
After the discovery of the location-based histone modifications throughout the genome, it has become vital that we revisit our research with all of the epigenetic, cis and trans elements related to SNCA in order to get a comprehensive picture of this gene regulation. We have seen that epigenetic structures such as DNA methylation and histone modifications of the same genetic region vary significantly across different cell types. It is now becoming evident that local chromatin structure of a gene plays a crucial role in its regulation. In order to study the gene regulation from SNpc dopaminergic neurons, it is highly recommended to isolate these neurons using precise laser capture microscopic techniques from the SN tissues without introducing confounding errors from neighboring cell types. We have seen that several active regulatory histone marks are present in the non-protein coding regions of SNCA; as such, it is of paramount importance to study the contribution of these non-traditional transcriptionally active areas in gene regulation under diseased conditions such as PD. Moreover, it is also vital to fully characterize the areas of the gene where H3K27ac and H3K4me1 signals have overlapped. It is already found that SNPs, which are strongly associated with PD, are mostly located in the intronic or UTR sequences or even further away from the gene. Armed with a true understanding of the epigenetic structure of SNCA, we could interpret the importance of these variants in disease pathogenesis. Simultaneously, it is also becoming increasingly necessary to study the transcriptional regulation of a gene via endogenous gene modification techniques like CRISPR/Cas9 that will preserve the gene’s epigenetic integrity and not reporter gene assays, where contributions of epigenetic modifications in gene regulation are largely ignored.
Highlights.
Epigenetic regulation of α-synuclein plays important role in Parkinson’s disease
Completion of ENCODE has revolutionized the present concept of SNCA regulation
Histone modifications at SNCA regulatory regions define cell-specific expression
Presence of intronic enhancer elements regulate SNCA expression
Non-coding SNCA variants alter gene expression by interfering epigenetic structure
Acknowledgments
The authors gratefully acknowledge the financial supports provided to YSK by the National Institute of Health (grant numbers 5R21NS088923), Michael J Fox Foundation for Rapid Response Awards 2014. The authors acknowledge Mr. James Holland from Burnett School of Biomedical Sciences for his contribution to professional editing and also Mr. Sambuddha Basu for careful proof-reading the article.
Abbreviations
- α-SYN
α-synuclein
- CGI
CpG island
- CTD
c-terminal domain
- DA
dopaminergic neurons
- DLB
Diffuse Lewy Body
- ENCODE
Encyclopedia of DNA Elements
- EOPD
early onset Parkinson’s disease
- GWAS
genome-wide association studies
- H3K4me1
Histone H3-lysine 4 monomethylation
- H3K4me3
Histone H3-lysine 4 trimethylation
- H3K27me3
histone H3 lysine 27 trimethylation
- H3K9ac
hitone lysine 9 acetylation
- H3K27ac
histone lysine 27 acetylation
- H3K36me3
histone H3 lysine 36 trimethylation
- L-DOPA
L-3,4-dihydroxyphenylalanine
- LD
linkage disequilibrium
- miRNA
micro RNA
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PD
Parkinson’s disease
- PARP-1
Poly (ADP Ribose) Polymerase-1
- PTM
post translational modification
- QTL
Quantitative Trait Locus
- SAM
S-adenosyl methionine
- SAH
S-adenosyl homocysteine
- SNCA
[synuclein, alpha (non A4 component of amyloid precursor)]
- SNpc
substantia nigra pars compacta
- SNP
single nucleotide polymorphisms
- UTR
untranslated region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflicts of interests
References
- Ai SX, Xu Q, Hu YC, Song CY, Guo JF, Shen L, Wang CR, Yu RL, Yan XX, Tang BS. Hypomethylation of SNCA in blood of patients with sporadic Parkinson’s disease. Journal of the neurological sciences. 2014;337:123–128. doi: 10.1016/j.jns.2013.11.033. [DOI] [PubMed] [Google Scholar]
- Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- Basu S, Adams L, Guhathakurta S, Kim YS. A novel tool for monitoring endogenous alpha-synuclein transcription by NanoLuciferase tag insertion at the 3′end using CRISPR-Cas9 genome editing technique. Sci Rep. 2017;8:45883. doi: 10.1038/srep45883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Je G, Kim YS. Transcriptional mutagenesis by 8-oxodG in alpha-synuclein aggregation and the pathogenesis of Parkinson’s disease. Exp Mol Med. 2015;47:e179. doi: 10.1038/emm.2015.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(Suppl 1):S4–11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Beitz JM. Parkinson’s disease: a review. Front Biosci (Schol Ed) 2014;6:65–74. doi: 10.2741/s415. [DOI] [PubMed] [Google Scholar]
- Beyer K. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta neuropathologica. 2006;112:237–251. doi: 10.1007/s00401-006-0104-6. [DOI] [PubMed] [Google Scholar]
- Beyer K, Ariza A. alpha-Synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol Neurobiol. 2013;47:509–524. doi: 10.1007/s12035-012-8330-5. [DOI] [PubMed] [Google Scholar]
- Beyer K, Domingo-Sabat M, Lao JI, Carrato C, Ferrer I, Ariza A. Identification and characterization of a new alpha-synuclein isoform and its role in Lewy body diseases. Neurogenetics. 2008;9:15–23. doi: 10.1007/s10048-007-0106-0. [DOI] [PubMed] [Google Scholar]
- Beyer K, Lao JI, Carrato C, Mate JL, Lopez D, Ferrer I, Ariza A. Differential expression of alpha-synuclein isoforms in dementia with Lewy bodies. Neuropathol Appl Neurobiol. 2004;30:601–607. doi: 10.1111/j.1365-2990.2004.00572.x. [DOI] [PubMed] [Google Scholar]
- Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic acids research. 1980;8:1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- Brenner S, Wersinger C, Gasser T. Transcriptional regulation of the alpha-synuclein gene in human brain tissue. Neuroscience letters. 2015;599:140–145. doi: 10.1016/j.neulet.2015.05.029. [DOI] [PubMed] [Google Scholar]
- Brookes E, Pombo A. Modifications of RNA polymerase II are pivotal in regulating gene expression states. EMBO reports. 2009;10:1213–1219. doi: 10.1038/embor.2009.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion D, Martin C, Heilig R, Charbonnier F, Moreau V, Flaman JM, Petit JL, Hannequin D, Brice A, Frebourg T. The NACP/synuclein gene: chromosomal assignment and screening for alterations in Alzheimer disease. Genomics. 1995;26:254–257. doi: 10.1016/0888-7543(95)80208-4. [DOI] [PubMed] [Google Scholar]
- Cardo LF, Coto E, de Mena L, Ribacoba R, Lorenzo-Betancor O, Pastor P, Samaranch L, Mata IF, Diaz M, Moris G, Menendez M, Corao AI, Alvarez V. A search for SNCA 3′ UTR variants identified SNP rs356165 as a determinant of disease risk and onset age in Parkinson’s disease. Journal of molecular neuroscience : MN. 2012;47:425–430. doi: 10.1007/s12031-011-9669-1. [DOI] [PubMed] [Google Scholar]
- Cardo LF, Coto E, de Mena L, Ribacoba R, Moris G, Menendez M, Alvarez V. Profile of microRNAs in the plasma of Parkinson’s disease patients and healthy controls. Journal of neurology. 2013;260:1420–1422. doi: 10.1007/s00415-013-6900-8. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Cheng X, Blumenthal RM. Introduction–Epiphanies in epigenetics. Prog Mol Biol Transl Sci. 2011;101:1–21. doi: 10.1016/B978-0-12-387685-0.00001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba-Falek O, Kowalak JA, Smulson ME, Nussbaum RL. Regulation of alpha-synuclein expression by poly (ADP ribose) polymerase-1 (PARP-1) binding to the NACP-Rep1 polymorphic site upstream of the SNCA gene. American journal of human genetics. 2005;76:478–492. doi: 10.1086/428655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba-Falek O, Lopez GJ, Nussbaum RL. Levels of alpha-synuclein mRNA in sporadic Parkinson disease patients. Movement disorders : official journal of the Movement Disorder Society. 2006;21:1703–1708. doi: 10.1002/mds.21007. [DOI] [PubMed] [Google Scholar]
- Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the alpha-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Human molecular genetics. 2001;10:3101–3109. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- Chiba-Falek O, Touchman JW, Nussbaum RL. Functional analysis of intra-allelic variation at NACP-Rep1 in the alpha-synuclein gene. Human genetics. 2003;113:426–431. doi: 10.1007/s00439-003-1002-9. [DOI] [PubMed] [Google Scholar]
- Chu Y, Kordower JH. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiology of disease. 2007;25:134–149. doi: 10.1016/j.nbd.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Clough RL, Dermentzaki G, Stefanis L. Functional dissection of the alpha-synuclein promoter: transcriptional regulation by ZSCAN21 and ZNF219. Journal of neurochemistry. 2009;110:1479–1490. doi: 10.1111/j.1471-4159.2009.06250.x. [DOI] [PubMed] [Google Scholar]
- Consortium, E.P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupland KG, Mellick GD, Silburn PA, Mather K, Armstrong NJ, Sachdev PS, Brodaty H, Huang Y, Halliday GM, Hallupp M, Kim WS, Dobson-Stone C, Kwok JB. DNA methylation of the MAPT gene in Parkinson’s disease cohorts and modulation by vitamin E in vitro. Movement disorders : official journal of the Movement Disorder Society. 2014;29:1606–1614. doi: 10.1002/mds.25784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristovao AC, Guhathakurta S, Bok E, Je G, Yoo SD, Choi DH, Kim YS. NADPH oxidase 1 mediates alpha-synucleinopathy in Parkinson’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:14465–14477. doi: 10.1523/JNEUROSCI.2246-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boni L, Riedel L, Schmitt I, Kraus TF, Kaut O, Piston D, Akbarian S, Wullner U. DNA methylation levels of alpha-synuclein intron 1 in the aging brain. Neurobiology of aging. 2015 doi: 10.1016/j.neurobiolaging.2015.08.028. [DOI] [PubMed] [Google Scholar]
- de Boni L, Tierling S, Roeber S, Walter J, Giese A, Kretzschmar HA. Next-generation sequencing reveals regional differences of the alpha-synuclein methylation state independent of Lewy body disease. Neuromolecular medicine. 2011;13:310–320. doi: 10.1007/s12017-011-8163-9. [DOI] [PubMed] [Google Scholar]
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes & development. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Spencer B, Coffee E, Patel P, Michael S, Patrick C, Adame A, Rockenstein E, Masliah E. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. The Journal of biological chemistry. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. The Journal of biological chemistry. 2010;285:12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxakis E. Principles of miRNA-target regulation in metazoan models. International journal of molecular sciences. 2013;14:16280–16302. doi: 10.3390/ijms140816280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duplan E, Giordano C, Checler F, Alves da Costa C. Direct alpha-synuclein promoter transactivation by the tumor suppressor p53. Mol Neurodegener. 2016;11:13. doi: 10.1186/s13024-016-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Annals of neurology. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, West A, de Silva R, Hardy J, Hernandez D. alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Human molecular genetics. 2001;10:1847–1851. doi: 10.1093/hmg/10.17.1847. [DOI] [PubMed] [Google Scholar]
- Feng J, Shao N, Szulwach KE, Vialou V, Huynh J, Zhong C, Le T, Ferguson D, Cahill ME, Li Y, Koo JW, Ribeiro E, Labonte B, Laitman BM, Estey D, Stockman V, Kennedy P, Courousse T, Mensah I, Turecki G, Faull KF, Ming GL, Song H, Fan G, Casaccia P, Shen L, Jin P, Nestler EJ. Role of Tet1 and 5-hydroxymethylcytosine in cocaine action. Nature neuroscience. 2015;18:536–544. doi: 10.1038/nn.3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs J, Tichopad A, Golub Y, Munz M, Schweitzer KJ, Wolf B, Berg D, Mueller JC, Gasser T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22:1327–1334. doi: 10.1096/fj.07-9348com. [DOI] [PubMed] [Google Scholar]
- Funahashi Y, Yoshino Y, Yamazaki K, Mori Y, Mori T, Ozaki Y, Sao T, Ochi S, Iga JI, Ueno SI. DNA methylation changes at SNCA intron 1 in patients with dementia with Lewy bodies. Psychiatry Clin Neurosci. 2017;71:28–35. doi: 10.1111/pcn.12462. [DOI] [PubMed] [Google Scholar]
- Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert ML, Schymick J, Okun MS, Mandel RJ, Fernandez HH, Foote KD, Rodriguez RL, Peckham E, De Vrieze FW, Gwinn-Hardy K, Hardy JA, Singleton A. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. The Lancet Neurology. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- Gallegos S, Pacheco C, Peters C, Opazo CM, Aguayo LG. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front Neurosci. 2015;9:59. doi: 10.3389/fnins.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert reviews in molecular medicine. 2009;11:e22. doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- Gasser T, Hardy J, Mizuno Y. Milestones in PD genetics. Movement disorders : official journal of the Movement Disorder Society. 2011;26:1042–1048. doi: 10.1002/mds.23637. [DOI] [PubMed] [Google Scholar]
- George JM. The synucleins. Genome Biol. 2002;3:REVIEWS3002. doi: 10.1186/gb-2001-3-1-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Santos C, Barrachina M, Gimenez-Xavier P, Dalfo E, Ferrer I, Ambrosio S. Induction of C/EBP beta and GADD153 expression by dopamine in human neuroblastoma cells. Relationship with alpha-synuclein increase and cell damage. Brain Res Bull. 2005;65:87–95. doi: 10.1016/j.brainresbull.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Grundemann J, Schlaudraff F, Haeckel O, Liss B. Elevated alpha-synuclein mRNA levels in individual UV-laser-microdissected dopaminergic substantia nigra neurons in idiopathic Parkinson’s disease. Nucleic acids research. 2008;36:e38. doi: 10.1093/nar/gkn084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guhathakurta S, Evangelista BA, Ghosh S, Basu S, Kim YS. Hypomethylation of intron1 of alpha-synuclein gene does not correlate with Parkinson’s disease. Mol Brain. 2017;10:6. doi: 10.1186/s13041-017-0285-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, Balazer JA, Eaves HL, Xie B, Ford E, Zhang K, Ming GL, Gao Y, Song H. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nature neuroscience. 2011;14:1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison IF, Dexter DT. Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacology & therapeutics. 2013;140:34–52. doi: 10.1016/j.pharmthera.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature genetics. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Heisters D. Parkinson’s: symptoms, treatments and research. Br J Nurs. 2011;20:548–554. doi: 10.12968/bjon.2011.20.9.548. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP. CpG islands–‘a rough guide’. FEBS letters. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing, C. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- International Parkinson Disease Genomics, C. Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S, Stefansson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Morino H, Oda M, Maruyama H, Udaka F, Kameyama M, Nakamura S, Kawakami H. Genetic studies in Parkinson’s disease with an alpha-synuclein/NACP gene polymorphism in Japan. Neuroscience letters. 2001;300:125–127. doi: 10.1016/s0304-3940(01)01557-9. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jakubowski JL, Labrie V. Epigenetic Biomarkers for Parkinson’s Disease: From Diagnostics to Therapeutics. J Parkinsons Dis. 2017;7:1–12. doi: 10.3233/JPD-160914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowaed A, Schmitt I, Kaut O, Wullner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E. Inhibition of miR-34b and miR-34c enhances alpha-synuclein expression in Parkinson’s disease. FEBS letters. 2015;589:319–325. doi: 10.1016/j.febslet.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivendi SV, Yedlapudi D, Hillard CJ, Kalyanaraman B. Oxidants induce alternative splicing of alpha-synuclein: Implications for Parkinson’s disease. Free Radic Biol Med. 2010;48:377–383. doi: 10.1016/j.freeradbiomed.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G. Alternative splicing and evolution: diversification, exon definition and function. Nature reviews Genetics. 2010;11:345–355. doi: 10.1038/nrg2776. [DOI] [PubMed] [Google Scholar]
- Khan N, Graham E, Dixon P, Morris C, Mander A, Clayton D, Vaughan J, Quinn N, Lees A, Daniel S, Wood N, de Silva R. Parkinson’s disease is not associated with the combined alpha-synuclein/apolipoprotein E susceptibility genotype. Annals of neurology. 2001;49:665–668. [PubMed] [Google Scholar]
- Kim HJ, Park G, Jeon BS, Park WY, Kim YE. A mir-153 binding site variation in SNCA in a patient with Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2013;28:1755–1756. doi: 10.1002/mds.25505. [DOI] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CM, Andrews RM, Flicek P, Dillon SC, Karaoz U, Clelland GK, Wilcox S, Beare DM, Fowler JC, Couttet P, James KD, Lefebvre GC, Bruce AW, Dovey OM, Ellis PD, Dhami P, Langford CF, Weng Z, Birney E, Carter NP, Vetrie D, Dunham I. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome research. 2007;17:691–707. doi: 10.1101/gr.5704207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Human molecular genetics. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, Bartus RT. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nature genetics. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kruger R, Vieira-Saecker AM, Kuhn W, Berg D, Muller T, Kuhnl N, Fuchs GA, Storch A, Hungs M, Woitalla D, Przuntek H, Epplen JT, Schols L, Riess O. Increased susceptibility to sporadic Parkinson’s disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Annals of neurology. 1999;45:611–617. doi: 10.1002/1531-8249(199905)45:5<611::aid-ana9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Latham JA, Dent SY. Cross-regulation of histone modifications. Nature structural & molecular biology. 2007;14:1017–1024. doi: 10.1038/nsmb1307. [DOI] [PubMed] [Google Scholar]
- Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, Verny C, Brice A, French Parkinson’s Disease Genetics Study, G. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Annals of neurology. 2013;73:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nature reviews Genetics. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, Liu T, Schilling M, Anderson KJ, Beecham G, Berg D, Biernacka JM, Brice A, DeStefano AL, Do CB, Eriksson N, Factor SA, Farrer MJ, Foroud T, Gasser T, Hamza T, Hardy JA, Heutink P, Hill-Burns EM, Klein C, Latourelle JC, Maraganore DM, Martin ER, Martinez M, Myers RH, Nalls MA, Pankratz N, Payami H, Satake W, Scott WK, Sharma M, Singleton AB, Stefansson K, Toda T, Tung JY, Vance J, Wood NW, Zabetian CP, Me Genetic Epidemiology of Parkinson’s Disease, C. International Parkinson’s Disease Genomics, C. Parkinson’s Disease, G.C. Wellcome Trust Case Control, C. Young P, Tanzi RE, Khoury MJ, Zipp F, Lehrach H, Ioannidis JP, Bertram L. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS genetics. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnertz C, Saucier L, Ge D, Cronin KD, Burke JR, Browndyke JN, Hulette CM, Welsh-Bohmer KA, Chiba-Falek O. Genetic regulation of alpha-synuclein mRNA expression in various human brain tissues. PloS one. 2009;4:e7480. doi: 10.1371/journal.pone.0007480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Sun L, Zhao XL, Zhang P, Zhao XM, Zhang J. PAR2-mediated epigenetic upregulation of alpha-synuclein contributes to the pathogenesis of Parkinsons disease. Brain research. 2014;1565:82–89. doi: 10.1016/j.brainres.2014.04.014. [DOI] [PubMed] [Google Scholar]
- Lu H, Liu X, Deng Y, Qing H. DNA methylation, a hand behind neurodegenerative diseases. Front Aging Neurosci. 2013;5:85. doi: 10.3389/fnagi.2013.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, Flavell RA, Lu B, Ming GL, Song H. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Wei L, Wu F, Hu Z, Liu Z, Yuan W. Advances with microRNAs in Parkinson’s disease research. Drug design, development and therapy. 2013;7:1103–1113. doi: 10.2147/DDDT.S48500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciotta S, Meregalli M, Torrente Y. The involvement of microRNAs in neurodegenerative diseases. Frontiers in cellular neuroscience. 2013;7:265. doi: 10.3389/fncel.2013.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, Greenberg ME. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nature neuroscience. 2014;17:1330–1339. doi: 10.1038/nn.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C, Genetic Epidemiology of Parkinson’s Disease, C. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA : the journal of the American Medical Association. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- Margis R, Margis R, Rieder CR. Identification of blood microRNAs associated to Parkinsonis disease. Journal of biotechnology. 2011;152:96–101. doi: 10.1016/j.jbiotec.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Matsumoto L, Takuma H, Tamaoka A, Kurisaki H, Date H, Tsuji S, Iwata A. CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PloS one. 2010;5:e15522. doi: 10.1371/journal.pone.0015522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazin P, Xiong J, Liu X, Yan Z, Zhang X, Li M, He L, Somel M, Yuan Y, Phoebe Chen YP, Li N, Hu Y, Fu N, Ning Z, Zeng R, Yang H, Chen W, Gelfand M, Khaitovich P. Widespread splicing changes in human brain development and aging. Mol Syst Biol. 2013;9:633. doi: 10.1038/msb.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Linnertz C, Saucier L, Burke JR, Hulette CM, Welsh-Bohmer KA, Chiba-Falek O. The effect of SNCA 3′ region on the levels of SNCA-112 splicing variant. Neurogenetics. 2011;12:59–64. doi: 10.1007/s10048-010-0263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD, Ginder GD. Specific DNA methylation sites in the vicinity of the chicken beta-globin genes. Nature. 1979;280:419–420. doi: 10.1038/280419a0. [DOI] [PubMed] [Google Scholar]
- McLean JR, Hallett PJ, Cooper O, Stanley M, Isacson O. Transcript expression levels of full-length alpha-synuclein and its three alternatively spliced variants in Parkinson’s disease brain regions and in a transgenic mouse model of alpha-synuclein overexpression. Mol Cell Neurosci. 2012;49:230–239. doi: 10.1016/j.mcn.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellick GD, Maraganore DM, Silburn PA. Australian data and meta-analysis lend support for alpha-synuclein (NACP-Rep1) as a risk factor for Parkinson’s disease. Neuroscience letters. 2005;375:112–116. doi: 10.1016/j.neulet.2004.10.078. [DOI] [PubMed] [Google Scholar]
- Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. 2004;62:1835–1838. doi: 10.1212/01.wnl.0000127517.33208.f4. [DOI] [PubMed] [Google Scholar]
- Minones-Moyano E, Porta S, Escaramis G, Rabionet R, Iraola S, Kagerbauer B, Espinosa-Parrilla Y, Ferrer I, Estivill X, Marti E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Human molecular genetics. 2011;20:3067–3078. doi: 10.1093/hmg/ddr210. [DOI] [PubMed] [Google Scholar]
- Miura P, Shenker S, Andreu-Agullo C, Westholm JO, Lai EC. Widespread and extensive lengthening of 3′ UTRs in the mammalian brain. Genome research. 2013;23:812–825. doi: 10.1101/gr.146886.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta I, Nishimura M, Mizuta E, Yamasaki S, Ohta M, Kuno S. Meta-analysis of alpha synuclein/NACP polymorphism in Parkinson’s disease in Japan. Journal of neurology, neurosurgery, and psychiatry. 2002;73:350. doi: 10.1136/jnnp.73.3.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish PK, Rakshi JS, Bailey DL, Sawle GV, Brooks DJ. Measuring the rate of progression and estimating the preclinical period of Parkinson’s disease with [18F]dopa PET. Journal of neurology, neurosurgery, and psychiatry. 1998;64:314–319. doi: 10.1136/jnnp.64.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouradian MM. MicroRNAs in Parkinson’s disease. Neurobiology of disease. 2012;46:279–284. doi: 10.1016/j.nbd.2011.12.046. [DOI] [PubMed] [Google Scholar]
- Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wullner U, Meitinger T, Gasser T. Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Annals of neurology. 2005;57:535–541. doi: 10.1002/ana.20438. [DOI] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, International Parkinson’s Disease Genomics, C. Parkinson’s Study Group Parkinson’s Research: The Organized, G.I. Me, GenePd. NeuroGenetics Research, C. Hussman Institute of Human, G. Ashkenazi Jewish Dataset, I. Cohorts for, H., Aging Research in Genetic, E. North American Brain Expression, C. United Kingdom Brain Expression, C. Greek Parkinson’s Disease, C. Alzheimer Genetic Analysis, G. Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nature genetics. 2014;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid R, Schadt A, Dillmann U, Kostopoulos P, Fassbender K, Herrmann W. Methylation status and neurodegenerative markers in Parkinson disease. Clinical chemistry. 2009;55:1852–1860. doi: 10.1373/clinchem.2009.125021. [DOI] [PubMed] [Google Scholar]
- Parsian A, Racette B, Zhang ZH, Chakraverty S, Rundle M, Goate A, Perlmutter JS. Mutation, sequence analysis, and association studies of alpha-synuclein in Parkinson’s disease. Neurology. 1998;51:1757–1759. doi: 10.1212/wnl.51.6.1757. [DOI] [PubMed] [Google Scholar]
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Poyhonen M, Paetau A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiology of aging. 2014;35:2180 e2181–2185. doi: 10.1016/j.neurobiolaging.2014.03.024. [DOI] [PubMed] [Google Scholar]
- Pihlstrom L, Berge V, Rengmark A, Toft M. Parkinson’s disease correlates with promoter methylation in the alpha-synuclein gene. Movement disorders : official journal of the Movement Disorder Society. 2014 doi: 10.1002/mds.26073. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Postuma RB, Gagnon JF, Montplaisir J. Clinical prediction of Parkinson’s disease: planning for the age of neuroprotection. Journal of neurology, neurosurgery, and psychiatry. 2010;81:1008–1013. doi: 10.1136/jnnp.2009.174748. [DOI] [PubMed] [Google Scholar]
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH. A novel alpha-synuclein missense mutation in Parkinson disease. Neurology. 2013;80:1062–1064. doi: 10.1212/WNL.0b013e31828727ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput A, Vilarino-Guell C, Rajput ML, Ross OA, Soto-Ortolaza AI, Lincoln SJ, Cobb SA, Heckman MG, Farrer MJ, Rajput A. Alpha-synuclein polymorphisms are associated with Parkinson’s disease in a Saskatchewan population. Movement disorders : official journal of the Movement Disorder Society. 2009;24:2411–2414. doi: 10.1002/mds.22795. [DOI] [PubMed] [Google Scholar]
- Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P. Alpha-synuclein and Parkinson’s disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2004;18:617–626. doi: 10.1096/fj.03-0338rev. [DOI] [PubMed] [Google Scholar]
- Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, Abeliovich A. Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nature communications. 2012;3:1084. doi: 10.1038/ncomms2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Appenzeller S, Ammerpohl O, Deuschl G, Paschen S, Bruggemann N, Klein C, Kuhlenbaumer G. No evidence for differential methylation of alpha-synuclein in leukocyte DNA of Parkinson’s disease patients. Movement disorders : official journal of the Movement Disorder Society. 2012;27:590–591. doi: 10.1002/mds.24907. [DOI] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nature genetics. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Scherzer CR, Grass JA, Liao Z, Pepivani I, Zheng B, Eklund AC, Ney PA, Ng J, McGoldrick M, Mollenhauer B, Bresnick EH, Schlossmacher MG. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10907–10912. doi: 10.1073/pnas.0802437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Chowdhury S, Peabody K, Brooks DW. Overcoming obstacles in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2012;27:1606–1611. doi: 10.1002/mds.25260. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature genetics. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R. Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature. 2016;533:95–99. doi: 10.1038/nature17939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Ding H, Yang J, Lin Q, Xue J, Zhang Y, Chan P, Cai Y. Pyrosequencing analysis of SNCA methylation levels in leukocytes from Parkinson’s disease patients. Neuroscience letters. 2014;569:85–88. doi: 10.1016/j.neulet.2014.03.076. [DOI] [PubMed] [Google Scholar]
- Sotiriou S, Gibney G, Baxevanis AD, Nussbaum RL. A single nucleotide polymorphism in the 3′UTR of the SNCA gene encoding alpha-synuclein is a new potential susceptibility locus for Parkinson disease. Neuroscience letters. 2009;461:196–201. doi: 10.1016/j.neulet.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadafora P, Annesi G, Pasqua AA, Serra P, Ciro Candiano IC, Carrideo S, Tarantino P, Civitelli D, De Marco EV, Nicoletti G, Annesi F, Quattrone A. NACP-REP1 polymorphism is not involved in Parkinson’s disease: a case-control study in a population sample from southern Italy. Neuroscience letters. 2003;351:75–78. doi: 10.1016/s0304-3940(03)00859-0. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Stefanis L. alpha-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöger R, Scaife PJ, Shephard F, Chakrabarti L. Elevated 5hmC levels characterize DNA of the cerebellum in Parkinson’s disease. Parkinson’s Disease. 2017;3 doi: 10.1038/s41531-017-0007-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugeno N, Jackel S, Voigt A, Wassouf Z, Schulze-Hentrich J, Kahle PJ. alpha-Synuclein enhances histone H3 lysine-9 dimethylation and H3K9me2-dependent transcriptional responses. Sci Rep. 2016;6:36328. doi: 10.1038/srep36328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nature reviews Genetics. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- Tan EK, Tan C, Shen H, Chai A, Lum SY, Teoh ML, Yih Y, Wong MC, Zhao Y. Alpha synuclein promoter and risk of Parkinson’s disease: microsatellite and allelic size variability. Neuroscience letters. 2003;336:70–72. doi: 10.1016/s0304-3940(02)01178-3. [DOI] [PubMed] [Google Scholar]
- Tan YY, Wu L, Zhao ZB, Wang Y, Xiao Q, Liu J, Wang G, Ma JF, Chen SD. Methylation of alpha-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson’s disease patients. Parkinsonism & related disorders. 2014;20:308–313. doi: 10.1016/j.parkreldis.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, Comyns K, Richards MB, Meng C, Priestley B, Fernandez HH, Cambi F, Umbach DM, Blair A, Sandler DP, Langston JW. Rotenone, paraquat, and Parkinson’s disease. Environmental health perspectives. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Molecular insights into Parkinson’s disease. F1000 Med Rep. 2011;3:7. doi: 10.3410/M3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchman JW, Dehejia A, Chiba-Falek O, Cabin DE, Schwartz JR, Orrison BM, Polymeropoulos MH, Nussbaum RL. Human and mouse alpha-synuclein genes: comparative genomic sequence analysis and identification of a novel gene regulatory element. Genome research. 2001;11:78–86. doi: 10.1101/gr.165801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Saitoh T, Mori H. Tissue-dependent alternative splicing of mRNA for NACP, the precursor of non-A beta component of Alzheimer’s disease amyloid. Biochem Biophys Res Commun. 1994;205:1366–1372. doi: 10.1006/bbrc.1994.2816. [DOI] [PubMed] [Google Scholar]
- Vaughan JR, Farrer MJ, Wszolek ZK, Gasser T, Durr A, Agid Y, Bonifati V, DeMichele G, Volpe G, Lincoln S, Breteler M, Meco G, Brice A, Marsden CD, Hardy J, Wood NW. Sequencing of the alpha-synuclein gene in a large series of cases of familial Parkinson’s disease fails to reveal any further mutations. The European Consortium on Genetic Susceptibility in Parkinson’s Disease (GSPD) Human molecular genetics. 1998;7:751–753. doi: 10.1093/hmg/7.4.751. [DOI] [PubMed] [Google Scholar]
- Venda LL, Cragg SJ, Buchman VL, Wade-Martins R. alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends in neurosciences. 2010;33:559–568. doi: 10.1016/j.tins.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes & development. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. The epigenotype. 1942. Int J Epidemiol. 2012;41:10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- Wang L, Yi R. 3′UTRs take a long shot in the brain. BioEssays : news and reviews in molecular, cellular and developmental biology. 2014;36:39–45. doi: 10.1002/bies.201300100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu J, Huang BO, Xu YM, Li J, Huang LF, Lin J, Zhang J, Min QH, Yang WM, Wang XZ. Mechanism of alternative splicing and its regulation. Biomed Rep. 2015;3:152–158. doi: 10.3892/br.2014.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wang X, Li R, Yang ZF, Wang YZ, Gong XL, Wang XM. A DNA methyltransferase inhibitor, 5-aza-2′-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS neuroscience & therapeutics. 2013;19:183–190. doi: 10.1111/cns.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Yang N, Xu Q, Sun Q, Guo J, Li K, Liu Z, Yan X, Zhu X, Tang B. The rs3756063 polymorphism is associated with SNCA methylation in the Chinese Han population. Journal of the neurological sciences. 2016;367:11–14. doi: 10.1016/j.jns.2016.05.037. [DOI] [PubMed] [Google Scholar]
- Xia Y, Rohan de Silva HA, Rosi BL, Yamaoka LH, Rimmler JB, Pericak-Vance MA, Roses AD, Chen X, Masliah E, DeTeresa R, Iwai A, Sundsmo M, Thomas RG, Hofstetter CR, Gregory E, Hansen LA, Katzman R, Thal LJ, Saitoh T. Genetic studies in Alzheimer’s disease with an NACP/alpha-synuclein polymorphism. Annals of neurology. 1996;40:207–215. doi: 10.1002/ana.410400212. [DOI] [PubMed] [Google Scholar]
- Xu Q, Modrek B, Lee C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic acids research. 2002;30:3754–3766. doi: 10.1093/nar/gkf492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of neurology. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]