

Abstract
Engineering and study of protein function by directed evolution has been limited by the requirement to introduce DNA libraries of defined size or to use global mutagenesis. Here, we develop a strategy to repurpose the somatic hypermutation machinery used in antibody affinity maturation to efficiently perform protein engineering in situ. Using catalytically inactive Cas9 (dCas9) to recruit variants of the deaminase AID (CRISPR-X), we can specifically mutagenize endogenous targets with limited off-target damage. This generates diverse libraries of localized point mutations, in contrast to insertions and deletions created by active Cas9, and can be used to mutagenize multiple genomic locations simultaneously. With this technology, we mutagenize GFP and select for spectrum-shifted variants, including EGFP. In addition, we mutate the target of the cancer therapeutic bortezomib, PSMB5, and identify known and novel mutations that confer resistance to treatment. Finally, we utilize a hyperactive AID variant with dramatically increased activity to mutagenize endogenous loci both upstream and downstream of transcriptional start sites. These experiments illustrate a powerful new approach to create highly complex libraries of genetic variants in native context, which can be broadly applied to investigate and improve protein function.
Directed evolution employs successive rounds of mutation and selection to engineer biomolecules with enhanced, novel or non-natural functions, such as improved antibodies1, more efficient enzymes2, or mutant proteins with altered activity3. A major limitation to these experiments is the generation and maintenance of a diverse mutant population. Radiation and chemically-induced DNA damage have been used to mutate the entire genome, but this requires maintaining a large number of cells since the majority of mutations are located outside the target of interest. Alternatively, diverse plasmid libraries can be introduced into cells; however, these proteins are often expressed at inappropriate levels and without normal regulation. Importantly, these libraries are of limited size, both in terms of total diversity and length of diversified region, which greatly restricts the potential for evolution experiments. Due to these limitations, the majority of these engineering experiments have been performed in bacteria, bacteriophage, and yeast due to the relative ease of generating diverse libraries in these organisms4–6. However, mammalian proteins engineered in these systems often change behavior in their native host environment. Hence, generating a diverse library of mutants in native context would have enormous advantages.
Nature has a built-in mechanism for generating diversity at a specific genetic locus, which is used with exquisite precision during the process of antibody maturation. After V-D-J recombination, B cells create point mutations in their immunoglobulin (Ig) regions through the process of somatic hypermutation (SHM) to perform affinity maturation on the antibody7,8. SHM is mediated by an enzyme called activation induced cytidine deaminase (AID), which deaminates cytosine (C) to a uracil (U) initiating a DNA repair response which causes errors in the Ig locus at a rate of 1/1000 bp−1 9, compared to the normal rate of mutation during cellular DNA replication of 1/109 bp−1 10. The process generates point mutations rather than insertions/deletions, and favors transition mutations (i.e. pyrimidine to pyrimidine or purine to purine) over transversions8. After deamination, mutations can be generated in three ways: the uracil-guanine (U-G) mismatch can be misread resulting in a (C>T) or (G>A) transition, the U can be removed by base excision repair and replaced by any base, or an error-prone translesion polymerase can be recruited through the mismatch repair pathway, generating transitions and transversions near the lesion7.
Although sequence elements flanking the immunoglobulin locus have been linked to SHM targeting11, the mechanisms by which SHM is regulated and targeted are not completely understood. It has been proposed that AID migrates with RNA polymerase II complex during transcription of the Ig locus and mutates specific hotspot sequence motifs12,13. Cell lines that misregulate or overexpress AID have the mutagenic capacity to evolve both fluorescent proteins14,15 and antibodies16, but these strategies create mutations throughout the genome.
With the recent advent of the CRISPR/Cas9 system, it has become possible to target functional proteins to specific genomic loci using catalytically dead Cas9 (dCas9). This approach has been used for both repression and activation of transcription17–20 as well as targeting fluorescent proteins21,22 and modifying enzymes23–26, and most recently for the efficient conversion of C>T as a means of therapeutic targeted editing23,26. Here, we use dCas9 to target hyperactive AID to induce localized, diverse point mutations (CRISPR-X). This process differs markedly from mutagenesis using active Cas927, which predominantly generates insertions and deletions28–30, or the introduction of mutations via externally generated oligonucleotide donor libraries by homologous recombination following Cas9 cleavage31,32. We show that AID-induced mutations can be generated in cells that express AID constitutively or transiently via electroporation, and can be targeted to multiple loci in the same cell. Furthermore, we show two proof of principle examples of protein engineering using CRISPR-X: the alteration of the absorption/emission spectrum of genomically integrated wild-type GFP, and the evolution of variants of PSMB5 that are resistant to bortezomib, a widely used chemotherapeutic drug. In the latter example, we not only generate mutations that have previously been observed in resistant cell lines, but also identify novel drug-resistant mutants that may reveal new properties of PSMB5 and its interaction with bortezomib. Finally, we demonstrate that a hyperactive AID enzyme introduces mutations at a higher rate and can generate variants in both protein coding regions as well as regulatory regions upstream of the transcription start site. Together, this work illustrates the potential of a novel targeted mutagenesis strategy for the engineering and evolution of new protein function in normal cellular context.
Results
Targeted mutagenesis through dCas9 recruitment of AID
In order to recruit the AID protein to a genetic locus, we used dCas930 combined with a single guide RNA (sgRNA) bearing an MS2 hairpin binding site (Fig. 1a) that has been previously used to recruit MS2 fused effector proteins to activate transcription20. In this system, the sgRNA contains two MS2 hairpins that each recruit two MS2 proteins (four in total) fused to AID. For our initial test, we generated MS2 fused to three AID variants (Supplementary Data Fig. 1a): wild-type AID, a truncated version without the last three amino acids (AIDΔ) which ablates its nuclear export signal (NES) while increasing SHM activity33, and a catalytically inactive truncated version (AIDΔDead)34. The deletion of the NES resulted in primarily nuclear localization of the MS2 fusion protein as observed by immunofluorescence staining in K562 cells (Supplementary Data Fig. 1b), with minimal change in protein expression (Supplementary Data Fig. 1c).
Figure 1. CRISPR-X generates targeted mutations.
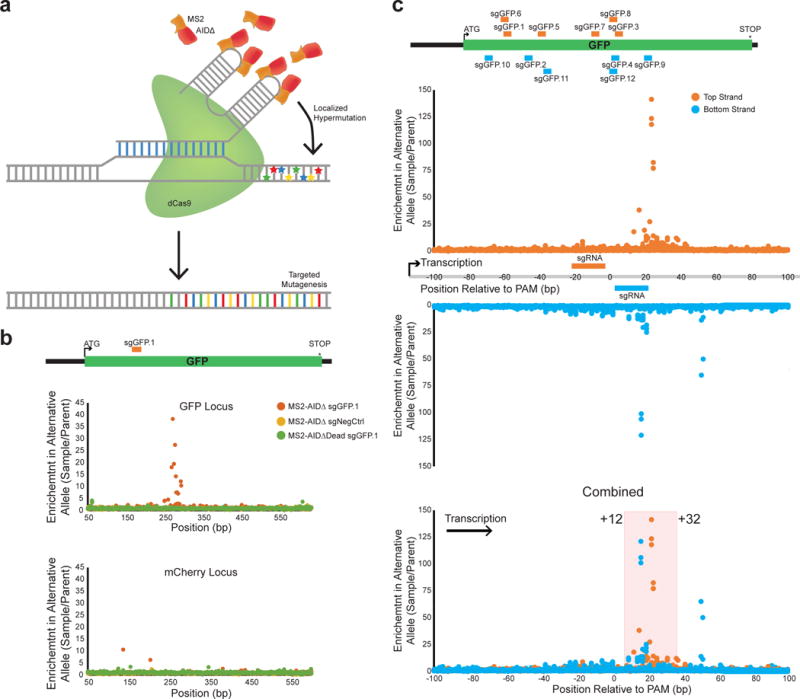
a) Schematic of CRISPR-X. dCas9 (160 kDa) complexes with an sgRNA containing MS2 hairpins in its stem loop, which recruit AIDΔ fused to MS2 binding protein (40 kDa). The deaminase induces local DNA damage which in turn introduces mutations. b) Cells expressing dCas9, GFP and mCherry were infected with indicated combinations of MS2-AIDΔ or MS2-AIDΔDead and sgGFP.1 or sgNegCtrl, and the GFP and mCherry loci were sequenced. Enrichment of mutations at each base position are shown for one replicate each. Additional replicates are shown in Supplementary Data Fig. 2b. c) 12 guides targeting GFP were infected into cells expressing dCas9, MS2-AIDΔ, GFP and mCherry. The targeting locations of the guides in the GFP locus are shown on the top panel. The GFP locus was sequenced for each sample. Enrichment of mutation relative to the position of the PAM of the sgRNAs is shown on the lower panel. The direction of transcription was defined as the positive direction as indicated by the arrow.
We generated K562 cells stably expressing dCas9 along with GFP and mCherry, which, when used together with sgRNAs targeting GFP, serve as phenotypic readout for on-target (GFP) and off-target mutations (mCherry). These cells were transiently electroporated with plasmids coding for either a GFP-targeting sgRNA (sgGFP.1) or a scrambled non-targeting sgRNA (sgNegCtrl) paired with plasmids coding for MS2-AID, MS2-AIDΔ, or MS2-AIDΔDead. After 10 days, cells were analyzed by flow cytometry to measure GFP and mCherry fluorescence (Supplementary Data Fig. 1d). As expected for on-target mutation resulting in non-fluorescent protein, we observed an increase in the GFP negative population for MS2-AIDΔ treatment when comparing sgGFP.1 to sgNegCtrl (1.64% vs. 0.55%). However, we did not see this effect with MS2-AID (0.71% vs. 0.78%). At the same time, the mCherry negative population showed little change (1.02% vs. 0.91%), indicating that targeting AIDΔ to GFP resulted in specific mutagenesis. Additional fluorescence measurements made beyond 10 days did not change, suggesting mutation had stabilized following electroporation (data not shown).
Based on the observed change in fluorescence, we performed a more detailed analysis of the population by sequencing the locus. To quantify mutations in the GFP negative population, we collected the GFP low population from the AIDΔ;sgGFP.1, AΓDΔ;sgNegCtrl, and AIDΔ-Dead:sgGFP.1 samples via FACS and sequenced the GFP locus. Enrichment of mutations was calculated by comparing collected samples to parental cells that had not been exposed to a mutagenic agent. We observed enrichment of mutations only in the AIDΔ:sgGFP.1 (Supplementary Data Fig. 1e). The most enriched position for mutations was base pair 280 which had over 500 fold enrichment in mutations and 41.2% of sequences at that base showed a G>A transition. This transition resulted in the introduction of a tyrosine in place of cysteine in GFP at amino acid 48. Reduced fluorescence of GFP due to this alteration is consistent with previous work showing that cysteine thiol binding by dTNB quenches GFP fluorescence35.
Given the superior performance of AIDΔ, we continued with this variant. To more accurately estimate the mutation rate without considering transient electroporation efficiency, we integrated the CRISPR-X system into our reporter cells. MS2-AIDΔ or MS2-AIDΔDead were stably integrated in cells together with sgGFP.1 or sgNegCtrl, and GFP and mCherry negative populations were monitored 14 days after infection (Supplementary Data Fig. 2a). As before, in the presence of MS2-AIDΔ, we observed an increase in the GFP negative population (1.88%) when compared to either the sgNegCtrl (0.75%) or MS2-AIDΔDead (0.47%). By contrast, the mCherry low population was minimally changed (0.67% MS2-AIDΔ:sgGFP.1, 0.34% MS2-AIDΔ:sgNegCtrl, 0.43% MS2-AIDΔDead:sgGFP.1) (Supplementary Data Fig. 2a). We sequenced both GFP and mCherry loci from these cells (Fig. 1b and Supplementary Data Fig. 2b), and observed an enrichment of mutations in the 270–290bp region of GFP only in cells expressing MS2-AIDΔ:sgGFP.1. We did not detect any enrichment of mutations in the mCherry locus.
Defining the region of mutagenesis for CRISPR-X
To determine the region of mutagenesis with respect to the sgRNA, we selected an additional 11 sgRNAs (sgGFP.2–12) tiling the GFP locus on both strands (Fig. 1c and Supplementary Data Fig. 3a). Since AID mutagenesis has been shown to require transcription12, we hypothesized the strand of the guide relative to the direction of transcription may change the targeting of mutations. We sequenced the GFP locus in each of these samples and mapped the mutations relative to the end of the PAM sequence of each sgRNA (Fig. 1c). While different sgRNAs exhibited a range of mutation efficiencies (Supplementary Data Fig. 3b), we observed a mutational hotspot region from +12 to +32 bp downstream of the PAM relative to the direction of transcription that was independent of the strand targeting (Fig. 1c). The mutational hotspot was defined to include any base with at least 10-fold increased mutation over all three biological replicates for a given sgRNA. Mutations in this region were measured for the 12 sgGFP guides, and a median mutation frequency of 0.0104 was observed (Supplementary Data Fig. 3c). This translates to a mutation rate of ~1/2000 bp−1, which is similar to that observed for somatic hypermutation8, and is an order of magnitude higher than the observed frequency of 0.0014 for a negative control sgRNA (MS2-AIDΔ:sgNegCtrl) and 0.0015 for catalytically inactive AID (MS2-AIDΔDead:sgGFP.1). Given the ability of this system to generate targeted point mutations, we sought to apply it for directed evolution experiments.
Evolution of wtGFP to EGFP using CRISPR-X
As an initial proof of principle experiment, we tested whether we could alter an integrated copy of wild-type GFP (wtGFP) from Aequorea victoria (excitation 395nm/emission 509nm) to EGFP (490/509nm)36. EGFP has two mutated residues from wtGFP: S65T, which shifts the ex/em spectrum, and F64L, which improves the folding kinetics of GFP36–38. We designed four guides (sgwtGFP.1–4) that target this region and introduced them via electroporation along with MS2-AIDΔ into K562 cells expressing dCas9 and wtGFP. As a negative control, we also electroporated four ‘safe harbor’ sgRNAs that target regions of the genome that are annotated as non-functional. Cells were grown for 10 days to allow for mutations to be introduced, and then sorted by FACS to collect cells expressing spectrum-shifted GFP (Fig. 2a). In biological replicate experiments, we observed a population with decreased signal in the Pacific blue channel and increased GFP signal (0.076% replicate 1, 0.025% replicate 2) (Fig. 2b and Supplementary Data Fig. 4a), which was not observed in the safe harbor samples (0.002%, 0.002%) (Fig. 2b and Supplementary Data Fig. 4a). After another round of sorting, the safe harbor samples did not have any cells pass the sorting gates, while the spectrum-shifted population had increased to 2.29% and 1.16% in the GFP-targeted replicates.
Figure 2. Evolution of wtGFP to EGFP using CRISPR-X.

a) Schematic of wtGFP evolution experiments. wtGFP expressing cells were transiently electroporated with MS2-AIDΔ and 4 sgRNAs either targeting GFP or safe harbor regions. Cells were sorted for spectrum shifted GFP bright cells followed by sequencing of the wtGFP locus. b) Cells were collected from unsorted populations and after each round of sorting, and the wtGFP locus was sequenced. (left) Enrichment of mutations at each base position for both wtGFP targeted and safe harbor targeted libraries are shown except after the Sort #2 condition where no safe harbor cells were recovered. Identified mutations are labeled. (right) Scatter plots of the flow cytometry and gating are shown for the wtGFP parent and pre-sorting populations. c) Lentiviral expression constructs were generated containing each of the S65T and Q80H mutations separately and together. Plasmids encoding these variants along with wtGFP and EGFP controls were lipid transfected into 293T cells and the GFP fluorescence was measured by flow cytometry.
The GFP locus was sequenced to identify mutations enriched by the sorting process (Fig. 2b and Supplementary Data Fig. 4a), revealing enrichment of mutations at positions 331 (G>C) and 377 (G>C). The former mutation introduces the known S65T mutation from EGFP (Fig. 2b). The latter mutation generated a Q80H substitution, which we suspected was a passenger mutation since the majority of sequences containing the mutation also showed the S65T transition. In order to determine the contribution of each individual mutation to changes in GFP fluorescence, we introduced each mutation into GFP separately, and confirmed that the S65T mutation alters the fluorescence spectrum of GFP while Q80H does not, either alone or in conjunction with S65T (Fig. 2c). We did not observe the F64L mutation in our selection, which was shown in an inducible expression system to affect protein stability36, but did not change fluorescence intensity under constitutive expression when coupled with the S65T mutation (Fig. 2c). We performed a similar selection experiment with the integrated CRISPR-X system and a single integrated guide (sgwtGFP.1 or sgSafe.2) and recovered the same S65T transition but did not observe the Q80H mutation (Supplementary Data Fig. 4b).
Identification of bortezomib-resistant PSMB5 variants
Another potential application of CRISPR-X is the investigation of mechanisms of drug resistance. Mutations are a common escape pathway for cancer cells to develop resistance to drug treatment39, and understanding which mutations can arise is important for the design of new drugs or drug combinations. To test this, we mutagenized PSMB5, a core subunit of the 20S proteasome, which is the target of the proteasome inhibitor bortezomib40. We generated a library of 143 guides tiling all coding exons of PSMB5, as well as a control library of 705 safe harbor guides (Extended Dataset 2). Both libraries were lentivirally integrated into K562 cells expressing dCas9 and MS2-AIDΔ, given 14 days to develop mutations, and pulsed with bortezomib three times (Fig. 3a). After selection, genomic DNA was extracted, the PSMB5 exonic loci of both libraries were sequenced, and variant frequencies were quantitated at each base (Fig. 3b–c and Supplementary Data Fig. 5a–b). The screen was performed in biological replicate, and mutants were selected for further analysis that showed enrichment of at least 20 fold in both replicates (Fig. 3c and Supplementary Data Fig. 5b). We identified 11 such mutations (Fig. 3d), including two (A108T/V) altering a residue known to be involved in binding bortezomib41. Novel mutations were identified near a threonine (residue 80) that also binds bortezomib (A74V, R78M/N, A79T/G, and G82D). We suspect these mutations disrupt the position of the threonine, destroying the binding pocket for bortezomib. Beyond mutations expected to affect the binding pocket, we identified two mutations in Exon 1 (L11L, G45G), an intronic mutation before Exon 2, and a mutation in Exon 4 (G242D) that is located on the side of the protein distal to the bortezomib binding pocket. No resistant mutations were identified in Exon 3, an alternate exon that is not expressed in K562 cells (data not shown). In the safe harbor control library, we identified one mutation (A79T) that was also found with the PSMB5 targeted library, and was likely present at undetectable levels in the parent K562 population (Fig. 3c and Supplementary Data Fig. 5b).
Figure 3. Directed evolution of bortezomib resistant mutations in PSMB5.
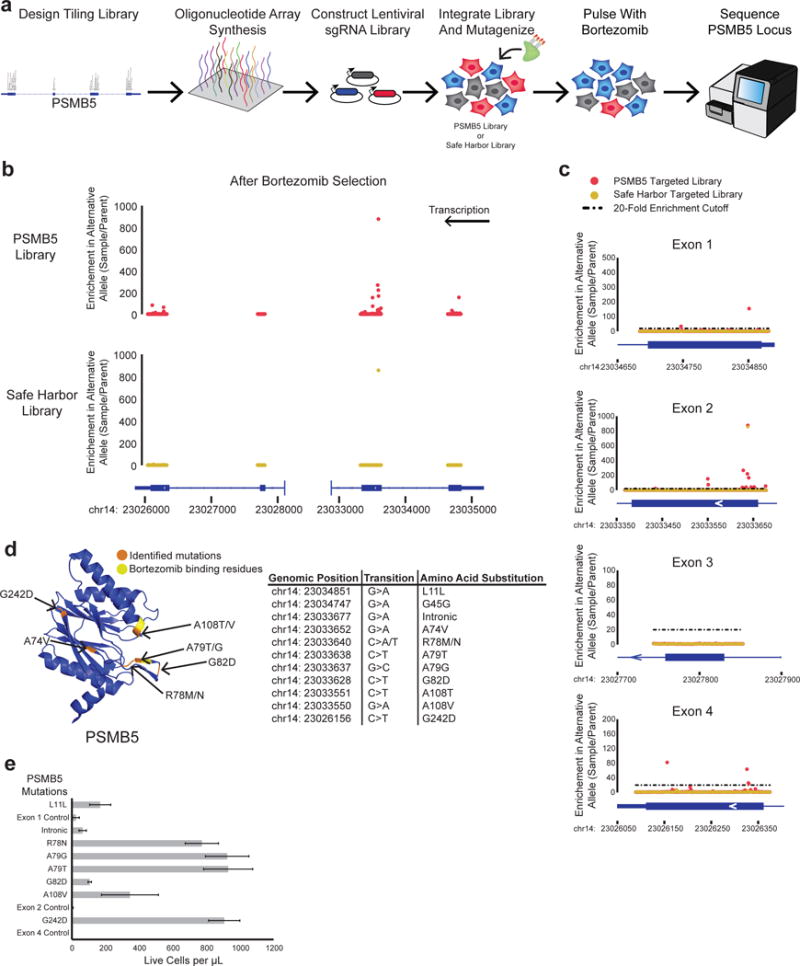
a) Schematic for PSMB5 mutagenesis and bortezomib selection. Libraries targeting the exons of PSMB5 or control safe harbor regions were designed and synthesized on an oligonucleotide array and cloned into an sgRNA expressing vector. This vector was integrated into cells expressing dCas9 and MS2-AIDΔ to generate mutations. Cells were pulsed with bortezomib, after which the PSMB5 exonic loci were sequenced. b) Graphs of the enrichment of mutation at each base position are shown for the PSMB5 locus in both PSMB5 and safe harbor targeted libraries for one biological replicate. c) Graphs of the enrichment of mutations are shown for individual PSMB5 exons. Positions that were above 20-fold enriched (black dashed line) in both replicates were identified as possible candidates. d) PSMB5 structure is shown. Identified mutations (orange) and residues involved in binding bortezomib (yellow) are indicated. A table summarizing the mutations is included. e) Mutations were installed into K562 cells and selected with bortezomib. A graph summarizing the density of live cells after selection is shown. Error bars indicate standard error.
We chose to functionally validate 8 of these mutations by knocking each one into the genome separately at the native PSMB5 locus using active Cas9 cutting followed by HDR mediated by a DNA donor oligo28,29. To control for the effect of Cas9 cutting and HDR, we knocked in a synonymous mutation not identified in our screen in each exon. We electroporated Cas9 expressing K562 cells with donor oligo and sgRNA and waited for six days followed by subsequent selection with bortezomib. After 14 days, the viability of the cells was measured (Fig. 3e). Five of the mutations (R78N, A79G, A79T, A108V, and G242D) were strongly protective against bortezomib-induced cell death, while the other three (L11L, Intronic, and G82D) showed more modest protection when compared to controls. For the most resistant mutations, the PSMB5 locus was sequenced following bortezomib selection and the presence of the expected mutation was verified in the majority of non-frameshifted sequences (Supplementary Data Fig. 6). Together, these experiments show that CRISPR-X can be used to selectively mutagenize an endogenously expressed protein target, identifying known and novel mutants that confer drug resistance.
Enhanced mutagenesis using a hyperactive AID mutant
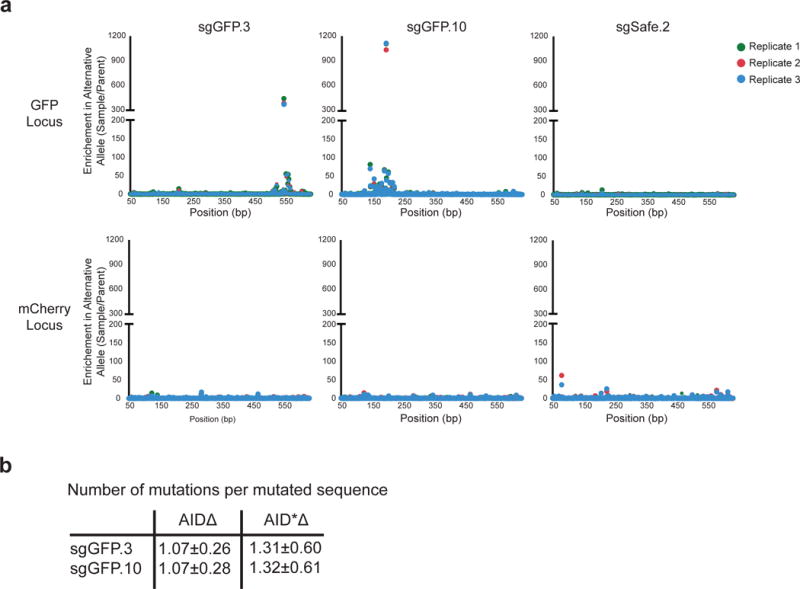
We observed variable mutation efficiency with AIDΔ, and therefore investigated whether this could be improved even further with AID variants previously shown to have increased SHM activity42. We selected one of the strongest mutants (AID*) and removed its NES as we had with wild-type AID (Supplementary Data Fig. 1a). AID*Δ was integrated along with one of three sgRNAs (sgGFP.3, sgGFP.10, and sgSafe.2), and the enrichment of mutation in GFP and mCherry loci was measured (Fig. 4a and Supplementary Data Fig. 7a). Despite lower expression of the protein (Supplementary Data Fig. 1c), we observed an approximate 10-fold increase in mutation at the most enriched base position for GFP-targeting sgRNAs when compared with AIDΔ, with no noticeable increase in mCherry off-target mutation (Fig. 4b). sgSafe.2 samples did not show mutation at either locus. We aligned these mutations relative to the PAM and observed an increase in the size of the hotspot to span from −50 to +50 bp (Fig. 4b). Within the hotspot window, we still observe the most highly mutated bases are located within the +12 to +32 region. This suggests that the targeting pattern of AID*Δ has not changed compared to AIDΔ, but that the increased activity of AID*Δ allows for detection of mutation over a larger window. Within this region, we observed a substantial increase in mutation rate (2.25 fold for sgGFP.3 and 6.52 fold for sgGFP.10) reaching over 20% of reads for sgGFP.10 (Fig. 4b), as well as a modest increase in sequences that contained multiple mutations per read (1.32 mutations/read for AID*Δ vs. 1.07 for AIDΔ, Supplementary Data Fig. S7b). Given that we can measure mutations in up to 10– 20% of sequences (which is ~ 1 mutation per 500–1000 bp within a hotspot), we estimate that CRISPR-X is capable of mutagenesis on par with somatic hypermutation (1/1000 bp−1)9.
Figure 4. Enhanced mutagenesis of genes, promoters, and multiple loci with hyperactive AID*Δ.
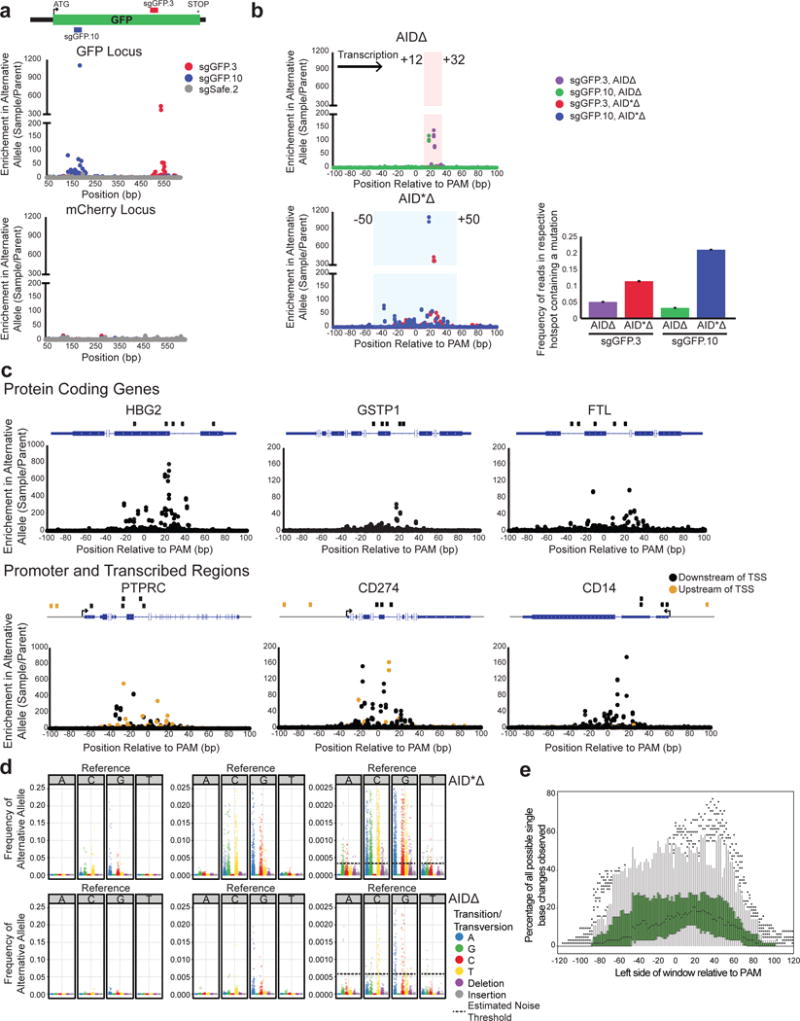
a) sgGFP.3, sgGFP.10, and sgSafe.2 were infected into cells expressing dCas9, MS2-AID*Δ, GFP, and mCherry. The GFP and mCherry loci were sequenced. Enrichment of mutations at each base position in both loci is shown. b) Enrichment of mutations at positions relative to the sgRNA PAM is shown for 2 GFP-targeting sgRNAs, sgGFP.3 and sgGFP.10, using either AIDΔ (top graph) or hyperactive AID*Δ (bottom graph). The shaded rectangles highlight the respective hotspot regions. (right) The frequencies of mutated sequences in the respective hotspots are shown. Error bars indicate standard error. c) sgRNAs were designed to target six endogenous loci. Gene diagrams for each locus are shown indicating the position of the respective guides. Cells expressing dCas9 and MS2-AID*Δ were infected with the sgRNAs, and the loci were sequenced. Shown are graphs of the enrichment of mutations at positions relative to the PAM at each of the loci. Samples with sgRNAs targeting upstream of the transcription start site are shown in orange. d) Transition and transversion mutations observed using AID*Δ and AIDΔ are shown at three different scales. At each base in the hotspot region, the frequency of each transition was calculated and normalized to the parent population. The AID*Δ transitions were tabulated from mutations generated with sgGFP.3, sgGFP.10, and sgRNAs targeting endogenous loci. The mutations induced by AIDΔ were tabulated from sgGFP.1–12. The standard deviation of alternative allele frequencies in the parental samples were calculated and indicated by the dashed black line. e) Graph of the percentage of all possible single base changes observed for AID*Δ targeted with sgRNAs (described in Fig. 4a,c) in a 21bp sliding window. Single base changes with a frequency above the estimated noise were counted over a 21bp window beginning at the indicated position relative to the PAM, and the measured fraction of all possible changes is reported for each window. Box plots at each position are shown summarizing the distribution observed over all sgRNAs. The box plot lines represent 1.5X the interquartile range.
To further explore the capacity of AID*Δ-induced mutagenesis, we targeted three classes of endogenous loci: protein coding genes, promoter regions, and safe-harbor regions. For the protein coding genes, we targeted five sgRNAs to 3 highly expressed genes, FTL, HBG2, and GSTP1, sequenced the respective loci, and quantitated mutation enrichment (Fig. 4c). Additionally, we quantified the frequency of mutations at each base position relative to the PAM site (Supplementary Data Fig. 8a and Fig. 4d). We observed mutated bases in each of the three genes with similar targeting in the −50 to +50 hotspot relative to the sgRNA PAM. To determine whether we could mutagenize genes with more moderate expression levels, as well as associated promoter regions, we targeted PTPRC, CD274, and CD14. For each gene, we targeted both the transcribed region as well as sequences upstream of the transcription start site (TSS). For each locus, we observed mutated bases for sgRNAs located both upstream and downstream of the TSS (Fig. 4c and Supplementary Data Fig. 8a). For CD274, we observe mutations targeted up to 3.2kb upstream of the TSS, suggesting some types of non-transcribed regions can be investigated using CRISPR-X. Lastly, we tested sgRNAs targeting four safe harbor regions (non-functional genomic regions) but we did not observe mutations in these samples (data not shown), although we cannot rule out that this was because of ineffective sgRNA choice or other factors.
We compared the mutation types observed for both AIDΔ and AID*Δ within their respective hotspots (Fig. 4b,d). The mutation rates were normalized by alternative allele frequencies observed in the parental samples within targeted hotspot regions. In addition, we calculated the standard deviation of the alternative allele frequency in the parent samples when compared to reference sequence (5.68·10−4 for AIDΔ and 3.74·10−4 for AID*Δ), and used this as a noise threshold for the transition/transversion frequencies. For both AID variants, we observe a preference for G>A and C>T transitions with the most highly mutated bases being G or C (Fig. 4d), consistent with the preference of AID deaminase activity. Importantly, we find a significant increase in mutation frequencies for all possible base changes except A>T for the AID*Δ treated samples. For both variants, low levels of insertions (maximum frequency of 1.98·10−3 for AID*Δ and 7.44·10−4 for AIDΔ) and deletions (maximum frequency of 5.15·10−4, 3.01·10−4) are observed, suggesting that mutation induced frame shifts are rare. Thus, the increased activity of AID*Δ can greatly expand the sequence space that can be mutagenized by a single sgRNA, including both coding and promoter regions of genes.
To quantitate the rate of mutation for AID*Δ over a range of sgRNAs, we tabulated mutation frequencies for each sgRNA over their respective 100bp hotspots. Consistent with previous observations for Cas9, certain sgRNAs did not show activity and we thus removed these (4/34 sgRNAs). To consider an sgRNA, we required that each replicate contained a base position in the hotspot that was enriched at least 10-fold over the parent population. For the remaining 30 sgRNAs, we calculated the percent of reads containing a mutation, and found that the median frequency was 0.0163, with ~25% of sgRNAs giving a frequency of >0.05 and up to 0.22 (Supplementary Data Fig. 8b).
To estimate the range of mutations that can be sampled using CRISPR-X in a population of cells, we quantified the diversity generated by AID*Δ. Using data from the sgRNAs targeting GFP and the endogenous loci (Fig. 4b–c), we scanned across the region next to the PAM with a 21bp window, which was the size of the AIDΔ hotspot. In each window, we calculated the percentage of all 63 possible single base variants (21 bases and 3 possible changes at each position) measured above the noise threshold in the population (Fig. 4e). While the efficiency of mutation varied with different sgRNAs, a window spanning from +20 to +40 from the PAM displayed the highest median percentage of possible variants (20.6%), and we observe up to 77.8% of all possible transitions in some cases. The +20 to +40 window is similar to the observed targeting hotspot for AIDΔ (Fig. 4b), suggesting that this region is the most highly mutagenized for both AIDΔ and AID*Δ.
Simultaneous mutation of multiple loci using CRISPR-X
Independent mutagenesis at multiple locations is typically not possible with traditional directed evolution experiments. However, the CRISPR/Cas9 system can target multiple loci using different sgRNAs28,29. We incorporated two guides, one targeting GFP (sgGFP.10) and the other targeting mCherry (sgmCherry.1), both individually and in combination. We measured GFP and mCherry fluorescence and observed ~15% GFP or mCherry low populations for each sgRNA individually (Supplementary Data Fig. 9), thereby demonstrating that these sgRNAs were effective in generating mutations that ablated fluorescence. Upon the addition of both sgRNAs, we observed a slight decrease in mutation of GFP or mCherry separately (~12%) perhaps due to sharing of CRISPR-X machinery, but an increase in cells with mutations at both loci to 1.92% compared to 0.26% or 0.30% for cells with either sgGFP.10 or sgmCherry.1 incorporated individually. These results demonstrate that CRISPR-X can be used to simultaneously mutagenize two sites within the same cell, suggesting that co-evolution of two genomic loci should be possible.
Discussion
Here we demonstrate that hyperactive AID targeted with dCas9 can be used to generate localized sequence diversity within the mammalian genome at a rate comparable to somatic hypermutation, and that these mutagenized populations can be subjected to selection to evolve new protein function. This system, CRISPR-X, can simultaneously mutagenize multiple genomic loci, and preserves reading frame by avoiding insertions/deletions observed with active Cas9. While the activity of AID in antibody maturation has been shown to require transcription12, we observed mutations above background for sgRNAs targeting both upstream and downstream of the TSS. Although regions upstream of the TSS may be transcribed at lower levels, these findings suggest that CRISPR-X is not bound to regions downstream of annotated transcription start sites and could allow for the engineering and investigation of promoters, enhancers, and other regulatory elements.
Using CRISPR-X, we highlight several examples of directed evolution. First, we show that GFP can be readily evolved to EGFP with the simple electroporation of an appropriately designed sgRNA and targeted AID. In addition, we demonstrate that mutagenesis of the target of the chemotherapeutic bortezomib (PSMB5) could reveal both known and novel mechanisms of resistance. In this experiment, we find the canonical A108V/T mutation, which was identified in bortezomib resistant cell lines41,43 and observed in colorectal cancer patient samples44, along with many others that are consistent with the disruption of the binding pocket of bortezomib. Interestingly, we uncover a mutation located in Exon 4 (G242D), which had not been previously connected to bortezomib resistance, and is located on the side of the protein opposite the bortezomib pocket (Fig. 3d). This could suggest additional mechanisms of resistance, and may inform study of PSMB5 function as well as future drug design. Additionally, we identified synonymous and intronic mutations which require further study.
CRISPR-X represents an efficient strategy to create a diverse library of point mutations in situ, which expands the repertoire of methods for genome engineering using Cas9. Mutagenesis using active Cas9 has been effective for inducing insertions and deletions, which can disrupt functional elements27,45,46, and inactivate protein function28–30,47. During the preparation of our manuscript, two elegant studies by Komor et al. and Nishida et al. demonstrated that dCas9 can be used to recruit deaminases for the remarkably precise conversion of C > T within a 5 bp window, as a way to correct single base changes observed in disease23,26. Here, we show that a hyperactive AID variant can create dense, highly variable point mutations within a region of 100bp surrounding an sgRNA target site at a rate of up to ~1/500–1/1000 bp−1 (Fig. 4b), compared to the normal mutation rate during cellular DNA replication of ~1/109 bp−1 10. As in antibody somatic hypermutation, we observe a large variety of transitions and transversions from C and G bases to all possible bases (rather than just C>T and G>A described in previous studies), and a low level of all base changes (Fig. 4d). Using this diverse population of mutants, we demonstrate that we are able to select for the evolution of new function.
CRISPR-X presents a number of significant advantages over existing methods used to engineer proteins or introduce diversity, which make it a highly complementary strategy for genome engineering. Previous work has demonstrated a powerful strategy by which active Cas9 can be used to introduce mutant oligonucleotide donor libraries by homologous recombination; the resulting cell populations can be used to study RNA and protein function in mammalian cells32 or select for improved fermentation in yeast31. However, this strategy requires the separate synthesis of a mutant donor library for each engineered site. In contrast, CRISPR-X repurposes the somatic hypermutation machinery, making it possible to generate a library of point mutations in situ using a single sgRNA, and even greater diversity through multiplexing. In addition, the targeting of AID should allow continuous mutagenesis and evolution of protein function as is observed in antibody affinity maturation, as opposed to introducing a synthetic library of defined size.
Previous efforts to use AID for mutagenesis used overexpression of both AID and the target protein. In those studies, the target is present at non-physiological levels, and cells have significant genome instability and potentially confounding off-target mutations due to promiscuous AID activity48,49. While elegant work has been done to understand the targeting of somatic hypermutation to the Ig locus11,50, the known control elements would be difficult to install systematically throughout the genome. CRISPR-X overcomes both of these limitations by using dCas9 to target somatic hypermutation, which should facilitate both engineering of new biomolecules as well as an understanding of the SHM process itself. Importantly, the ability to introduce the CRISPR-X system by electroporation, use multiple sgRNAs, and potentially induce repeated rounds of mutagenesis should allow exploration of a virtually limitless sequence space, since combinations of mutations observed with single sgRNAs can be multiplied by simultaneously targeting multiple genomic locations. We envision that this system should make it possible to study the co-evolution of two interacting proteins expressed at endogenous levels, and could provide a streamlined strategy for selection of enhanced antibody and enzyme function via mutagenesis in native context.
Methods
Design and construction of CRISPR-X and fluorescent protein plasniids
A list of the plasmids and primers used are listed in Extended Dataset 1. Lenti dCAS-VP64_Blast, lenti MS2-P65-HSF1_Hygro, and lenti sgRNA(MS2)_zeo backbone were a gift from Feng Zhang (Addgene plasmids #61425–61427). The VP64 effector was removed from the dCas9 construct by digesting with BamHI and EcoRI followed by Gibson assembly to re-insert PCR amplified blasticidin resistance marker (pGH125). For the MS2 fusions, the P65-HSF1 was removed using restriction digest with BamHI and BsrGI. AID (pGH156) and AIDΔ (pGH153) were PCR amplified from a FLAG-AID expressing plasmid, courtesy of the Cimprich Lab, and Gibson assembled into the digested vector. Catalytically inactive (pGH183) and hyperactive mutants (pGH335) were generated using PCR primers containing the desired mutations. Subunits of AID were amplified using those primers and then joined using overlapping PCR. The mutant AID PCR product was Gibson assembled into the digested MS2 expression vector. GFP, mCherry, and wtGFP expressing plasmids driven by an Ef1α promoter were generated using pMCB246 which was digested with NheI and XbaI, removing a puromycin resistance-T2A-mCherry cassette. GFP (pGH045) and mCherry (pGH044) were PCR amplified and inserted into the digested vector using Gibson assembly. Variants of GFP (wtGFP (pGH220) and identified mutants (pGH311-S65T, pGH312-Q80H, pGH314-S65T + Q80H) were constructed using the previously described overlapping PCR method followed by Gibson assembly. Plasmids maps of these constructs are available upon request. For dual guide experiments, a second sgRNA expressing plasmid was constructed by removing the zeocin resistance (digestion of lenti sgRNA(MS2)_zeo with BsrGI and EcoRI) and replaced with puromycin resistance with a removed BsmBI cut site by Gibson assembly (pGH224). sgRNA vectors were generated by digesting the either lenti sgRNA(MS2)_zeo or pGH224 with BsmBI. Oligonucleotides with overhangs compatible with subsequent ligation were designed and annealed followed by ligation into the digested vector. The sequence for the sgRNAs are listed in the Extended Dataset 1. All plasmid sequences were verified using Sanger sequencing. All oligonucleotides were ordered from Integrated DNA Technologies (IDT).
Cell Culture and generating parent cell lines
Lentiviral production as well as infection and culturing of K562 cells (ATCC) were performed as described51. Parental K562 cell lines were generated by infecting dCas9-Blast (pGH125) followed by blasticidin selection (10μg/mL, Gibco) for 7 days. Cells were subsequently infected with both GFP (pGH045) and mCherry (pGH044) expression vectors or with a wtGFP (pGH220) expression vector and sorted via FACS for fluorescence. These cell lines were used as the parental samples in the sequencing assays. For integrated CRISPR-X experiments, these cells were infected with MS2-AID (pGH153, 156, 183, and 335) expressing vectors followed by selection with hygromycin B (200μg/mL, Life Technologies) for 7 days. All cell lines were maintained in a humidified incubator (37°C, 5% CO2), and checked regularly for mycoplasma contamination.
Fluorescence Microscopy of MS2-AID localization
K562 cells were lentivirally infected by constructs expressing an MS2-AIDΔ (pGH153) and MS2-AID (pGH156) and selected with hygromycin B for 7 days. 1 million cells were harvested and fixed in 4% paraformaldehyde for 15 min at room temperature. Cells were washed 3 times with PBS and then permeabilized with 0.1% Triton-X in PBS for 10 min at 4°C. Cells were incubated in blocking solution (3% BSA in PBS) for 1h at room temperature. They were centrifuged at 500 g for 5 minutes and resuspended in 1:500 dilution of rabbit anti-MS2 antibody (Millipore, cat no. ABE76) in blocking solution for 2h at room temperature. The cells were washed 3 times with PBS and resuspended in 1:1000 dilution of Alexa Fluor 488 conjugated goat anti-rabbit antibody (Life Technologies) in blocking solution and incubated for 2h at room temperature. Cells were washed in PBS 3 times and resuspended in Vectashield (Vector Laboratories) containing DAPI. The samples were deposited on a glass coverslip and imaged using an inverted Nikon Eclipse Ti confocal microscope with 488nm (AlexaFluor488) and 405nm (DAPI) lasers, an oil immersion objective (Plan Apo λ, N.A. = 1.5, 100X, Nikon), and an Andor Ixon3 EMCCD camera. Images were processed using ImageJ (National Institutes of Health).
Comparison of MS2-AID variant expression
K562 cells were infected with constructs expressing MS2-AID (pGH156), MS2-AIDΔ (pGH153), and MS2-AID*Δ (pGH335) and selected with hygromycin B for 7 days. 1.2 million cells were harvested and rinsed once with ice cold PBS before being lysed in lysis buffer (1% Triton X-100, 150mM NaCl, 50mM Tris pH 7.5, and 1mM EDTA) for 20 minutes on ice. Debris was removed by centrifugation for 10min at 21,000g at 4°C. The supernatant was collected and protein was quantified for each sample using DC Protein Assay (Bio-Rad). For each sample, 100μg of protein was denatured under reducing conditions (NuPAGE® LDS Sample Buffer (4X), Life Technologies, cat no. NP0007, and 100mM DTT), loaded on a 4–12% Novex BisTris SDS-PAGE gel (Life Technologies), and analyzed by immunoblot using a rabbit anti-MS2 antibody (1:1000 dilution, Millipore, cat no. ABE76) and mouse anti-GAPDH antibody (1:4000 dilution, Life Technologies, cat no. AM4300). Donkey anti-mouse IRDye 680 LT and goat anti-rabbit IRDye 800CW (1:20000 dilution, LI-COR Biosciences, product nos: 925–68022 and 925–32211) were used as secondary antibodies. Immunoblots were imaged using an Odyssey infrared imaging system (LI-COR Biosciences).
Transient electroporation of K562 cells and testing MS2-AID variants
Nucleofection of K562 cells were performed as described52. 1 million K562 cells were harvested for each electroporation. Cells were centrifuged at 300 g for 5 min and resuspended in 100μL of nucleofection solution and mixed with plasmid DNA (5μg MS2-AID expressing plasmid and 5μg sgRNA expression vector) and loaded into a 2mM cuvette (VWR). Electroporations were performed using the T-016 program on the Lonza Nucleofector 2b. After electroporation, cells were rescued in warm supplemented RPMI media. Cells were grown for 10 days and the GFP and mCherry fluorescence was measured using the BD Accuri C6 flow cytometer. The scatter plots shown were generated in FlowJo. The cells were sorted for low GFP fluorescence and the cells were grown before preparation of sequencing.
Generating mutations from individual and dual sgRNA experiments
For integrated CRISPR-X experiments, three days after infection, selection was applied and continued for 11 days using blasticidin for dCas9, hygromycin B for MS2-AID variants, and zeocin (200μg/mL, Life Technologies) for sgRNA. For dual sgRNA experiments, the sgGFP.10 plasmid was further selected using puromycin (1μg/mL, Sigma-Aldrich). For GFP and mCherry targeting sgRNAs, the GFP and mCherry fluorescence were measured after selection using a BD Accuri C6 flow cytometer. Scatter plots shown were generated in FlowJo. Experiments targeting GFP or mCherry were performed with 3 biological replicates while endogenous loci were performed with 2 biological replicates.
Preparation of sequencing samples
To sequence the targeted loci, genomic DNA was extracted from 0.5–1.5 million cells using the QiaAmp DNA mini kit (Qiagen). The targeted loci were PCR amplified from 0.5–1μg of genomic DNA using primers shown in Extended Dataset 1. The product was purified on a 0.8–1% TAE agarose gel. The concentration was measured by Qubit (Life Technologies) and then prepared for sequencing following the Nextera XT kit protocol (Illumina). For PSMB5 experiments, DNA was extracted from 20 million cells and PCR amplification was performed on 5μg of genomic DNA. After individual gel purification of PCR product from each exon, PCR products were mixed in equimolar amounts before beginning the Nextera XT preparation.
Sequences were measured on a NextSeq 500 (Illumina) with paired end reads of length 76 or 151bp. Every sequencing run included a parental sample for each locus that was being sequenced.
Analysis of Sequencing data
Sample sequencing and Alignment
Over all sequenced samples, 4.5 million reads were produced on average. Sequencing adapters (5′ adapter: CTGTCTCTTATACACATCTCCGAGCCCACGAGAC; 3′ adapter: CTGTCTCTTATACACATCTGACGCTGCCGACGA) were trimmed using cutadapt (version 1.8.153), also discarding reads under 30 bp and nucleotides flanking the adapters with Illumina quality score lower than 30 (leaving only flanking sequences for which the base call accuracy is over 99.9%). Alignment on respective reference loci was performed using bwa aln (v0.7.7) and bwa samse54). A maximum number of 3 or 5 mismatches was allowed for samples with read length of 76 bp and 151 bp respectively. Aligned files were then sorted using samtools (v0.1.1955)
Only reads aligned to their respective references with mapping quality over 30 were kept for further analysis. On average, 90% of sequenced reads (Standard Deviation 16%) were successfully mapped to the provided reference genome. From these aligned reads, 96% (Standard Deviation 5.7%) were remaining after filtering on mapping quality.
Tabulation of mutations per base
We computed allelic counts at each position with a custom script, after filtering for nucleotides with Illumina base quality score over 30 using samtools mpileup (version 1.2). The parental sample was used to estimate the mutations introduced through sample preparation and sequencing. Using the parental as a reference, we calculated the mutation enrichment at each base by taking the percentage of reads with alternative alleles in comparison to the same proportion calculated in the parental sample. For frequency of mutation calculated at each base, we subtracted the frequency of alternative alleles in the parental sample from the frequency calculated for the mutated sample. The first and last 50 bases of each locus were excluded from these enrichments given the ends had lower read coverage that was a byproduct of the Nextera XT preparation. We calculated the transitions/transversions/indels observed in the hotspots by looking at the distribution of frequencies of every possible alternative nucleotide at each position. We then subtracted the parental cell line respective frequencies in the hotspots to take into account the background noise. Negative values were set to 0. To estimate the remaining noise resulting from sequencing and variability between samples, we calculated the standard deviation of the frequency of alternative alleles in all parental samples from the studied batch (Fig. 4d). Reported medians, maximums, and distributions result from this calculation.
Calculation of mutation frequency in hotspot regions
The number of mutations per read was limited during the alignment step (see above). We performed mutation counts from the filtered aligned data to compute the enrichment of reads carrying mutations within the hotspot. After selecting all reads overlapping the hotspot using samtools view (version 1.255), each read was screened for mutations with their respective positions. These results were then summarized for each sample by calculating the ratio between the number of reads with mutations spanning the hotspot and the total number of reads spanning the hotspot. The frequency of mutations enrichment was calculated by subtracting the results from the parental cell line as background.
Calculation of the observed percentage of possible transitions
To estimate mutant diversity in a population of cells, we analyzed the hyperactive AID*Δ mutant samples (Fig. 4b,c) with a custom R script. For each sgRNA-targeted sample, we selected the mutation hotspot (+/− 50bp with respect to the PAM) and computed the frequency of each observed alternate allele. At each position we subtracted the respective allelic frequency observed in the corresponding parent sample. Using a sliding window of 21 bp over the hotspot and the 20bp flanking each end, we calculated the percentage of all 63 possible transitions in the window that were observed above noise. Noise was defined as the standard deviation of the alternative allele frequency among all parent samples. Results were then output by window as a boxplot representing the combination of all considered sgRNA-targeted samples.
Evolution of wtGFP to EGFP using CRISPR-X
For transient electroporation wtGFP experiments, K562 cells expressing dCas9 and wtGFP were electroporated as described earlier with 5μg of MS2-AIDΔ and either 1.25μg for each of wtGFP.1–4 or Safe.2,4–6 sgRNA expressing vectors. Cells were grown for 10 days after electroporation before sorting. For integrated experiments, K562 cells expressing dCas9, MS2-AIDΔ, and wtGFP were infected with either wtGFP.1 or Safe.2 sgRNA expressing vectors. After 3 days, cells were selected with blasticidin, hygromycin B, and zeocin for 11 days. Cells were sorted via FACS to obtain spectrum-shifted GFP variants. For the electroporation experiments, cells were grown for 7 days between sorting rounds. Samples were prepared for sequencing as described previously.
Flow cytometry of wtGFP variants
HEK293T (ATCC) cells were cultured in DMEM with 10% FBS, penicillin/streptomycin, and L-glutamine. For each transfection, 1 million HEK293T cells were plated in 2 mL of supplemented DMEM media. 1.5μg of wtGFP expressing plasmid (pGH045, 220, 311, 312, and 314) was mixed with 200μL serum-free DMEM and 10μL of polyethylenimine (PEI, 1mg/mL, pH 7.0, PolySciences Inc.) and incubated at room temperature for 30 minutes. The mixture was added to the cells and grown for 72 h with an additional 3 mL of DMEM supplemented media added after 24 h. The samples were trypsinized and analyzed using a FACScan flow cytometer (BD Biosciences). Additional analysis of the data was performed using FlowJo.
Design and construction of PSMB5 Tiling libraries
The PSMB5 tiling library was generated using CHOPCHOP online tool56 for the three PSMB5 isoforms (NM_0011449632, NM_00130725, and NM_002797). sgRNAs for each isoform were combined. sgRNAs having any genomic off-target matches, more than 1 off-target when allowing one mismatch in the sgRNA sequence, or 5 or more off-targets when allowing one or two mismatches within the sgRNA sequence were removed. The sgRNAs were further filtered by removing any containing a BsmBI cut site, which interferes with the library cloning strategy. The final library contained 143 sgRNAs. Safe harbor sgRNAs were designed to target genomic loci that have not been annotated to include gene exons or UTRs, have signal in biochemical assays (DNaseI, CHIP-Seq, etc.) or have signal in sequence-based analyses (conserved elements, transcription factor motif searches, etc). The design and selection of these sgRNAs will be discussed in more detail in future work. 705 sgRNAs targeting safe harbor regions were selected to serve as a control library. The sgRNA sequences for both libraries are included in Extended Dataset 2.
Oligonucleotide libraries were synthesized by Agilent and cloned into the sgRNA expression vector as previously described57–59. Vector and sgRNA inserts were digested with BsmBI. Large scale lentivirus production and infection of K562 cells were performed as described57,59. Three days after infection, selection began with blasticidin, hygromycin B, and zeocin for 11 days. Cells were expanded to 20 million cells for each treatment (safe harbor and PSMB5 libraries in duplicate) and were pulsed with 20nM bortezomib (Fisher Scientific) for three days followed by recovery until log growth was restored (5–10 days) before the next pulse. The cells were pulsed a total of three times. After the final pulse, cells were harvested and prepared for sequencing as described earlier.
Installation and validation of bortezomib resistant PSMB5 mutations
sgRNAs were designed to target near the location of the installed SNP and 101nt donor oligos were designed to be centered around the installed mutation. Oligonucleotides with proper overhangs were ordered from IDT and annealed before ligation into BbsI digested pGH020, a hu6 driven sgRNA expression vector. All plasmids were verified by Sanger sequencing. The sgRNA and ssDNA donor oligo sequences are listed in Extended Dataset 1, respectively.
K562 cells expressing Cas9 were electroporated with 5μg of sgRNA expressing vector and 100 picomoles of donor oligo. Cells were grown for 6 days before 300,000 cells were placed under selection with 20nM bortezomib for 14 days. The viability of the cells was measured by flow cytometry using a live cell gate (FSC/SSC). After selection, 750,000 cells were harvested and genomic DNA was extracted using the QiaAmp DNA Mini Kit (Qiagen). The PSMB5 exonic locus containing the mutation was PCR amplified, gel purified, and ligated into the pCR-Blunt vector using the Zero-Blunt cloning kit (Life Technologies). 8–15 colonies were Sanger sequenced for each sample.
Extended Data
Extended Data Figure 1. Characterization of AID variants.

a) Diagram of AID variants. NLS, NES, deaminase domain, truncations, and activity-altering mutations are indicated. b) Fluorescence microscopy of MS2-AID and MS2-AIDΔ constructs in K562 cells is shown. Cells were fixed and stained with an MS2 antibody (green) and the nuclear stain DAPI (blue). c) A comparison of the expression of different MS2-AID variants is shown. K562 cells expressing the variants were lysed and analyzed on an SDS-PAGE gel followed by immunoblotting with an MS2 antibody (top) or GAPDH antibody (bottom). d) K562 cells containing dCas9, GFP, and mCherry were transiently electroporated with indicated combinations of MS2-AID, MS2-AIDΔ, or MS2-AIDΔDead and either sgGFP.1 or sgNegCtrl. GFP and mCherry fluorescence of the cells were measured by flow cytometry as a proxy for mutation rate. Shown are the scatter plots from the flow cytometry and a graph summarizing the non-fluorescent populations. e) Cells were sorted for low GFP expression and the GFP locus was sequenced. A graph of the enrichment of mutation at each base is shown here.
Extended Data Figure 2. On-target mutagenesis using CRISPR-X with limited off-target effect.

a) Cells were infected with indicated combinations of MS2-AIDΔ or MS2-AIDΔDead and sgGFP.1 or sgNegCtrl and the GFP and mCherry fluorescence of the cells was measured by flow cytometry as a proxy for mutation rate. Shown are the scatter plots from flow cytometry and graphs summarizing the non-fluorescent populations. Error bars represent standard error. b) GFP and mCherry loci of the infected cells were sequenced and enrichment of mutation was calculated at each base position for three replicate experiments.
Extended Data Figure 3. CRISPR-X tiling of GFP locus.

a) Map of sgRNAs tiling the GFP locus. b) sgRNAs targeting GFP were integrated into cells expressing dCas9, MS2-AIDΔ, GFP, and mCherry, and the GFP locus was sequenced. Enrichment of mutations at each base position is shown for three replicates of each sgRNA. c) A box plot indicating the frequency of mutated reads observed in the respective hotspot of each sgRNA is shown. The median value for the conditions is listed above each sample. The box plot lines represent the 1.5 of the interquartile range.
Extended Data Figure 4. Directed evolution of wtGFP to EGFP using CRISPR-X.

a) A replicate of the wtGFP evolution experiment (Fig. 2a) was performed using electroporated sgRNAs and MS2-AIDΔ. Flow cytometry scatter plots are shown for the wtGFP parent and samples before each round of sorting. The wtGFP locus was sequenced for the unsorted condition and after both sorting rounds. Enrichment of mutation was calculated at each base position. The graphs of enrichment are shown for both wtGFP targeted and safe harbor targeted libraries except after Sort #2 where no safe harbor cells were recovered after sorting. Identified mutations are labeled in the graphs. b) wtGFP cells expressing dCas9, MS2-AIDΔ, and wtGFP were lentivirally infected with sgwtGFP.1 or sgSafe.2 in replicate and sorted once, enriching for spectrum-shifted GFP cells. Scatter plots for the parent and unsorted populations are shown for both replicates. The wtGFP locus was sequenced pre- and post-sorting, and enrichment of mutations at each base position is shown. The S65T mutation is labeled in the graph for the sorted condition.
Extended Data Figure 5. Identifying bortezomib resistant mutations in PSMB5.
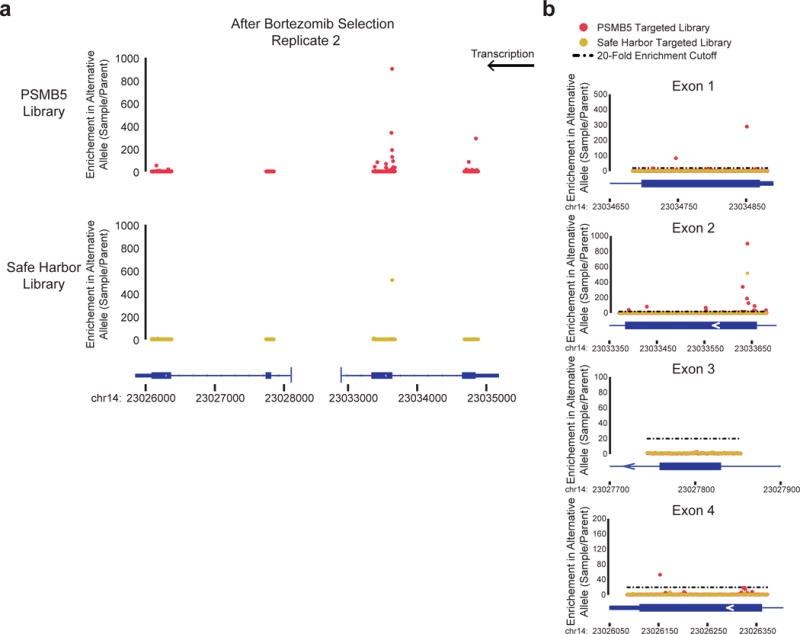
a) A replicate experiment was performed for directed evolution of bortezomib-resistant PSMB5 mutations (see Fig. 3). The PSMB5 exonic loci were sequenced after selection with bortezomib for both the PSMB5 and Safe Harbor libraries and enrichment of mutations at each base position is shown. b) Graphs of mutation enrichment are shown for individual exonic loci of PSMB5. Mutations that were enriched beyond the 20-fold cutoff (dashed black line) are observed in Exons 1, 2, and 4.
Extended Data Figure 6. Knock-in and validation of novel bortezomib-resistant PSMB5 variants.

Bortezomib resistant mutations observed in PSMB5 (Fig. 3d) were knocked-in to K562 cells and populations were selected with bortezomib. The corresponding PSMB5 exons for the five most viable mutations were amplified, cloned into pCR-Blunt, and sequenced individually. Shown is a table summarizing the sequences of individual colonies with mutations or insertions/deletions shown in red; the targeted base is in bold.
Extended Data Figure 7. Improved mutagenesis using AID*Δ.
a) sgRNAs targeting either GFP (sgGFP.3 and sgGFP.10) or a safe harbor locus (sgSafe.2) were integrated into cells expressing dCas9, MS2-AID*Δ, GFP, and mCherry. The GFP and mCherry loci were sequenced. Enrichment of mutation at each base position is shown. b) For sgGFP.3 and sgGFP.10 paired with either AIDΔ or AID*Δ, sequences were filtered for those containing a mutation, and the average number of mutations per sequence was calculated. The average and standard error are shown.
Extended Data Figure 8.

a) sgRNAs targeting either GFP or endogenous loci were integrated into cells expressing dCas9, MS2-AID*Δ, GFP, and mCherry. The respective targeted loci were sequenced. Graphs showing the frequency of alternative alleles at each base position relative to the PAM of the sgRNA are shown. b) Box plot indicating the range of frequency of mutated reads over the 100bp region for 30 sgRNAs is shown. The lines represent 1.5 times the interquartile range. Median value is indicated above graph.
Extended Data Figure 9.

sgGFP.10 and sgmCherry.1 were integrated separately or in combination into cells expressing dCas9, MS2-AID*Δ, GFP, and mCherry. The GFP and mCherry fluorescence of the cells were measured. The scatter plots of the flow cytometry for each of the samples are shown (left). A graph summarizing the percentage of GFP negative or mCherry negative cells is shown (top left). In the bottom left panel, a graph displaying the percentage of cells that have neither GFP nor mCherry is shown. Error bars indicate standard error.
Extended Dataset 1.
Complete list of plasmids, oligonucleotides, and sgRNA sequences used.
| PLASMIDS | |
|---|---|
| Name | Description |
| pGH125 | dCas9-Blast |
| pGH153 | MS2-AIDΔ-Hygro |
| pGH156 | MS2-AID-Hygro |
| pGH183 | MS2-AIDΔDead-Hygro |
| pGH224 | sgRNA_2xMS2_Puro |
| pGH044 | mCherry |
| pGH045 | GFP |
| pGH220 | wtGFP |
| pGH311 | wtGFP S65T |
| pGH312 | wtGFP Q80H |
| pGH314 | wtGFP S65T, Q80H |
| pGH335 | MS2-AID*Δ-Hygro |
| pGH020 | sgRNA_G418-GFP |
| OLIGONUCLEOTIDES | ||
|---|---|---|
| Vector | Name | Sequence (5′–3′) |
| dCas9 | dCas9-Blast For (oGH255) | AAAAAGAGGAAGGTGGCGGCCGCTGGATCCGAGGGCAGAGGAAGTCTGCTAACAT |
| dCas9-Blast Rev (oGH256) | AGGTTGATTACCGATAAGCTTGATATCGAATTC | |
| MS2-AID | MS2-AID For (oGH272) | AAGAGGAAGGTGGCGGCCGCTGGATCCATGGACAGCCTCTTGATGAACCG |
| MS2-AID Rev (oGH273) | TTCCTCTGCCCTCTCCACTGCCTGTACAAAGTCCCAAAGTACGAAATGCGTC | |
| MS2-AIDΔ Rev (oGH274) | TTCCTCTGCCCTCTCCACTGCCTGTACAAGTACGAAATGCGTCTCGTAAGTC | |
| AIDΔDead Mut For (oGH315) | GAACGGCTGCCGCGTGCAATTGCTCTTCCTCCGCTACATCTCG | |
| AIDΔDead Mut Rev (oGH316) | AAGAGCAATTGCACGCGGCAGCCGTTCTTATTGCGAAGATAAC | |
| AID*Δ K10E For (oGH456) | AAGAGGAAGGTGGCGGCCGCTGGATCCATGGACAGCCTCTTGATGAACCGGAGGGAGTTTCTTTACCAA | |
| AID*Δ E156G For (oGH457) | TACTGCTGGAATACTTTTGTAGAAAACCACGGAAGAACTTTCAAAGCCTGGGAAGG | |
| AID*Δ E156G Rev (oGH458) | CCTTCCCAGGCTTTGAAAGTTCTTCCGTGGTTTTCTACAAAAGTATTCCAGCAGTA | |
| AID*Δ T82I For (oGH459) | GCTGCTACCGCGTCACCTGGTTCATCTCCTGGAGCCCCTGCTACGAC | |
| AID*Δ T82I Rev (oGH460) | GTCGTAGCAGGGGCTCCAGGAGATGAACCAGGTGACGCGGTAGCAGC | |
| Fluorescent Proteins | GFP/mCherry For (oGH144) | CATTTCAGGTGTCGTGAGCTAGCCCACCATGGTGAGCAAGGGCGAGGAG |
| GFP/mCherry Rev (oGH146) | CTGGCTTACTAGTCGGTTCAACTCTAGATTACTTGTACAGCTCGTCCATGCCG | |
| wtGFP Mut For (oGH363) | GTGACCACCTTCAGCTACGGCGTGCAGTGC | |
| wtGFP Mut Rev (oGH364) | GCACTGCACGCCGTAGCTGAAGGTGGTCAC | |
| wtGFP Q80H For (oGH447) | ACCCCGACCACATGAAGCACCACGACTTCTTCAAGTCC | |
| wtGFP Q80H Rev (oGH448) | GGACTTGAAGAAGTCGTGGTGCTTCATGTGGTCGGGGT | |
| wtGFP S65T For (oGH449) | CCTCGTGACCACCTTCACCTACGGCGTGCAGTGCT | |
| wtGFP S65T Rev (oGH450) | AGCACTGCACGCCGTAGGTGAAGGTGGTCACGAGG | |
| Puromycin Resistance | Puro For (oGH375) | TTTCTTCCATTTCAGGTGTCGTGATGTACAATGACCGAGTACAAGCCCACGG |
| Puro Rev (oGH376) | ATTACCGATAAGCTTGATATCGAATTCTCAGGCACCGGGCTTGCGGGTCATG | |
| Puro BsmBI For (oGH377) | TCCTGGCCACCGTCGGCGTATCGCCCGACC | |
| Puro BsmBI Rev (oGH378) | GGTCGGGCGATACGCCGACGGTGGCCAGGA | |
| sgRNA Sequences | ||
|---|---|---|
| Name | sgRNA Sequence (5′–3′) | Genomic Position |
| sgGFP.1 | GGCGAGGGCGATGCCACCTA | |
| sgNegCtrl | GCTCAAGAACGCCTTCCCCAGTC | |
| sgGFP.2 | GGCACGGGCAGCTTGCCGG | |
| sgGFP.3 | AAGGGCATCGACTTCAAGG | |
| sgGFP.4 | CGATGCCCTTCAGCTCGATG | |
| sgGFP.5 | CTCGTGACCACCCTGACCTA | |
| sgGFP.6 | CAAGTTCAGCGTGTCTGGCG | |
| sgGFP.7 | CAACTACAAGACCCGCGCCG | |
| sgGFP.8 | GGTGAACCGCATCGAGCTGA | |
| sgGFP.9 | CGGCCATGATATAGACGTTG | |
| sgGFP.10 | CGTCGCCGTCCAGCTCGACC | |
| sgGFP.11 | AGCACTGCACGCCGTAGGTC | |
| sgGFP.12 | TCAGCTCGATGCGGTTCACC | |
| sgwtGFP.1 | CCGGCAAGCTGCCCGTGCCC | |
| sgwtGFP.2 | GCTTCATGTGGTCGGGGTAG | |
| sgwtGFP.3 | CGTGCTGCTTCATGTGGTCG | |
| sgwtGFP.4 | GTCGTGCTGCTTCATGTGGT | |
| sgSafe.2 | TCCCCCTCAGCCGTATT | chr12: 114129110-114129129 |
| sgSafe.4 | GATTGATATTGCCTTCT | chr12: 17350231-17350250 |
| sgSafe.5 | TCTGACTCCTAATGGAG | chr12: 114127368-114127387 |
| sgSafe.6 | ATTACTTTAGAGTAAGA | chr13: 105390313-105390332 |
| sgHBG2.1 | GGTCCATGGGTAGACAACC | chr11: 5249566-5249584 |
| sgHBG2.2 | GTGAGATTGACAAGAACAGT | chr11: 5249593-5249612 |
| sgHBG2.3 | AGGTCGCTTCTCAGGATTTG | chr11: 5249633-5249652 |
| sgHBG2.4 | GAGATCATCCAGGTGCTTTG | chr11: 5249437-5249456 |
| sgHBG2.5 | GCTACTATCACAAGCCTGTG | chr11: 5249758-5249777 |
| sgGSTP1.1 | GGAGATGTATTTGCAGCGG | chr11: 67585205-67585223 |
| sgGSTP1.2 | GGACATGGTGAATGACGGCG | chr11: 67585175-67585194 |
| sgGSTP1.3 | AGCCACCTGAGGGGTAAGGG | chr11: 67585310-67585329 |
| sgGSTP1.4 | CTGCACCCTGACCCAAGAAG | chr11: 67585341-67585360 |
| sgGSTP1.5 | TGATCAGGCGCCCAGTCACG | chr11: 67585090-67585109 |
| sgFTL.1 | GCCGAGGAGAAGCGCGA | chr19: 48965833-48965849 |
| sgFTL.2 | GCGCGAGGAGCCTTGATTTG | chr19: 48965963-48965982 |
| sgFTL.3 | CTCTATTTCCAGCGGTTAAG | chr19: 48966038-48966057 |
| sgFTL.4 | TAGCGGGAGGCGAGGCCAAG | chr19: 48965721-48965740 |
| sgFTL.5 | ACGCGCCAGCCTTCTTTGTG | chr19: 48965673-48965692 |
| sgPTPRC.1 | GTTTGTTCTTAGGGTAACAG | chr1: 198639077-198639096 |
| sgPTPRC.2 | TATCCTTGTGAAGCTAGGAG | chr1: 198638504-198638523 |
| sgPTPRC.3 | TGTTCTTGGCGCTACTGATG | chr1: 198638409-198638428 |
| sgPTPRC.4 | GGCGAGTGTGTATAGATCAG | chr1: 198697174-198697193 |
| sgPTPRC.5 | TAATGCATGTTGTTAGGGAG | chr1: 198697085-198697104 |
| sgPTPRC.6 | TGGGGAGTTAGTATACTGGG | chr1: 198696623-198696642 |
| sgPTPRC.7 | ATACACACTATAGTGGACTG | chr1: 198696605-198696624 |
| sgCD274.1 | AACTCCCACAGCATTTATCC | chr9: 5447248-5447267 |
| sgCD274.2 | ATGGGAAAATGAATGGCTGA | chr9: 5448598-5448617 |
| sgCD274.3 | CACCACCAATTCCAAGAGAG | chr9: 5462979-5462998 |
| sgCD274.4 | CAATGCAGGCTGGTTCTCAG | chr9: 5462727-5462746 |
| sgCD274.5 | TTTCATAGCCGGGAAACCTG | chr9: 5463466-5463485 |
| sgCD14.1 | TCAGGGAGGGGGACCGTAAC | chr5: 140633319-140633338 |
| sgCD14.2 | GGAGGGGGACCGTAACAGGA | chr5: 140633323-140633342 |
| sgCD14.3 | ATTCAGGGACTTGGATTTGG | chr5: 140633606-140633625 |
| sgCD14.4 | CCTCATCTGTTGGCACCAAG | chr5: 140633670-140633689 |
| sgCD14.5 | AGGAGAGAGCAACGTGCAAG | chr5: 140634212-140634231 |
| sgmCherry.1 | GCGGTCTGGGTGCCCTCGTA |
| Genomic Amplification Primers | ||
|---|---|---|
| Locus | Direction | Sequence (5′–3′) |
| GFP | For (oGH072) | AGGCCAGCTTGGCACTTGATGT |
| Rev (oGH046) | TGTTGTGGCGGATCTTGAAGTTC | |
| mCherry | For (oGH072) | AGGCCAGCTTGGCACTTGATGT |
| Rev (oGH343) | GCTTCAGCCTCTGCTTGATCTC | |
| Safe.2 | For (oGH371) | CACTATGACCACAGCCACTCAC |
| Rev (oGH372) | CTTTCTGAAAAGTAACCCAGCCTCA | |
| Safe.4 | For (oGH397) | GAACTGTGAATAATAAGCAATCATCCAG |
| Rev (oGH398) | GCTTGCCAAAAATTGTGTACCCTTTCC | |
| Safe.5 | For (oGH399) | TAGGTAACCCATCTGAGGTTTTCAAATAT |
| Rev (oGH400) | GAGAAAAGAACATGACTTCCAGCAGC | |
| Safe.6 | For (oGH401) | CCAAATTGCAGCCACACTTGAAAACC |
| Rev (oGH402) | TAGGAAGCAGTGTAGGAGGATTGG | |
| wtGFP | For (oGH072) | AGGCCAGCTTGGCACTTGATGT |
| Rev (oGH029) | AAGCAGCGTATCCACATAGCGT | |
| PSMB5 Exon 1 | For (oGH468) | GCAAGGGGGCTGGCTCCACAC |
| Rev (oGH469) | TTAGTTCTTTCTGCCCACACTAGAC | |
| PSBM5 Exon 2 | For (oGH470) | CATGTGGTTGCAGCTTAACTCAC |
| Rev (oGH471) | GTGTTTTTGTGGTCTTATGTGGCC | |
| PSMB5 Exon 3 | For (oGH472) | ACAACATACCACCCCATCTCACC |
| Rev (oGH473) | CAAAGTGCTGGGATTACGGGTTTG | |
| PSMB5 Exon 4 | For (oGH474) | CAAGCAGCTGCATCCACCCTCTT |
| Rev (oGH475) | CTGCTAACCTCATCTCCCTTTCCAG | |
| HBG2 | For (oGH440) | GTATCTTCAAACAGCTCACACCC |
| Rev (oGH441) | GTCTTAGAGTATCCAGTGAGGCC | |
| GSTP1 | For (oGH442) | CACTGAGGTTACGTAGTTTGCCC |
| Rev (oGH443) | CGACAAATCCTCCTCCACCTCT | |
| FTL | For (oGH454) | TTCCTCTCCGCTTGCAACCTCC |
| Rev (oGH455) | CGGCACATAGAACTAAACCTACATTTC | |
| PTPRC Locus 1 | For (oGH500) | GCCAGTAAGCATTTTCCTAATAGATGGAC |
| Rev (oGH501) | GCCAAATGCCAAGAGTTTAAGCC | |
| PTPRC Locus 2 | For (oGH502) | TCATCCTTCTGAACTCAATTGCTTTG |
| Rev (oGH503) | CAATGATGCAAATGCTCTTAAAAGAAACTC | |
| CD274 Locus 1 | For (oGH504) | GGTGACTATTTCATTTGTGTGACACTC |
| Rev (oGH505) | GAAAGCAGTGTTCAGGGTCTACC | |
| CD274 Locus 2 | For (oGH508) | GAAAACCTGAACAAATGGAGAGGG |
| Rev (oGH509) | GCTTGCTCAGTAGATTATAATCCTACAGG | |
| CD14 | For (oGH510) | GGTCGATAAGTCTTCCGAACCTC |
| Rev (oGH511) | GCGAAACTGGTGAGTTACTAATTAATCC |
| Reagents for PSMB5 Variant Installation by HDR | |
|---|---|
| sgRNAs | |
| Mutation | sgRNA Sequence (5′–3′) |
| L11L, Exon 1 Control | CCGCGCTGGTTCACCGGTAG |
| Intronic | CTGCAACTATGACTCCATGG |
| R78N, A79TG, Exon 2 Control | TCATAGTTGCAGCTGACTCC |
| G82D | AGCTGACTCCAGGGCTACAG |
| A108V | CTGCTAGGCACCATGGCTGG |
| G242D | CAACCTCTACCACGTGCGGG |
| Exon 4 Control | TGAAGGGAACCGGATTTCAG |
| ssDNA donor oligonucleotides | |
|---|---|
| Mutation | Sequence (5′–3′) |
| L11L (oGH512) | CAGATCTGCACGACCCCCAAGTCCGAAAAACCCGCGCTGGTTCACCGGTAACGGTCTCTCCAACACGCTGGCAAGCGCCATGTCTAGTGTGGGCAGAAAG |
| Exon 1 Control (oGH513) | CTCCCTGGACCTAGATCCAGCAGATCTGCACGACCCCCAAGTCCGAAAAATCCGCGCTGGTTCACCGGTAGCGGTCTCTCCAACACGCTGGCAAGCGCCAT |
| Intronic (oGH520) | ACCCGCTGTAGCCCTGGAGTCAGCTGCAACTATGACTCCATGGCGGAACTATTAAGATCAGAGGAAAACACAAAACAGGCCACATAAGACCACAAAAACAC |
| R78N (oGH518) | CTATCACCTTCTTCACCGTCTGGGAGGCAATGTAAGCACCCGCTGTAGCCTTGGAGTCAGCTGCAACTATGACTCCATGGCGGAACTGTTAAGATCAGAGG |
| A79T (oGH517) | CTCTATCACCTTCTTCACCGTCTGGGAGGCAATGTAAGCACCCGCTGTAGTCCTGGAGTCAGCTGCAACTATGACTCCATGGCGGAACTGTTAAGATCAGA |
| A79G (oGH516) | TCTCTATCACCTTCTTCACCGTCTGGGAGGCAATGTAAGCACCCGCTGTACCCCTGGAGTCAGCTGCAACTATGACTCCATGGCGGAACTGTTAAGATCAG |
| G82D (oGH515) | ATGGGTTGATCTCTATCACCTTCTTCACCGTCTGGGAGGCAATGTAAGCATCCGCTGTAGCCCTGGAGTCAGCTGCAACTATGACTCCATGGCGGAACTGT |
| A108V (oGH514) | AGATTCGACATTGCCGAGCCAACAGCCGTTCCCAGAAGCTGCAATCCGCTACGCCCCCAGCCATGGTGCCTAGCAGGTATGGGTTGATCTCTATCACCTTC |
| Exon 2 Control (oGH519) | ATCTCTATCACCTTCTTCACCGTCTGGGAGGCAATGTAAGCACCCGCTGTCGCCCTGGAGTCAGCTGCAACTATGACTCCATGGCGGAACTGTTAAGATCA |
| G242D (oGH521) | TATACTTCTCATGTAGATCAGCCACATTGTCACTGGAGACTCGGATCCAGTCATCCTCCCGCACGTGGTAGAGGTTGACTGCACCTCCTGAGTAGGCATCT |
| Exon 4 Control (oGH523) | TCCATGACCCCATATGCATACACAGAGCCAGAACCTACAGAGAAGGTGGCACCTGAAATCCGGTTCCCTTCACTGTCCACGTAGTAGAGGCCTGGAAAGGG |
Extended Dataset 2.
Complete list of sgRNA sequences of PSMB5 Tiling and Safe Harbor libraries (Microsoft Excel file available online).
| PSMB5 Tiling Library | |
|---|---|
| sgRNA Name | sgRNA sequence |
| PSMB5_001144932.23 | AAAAACCCGCGCTGGTTCAC |
| PSMB5_001144932.36 | AACAACCACCCTGGCCTTCA |
| PSMB5_00130725.83 | AACATGGTGTATCAGTACAA |
| PSMB5_001144932.101 | AAGGTAGTTATTATAATATA |
| PSMB5_001144932.107 | AAGTACATTCCAAATGACTT |
| PSMB5_00130725.84 | AATCTATGAGCTTCGAAATA |
| PSMB5_00130725.60 | ACCACGTGCGGGAGGATGGC |
| PSMB5_00130725.47 | ACCTGCTAGGCACCATGGCT |
| PSMB5_00130725.29 | ACGTAGTAGAGGCCTGGAAA |
| PSMB5_00130725.52 | ACGTGGACAGTGAAGGGAAC |
| PSMB5_00130725.36 | AGAAGGTGGCCCCTGAAATC |
| PSMB5_001144932.29 | AGACCATCACTGAGACTCCC |
| PSMB5_00130725.78 | AGAGCCAGAACCTACAGAGA |
| PSMB5_001144932.59 | AGAGGATCGGCAACATGGCA |
| PSMB5_001144932.97 | AGCCTGGCCGCGCCAGGCTG |
| PSMB5_001144932.27 | AGCGCGGGTTTTTCGGACTT |
| PSMB5_001144932.9 | AGCTGACTCCAGGGCTACAG |
| PSMB5_00130725.61 | AGCTGCATCCACCCTCTTTC |
| PSMB5_00130725.67 | AGGCATCTCTGTAGGTGGCT |
| PSMB5_00130725.44 | AGTCAACCTCTACCACGTGC |
| PSMB5_00130725.34 | AGTGAAGGGAACCGGATTTC |
| PSMB5_00130725.80 | AGTGGAGCAGGCCTATGATC |
| PSMB5_00130725.19 | ATCCGCTGCGCCCCCAGCCA |
| PSMB5_001144932.90 | ATCTGCTGGATCTAGGTCCA |
| PSMB5_00130725.70 | ATCTGTGGCTGGGATAAGAG |
| PSMB5_00130725.39 | ATGCATATGGGGTCATGGAT |
| PSMB5_001144932.33 | ATTTCGATTCCTGGCTCTTC |
| PSMB5_00130725.24 | CAAAGGCATGGGGCTGTCCA |
| PSMB5_00130725.9 | CAACCTCTACCACGTGCGGG |
| PSMB5_001144932.25 | CAAGTCCGAAAAACCCGCGC |
| PSMB5_00130725.2 | CACCATGGCTGGGGGCGCAG |
| PSMB5_00130725.50 | CACCATGTTGGCAAGCAGTT |
| PSMB5_001144932.99 | CACCCCAGCCTGGCGCGGCC |
| PSMB5_001144932.10 | CACCTTCTTCACCGTCTGGG |
| PSMB5_00130725.30 | CACGTAGTAGAGGCCTGGAA |
| PSMB5_001144932.26 | CAGCGCGGGTTTTTCGGACT |
| PSMB5_001144932.39 | CAGCTGCAACTATGACTCCA |
| PSMB5_00130725.23 | CAGCTTCTGGGAACGGCTGT |
| PSMB5_00130725.8 | CAGTCAACCTCTACCACGTG |
| PSMB5_00130725.79 | CATAGGCCTGCTCCACTTCC |
| PSMB5_001144932.70 | CATAGTTGCAGCTGACTCCA |
| PSMB5_00130725.16 | CATCCTCCCGCACGTGGTAG |
| PSMB5_001144932.19 | CATGGCGCTTGCCAGCGTGT |
| PSMB5_00130725.3 | CATGTTGGCAAGCAGTTTGG |
| PSMB5_001144932.6 | CCACACCTTGAAGGCCAGGG |
| PSMB5_00130725.76 | CCACATTGTCACTGGAGACT |
| PSMB5_001144932.34 | CCATGAAGCATTTCGATTCC |
| PSMB5_00130725.18 | CCATGGTGCCTAGCAGGTAT |
| PSMB5_00130725.48 | CCCCAGCCATGGTGCCTAGC |
| PSMB5_001144932.2 | CCGCGCTGGTTCACCGGTAG |
| PSMB5_00130725.21 | CGCAGCGGATTGCAGCTTCT |
| PSMB5_001144932.4 | CGCGGGTTTTTCGGACTTGG |
| PSMB5_001144932.22 | CGCTACCGGTGAACCAGCGC |
| PSMB5_00130725.22 | CGGATTGCAGCTTCTGGGAA |
| PSMB5_001144932.28 | CGTGCAGATCTGCTGGATCT |
| PSMB5_001144932.21 | CGTGTTGGAGAGACCGCTAC |
| PSMB5_00130725.64 | CTAACCTCATCTCCCTTTCC |
| PSMB5_001144932.45 | CTATCACCTTCTTCACCGTC |
| PSMB5_00130725.56 | CTATGACCTGGAAGTGGAGC |
| PSMB5_00130725.14 | CTATTCCTATGACCTGGAAG |
| PSMB5_00130725.59 | CTCTACCACGTGCGGGAGGA |
| PSMB5_00130725.11 | CTCTACCCCCTGAAAGAGGG |
| PSMB5_00130725.32 | CTCTACTACGTGGACAGTGA |
| PSMB5_001144932.8 | CTGCAACTATGACTCCATGG |
| PSMB5_00130725.13 | CTGCATCCACCCTCTTTCAG |
| PSMB5_00130725.1 | CTGCTAGGCACCATGGCTGG |
| PSMB5_00130725.55 | CTGCTCCACTTCCAGGTCAT |
| PSMB5_00130725.65 | CTGGCTCTGTGTATGCATAT |
| PSMB5_00130725.31 | CTGTCCACGTAGTAGAGGCC |
| PSMB5_00130725.26 | CTTATCCCAGCCACAGATCA |
| PSMB5_00130725.5 | CTTCACTGTCCACGTAGTAG |
| PSMB5_00130725.4 | CTTTCCAGGCCTCTACTACG |
| PSMB5_001144932.17 | CTTTCTGCCCACACTAGACA |
| PSMB5_001144932.72 | GAGATCAACCCATACCTGCT |
| PSMB5_001144932.102 | GAGCCTGGCCGCGCCAGGCT |
| PSMB5_00130725.85 | GATCTACATGAGAAGTATAG |
| PSMB5_001144932.94 | GATCTGCTGGATCTAGGTCC |
| PSMB5_001144932.18 | GCAAGCGCCATGTCTAGTGT |
| PSMB5_00130725.7 | GCATATGGGGTCATGGATCG |
| PSMB5_00130725.63 | GCCACAGATCATGGTGCCCA |
| PSMB5_00130725.37 | GCCACCTTCTCTGTAGGTTC |
| PSMB5_00130725.71 | GCCAGAACCTACAGAGAAGG |
| PSMB5_00130725.62 | GCCATGGTGCCTAGCAGGTA |
| PSMB5_00130725.20 | GCGCAGCGGATTGCAGCTTC |
| PSMB5_001144932.3 | GCGCGGGTTTTTCGGACTTG |
| PSMB5_001144932.69 | GCTCCACACCTTGAAGGCCA |
| PSMB5_001144932.71 | GCTGACTCCAGGGCTACAGC |
| PSMB5_00130725.46 | GCTGCATCCACCCTCTTTCA |
| PSMB5_001144932.35 | GCTTCATGGAACAACCACCC |
| PSMB5_001144932.1 | GGCAAGCGCCATGTCTAGTG |
| PSMB5_001144932.7 | GGCGGAACTGTTAAGATCAG |
| PSMB5_001144932.95 | GGCTCCACACCTTGAAGGCC |
| PSMB5_00130725.41 | GGCTCGACGGGCCAGATCAT |
| PSMB5_00130725.75 | GGCTGGGATAAGAGAGGCCC |
| PSMB5_00130725.42 | GGCTTGGTAGATGGCTCGAC |
| PSMB5_001144932.37 | GGGCTGGCTCCACACCTTGA |
| PSMB5_001144932.67 | GGTCCAGGGAGTCTCAGTGA |
| PSMB5_001144932.30 | GGTCTGAGCCTGGCCGCGCC |
| PSMB5_00130725.51 | GGTGTATCAGTACAAAGGCA |
| PSMB5_00130725.27 | GGTTGCAGCTTAACTCACCA |
| PSMB5_001144932.41 | GTAAGCACCCGCTGTAGCCC |
| PSMB5_001144932.24 | GTGAACCAGCGCGGGTTTTT |
| PSMB5_00130725.35 | GTGAAGGGAACCGGATTTCA |
| PSMB5_00130725.10 | GTGGCTCTACCCCCTGAAAG |
| PSMB5_00130725.73 | GTGTATCAGTACAAAGGCAT |
| PSMB5_00130725.58 | GTTGACTGCACCTCCTGAGT |
| PSMB5_00130725.77 | TAGATCAGCCACATTGTCAC |
| PSMB5_001144932.20 | TAGCGGTCTCTCCAACACGC |
| PSMB5_001144932.44 | TATCACCTTCTTCACCGTCT |
| PSMB5_001144932.40 | TCATAGTTGCAGCTGACTCC |
| PSMB5_00130725.17 | TCCAGCCATCCTCCCGCACG |
| PSMB5_00130725.25 | TCCATGGGCACCATGATCTG |
| PSMB5_00130725.54 | TCGGGGCTATTCCTATGACC |
| PSMB5_00130725.33 | TCTACTACGTGGACAGTGAA |
| PSMB5_001144932.81 | TCTCAGTGATGGTCTGAGCC |
| PSMB5_00130725.53 | TCTGGCTCTGTGTATGCATA |
| PSMB5_00130725.49 | TCTGGGAACGGCTGTTGGCT |
| PSMB5_00130725.57 | TCTGTAGGTGGCTTGGTAGA |
| PSMB5_001144932.31 | TCTTCTGGGACACCCCAGCC |
| PSMB5_00130725.6 | TGAAGGGAACCGGATTTCAG |
| PSMB5_001144932.68 | TGAGCCTGGCCGCGCCAGGC |
| PSMB5_00130725.15 | TGAGTAGGCATCTCTGTAGG |
| PSMB5_001144932.38 | TGATCTTAACAGTTCCGCCA |
| PSMB5_00130725.40 | TGCATATGGGGTCATGGATC |
| PSMB5_00130725.12 | TGCATCCACCCTCTTTCAGG |
| PSMB5_001144932.43 | TGCCTCCCAGACGGTGAAGA |
| PSMB5_001144932.58 | TGCTGAGAGGATCGGCAACA |
| PSMB5_001144932.42 | TGCTTACATTGCCTCCCAGA |
| PSMB5_001144932.104 | TGCTTGAAACCTAAGTCATT |
| PSMB5_00130725.45 | TGGCTCTACCCCCTGAAAGA |
| PSMB5_00130725.38 | TGGCTCTGTGTATGCATATG |
| PSMB5_00130725.43 | TGGCTTGGTAGATGGCTCGA |
| PSMB5_001144932.5 | TGGGACACCCCAGCCTGGCG |
| PSMB5_001144932.80 | TGGGGGTCGTGCAGATCTGC |
| PSMB5_001144932.82 | TGGGGTGTCCCAGAAGAGCC |
| PSMB5_00130725.28 | TGGTTGCAGCTTAACTCACC |
| PSMB5_001144932.57 | TGTGGGTGTGCTGAGAGGAT |
| PSMB5_00130725.66 | TGTGTATGCATATGGGGTCA |
| PSMB5_001144932.78 | TGTTTTGTGGGTGTGCTGAG |
| PSMB5_001144932.105 | TTGGAATGTACTTGTTTTGT |
| PSMB5_001144932.32 | TTTCGATTCCTGGCTCTTCT |
| PSMB5_001144932.98 | TTTGGAATGTACTTGTTTTG |
| PSMB5_00130725.82 | TTTGTACTGATACACCATGT |
| Safe Harbor Library | |
|---|---|
| sgRNA Name | sgRNA sequence |
| SafeHarbor.1 | GGCTAAATTCCTCTTATTCA |
| SafeHarbor.2 | GTAACCAAGAGTCAGGACTG |
| SafeHarbor.3 | GGGATAATATAAGGCATTCT |
| SafeHarbor.4 | GGATCTTATAATCTAGTTAT |
| SafeHarbor.5 | GTTAATGCCTTGGTCAAATG |
| SafeHarbor.6 | GTGTAAACTAAGACCTAAGT |
| SafeHarbor.7 | GCTAAAGTTGTCATTGATTT |
| SafeHarbor.8 | GTGCTTCCGACAAACTACAA |
| SafeHarbor.9 | GGAACGTAGGTAATAAGGTC |
| SafeHarbor.10 | GATTCTTCATATCTTTCTCA |
| SafeHarbor.11 | GCTCATGAGACACTTCACAG |
| SafeHarbor.12 | GTCAGCATTAAACATGCTTA |
| SafeHarbor.13 | GTGAAAGTTCTCATCTTCTT |
| SafeHarbor.14 | GCATGAGAAGAGGAGATTGA |
| SafeHarbor.15 | GACTGTTCATAGGACCCTAA |
| SafeHarbor.16 | GCCCTGTCTGTATCCAGTCC |
| SafeHarbor.17 | GGGATCTTTCAGTGTAGGTA |
| SafeHarbor.18 | GATTCTGTATAATGGAAATC |
| SafeHarbor.19 | GACATGTCCTAATTGTATGG |
| SafeHarbor.20 | GTGTGCTTTGAAGAATAATG |
| SafeHarbor.21 | GCAATATGATCTCATTTGTG |
| SafeHarbor.22 | GAGTTTAGAGGTTTGAGATT |
| SafeHarbor.23 | GTGGTCCTGGACTGGTCTCA |
| SafeHarbor.24 | GTTATGCCAACACATTTGTA |
| SafeHarbor.25 | GTTACATACAAAAATTGGAT |
| SafeHarbor.26 | GCATATTATCACTCCAGTGA |
| SafeHarbor.27 | GACATTGGGATTAAATTTGG |
| SafeHarbor.28 | GGTGGCCGCCATCATGGCTG |
| SafeHarbor.29 | GGCAGATCAGAATGTGAGCT |
| SafeHarbor.30 | GAGGAAGGAGTTATATTGAC |
| SafeHarbor.31 | GAGCCAAAGATAAGCATGAG |
| SafeHarbor.32 | GGCTACTCAGATATAGTCAT |
| SafeHarbor.33 | GTTATTTGATGAGCAGCTAT |
| SafeHarbor.34 | GACGTAGTAAGGTAGAGACA |
| SafeHarbor.35 | GTGATGAAGAGTGCTACAGC |
| SafeHarbor.36 | GCTAGGGACTTCAAAGTTAT |
| SafeHarbor.37 | GATATCTTCCCAATGATGAC |
| SafeHarbor.38 | GAGTAGTTTCTGACGTCCGA |
| SafeHarbor.39 | GAGCATAATGAAGGTTCTTG |
| SafeHarbor.40 | GCGTTTCCAATCCCAGAGAG |
| SafeHarbor.41 | GGCCTAATAGCTTTGGTAGA |
| SafeHarbor.42 | GACAGGAGGAACTTGTAACC |
| SafeHarbor.43 | GAGAGCACTCAGCAAAATCA |
| SafeHarbor.44 | GCGTTGGTGAAATTACAATT |
| SafeHarbor.45 | GTTAATGATCAAAAGTTACA |
| SafeHarbor.46 | GAGAGAATTGCTATTCTGAG |
| SafeHarbor.47 | GATTGTATGAAAACATAGAT |
| SafeHarbor.48 | GGCTACCTGTCTATTGGCAC |
| SafeHarbor.49 | GGCATGTGTGTCTGAATACA |
| SafeHarbor.50 | GCTGAAGCTCTGGCAAGAGC |
| SafeHarbor.51 | GTACCTTAATCACACCTTTG |
| SafeHarbor.52 | GTTCACATAGCAGTACTTGT |
| SafeHarbor.53 | GACTGACCTTTCTTTGAGAG |
| SafeHarbor.54 | GACTTGAATGATCAATTACT |
| SafeHarbor.55 | GTTCTGAGTTACTGGAACCC |
| SafeHarbor.56 | GCAAGATCAGGTAAGTATCT |
| SafeHarbor.57 | GTCGTGAAGCTGTGTTTGAC |
| SafeHarbor.58 | GGTCTTGAAATAAAATTTAG |
| SafeHarbor.59 | GACTGCTTCTTAGTTAGGTA |
| SafeHarbor.60 | GGAAATCCTTGAGTTTCAGG |
| SafeHarbor.61 | GCCCAAGCAGGCTACATTGC |
| SafeHarbor.62 | GAGGTGGCAAAGAATGTGCC |
| SafeHarbor.63 | GTTCAAATAATAGGGTGCAT |
| SafeHarbor.64 | GAGGGGATACTCAAGCTAGG |
| SafeHarbor.65 | GGGTATCAGCTCACCTCCTC |
| SafeHarbor.66 | GAAGTACTGGCAATGCAACT |
| SafeHarbor.67 | GACATAGCCTGCAATTGTTT |
| SafeHarbor.68 | GGGCAGATTGGAAGAGCCCT |
| SafeHarbor.69 | GTGTACAACATCACAGCATA |
| SafeHarbor.70 | GGGTGGTTCTGAATGGGAGC |
| SafeHarbor.71 | GCTATCCTTAAATTGGCCTG |
| SafeHarbor.72 | GCCTGAATATAGTGAAAGTC |
| SafeHarbor.73 | GGGAAGTCCTGGGGTTTGAT |
| SafeHarbor.74 | GTCAGTTATTCTTTCCTCTA |
| SafeHarbor.75 | GCATGGTCACAATAATCTTG |
| SafeHarbor.76 | GGGAGGATAAGAGACACTTT |
| SafeHarbor.77 | GCTTATTTAGTTTGGTTCAA |
| SafeHarbor.78 | GTCTCTACTAGAACTCAATC |
| SafeHarbor.79 | GGAGCTTGGTATCTAAAATT |
| SafeHarbor.80 | GATGTTCACTGTTAATTGAT |
| SafeHarbor.81 | GCTACTTAAATCATTGCCAT |
| SafeHarbor.82 | GCACTTCACCTGAGAAAAAC |
| SafeHarbor.83 | GCTTGCTTGTCTCTGTTTCG |
| SafeHarbor.84 | GTCAACAGCAAGGCTACTGA |
| SafeHarbor.85 | GACAGAAGAAGCTAGAAGTC |
| SafeHarbor.86 | GTACAACCCAAAGTATATGG |
| SafeHarbor.87 | GAATCCCGGGCTTTCTCTGT |
| SafeHarbor.88 | GATAATTTCAGGAGTGAGAT |
| SafeHarbor.89 | GTATTGTGATCAAGTAATTT |
| SafeHarbor.90 | GAACCTAAAAATATAGTTGT |
| SafeHarbor.91 | GCATTGGTGCCCAGTAGGAG |
| SafeHarbor.92 | GAATACTGTGAGAAATTTCA |
| SafeHarbor.93 | GTCAAGATATACCTAGCAAA |
| SafeHarbor.94 | GACCTCACTTACTGTTGCCA |
| SafeHarbor.95 | GCATACCATAGGGTAAAGGC |
| SafeHarbor.96 | GGTGACAATCAAACTGGCAA |
| SafeHarbor.97 | GGTATTGTCAATGTAAAAAG |
| SafeHarbor.98 | GCACAGTAAATATACGTGTG |
| SafeHarbor.99 | GTGTGCCCCTCCAAAAGAGA |
| SafeHarbor.100 | GACATATGCTATGCAGAGTT |
| SafeHarbor.101 | GTAAGAATCAAATCATCATG |
| SafeHarbor.102 | GGAAATTGCTTCTGGTTTAT |
| SafeHarbor.103 | GTAGATGAGCTCTTATCAGT |
| SafeHarbor.104 | GGCTTTGTTCATGACTTTGA |
| SafeHarbor.105 | GCACCAGTCTATGCCACCAC |
| SafeHarbor.106 | GTAATGACTTGGGGGAGATA |
| SafeHarbor.107 | GAGTCTGTCTCTAATGAGAC |
| SafeHarbor.108 | GTGGTCCACAGACAATGCAT |
| SafeHarbor.109 | GGTTAAGAAAAGACACTCAG |
| SafeHarbor.110 | GGTAATCATAAGTTGTATAA |
| SafeHarbor.111 | GGCCCTCCTTAGAAGTTGCA |
| SafeHarbor.112 | GAAATTGGTCCCCACCTTCA |
| SafeHarbor.113 | GTCCAAGAACAAAGCAAAGA |
| SafeHarbor.114 | GATGAGCCAATCTTTAGCAA |
| SafeHarbor.115 | GTGAATCAAGAAGCAATGTC |
| SafeHarbor.116 | GAAAGGCAGACATGGCTAAA |
| SafeHarbor.117 | GACAAAAGCAGAATACCAGA |
| SafeHarbor.118 | GCACACAAAATATCGTTATT |
| SafeHarbor.119 | GAGAAAGGCCCAGCTCTGAT |
| SafeHarbor.120 | GCCAGTCTACCCACTGTCCC |
| SafeHarbor.121 | GCAGGGTGAAGGTCCTCCTC |
| SafeHarbor.122 | GAAGAGACTACAATTATTCT |
| SafeHarbor.123 | GATATCCTTTGTGTTAACTT |
| SafeHarbor.124 | GAATGACTCGCATGACTTTA |
| SafeHarbor.125 | GGATGTTCAAACCTTCAAAA |
| SafeHarbor.126 | GAGAATATATGTTTCCATTA |
| SafeHarbor.127 | GGAAAAGTAATGAATCATAC |
| SafeHarbor.128 | GTTACACGAAGCACAGGGTG |
| SafeHarbor.129 | GAACTAGGTGCTCAAGGAAT |
| SafeHarbor.130 | GGCAAAGACCAGTCTGATAC |
| SafeHarbor.131 | GTCTAGTTTCACAATAATTT |
| SafeHarbor.132 | GCTTTATATAAGATATGAGA |
| SafeHarbor.133 | GCATAGGATATTATATTTCG |
| SafeHarbor.134 | GACCTTGACTGCTCCTGAAC |
| SafeHarbor.135 | GCAGCTCCCTAGTTCACAGA |
| SafeHarbor.136 | GTCTGACCAGAGGTGGAGAG |
| SafeHarbor.137 | GAATCACATTGTACCACAAA |
| SafeHarbor.138 | GACAAAATTGATACAACAGC |
| SafeHarbor.139 | GAATTCCAAGACTTCACATT |
| SafeHarbor.140 | GACAGGGACCGCCATCCACT |
| SafeHarbor.141 | GTTGTATGGTTCCTAAGGAT |
| SafeHarbor.142 | GAATATCCACTACTAGCTTT |
| SafeHarbor.143 | GCCATTAATCATGATCTGGA |
| SafeHarbor.144 | GGTGAATAGGTAGGTATTGA |
| SafeHarbor.145 | GCTCATCAAAGGTAGTAAAC |
| SafeHarbor.146 | GGGACCCAGCCCTTGGGCTG |
| SafeHarbor.147 | GTGCACCTTTCTATAAATGT |
| SafeHarbor.148 | GACTTCATTAAAAGCAGTCT |
| SafeHarbor.149 | GTTGAACTTGTGAACACAAA |
| SafeHarbor.150 | GGGTCCTCACCAGGAAATTT |
| SafeHarbor.151 | GTAGCCTATTGGCAATTGGC |
| SafeHarbor.152 | GCATAAATAAAATCGATTCC |
| SafeHarbor.153 | GAAGGGCAATAATTGGTACA |
| SafeHarbor.154 | GAGTTCTTAATAACATTCTA |
| SafeHarbor.155 | GCTTTCTACTTGCCTTAGAT |
| SafeHarbor.156 | GCTTCTTATTTCTCTCCAGT |
| SafeHarbor.157 | GCATTCTGTCCTAATAAGAA |
| SafeHarbor.158 | GCTTAAGCTAGTTTAAAGAA |
| SafeHarbor.159 | GGTTTCCAGTGTTTATCTGT |
| SafeHarbor.160 | GAGAGTCTAGGTACGTTCTC |
| SafeHarbor.161 | GCTTTCAAGTTAACATAGCT |
| SafeHarbor.162 | GTAAAATGAACCGAGCTTTA |
| SafeHarbor.163 | GTAAGATTATTAACCCCTTC |
| SafeHarbor.164 | GGGTCCTCACGATAGAAGAA |
| SafeHarbor.165 | GATTACACTCAAGAAAGCGA |
| SafeHarbor.166 | GATGTAGACGTAGAAGTGAT |
| SafeHarbor.167 | GTGAGTTACAGAAATTAGCA |
| SafeHarbor.168 | GCAGGGGGACACGGGCACAT |
| SafeHarbor.169 | GACAATTGTGTTGCAGACAA |
| SafeHarbor.170 | GTCAATGGGAAATTATAAAC |
| SafeHarbor.171 | GAGTTATAGCACACTTAGAA |
| SafeHarbor.172 | GATTGAAACCAGAAAATAAG |
| SafeHarbor.173 | GGAGTCTAGTGATAGGGGTA |
| SafeHarbor.174 | GGGATAGTCTTAGAAGGCTT |
| SafeHarbor.175 | GTCAATTGATTCACTGGAAT |
| SafeHarbor.176 | GTATTCCTGCAAGATAATTC |
| SafeHarbor.177 | GGTCAAGCAACAGGCATAAT |
| SafeHarbor.178 | GACATCCATAACTTCCTAAC |
| SafeHarbor.179 | GTCAAACAAAAGCGTCTATA |
| SafeHarbor.180 | GCTAGATTAATATGAATGAG |
| SafeHarbor.181 | GAACCCCATAGGAGGTTTAG |
| SafeHarbor.182 | GCCTCTTTCCCCTGCCGGCA |
| SafeHarbor.183 | GGTAAGGGCTGCTTATCTTT |
| SafeHarbor.184 | GTATTCAGTATAATCAAGGA |
| SafeHarbor.185 | GTTGTCTTATGGGACTGCAT |
| SafeHarbor.186 | GTATACGATATGATTGACTC |
| SafeHarbor.187 | GGTAGAGACAAAATATATTT |
| SafeHarbor.188 | GTACCTATGTCCTTGAGGCT |
| SafeHarbor.189 | GGCAAAAGAACGTCTGTAAT |
| SafeHarbor.190 | GGACTAGTTTACCTAGGGAG |
| SafeHarbor.191 | GGAGGGTGGAGCAAAGAAAG |
| SafeHarbor.192 | GAGCCATATTATGTCCTTTA |
| SafeHarbor.193 | GTGCACTCTATGCACCAAAG |
| SafeHarbor.194 | GGTCTCCCGAGTCATTGTTG |
| SafeHarbor.195 | GCAATCATTCTGGTTCAGGC |
| SafeHarbor.196 | GCACAGGTTCCCCTCCTAAC |
| SafeHarbor.197 | GATCAGGGAATCTTTGAGAA |
| SafeHarbor.198 | GAACCCAGCTGTCCTCGCTG |
| SafeHarbor.199 | GCTAACTGTGTTACAAGCAG |
| SafeHarbor.200 | GTGATCAAAGAGAGAGGTGT |
| SafeHarbor.201 | GGAAAGCCCGTTGTATTTAT |
| SafeHarbor.202 | GGTCCCCCACTTTCTCCTTG |
| SafeHarbor.203 | GCCAGATGACCATAGAAACT |
| SafeHarbor.204 | GGTGCAATCCAAAGGTGGGC |
| SafeHarbor.205 | GTGTAAAATCACTTTAAACT |
| SafeHarbor.206 | GTCACATGTTCAAGTTTAAC |
| SafeHarbor.207 | GAAGCTTAGTCCTGAATTGT |
| SafeHarbor.208 | GGGTCTGTTTCCTTGTGTTA |
| SafeHarbor.209 | GATAGAGACTGGATGAAGTT |
| SafeHarbor.210 | GCAACAAGGCAAATGTGGTA |
| SafeHarbor.211 | GCTATTTAGCTCAACCTTGT |
| SafeHarbor.212 | GTGCCATTATCATTTCCTCA |
| SafeHarbor.213 | GCAAATAGAAGAGACAATCT |
| SafeHarbor.214 | GAAAATATATGGACTGGGAT |
| SafeHarbor.215 | GAATAGAACTCCTGCCATCA |
| SafeHarbor.216 | GCTTTCTACCTGGATGTTTA |
| SafeHarbor.217 | GCTAACTTGAGGGCAAAAGA |
| SafeHarbor.218 | GTGGTAAAAATGTGCTTTGT |
| SafeHarbor.219 | GAGCCTCAGCTGGTGCATGG |
| SafeHarbor.220 | GCCTATGCCGCAATACCCTC |
| SafeHarbor.221 | GACCTGTGTAAACCAGCTAA |
| SafeHarbor.222 | GACCTCATTCCTGAGTGTGT |
| SafeHarbor.223 | GTGTTTGCCTCATAATAACC |
| SafeHarbor.224 | GACTGGGCATACAGCCATTT |
| SafeHarbor.225 | GGCATACTACATTGGCTTTA |
| SafeHarbor.226 | GCAAACATATTGGAGTACTG |
| SafeHarbor.227 | GGGGAGTAGGGAAGAGCTTA |
| SafeHarbor.228 | GGGCTCGTATGTCGTTCTTC |
| SafeHarbor.229 | GTGCCTTATCTATTTCCACA |
| SafeHarbor.230 | GGTAATTACCTGCTCTCTGC |
| SafeHarbor.231 | GTCTGATAACTTGTGTTACT |
| SafeHarbor.232 | GACTGAGTTAATAATAGCGG |
| SafeHarbor.233 | GAATATTGTGCACTGTATTT |
| SafeHarbor.234 | GTTTCTAAATGTGATCTGTG |
| SafeHarbor.235 | GCACACTGGCTAGTTAAGGA |
| SafeHarbor.236 | GGAGGAGTGTGCAATGAAGC |
| SafeHarbor.237 | GAGGACGGGTGGGAAGTTAG |
| SafeHarbor.238 | GATACTGTAGCAGTTACTGA |
| SafeHarbor.239 | GATTCTAAGCAAAGGACAGA |
| SafeHarbor.240 | GGAGCTTAGACCATATTTGG |
| SafeHarbor.241 | GTGTCCGTGGGTCTGTTCCC |
| SafeHarbor.242 | GCAATAGCTGTGAGCTCATA |
| SafeHarbor.243 | GGGATGGGCCATCCAGCTGT |
| SafeHarbor.244 | GACAGATTACTTAATAAAAG |
| SafeHarbor.245 | GTGGCAAGGTTAAGTACAAT |
| SafeHarbor.246 | GGAGGAAACAGAATAATGGC |
| SafeHarbor.247 | GTGAATTAATGTCATTTCAC |
| SafeHarbor.248 | GTGAACTAGAACACTGAGAG |
| SafeHarbor.249 | GATGCTGTGGCCAATGTGCA |
| SafeHarbor.250 | GACTGTAAGCATTCCTGACA |
| SafeHarbor.251 | GTCCTAATTCCATGCCTAAA |
| SafeHarbor.252 | GTGGGTTCGTTGTCTACTAC |
| SafeHarbor.253 | GAGACTATTAGATCGTATGT |
| SafeHarbor.254 | GGTGTAGTATCAAAAATTGA |
| SafeHarbor.255 | GATAGCTCTTAAGGATAAAT |
| SafeHarbor.256 | GATTCAGTCACATCACAATA |
| SafeHarbor.257 | GTCTAAGAAAGACTTCTAGG |
| SafeHarbor.258 | GATTTGGGTCTTTGCGCATC |
| SafeHarbor.259 | GACCTTAAAGTTATAGTTAA |
| SafeHarbor.260 | GCTCTGCATCTTTCCCCAGG |
| SafeHarbor.261 | GACCTAAGTTTGAGAATGAG |
| SafeHarbor.262 | GAAAGTACATTCATTAGCAT |
| SafeHarbor.263 | GGAGAACGTGGTGATAAAGC |
| SafeHarbor.264 | GGCAACATGGCAAAATAGTT |
| SafeHarbor.265 | GATAATAGCAGAGAGAGGTG |
| SafeHarbor.266 | GGACTTTAAGGAATTCAGCT |
| SafeHarbor.267 | GAATATTGGGGGGTGGATGG |
| SafeHarbor.268 | GGAGTAAGTATGTGTGTTGA |
| SafeHarbor.269 | GTATTGGATAAGGGAGCTCA |
| SafeHarbor.270 | GTGAGTTGGGAGATGTACTG |
| SafeHarbor.271 | GTTTACAATTTCATTTGTAC |
| SafeHarbor.272 | GTCCATTCAATTTGGACATG |
| SafeHarbor.273 | GAGTGCTTACTGGGAATGAG |
| SafeHarbor.274 | GCTAATTGTTCAAAAAGCCC |
| SafeHarbor.275 | GCTTTCAAGAGTTTATTTGA |
| SafeHarbor.276 | GATATTCTGTGCAATCTGTT |
| SafeHarbor.277 | GTGTAGGACTACGCTGGCAC |
| SafeHarbor.278 | GTCTTAAAGAGTAAAGTACA |
| SafeHarbor.279 | GTTAGACTGCAAACACCCAC |
| SafeHarbor.280 | GCCTAGGAGAAGCCCTGGCA |
| SafeHarbor.281 | GTCGAGTATTTCTAATCTTT |
| SafeHarbor.282 | GAATCTGAGACATCATTCAT |
| SafeHarbor.283 | GACAAAAGATTATGCTTCCC |
| SafeHarbor.284 | GAGAATTACATTCATGATCT |
| SafeHarbor.285 | GAACTGAGCTTCTACCATGC |
| SafeHarbor.286 | GGTAAGATTGTAATAGCTTG |
| SafeHarbor.287 | GTCAGAAATGATCTCGTCCT |
| SafeHarbor.288 | GACATATCTAAGAACTGAGC |
| SafeHarbor.289 | GCTTCAATATGACAGAACTC |
| SafeHarbor.290 | GGAGAGCAAATCAGCATATC |
| SafeHarbor.291 | GCAAAATAGCCGCACAGAAA |
| SafeHarbor.292 | GCATATTTCTATACAATACA |
| SafeHarbor.293 | GATGCAAATTCATGGTGGTA |
| SafeHarbor.294 | GAACTGTAATAGTCTTGAGC |
| SafeHarbor.295 | GAACTCACTACATTAAGGCT |
| SafeHarbor.296 | GAGGTAAATCAGTACAAACA |
| SafeHarbor.297 | GTTGTTTCTAAGATTAAAAG |
| SafeHarbor.298 | GTGGTAGTCAGTTTCACAAA |
| SafeHarbor.299 | GGTTTCAAATAGTTGGATCA |
| SafeHarbor.300 | GAATATGAAAGACATCATAA |
| SafeHarbor.301 | GAAGTAGGAAGGAGATTGCC |
| SafeHarbor.302 | GGAAAAGTGCTGTTTGCATT |
| SafeHarbor.303 | GAGCATTAGGCTGGGGCCTT |
| SafeHarbor.304 | GTCTAGGTATGATTAGAAGA |
| SafeHarbor.305 | GAGTTATAATCTTCAGAAAA |
| SafeHarbor.306 | GCTGTAATGAGACTTCAGCT |
| SafeHarbor.307 | GTGTGCAATCTGAAGGAAAT |
| SafeHarbor.308 | GTGATGAGGTCGCTGAAGTT |
| SafeHarbor.309 | GTGGAGCCCTTATAACCCTG |
| SafeHarbor.310 | GTTGGATTATTTCTTCTATA |
| SafeHarbor.311 | GGATTTCTACATTATATACT |
| SafeHarbor.312 | GCTAATGTAGATCAAGTTAT |
| SafeHarbor.313 | GATTGCAAGAGACTGAACTC |
| SafeHarbor.314 | GGGTGAACTTGAGTGAACTT |
| SafeHarbor.315 | GGGCTCAAATCCCTATAATT |
| SafeHarbor.316 | GATAGAAGGTATTAACTCCC |
| SafeHarbor.317 | GGCTATAAGCACAAATGTAA |
| SafeHarbor.318 | GATTCCCATTGCATGCCAGT |
| SafeHarbor.319 | GCAAATTACAATTATGTTTC |
| SafeHarbor.320 | GAATTAAATTCACTTTGAAC |
| SafeHarbor.321 | GAGCAGACAGGAAATAAAGC |
| SafeHarbor.322 | GCCCACCAGTCCTTCTCACT |
| SafeHarbor.323 | GTTAAGAAGTGAAAGAAATT |
| SafeHarbor.324 | GTTGAATTGAATGGGTCATT |
| SafeHarbor.325 | GTAGACACAAACTTGTGTAA |
| SafeHarbor.326 | GAGCGTACTATATTCTTAAA |
| SafeHarbor.327 | GGTGGTACATCGTTGAAGGA |
| SafeHarbor.328 | GATGAACTCCCAATCACAGG |
| SafeHarbor.329 | GTATAAATAAGGATAAGGTA |
| SafeHarbor.330 | GGAAATAATCTTGGAACATA |
| SafeHarbor.331 | GGTAGTTAATCTTCTACTTT |
| SafeHarbor.332 | GAGAAGAGAACATTCTAGTT |
| SafeHarbor.333 | GTCGGAGCTCAGTGTTGCAT |
| SafeHarbor.334 | GAAGAGACATGTTTCAGTGA |
| SafeHarbor.335 | GTCATATCTGACTTAAATTG |
| SafeHarbor.336 | GGAGAATATGCTAAAAGCGT |
| SafeHarbor.337 | GATTGTTGTAGTAGAATAAA |
| SafeHarbor.338 | GTAAGCAGCACCACCACTTA |
| SafeHarbor.339 | GTCTTGTGCTGACATGCTCA |
| SafeHarbor.340 | GCAGACTTTATTAGCTAGTG |
| SafeHarbor.341 | GAGGTATTTGATATGACTCA |
| SafeHarbor.342 | GCAGGTTGCCCATTCTCCCA |
| SafeHarbor.343 | GAGGGGACGTTGACCTGTGG |
| SafeHarbor.344 | GAACCCAAGGATTTATAAAG |
| SafeHarbor.345 | GTGTTCAGGACATGTACTCA |
| SafeHarbor.346 | GGTGATGATAGTCAAATACC |
| SafeHarbor.347 | GCTTTACAGCTAATTTCTAA |
| SafeHarbor.348 | GGTATCTACATTAACACTCA |
| SafeHarbor.349 | GACAGTTTGCTTACTATGGA |
| SafeHarbor.350 | GAAAAACTCTTAGCTTAATG |
| SafeHarbor.351 | GTCATCTTAACTTCAGTAGA |
| SafeHarbor.352 | GATCACTGGTAGGCCACAGT |
| SafeHarbor.353 | GAGAAAGGCAAGTGCATCAA |
| SafeHarbor.354 | GAACTGATAAAGATTCAGTA |
| SafeHarbor.355 | GCCATTCAAAAGCAGCTATA |
| SafeHarbor.356 | GACAGAACTTCTTTGAGCTA |
| SafeHarbor.357 | GGGTGACATTGAAATTTAAC |
| SafeHarbor.358 | GACTATAAACTGCACACTAT |
| SafeHarbor.359 | GCTATGGTGGGAAAGCTCAT |
| SafeHarbor.360 | GACTAACTTGCTAATGGCTA |
| SafeHarbor.361 | GAGAGTCACTTCAAAGTGTG |
| SafeHarbor.362 | GAGTGTATTTGTGGACAATA |
| SafeHarbor.363 | GAAGAATTAGGGTTCCATTT |
| SafeHarbor.364 | GAGGAGTGGCACTTTATACT |
| SafeHarbor.365 | GAAGGATGCAGTAGCCATTG |
| SafeHarbor.366 | GTGCATTGTTGGTGGTTGTG |
| SafeHarbor.367 | GAGAAGTTATGCAAATTTAT |
| SafeHarbor.368 | GAAATAGATTGGCAGAGTGT |
| SafeHarbor.369 | GTGGGGTGGGCTCCCTGCCT |
| SafeHarbor.370 | GTCTCTAACAAGACTGAAAT |
| SafeHarbor.371 | GCAGAGTAGATCTACATCTT |
| SafeHarbor.372 | GTGCCAGCTAAGATGAAATT |
| SafeHarbor.373 | GATGGTGATGCACCAACTTT |
| SafeHarbor.374 | GAAGTGTTGCCATTCAATTC |
| SafeHarbor.375 | GAGAGAGTTGGAATAAGCTA |
| SafeHarbor.376 | GAGGGTACTTATTTCAACTT |
| SafeHarbor.377 | GCTACATGTTCTAGAATACA |
| SafeHarbor.378 | GAGAAATCTCTTTGAGCTGG |
| SafeHarbor.379 | GGCTTTGTGTCTGACTTTCC |
| SafeHarbor.380 | GGATTAGATCAATTATTCTA |
| SafeHarbor.381 | GATTCTGGAAATAAGTACCT |
| SafeHarbor.382 | GAGATAAAATTGCGAGACCA |
| SafeHarbor.383 | GACAAAATTTAGCAACTCAG |
| SafeHarbor.384 | GCAGATACTCACCATTACCC |
| SafeHarbor.385 | GGTGATTGTTGCAGCTGTCA |
| SafeHarbor.386 | GATAGACTTGTGAAGGAAAC |
| SafeHarbor.387 | GAGTCACTGGATTGTTGTCC |
| SafeHarbor.388 | GGATTATATGGGAGGTACAC |
| SafeHarbor.389 | GCTTAAAAATACTATCTGCT |
| SafeHarbor.390 | GACAAGGAGGACCAAAGTTG |
| SafeHarbor.391 | GGCAGTGATTTACTCCTATC |
| SafeHarbor.392 | GATCTTCCAGGACTGTTAGA |
| SafeHarbor.393 | GAAACAAGCTAATATTATCA |
| SafeHarbor.394 | GTCAGTCTTTACAAATCACT |
| SafeHarbor.395 | GGCAGTTGAGTAAACGTAAG |
| SafeHarbor.396 | GCCTCTACTGCTAACTCTAT |
| SafeHarbor.397 | GTTGTAATTTAAAGCACTCA |
| SafeHarbor.398 | GCATAAAGAGAACAAGCAAT |
| SafeHarbor.399 | GGTAGTTGGTCTAATCAGTA |
| SafeHarbor.400 | GGCTAACACCTGCCAACTTT |
| SafeHarbor.401 | GTCTAATCTAGCATCAAACT |
| SafeHarbor.402 | GAGAGAGACTATTTCAGGAT |
| SafeHarbor.403 | GACCTAGACCAAGCTACGAA |
| SafeHarbor.404 | GTTACTGATACCAGTCCCTG |
| SafeHarbor.405 | GCCCTACTGTGGTAACTTTG |
| SafeHarbor.406 | GTGTAAAGGAATCTTAGCTT |
| SafeHarbor.407 | GGTGAGACTATTATATTTAT |
| SafeHarbor.408 | GCTTCAGAGAACTATTTGGT |
| SafeHarbor.409 | GATGTGTTCGTTGAGGCATA |
| SafeHarbor.410 | GTTGACTCTAACTATAGAGT |
| SafeHarbor.411 | GGACAGCCATTGAAGATATG |
| SafeHarbor.412 | GATGGAGAGCCTGGAGCATA |
| SafeHarbor.413 | GCATGATTAAAGGTGAGCAT |
| SafeHarbor.414 | GGAACCCACAGATATAGCTA |
| SafeHarbor.415 | GCATAGCTTCAGAGTTCAGA |
| SafeHarbor.416 | GAGAAAAGACGTGTATTTCC |
| SafeHarbor.417 | GCTAGAGCTTCCTTATGTTT |
| SafeHarbor.418 | GATGGGCAGTCAGGACTACG |
| SafeHarbor.419 | GTTCTGCATGAGAAGCACTA |
| SafeHarbor.420 | GACTCCACCTATCTCAAAAT |
| SafeHarbor.421 | GATATTTGACAGTGGATAAA |
| SafeHarbor.422 | GAAAGATTATGGATCATAGT |
| SafeHarbor.423 | GCATCAATGTACACTGTGGC |
| SafeHarbor.424 | GCAGCAAGCTATGGTCCATG |
| SafeHarbor.425 | GGTTGTTTGAATTAAAGACT |
| SafeHarbor.426 | GAACCCCTGGCTAGTTTCCC |
| SafeHarbor.427 | GGATAAAGAGTGAACCTGTA |
| SafeHarbor.428 | GTAGATTTCACTAAATTGTT |
| SafeHarbor.429 | GTGTAGTTAGAATAAGAAGG |
| SafeHarbor.430 | GTGGCAATGTCCTGGAGAAA |
| SafeHarbor.431 | GTGAAGTGCTTTATCTGTAC |
| SafeHarbor.432 | GAGTTTATATAGGTATGAAA |
| SafeHarbor.433 | GACCTCATAAACAAATCACT |
| SafeHarbor.434 | GAAACGTCTGTATGCAAAGC |
| SafeHarbor.435 | GGTGTGGTGCAAGGGTGAGT |
| SafeHarbor.436 | GAGAATCTGCTATTGCCAAT |
| SafeHarbor.437 | GTACTAAGTATCTTGAAATG |
| SafeHarbor.438 | GTCATGACATGAGTTGCATG |
| SafeHarbor.439 | GCAGTGATCAGAGACAGTTG |
| SafeHarbor.440 | GGCAAAATAACTTCATCTAT |
| SafeHarbor.441 | GCCTGGCCTTCTGTGGAATT |
| SafeHarbor.442 | GGTGGCCTTTGTTTGCAGGC |
| SafeHarbor.443 | GAGATGGTATATTTGTCAGA |
| SafeHarbor.444 | GGGACACCCAGCATCTCAAC |
| SafeHarbor.445 | GTATATGACAGTAGGGTTGG |
| SafeHarbor.446 | GGACCCCAGAACTGAAATCA |
| SafeHarbor.447 | GGGCACCACTGAGAATGTAT |
| SafeHarbor.448 | GGGACTACAAATATGAAAAA |
| SafeHarbor.449 | GTAAAATTATGAGCTCCAGT |
| SafeHarbor.450 | GATTGTGAGTGATGAGAATC |
| SafeHarbor.451 | GAGACTGAGGGTTGCTCTTA |
| SafeHarbor.452 | GCATAGAGTGAACACTTTGG |
| SafeHarbor.453 | GAAGTTCTCCTTTAACCAAT |
| SafeHarbor.454 | GACCTTGACCAAAGATATTA |
| SafeHarbor.455 | GTGTGGGCAAGAGACAGTCC |
| SafeHarbor.456 | GTTGGGGGCTCTCTTGCCAC |
| SafeHarbor.457 | GGATAAAACTCTAACAGAAC |
| SafeHarbor.458 | GGAAACATATTACCCCTCCA |
| SafeHarbor.459 | GCACTATTACTCCACTGAGA |
| SafeHarbor.460 | GTGAGCAGAGATCACCTTAG |
| SafeHarbor.461 | GGGTTCATATAGGTCGGAAT |
| SafeHarbor.462 | GTGCCCCCGATTCTTCCATG |
| SafeHarbor.463 | GGAACAAAATTTGCACATAA |
| SafeHarbor.464 | GAGAAAGTCCAAGGGTAAAA |
| SafeHarbor.465 | GCAATTAACTCTACAAGGAA |
| SafeHarbor.466 | GTTTCAACCATTAGGGGGCT |
| SafeHarbor.467 | GGCAGGGGTAGTAAGCTTAG |
| SafeHarbor.468 | GTACACATCTTCCCAATCAG |
| SafeHarbor.469 | GTTACTTGGAAAAATGACCA |
| SafeHarbor.470 | GTACCCGGTAAATCATAGAG |
| SafeHarbor.471 | GTGTATTATCCTGCATTCCA |
| SafeHarbor.472 | GGGTAAAACAAATGCATCAT |
| SafeHarbor.473 | GTGTGTTGGCCTAGGGATGA |
| SafeHarbor.474 | GGTGTGATAAAACCTCAGAG |
| SafeHarbor.475 | GAGCTAATTGGTCAGATTCT |
| SafeHarbor.476 | GTACCAGAGTACAGTGTCCG |
| SafeHarbor.477 | GGTCAGTGCTCTATCATTTA |
| SafeHarbor.478 | GTTGCCTATCTTCAGAGTAC |
| SafeHarbor.479 | GAAGATGCATGGACCTACCA |
| SafeHarbor.480 | GAATAGACACTGGTTCTCTG |
| SafeHarbor.481 | GTCAGCTCTTAACATCTGGT |
| SafeHarbor.482 | GATAACAAGGCTCAGAAGGC |
| SafeHarbor.483 | GTCAAAACACAGTGAGCTGT |
| SafeHarbor.484 | GAGAATATAGCTGAAGGTGG |
| SafeHarbor.485 | GGGATTGACCATCAATACAG |
| SafeHarbor.486 | GAAACCCCCATCTCAGTCTT |
| SafeHarbor.487 | GTACAGATACCACTATTTGG |
| SafeHarbor.488 | GAGTAGCTAGAGGCACTCTT |
| SafeHarbor.489 | GAGATTTGCAGTGCATGAAT |
| SafeHarbor.490 | GTTCAACTAAAGGTCTTATG |
| SafeHarbor.491 | GTGTTTCACTGTTCTCTTCA |
| SafeHarbor.492 | GTGAAGTAGAGATTATGTAA |
| SafeHarbor.493 | GTCAAACCAAGTTGAATTCA |
| SafeHarbor.494 | GATGCTAAAAATCTAAACCT |
| SafeHarbor.495 | GGCCCTTATTACCAGATTTG |
| SafeHarbor.496 | GTGGAGATTTGCTTACGAGC |
| SafeHarbor.497 | GAACCTTGGAGAATTGAATA |
| SafeHarbor.498 | GATAGAAAAGAGCAGCTACA |
| SafeHarbor.499 | GCAAGAAGAAACTGCTATTA |
| SafeHarbor.500 | GTAATGTTGCCGAAGCAATT |
| SafeHarbor.501 | GAATTTCATTACAGGAAGTA |
| SafeHarbor.502 | GAAAACACACCTTATCACAG |
| SafeHarbor.503 | GTTATCTTTGAGAGAACATT |
| SafeHarbor.504 | GAACTCTTAAGGTTAATAAG |
| SafeHarbor.505 | GAACCATCCATCCTCACCTG |
| SafeHarbor.506 | GGAGATGCACTGGTAAAAAG |
| SafeHarbor.507 | GCTCATCTCCACAGCCATCC |
| SafeHarbor.508 | GAGTGGCCGGTGCCATTTCT |
| SafeHarbor.509 | GCTACTAGCGAAGAAGAAGG |
| SafeHarbor.510 | GTAAGCTTAAAACATTAGTA |
| SafeHarbor.511 | GTTTACAGGAAGGAGAAGGA |
| SafeHarbor.512 | GTAATATTTGAGGTATGAAT |
| SafeHarbor.513 | GATGGCTCACACTTGCTGTA |
| SafeHarbor.514 | GAAACTGGGAACAAGCTTTA |
| SafeHarbor.515 | GCTAATGCTTTGCCTACCCC |
| SafeHarbor.516 | GCCTTACCCTCAGTAGTGAA |
| SafeHarbor.517 | GAACTGAAGTTTAGAAGTAA |
| SafeHarbor.518 | GAAATATCATGATGGTGAAG |
| SafeHarbor.519 | GTGTTGATTCTGAACAAGTT |
| SafeHarbor.520 | GGCCCTGTCCTGGACATAAA |
| SafeHarbor.521 | GCACATTCTAATTTGTGGAT |
| SafeHarbor.522 | GAAGTTAACATGGAATTAAA |
| SafeHarbor.523 | GTCCTTAGGCTTGCAATGCT |
| SafeHarbor.524 | GAGAGACAATTTGGGTCTAG |
| SafeHarbor.525 | GTTAAATCCAATGGATTCCT |
| SafeHarbor.526 | GTTCTCAATTTACTGGGATT |
| SafeHarbor.527 | GCAGCTGTGCTCAAAAGACC |
| SafeHarbor.528 | GAGGCTTAGTTGTAATAATG |
| SafeHarbor.529 | GCCCCTCAATTCCAGTGTAA |
| SafeHarbor.530 | GACTGGCAAATACAATTTGC |
| SafeHarbor.531 | GAATGCAATATAGTGATCTT |
| SafeHarbor.532 | GGAGAGGGTGGTTTAAAAGC |
| SafeHarbor.533 | GGGTATACCTTAGGAAAGCT |
| SafeHarbor.534 | GATGCATTCAATAGCTCTGT |
| SafeHarbor.535 | GGGCTAAATAAAGCAATGTT |
| SafeHarbor.536 | GTTATTCATAAATTGTAAGC |
| SafeHarbor.537 | GTGACATAGTGGGATAGCCC |
| SafeHarbor.538 | GGGAACATTTCTTCATAGGG |
| SafeHarbor.539 | GGTATGTGTCCATATGTGTC |
| SafeHarbor.540 | GAAGAATTAACACATTGTCT |
| SafeHarbor.541 | GATGCCTGGTTAACAATTCA |
| SafeHarbor.542 | GCCTTAAAGCTCCTATAGAA |
| SafeHarbor.543 | GGGCCCACATTTATCTCTAT |
| SafeHarbor.544 | GCAGGTGTCTAAATTCACTC |
| SafeHarbor.545 | GAACAATAAGTCAAGCAAGT |
| SafeHarbor.546 | GGGACAATCTAAATGTCCTA |
| SafeHarbor.547 | GGATATAAAAGCATACAAAA |
| SafeHarbor.548 | GAGTCACCCCAGGGACAAAC |
| SafeHarbor.549 | GGACCCTAAGGGAAGCTTGA |
| SafeHarbor.550 | GTACTCACTGATACACAGCT |
| SafeHarbor.551 | GTTTATAAATATTCCGACTA |
| SafeHarbor.552 | GGTGACTAGGAAGTTTCTGC |
| SafeHarbor.553 | GACTTAGAAACAGTTAATAA |
| SafeHarbor.554 | GTTATTATTGAGTTGGTATA |
| SafeHarbor.555 | GAACACTTTCACTGGGAATA |
| SafeHarbor.556 | GGGATTCTCCTAGAATAAAT |
| SafeHarbor.557 | GCCCACTTATGCAGTATAAG |
| SafeHarbor.558 | GTGCATACCAAATTAGTGTC |
| SafeHarbor.559 | GTATTCACAGCCAAAAAGTA |
| SafeHarbor.560 | GTTCTGCTTCTAACATAGTA |
| SafeHarbor.561 | GGAAAAGCTATGTTAAACCT |
| SafeHarbor.562 | GTATCTGCATATTAAACACA |
| SafeHarbor.563 | GGCCCTTAAAACATGGAACC |
| SafeHarbor.564 | GTAGCCTATGTCAGAATGAG |
| SafeHarbor.565 | GAGTTGCTAGACAGCTACCA |
| SafeHarbor.566 | GAAGCAACACAGATTCTCAC |
| SafeHarbor.567 | GGTTAGCAAAATTGCAAGAG |
| SafeHarbor.568 | GGAACCTGGAGAATGTTAAG |
| SafeHarbor.569 | GTGTTCTCATTCTTCACTCA |
| SafeHarbor.570 | GAGTCACGGTCAAACAGTCG |
| SafeHarbor.571 | GAGAACATACACATAATGAC |
| SafeHarbor.572 | GCTTCAAATGTGTGTGCTTC |
| SafeHarbor.573 | GAGAAATTAACTCACTTTAT |
| SafeHarbor.574 | GTATTTAGGCTATGCTTGAA |
| SafeHarbor.575 | GTCTTTGGAAACAACCATGT |
| SafeHarbor.576 | GCCCATCATGACAGGACAGG |
| SafeHarbor.577 | GGTAGAGCAGGGGTATTACT |
| SafeHarbor.578 | GGAAGTGCATGCATGACCTT |
| SafeHarbor.579 | GTTGAAATCAACATAAGGAA |
| SafeHarbor.580 | GGGGTGGCACTGGGTTAATT |
| SafeHarbor.581 | GGGCAGATCGACAACTGCCG |
| SafeHarbor.582 | GTTGAATTATGTTACCTCCA |
| SafeHarbor.583 | GAAAAATGACCCATGATTAA |
| SafeHarbor.584 | GGTAGAGGGATAATGCACTG |
| SafeHarbor.585 | GAAAGTCAAGCAGAGGGGCA |
| SafeHarbor.586 | GGAGAGAATTAATCTTATTT |
| SafeHarbor.587 | GGAGACACCAGTCACGGAGT |
| SafeHarbor.588 | GAGCCAAAGTGGCAAAGTGG |
| SafeHarbor.589 | GTGGGAGGACAGGCAGCAGA |
| SafeHarbor.590 | GATTAAAGACTTGCTTAGTT |
| SafeHarbor.591 | GAGCTTATTTGACATGTTAG |
| SafeHarbor.592 | GGATTAATGTAGCTGTAAAT |
| SafeHarbor.593 | GTAAGAGACCAAGCCCAAGT |
| SafeHarbor.594 | GGTTCACTGAGTATGTGCCC |
| SafeHarbor.595 | GGATGCAGCCACTCTCAGAG |
| SafeHarbor.596 | GAGGTACCTCACAATTTGAA |
| SafeHarbor.597 | GTATCAACAGAGTGTCAGAT |
| SafeHarbor.598 | GTACCTCAAAGTGTTCCCTG |
| SafeHarbor.599 | GGCCTCTGTAAGAGGGGAGT |
| SafeHarbor.600 | GATATATAAAGTAAGTGGAG |
| SafeHarbor.601 | GATCCTTATTGCTCCATTCT |
| SafeHarbor.602 | GAACTTATAAAGTGCCCACA |
| SafeHarbor.603 | GGTAGGGTTGGAAGGGTAAC |
| SafeHarbor.604 | GTGATGCATAGCATAGTTTC |
| SafeHarbor.605 | GGGAGGCAACCTGTCCCTGC |
| SafeHarbor.606 | GGTACAATAGATGCCTGAAA |
| SafeHarbor.607 | GGGAGTGACTCAGCTACATG |
| SafeHarbor.608 | GGTCATGATGCCACTGGGAG |
| SafeHarbor.609 | GACCAGTAAGATTAAAAATG |
| SafeHarbor.610 | GGCACTGGTTTGTGCACTTC |
| SafeHarbor.611 | GAAATATTCAAGTTTATGAG |
| SafeHarbor.612 | GTTTGCAGCACACAGGTAGA |
| SafeHarbor.613 | GTTTGGTACAGTATAACCAA |
| SafeHarbor.614 | GATCATAACAGAAGCTCCAA |
| SafeHarbor.615 | GCAAGAGCAATTCTCAGGCT |
| SafeHarbor.616 | GGGCCATGGAAAACAGCCCA |
| SafeHarbor.617 | GTGTTATGACTTTAAAGTTA |
| SafeHarbor.618 | GCAGGTCAAAAGCTCTAGAC |
| SafeHarbor.619 | GAAACCTAAACAATAGCTCC |
| SafeHarbor.620 | GCCAAGTGGACTAGAAGCCG |
| SafeHarbor.621 | GTGTCATCATGCTAAGTAAT |
| SafeHarbor.622 | GCTCTAGATTAGTTGGCTTA |
| SafeHarbor.623 | GACCTCTAATTCACAGAGAG |
| SafeHarbor.624 | GACTGAGGGTGGATAATCCA |
| SafeHarbor.625 | GAGTCGAATGTAAGAAATTC |
| SafeHarbor.626 | GATATGAGAGATAATTAAAG |
| SafeHarbor.627 | GAATACCTACCCATTAGTGA |
| SafeHarbor.628 | GTGTTAAGTAGGGAATATAC |
| SafeHarbor.629 | GAGAAATGAGGCGCTTGTTA |
| SafeHarbor.630 | GATTCACTTAGTTGCTCCCC |
| SafeHarbor.631 | GAATATGAGCTCCTAACATA |
| SafeHarbor.632 | GTACTCAGCAGAAACAAAGG |
| SafeHarbor.633 | GTGTACATAAACAAAAAGTT |
| SafeHarbor.634 | GCAGGTGCAATATTTAGTAG |
| SafeHarbor.635 | GTAAGGCCATGACACCAATT |
| SafeHarbor.636 | GTCTTAGGTGCACAATTCCC |
| SafeHarbor.637 | GTGTTATCTTTCACTCATAT |
| SafeHarbor.638 | GATTTAAGTCCTCCATGCTT |
| SafeHarbor.639 | GATTTGACATGCTTTAATAA |
| SafeHarbor.640 | GTTTCCAGGTGACTCAGTTA |
| SafeHarbor.641 | GGTCTGTGTGTGGATTTCCA |
| SafeHarbor.642 | GTCAAGCCTTATGCAATTTC |
| SafeHarbor.643 | GTCACTGGAGAAGCAACTTC |
| SafeHarbor.644 | GAGACTAAATGCGGGAAAGA |
| SafeHarbor.645 | GAACTAATCAATGTGCATCA |
| SafeHarbor.646 | GGCAGCCCTAAGGCAGTCAC |
| SafeHarbor.647 | GGGATTGTTAATGTCCAAGC |
| SafeHarbor.648 | GCATAAACATTCATGAGTTT |
| SafeHarbor.649 | GCACTCACGGAGTGCTAGGG |
| SafeHarbor.650 | GTGCTTAATATGAATGCTGG |
| SafeHarbor.651 | GGAACATGAAAATAACGTTG |
| SafeHarbor.652 | GTGACTTCATTTGATTTCAC |
| SafeHarbor.653 | GCCATCCACCATGCTATCAA |
| SafeHarbor.654 | GAGAATGGAGCTGAAAATAC |
| SafeHarbor.655 | GCTTGCTCTGTATGACTGTC |
| SafeHarbor.656 | GTCATCAGGATAAATCAGCG |
| SafeHarbor.657 | GTCTTAGTCAGGGAAGGAGT |
| SafeHarbor.658 | GGATCTCAAGAGCTACCTAA |
| SafeHarbor.659 | GAAATTACATCCCTAGATAG |
| SafeHarbor.660 | GAAGCAAAACTACCTTTGTT |
| SafeHarbor.661 | GCTTCATCTGGGGTGAAACC |
| SafeHarbor.662 | GCATTACTAACCATGGAAAG |
| SafeHarbor.663 | GTGGGTCATTCAAGTGGAGC |
| SafeHarbor.664 | GTTCCATAAGTGGAAGCGTT |
| SafeHarbor.665 | GAAATAGGAAGGGAATATAA |
| SafeHarbor.666 | GTAACACTCAGCAGCTGAGA |
| SafeHarbor.667 | GCTATTCCAGGAGAACACAT |
| SafeHarbor.668 | GTGTTGATAACAGAAGATCC |
| SafeHarbor.669 | GGATCACATATACATGCCTG |
| SafeHarbor.670 | GTCAAACTCTTCAATATTCT |
| SafeHarbor.671 | GCAACTTGAACTCCAACTTA |
| SafeHarbor.672 | GAGACTGAATATAAGATGTA |
| SafeHarbor.673 | GTGTCAAAAAACCTCAGAAA |
| SafeHarbor.674 | GTTAGGAAGTATTCGGAGTT |
| SafeHarbor.675 | GTATCAAGTAAATAGGTGGA |
| SafeHarbor.676 | GTAAAGCAACAGGTAATTAA |
| SafeHarbor.677 | GATGTTTATTGTAGGGCATG |
| SafeHarbor.678 | GACCACTCAATTTATATATT |
| SafeHarbor.679 | GGCCATTATTTGTTGATCAT |
| SafeHarbor.680 | GGAGAAACTGGATTTAAAGA |
| SafeHarbor.681 | GTCTACAGACCACAGAAGAA |
| SafeHarbor.682 | GGTATCCCTTAAGAATTTAA |
| SafeHarbor.683 | GGTAGATTAATATTCTGGAA |
| SafeHarbor.684 | GTAGTTATCCAAGGTAACAG |
| SafeHarbor.685 | GGATTTGCGCAGGTCCCTCT |
| SafeHarbor.686 | GCATGTTAGCCAGCAGAACA |
| SafeHarbor.687 | GTCACCTAAAACGATGTATG |
| SafeHarbor.688 | GATACTAATCAATAAGTGGG |
| SafeHarbor.689 | GAAGGTTATGGGAGGGGTAC |
| SafeHarbor.690 | GCAGAAAGTGATCTTTACAT |
| SafeHarbor.691 | GAAGAGGTTTAGGTTGTCAG |
| SafeHarbor.692 | GAGCCACAGTTAGAGTAACT |
| SafeHarbor.693 | GTATTGGCTAGTTAAGTGCA |
| SafeHarbor.694 | GGTCACCTTAAAAACATCTA |
| SafeHarbor.695 | GTGCATTTGGGTATTAGATT |
| SafeHarbor.696 | GAATAATAGCTATGGCTGCT |
| SafeHarbor.697 | GGGCATTGCCTGTTTAATCT |
| SafeHarbor.698 | GACTTTGTCACTAACACGCA |
| SafeHarbor.699 | GTAAGCATGTACGAAGTAAC |
| SafeHarbor.700 | GTTTGCCTTCCAGATAGGAG |
| SafeHarbor.701 | GGGAGTGTATGTTCATTGGA |
| SafeHarbor.702 | GGGTGACTACTGGTTGCTTT |
| SafeHarbor.703 | GTTAAACCTGTTTATGCTCT |
| SafeHarbor.704 | GGATTCTGAATTAATTGTAG |
| SafeHarbor.705 | GATTCTATAGTCTATAGTTA |
Supplementary Material
Acknowledgments
We thank J. Sage, A. Brunet, A. Fire, and members of the Bassik lab for critical reading of the manuscript and helpful discussions. We thank J. Sollier for the FLAG-AID plasmid, and A. Sockell for help with sequencing. We thank O. Ursu, D. Morgens, C. Araya, and A. Kundaje for their design of safe harbor sgRNAs. Cell sorting/flow cytometry analysis was performed on an instrument in the Stanford Shared FACS Facility obtained using NIH S10 Shared Instrument Grant (S10RR025518-01). This work was funded by NIH T32HG000044 (G.T.H.), CEHG Fellowship (L.F.), the Walter V. and Idun Berry postdoctoral fellowship (K.H.), NSF DGE-114747 (C.H.L.), NIH ES016486 (K.A.C.), NIH R01HG008150 (S.B.M. and M.C.B.) and NIH 1DP2HD084069-01 (M.C.B.).
Footnotes
Author Contributions
The research was conceived by G.T.H. and M.C.B. G.T.H. conducted experiments with aid of A.L. L.F. performed sequencing data analyses with the aid of G.T.H. K.H. aided in the fluorescence microscopy. C.H.L. aided in the design of PSMB5 mutation validation experiments. K.A.C. aided in design of mutagenic approach. S.B.M. aided in developing analysis methods. G.T.H., L.F., and M.C.B. wrote the paper with help from all authors.
References
- 1.Doerner A, Rhiel L, Zielonka S, Kolmar H. Therapeutic antibody engineering by high efficiency cell screening. FEBS letters. 2014;588:278–287. doi: 10.1016/j.febslet.2013.11.025. [DOI] [PubMed] [Google Scholar]
- 2.Bornscheuer UT, et al. Engineering the third wave of biocatalysis. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 3.Soskine M, Tawfik DS. Mutational effects and the evolution of new protein functions. Nature reviews Genetics. 2010;11:572–582. doi: 10.1038/nrg2808. [DOI] [PubMed] [Google Scholar]
- 4.Lienert F, Lohmueller JJ, Garg A, Silver PA. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nature reviews Molecular cell biology. 2014;15:95–107. doi: 10.1038/nrm3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nature biotechnology. 2005;23:1105–1116. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 6.Liu W, Brock A, Chen S, Chen S, Schultz PG. Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nature methods. 2007;4:239–244. doi: 10.1038/nmeth1016. [DOI] [PubMed] [Google Scholar]
- 7.Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nature reviews Immunology. 2006;6:573–583. doi: 10.1038/nri1896. [DOI] [PubMed] [Google Scholar]
- 8.Di Noia JM, Neuberger MS, Molecular MS. mechanisms of antibody somatic hypermutation. Annual review of biochemistry. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 9.Rajewsky K, Forster I, Cumano A. Evolutionary and somatic selection of the antibody repertoire in the mouse. Science. 1987;238:1088–1094. doi: 10.1126/science.3317826. [DOI] [PubMed] [Google Scholar]
- 10.McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell research. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeap LS, et al. Sequence-Intrinsic Mechanisms that Target AID Mutational Outcomes on Antibody Genes. Cell. 2015;163:1124–1137. doi: 10.1016/j.cell.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaudhuri J, et al. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 13.Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation-induced deaminase activity at cytidine. The Journal of biological chemistry. 2004;279:6496–6500. doi: 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- 14.Arakawa H, et al. Protein evolution by hypermutation and selection in the B cell line DT40. Nucleic acids research. 2008;36:e1. doi: 10.1093/nar/gkm616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Jackson WC, Steinbach PA, Tsien RY. Evolution of new nonantibody proteins via iterative somatic hypermutation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:16745–16749. doi: 10.1073/pnas.0407752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bowers PM, et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:20455–20460. doi: 10.1073/pnas.1114010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chavez A, et al. Highly efficient Cas9-mediated transcriptional programming. Nature methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert LA, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen B, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma H, et al. Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nature biotechnology. 2016;34:528–530. doi: 10.1038/nbt.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kearns NA, et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nature methods. 2015;12:401–403. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai SQ, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature biotechnology. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishida K, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016 doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- 27.Canver MC, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan OW, et al. Selection of chromosomal DNA libraries using a multiplex CRISPR system. eLife. 2014;3 doi: 10.7554/eLife.03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014;513:120–123. doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito S, et al. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1975–1980. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Papavasiliou FN, Schatz DG. The activation-induced deaminase functions in a postcleavage step of the somatic hypermutation process. The Journal of experimental medicine. 2002;195:1193–1198. doi: 10.1084/jem.20011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inouye S, Tsuji FI. Evidence for redox forms of the Aequorea green fluorescent protein. FEBS letters. 1994;351:211–214. doi: 10.1016/0014-5793(94)00859-0. [DOI] [PubMed] [Google Scholar]
- 36.Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- 37.Tsien RY. The green fluorescent protein. Annual review of biochemistry. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 38.Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 39.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 40.Hideshima T, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer research. 2001;61:3071–3076. [PubMed] [Google Scholar]
- 41.Lu S, Wang J. The resistance mechanisms of proteasome inhibitor bortezomib. Biomarker research. 2013;1:13. doi: 10.1186/2050-7771-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang M, Yang Z, Rada C, Neuberger MS. AID upmutants isolated using a high-throughput screen highlight the immunity/cancer balance limiting DNA deaminase activity. Nature structural & molecular biology. 2009;16:769–776. doi: 10.1038/nsmb.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu S, et al. Different mutants of PSMB5 confer varying bortezomib resistance in T lymphoblastic lymphoma/leukemia cells derived from the Jurkat cell line. Experimental hematology. 2009;37:831–837. doi: 10.1016/j.exphem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajagopal N, et al. High-throughput mapping of regulatory DNA. Nature biotechnology. 2016;34:167–174. doi: 10.1038/nbt.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korkmaz G, et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nature biotechnology. 2016;34:192–198. doi: 10.1038/nbt.3450. [DOI] [PubMed] [Google Scholar]
- 47.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. The New England journal of medicine. 1999;341:1520–1529. doi: 10.1056/NEJM199911113412007. [DOI] [PubMed] [Google Scholar]
- 49.Unniraman S, Schatz DG. AID and Igh switch region-Myc chromosomal translocations. DNA repair. 2006;5:1259–1264. doi: 10.1016/j.dnarep.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 50.Blagodatski A, et al. A cis-acting diversification activator both necessary and sufficient for AID-mediated hypermutation. PLoS genetics. 2009;5:e1000332. doi: 10.1371/journal.pgen.1000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deans RM, et al. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nature chemical biology. 2016;12:361–366. doi: 10.1038/nchembio.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hendel A, et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature biotechnology. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–12. [Google Scholar]
- 54.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic acids research. 2014;42:W401–407. doi: 10.1093/nar/gku410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bassik MC, et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013;152:909–922. doi: 10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bassik MC, et al. Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nature methods. 2009;6:443–445. doi: 10.1038/nmeth.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kampmann M, Bassik MC, Weissman JS. Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2317–2326. doi: 10.1073/pnas.1307002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.