

Abstract
Chemical penetration enhancers (CPEs) are present in a large number of transdermal, dermatological, and cosmetic products to aid dermal absorption of curatives and aesthetics. This wide spectrum of use is based on only a handful of molecules, the majority of which belong to three to four typical chemical functionalities, sporadically introduced as CPEs in the last 50 years. Using >100 CPEs representing several chemical functionalities, we report on the fundamental mechanisms that determine the barrier disruption potential of CPEs and skin safety in their presence. Fourier transform infrared spectroscopy studies revealed that regardless of their chemical make-up, CPEs perturb the skin barrier via extraction or fluidization of lipid bilayers. Irritation response of CPEs, on the other hand, correlated with the denaturation of stratum corneum proteins, making it feasible to use protein conformation changes to map CPE safety in vitro. Most interestingly, the understanding of underlying molecular forces responsible for CPE safety and potency reveals inherent constraints that limit CPE performance. Reengineering this knowledge back into molecular structure, we designed >300 potential CPEs. These molecules were screened in silico and subsequently tested in vitro for molecular delivery. These molecules significantly broaden the repertoire of CPEs that can aid the design of optimized transdermal, dermatological, and cosmetic formulations in the future.
Keywords: stratum corneum, spectroscopy, skin irritation, lipid
Currently, hypodermic needles are the only available mode for systemic delivery of macromolecular drugs into humans. Transdermal delivery offers an attractive alternative to needle-based drug administration. However, evolved to impede the flux of exogenous molecules, stratum corneum (SC), the topmost layer of the skin, provides a strong barrier to molecular delivery. This is especially problematic for relatively large drugs (molecular mass > 500 Da), which represent a large majority of active agents for therapeutic applications (1). Over 350 molecules, termed chemical penetration enhancers (CPEs), have been identified to perturb the SC barrier to facilitate molecular delivery. However, incorporation of CPEs into products has been mitigated by safety concerns related to the health of the skin membrane (2-4). Accordingly, overcoming the skin barrier in a safe and effective way still remains the bottleneck of transdermal and topical therapies.
Identification of chemicals to increase skin permeability has been an area of high activity in the last three decades (5-7). After an initial rise in the number of CPEs in the 1980s, the active pool of CPEs has reached a plateau in the last decade. In an era where new chemical entities are being discovered at an exponential rate (as indicated by the entries in the Chemical Abstract Service), the plateau in the number of CPE molecules is rather surprising (only 1 in 100,000 known molecules represents a CPE). This anomaly originates from the slow rates of syntheses of CPEs when compared with other chemical families representing small molecules and sequences. Lack of a fundamental understanding of mechanistic principles that define CPE performance has also added to the slow rate of CPE discovery. In addition, low throughput of experimental tools to probe CPE potency [O(10) per day for typical diffusion measurements] has forced researchers to explore narrow search spaces bounded by a handful of chemical functionalities identified in the past. Not surprisingly, CPEs have not reached their full potential in transdermal or topical systems so far.
We report, first, on the mechanisms of barrier disruption and safety in presence of CPEs based on the structural changes in the microscopic domains of the SC membrane. Next, we identify dominant molecular features that govern changes in the microscopic organization of the SC and hence the macroscopic endpoints of CPE potency and membrane safety. These studies reveal fundamental constraints in optimizing the balance between CPE potency and membrane safety. Nevertheless, identifying basic principles of CPE performance enabled the discovery of additional molecules that provide safe and effective permeabilization of the SC. Additionally, this understanding also enabled the establishment of in vitro spectroscopic and in silico computational methods to accelerate the evaluation of CPEs in the future.
Materials and Methods
Library of Chemical Penetration Enhancers. One hundred and two CPEs, chosen from 10 categories [(i) anionic surfactants (AI), (ii) cationic surfactants (CI), (iii) zwitterionic surfactants (ZI), (iv) nonionic surfactants (NI), (v) fatty acids (FA), (vi) fatty esters (FE), (vii) fatty amines (FM), (viii) Azone-like compounds (AZ), (ix) sodium salts of fatty acids (SS), and (x) others (OT)] were used in this study (see Supporting Text, which is published as supporting information on the PNAS web site). These chemicals were chosen to represent a diverse library of CPEs while still including the more conventional penetration enhancers from the existing transdermal literature. Formulations containing 1.5% (wt/vol) of each CPE were prepared in a 1:1 EtOH:PBS (ethanol:PBS) solvent.
Measurement of Enhancement Ratio (ER) and Irritation Potential (IP). Potencies of CPEs were quantified in terms of skin conductivity ER. ER is a good measure of the extent of SC disruption and, hence, permeability enhancement (8). These experiments were performed by using porcine skin. Quantitatively, ER was defined as skin conductivity at the end of 24 h, when incubated with a particular formulation, normalized to conductivity at time 0. ER was measured using methods in ref. 8. The IP of CPEs was estimated from cell viability of normal human-derived epidermal keratinocytes in a MTT (methyl thiazol tetrazolium) assay, using Epiderm (MatTek) as described in ref. 8.
Fourier Transform Infrared (FTIR) Spectroscopy Measurements. Porcine epidermis was isolated from the dermis in full thickness skin by heat stripping (9, 10). Isolated epidermis was then floated over 0.25% (wt/vol) trypsin solution overnight at room temperature to digest the epidermis (11). These sample preparation techniques are not expected to cause changes in the SC ultrastructure (12, 13). The residual SC film was then washed with PBS and dried at room temperature for 48 h. At this point, the SC is free of hair and other epidermal debris. The SC was then cut into square pieces of 1.5 × 1.5 cm. Each SC piece was used as its own control, and, accordingly, we recorded an interferogram of the SC samples before and after treatment. Each SC sample was incubated with 2 ml of CPE formulation for 24 h. The CPE formulations were prepared in 1:1 EtOD:D2O (deuterated ethanol:deuterated water). At the end of incubation, the SC samples were removed from the CPE formulation and rinsed thoroughly with 1:1 EtOD:D2O to remove any excess enhancer on the SC surface. The SC pieces were then dried at room temperature for 48 h at the end of which interferograms were recorded again. Spectra were recorded by using a Nicolet Magna 850 spectrometer setup at a resolution of 2 cm-1 and were averaged over 100 scans. The spectra were smoothed, baseline-corrected, and saved in the comma separated value format for further analysis in origin (14, 15). Each CPE formulation was studied in triplicate to assess its effect on the SC.
Molecular Descriptors. Molecular descriptors were calculated for all of the CPEs used in this study by using molecular modeling pro (ChemSW). To determine physicochemical properties, molecular structures were drawn by using the interface provided by molecular modeling pro. Molecules were relaxed to represent the 3D structure in the lowest energy conformation. The octanol water partition coefficient was calculated by using four independent methods: atom-based log P (16), fragment addition log P (17), Q log P (18), and Moriguchi's method (19, 20). The average of three closest values was used for further analysis. Components of solubility parameters related to hydrogen bonding, polarity, and dispersion were calculated by using the methods outlined by Hansen (proprietary algorithm of ChemSW), van Krevelen and Hoftyzer (21), and Hoy (22). Again, instead of using any one independent method for calculations, an average of the two closest values was used.
Selection of Wild-Type CPE and Mutation. Ten molecules, one from each of the 10 chemical classes, were selected as wild-type enhancers. These wild-type enhancers were then mutated by making substitutions in the functional groups, giving rise to a library of 325 mutants. The substitutions included varying the length of carbon chains, adding heterocyclic rings and secondary functional groups. The aim of this exercise was to illustrate the utility and practical effectiveness of the “parametrical tools” for screening CPEs in silico. The list of mutants can be further stretched by inclusion of a wider set of rules for mutations. ER/IP was calculated for each mutant assuming it to be a fluidizer as well as an extractor (leading to 650 predictions) based on parametrical equations describing ER and IP discussed in the results section. These values were compared with those in the pool of 102 experimentally tested CPEs. Mutants with ER/IP comparable to or better than the best CPEs in the experimental pool were then selected for further studies with inulin permeation. Some of the mutants with desirable ER/IP in our library could not be procured commercially and were dropped from further consideration. The mutant CPEs that were available commercially were obtained from Sigma.
Inulin Permeability Measurement. Permeation experiments were performed with Franz diffusion cells (FDC), using 3H-labeled inulin. 1.5% wt/vol formulations of test CPEs were prepared in 1:1 EtOH:PBS. 3H-labeled inulin (10 μCi/ml; 1 Ci = 37 GBq) was added to each formulation and placed on full thickness skin in FDCs. Skin samples were incubated with the CPE formulations for 96 h, during which receiver samples were collected and analyzed in a liquid scintillation counter to detect radioactivity (Tri-Carb 2100TR, Packard). Permeability was calculated by using standard equations (23). It was verified in an independent study that all detected radioactivity came from the model solute and not from tritiated water that may have resulted from tritium exchange. Specifically, receiver samples were desiccated and analyzed for radioactivity, and no significant difference was observed between native and desiccated receiver samples.
Results
ER and IP of CPEs. One hundred and two enhancers representing 10 diverse chemical functionalities fell into two classes depending on the relationship between ER and IP. The first class (Fig. 1A), where ER increases proportionately with IP, includes NI, ZI, AZ, and SS. The second class (Fig. 1B), where ER did not correlate particularly well with IP, comprises FA, FM, AI, FE, CI, and OT. The best CPEs in each category (as judged by ER/IP ratio) are shown in Fig. 1C.
Fig. 1.

ER-IP correlations of CPEs in 10 different categories. (A) ER is proportional to IP for CPEs belonging to NI (blue circles), ZI (red circles), AZ (black circles), and SS (green circles). (B) ER does not show a correlation with IP for CPEs belonging to FM (blue circles), FE (red circles), AI (black circles), FA (green circles), CI (brown circles), or OT (gray circles). (C) Bar graph showing ER/IP for the best CPE in each chemical class (for complete names refer to Table 1, which is published as supporting information on the PNAS web site). Error bars correspond to SD (n = 3).
FTIR Spectroscopy of SC. Two primary signals of the SC IR interferogram were mapped: CH2 symmetric stretching mode, νsymCH2, at 2,850 cm-1 arising from the lipids in the SC bilayers, and amide I band (1,700-1,600 cm-1) of the SC (Fig. 2B) arising from the corneocyte proteins.
Fig. 2.

Spectroscopic studies of the SC. (A) Plot of conductivity ER against change in integrated absorbance of methylene stretching, Δ(νsymCH2). Extractors show a reduction in absorbance, whereas fluidizers show an increase in absorbance. In each class, ER correlates well with Δ(νsymCH2). Error bars correspond to SD (n = 3). (B) Deconvoluted peaks of the amide I band of an SC IR spectrum before (blue curves) and after (red curves) the treatment with a formulation containing 1.5% wt/vol lauric acid in 1:1 EtOD:D2O. Treatment with lauric acid decreases the relative contribution of the α-helical structures to the amide I band compared to the untreated region of the same sample. Contributions from other secondary structures, β-sheets, random coils, and antiparallel β-sheets and turns, in contrast, increase compared with the corresponding regions of the untreated sample. (C) Plot of IP against change in integrated absorbance of carbonyl stretching mode at 1,650 cm-1, Δ(νC═O). Δ(νC═O) correlates well with IP. Error bars correspond to SD (n = 3).
Barrier disruption and ER. ER values correlated with the changes in the integrated absorbance of νsymCH2 (Fig. 2 A). A reduction in methylene stretching modes, indicated by a negative peak area change [Δ(νsymCH2) = area after treatment - area before treatment], indicates a decrease in the lipid content or lipid extraction from the bilayers. An increase in the peak area, or a positive value of Δ(νsymCH2), indicates partitioning of enhancer molecules in the bilayer. Regardless of their chemical nature, CPEs fell into two categories (Fig. 2A): ones that extract the lipids from the SC, labeled “extractors,” and ones that partition into the SC lipid bilayers exhibiting a fluidizing effect on the lipid bilayers. Fluidization was confirmed by following the wagging progressions of the CH2 groups in the 1,180- to 1,350-cm-1 region of the SC IR spectrum (data not shown). Specifically, we followed the change in the number of peak assignments in this region before and after the treatment with CPE. Reduction in the number of peak assignments is an indication of fluidization (24). All CPEs that partitioned into the SC showed this behavior and were labeled as “fluidizers.” The change in the 2,850-cm-1 peak area for fluidizers and extractors correlated with ER values (Fig. 2A)(r2 = 0.67 for fluidizers; r2 = 0.53 for extractors). A 1:1 solution of PBS:EtOH, the base solvent in all formulations, acts as a very weak extractor with a slightly negative value of Δ(νsymCH2) and results in an ER of ≈3. Normalizing ER and Δ(νsymCH2) of all CPEs with respect to PBS:EtOH would shift the origin in Fig. 2 A to the point corresponding to PBS:EtOH, thereby negating the solvent effects on ER and bilayer organization.
Safety and IP. Irritation response of CPEs correlated with the denaturation of the SC proteins, investigated by characterizing the amide I band (1,700-1,600 cm-1) in the IR absorption spectrum of the SC. The amide I band corresponds to the most intense band in protein IR spectra and is sensitive to the protein conformation. This band can be deconvoluted to obtain contributions from the four broad secondary protein structures: β-sheets (1,640-1,620 cm-1), random coils (1650-1640 cm-1), α-helices (1,660-1,650 cm-1), and antiparallel β-sheets and β-turns (1,695-1,660 cm-1). In the IR spectrum of untreated SC, the absorbance in the 1,650- to 1,660-cm-1 region is at a maximum indicating the abundance of α-helical conformations in the SC proteins (Fig. 2B). Formulation constituents that gain access to the interior of the corneocytes may prompt unfolding of the SC proteins, thereby changing their conformation to other less rigid secondary structures. Such conformational changes are characteristic of protein unfolding or denaturation (14). The change in the integrated absorbance of the deconvoluted spectrum in the region of 1,660-1,650 cm-1, Δ(νC═O), was used as a quantitative parameter describing the extent of changes in the SC protein structure. Δ(νC═O) correlated well with the IP values assessed using EpiDerm (Fig. 2C; r2 = 0.7). A decrease in the C═O peak intensity (1,650 cm-1) was proportionally accompanied by an increase in the intensity of N-D bending vibrations peak (1,440-1,450 cm-1) in the amide II band of the IR spectrum (data not shown). This peak, arising from hydrogen-deuterium (H-D) exchange between proteins and formulations, indicates that irritating chemicals breach the cornified envelope and expose the amide bonds in the SC proteins to EtOD:D2O. Extensive H-D exchange between proteins and solvents has been typically associated with unfolding (25).
Origin of Potency and Safety in Molecular Structure. To understand the molecular attributes of CPEs that dictate their potency and safety, a total of 35 different parameters were calculated for each molecule (see Supporting Text). A correlation matrix was run on all variables to eliminate redundant variables (data not shown). The fluidization potential of CPEs (as judged by an increase in the integrated absorbance of νsymCH2 peak) correlated with their hydrophobicity quantified in terms of log P, the octanol-water partition coefficient (Fig. 3A, red circles, r2 = 0.86). On the other hand, the extraction potential of CPEs (as judged by a decrease in νsymCH2 peak) correlated with the ratio of the hydrogen bonding component of solubility parameter (δh) to the square root of cohesive energy density. Cohesive energy density is the sum of squares of polar (δp), dispersive (δd), and hydrogen bonding (δh) components of solubility parameter (EC = δ2p + δ2h + δ2d) (Fig. 3A, green circles, r2 = 0.54). Finally, the IP of CPEs [as judged by Δ(νC═O)] correlated with the ratio of δh to δp for extractors and fluidizers (Fig. 3B, r2 = 0.78).
Fig. 3.

Molecular descriptors of ER and IP. (A) Plot of Δ(νsymCH2) against molecular descriptors for fluidizers (red circles) and extractors (green circles). Change in the absorbance of the methylene stretching peak correlates with logarithm of octanol-water partition coefficient (log P) for fluidizers and the ratio of hydrogen bonding (δh) to square root of cohesive energy density (EC) for extractors. Error bars correspond to SD (n = 3). (B) IP correlates with the ratio of hydrogen bonding interactions (δh) to polar interactions (δp).
Having defined the molecular descriptors for ER and IP, one can now define a descriptor for the overall quality of a CPE (ER/IP) as follows.
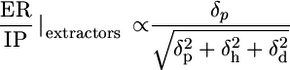 |
[1] |
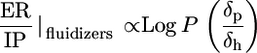 |
[2] |
Experimental measurements of ER/IP correlated well with these molecular descriptors for all fluidizers (r2 = 0.84) and extractors (r2 = 0.73) (Fig. 4 A and B).
Fig. 4.

Plot of experimental ER/IP vs. ER/IP predicted from molecular descriptors for extractors (A) and fluidizers (B). For extractors and fluidizers, ER/IP is predicted from Eqs. 1 and 2, respectively.
Eqs. 1 and 2 confirm the existence of a fundamental limitation in designing potent and safe enhancers. In case of extractors, ER/IP is bound by a theoretical limit of one. However, in practice, a limit much smaller than one is likely because δh is typically comparable to δp in magnitude. Assuming a simplified scenario where δd and δh are comparable to δp, we can put a theoretical limit of 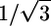 on ER/IP. Further assuming that δh is negligible yields another theoretical limit of
on ER/IP. Further assuming that δh is negligible yields another theoretical limit of 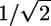 . In the case of fluidizers, the existence of an inverse relation between log P and δp also puts an upper bound on ER/IP. Furthermore, δp typically varies proportionally with δh, providing additional reasons for the existence of an upper limit. However, no theoretical upper limit is evident in the case of fluidizers.
. In the case of fluidizers, the existence of an inverse relation between log P and δp also puts an upper bound on ER/IP. Furthermore, δp typically varies proportionally with δh, providing additional reasons for the existence of an upper limit. However, no theoretical upper limit is evident in the case of fluidizers.
Design and Assessment of New CPEs. It is evident from Eqs. 1 and 2 that there are fundamental limitations in CPE optimization. However, parametrical design rules such as those previously mentioned open up the possibility of identifying a plethora of new molecules, especially fluidizers that perform better than the current CPEs (as judged by ER/IP). To demonstrate this principle, 10 wild-type CPEs (one representing the best molecule in each category; Fig. 1C) were selected and mutated in silico by substituting one of their chemical functional groups as discussed in Materials and Methods to generate a library of mutants. These mutants, along with their wild-type CPEs, were screened by using Eqs. 1 and 2 to identify safe and potent extractors as well as fluidizers (Fig. 5A: filled green circles, existing extractors; open green circles, mutant extractors; filled red circles, existing fluidizers; open red circles, mutant fluidizers). As predicted, ER/IP of mutant extractors could not be pushed significantly over 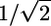 . However, several fluidizers that outperform their wild-type analogs were found (Fig. 5B). In the original pool of 102 enhancers, 9 fluidizers exhibited ER/IP better than 3.8 [the value for oleic acid (OA), a commonly used fluidizer in transdermal literature]. This number was improved by a factor of 12 based on newly designed mutant fluidizers (110 of 325 mutant fluidizers showed ER/IP >3.8). Chemical structures of some of the lead mutants are shown in Fig. 6. The predicted ER/IP value of the best mutant from among those in Fig. 6 that are commercially available (SM, stearyl methacrylate) was about three times higher than OA (Fig. 5C, gray bars). SM also enhanced the delivery of a model macromolecule, inulin (Fig. 5C, black bars), across the skin in vitro.
. However, several fluidizers that outperform their wild-type analogs were found (Fig. 5B). In the original pool of 102 enhancers, 9 fluidizers exhibited ER/IP better than 3.8 [the value for oleic acid (OA), a commonly used fluidizer in transdermal literature]. This number was improved by a factor of 12 based on newly designed mutant fluidizers (110 of 325 mutant fluidizers showed ER/IP >3.8). Chemical structures of some of the lead mutants are shown in Fig. 6. The predicted ER/IP value of the best mutant from among those in Fig. 6 that are commercially available (SM, stearyl methacrylate) was about three times higher than OA (Fig. 5C, gray bars). SM also enhanced the delivery of a model macromolecule, inulin (Fig. 5C, black bars), across the skin in vitro.
Fig. 5.

Design of CPEs. (A)ER/IP predictions for mutant CPEs calculated using Eqs. 1 and 2. The x axis lists the number of mutant and original CPEs studied and y axis shows the corresponding ER/IP values for mutant extractors (green open circles) and fluidizers (red open circles). Filled green (extractors) and red (fluidizers) circles show ER/IP values for CPEs in the original pool. (B) Pool size of mutant and original fluidizers as a function of ER/IP. Mutant fluidizers clearly outperform the original CPEs. (C) Predicted ER/IP for a mutant CPE, SM, and a commonly used CPE in transdermal literature, OA (gray bars), and comparison of inulin permeability enhancement for SM and OA (black bars).
Fig. 6.

Molecular structures of some of the best mutant fluidizers. I, stearyl methacrylate; II, 1-(2-hydroxy-phenoxy), 1-(4-hydroxy-phenoxy) tetradecane; III, 1-(8-octyl-8-(1,1-dimethylhexyl)heptadecane)-1,3,5-triazine-2,4,6-trione; IV, 1-benzyl-4-(2-((1,1′-biphenyl)-4-yloxy)ethyl)piperazine; V, 1,4-bis-((2-chlorophenyl)-phenyl-methyl)-piperazine; VI, 2,3,6,7-tetrakis(chloromethyl)-1,4,5,8-tetramethylbiphenylene.
Discussion
A large body of transdermal literature has been devoted to studying and predicting the effect of chemical enhancers on skin permeation (7, 26, 27). It is common in skin permeation literature to assess the effect of chemical enhancers on skin in conjunction with a solute (drug) of interest. These methods have inherent limitations because they cannot decouple the contribution of drug physicochemistry to enhancer activity. In contrast, the use of conductivity ER, in the absence of a drug provides an independent evaluation of enhancer potencies.
The studies presented here uncover the fundamental mechanisms that define the potency and irritation of chemical penetration enhancers. It is commonly perceived in the literature that potency and irritation are tied together. This is indeed true for CPEs falling in the chemical classes shown in Fig. 1 A. (AZ, SS, ZI, and NI). However, Fig. 1B demonstrates that there exist classes of enhancers for which potency and irritation are not particularly well related (AI, FE, FM, FA, CI, and OT), although the trends are clearer within each category. For molecules in these classes, the very act of barrier disruption does not lead to irritation, and these molecules can potentially be exploited for the design of potent and safe CPEs.
To understand the ER-IP relations, we explored the morphological changes in the skin microenvironment in the presence of CPEs using FTIR spectroscopy. Data in Fig. 3A reveal that two independent mechanisms are responsible for CPE action (fluidization and extraction) and that these stem from completely independent molecular forces. In the case of fluidizers, membrane partitioning is of prime importance, whereas for extractors, intermolecular ionic forces (especially hydrogen bonding) are important. Hydrogen bonding interactions (δh) in CPEs originate from highly electronegative atoms such as S, N, and O. These interactions compete with water-mediated intermolecular hydrogen bonding between the lipid molecules that is responsible for the structural stability of bilayer lamellae. Extraction capacity of CPEs varies inversely with the square root of cohesive energy density (EC), which is determined by a combination of dispersive interactions (δd) arising from temporary induced dipoles, polar interactions arising from dipole-dipole interactions (δp), and hydrogen bonding interactions (δh)(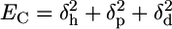 ). At large values of EC, the CPE molecules are unlikely to participate in the solvation of lipids.
). At large values of EC, the CPE molecules are unlikely to participate in the solvation of lipids.
The irritation behavior of CPEs is related to the ratio of hydrogen bonding to polar interactions. Hydrogen bonds are of significant importance in holding the proteins in their native structures. Competitive hydrogen bonding from CPEs can potentially change the native hydrogen bonding in proteins leading to unfolding. Consequently, IP scales directly with hydrogen bonding ability. Polar interactions scale inversely with the hydrophobicity of a molecule. Hydrophobic molecules may promote partitioning of CPEs in the hydrophobic protein core resulting in the loss of structural conformations and, hence, the inverse dependence of IP on polarity. Much of the prior work on skin irritation is based on elucidating cellular responses and signaling pathways involved in skin irritation due to chemical assault. Several different mechanisms specific to certain classes of chemicals have been speculated (2, 28). The studies reported here show the existence of a general correlation between the molecular descriptors and the IP of CPEs. In addition, these studies also show that IR spectroscopic analysis of protein conformation in the SC can be used to predict the irritation response. Such methods provide a superior alternative to cell culture or animal-based methods that are resource-intensive, expensive, and exhibit higher variability (28-30).
Molecular interactions described in Fig. 3 can be used to explain the observed ER-IP relations in Fig. 1 A and B. Molecules represented in Fig. 1 A are typically extractors and show direct correlation between ER and IP. FTIR spectroscopy and molecular modeling studies revealed that the forces responsible for lipid extraction and irritation are both proportional to hydrogen bonding, leading to a stronger tie between irritation and potency for these molecules. On the other hand, the molecular forces responsible for potency and irritation of fluidizers do not fundamentally contradict each other. The lack of correlation between ER and IP for molecules in Fig. 1B, typically fluidizers, is then understandable.
Using Eqs. 1 and 2, we demonstrate that there is a fundamental constraint in designing CPEs that are both highly potent and safe. This constraint may partly explain the limited success of CPE discovery in the past. Beyond revealing the limitations in CPE performance, Eqs. 1 and 2 can be used to rapidly screen a large pool of candidate CPEs in silico to broaden the current repertoire of CPEs. We expect that such newly identified chemical enhancer molecules will provide added flexibility in formulation design in the future. To demonstrate the ability of this approach, we selected the 10 best CPEs from the 10 different chemical classes and mutated them to generate their variants. In general, the mutant extractors exhibited ER/IP values comparable to the original extractors. However, a significant number of the mutant fluidizers were better than those in the original pool (Fig. 5B). The lead candidate (SM), among the commercially available subset of structures in Fig. 6, was further tested experimentally. SM yielded a predicted ER/IP value of ≈12, which is substantially higher than OA (3.8) (Fig. 5C). The enhancement in ER/IP originated partly from the enhancement in ER but largely from a reduction in IP. SM also significantly enhanced skin permeability to a macromolecule, inulin. More importantly, the data in Fig. 5 open up the possibility of generating thousands of additional mutants that can be designed and tested by using the principles described here. With further studies focused on skin safety, systemic toxicity, and potency, these molecules may assist in developing the next generation of transdermal, dermatological, and cosmetic products.
Supplementary Material
Acknowledgments
We thank MatTek for the gift of Epiderm samples. This work was supported by Materials Research Science and Engineering Center (National Science Foundation) Grant DMR00-80034.
Author contributions: P.K., A.J., and S.M. designed research; P.K., A.J., K.E., and V.K. performed research; P.K. and A.J. analyzed data; and P.K. and S.M. wrote the paper.
Abbreviations: AI, anionic surfactants; AZ, azone-like compounds; CI, cationic surfactants; CPE, chemical penetration enhancer; ER, enhancement ratio; FA, fatty acids; FE, fatty esters; FM, fatty amines; IP, irritation potential; FTIR, Fourier transform infrared; NI, nonionic surfactants; OA, oleic acid; PBS, phosphate buffered saline; SC, stratum corneum; ZI, zwitterionic surfactants.
References
- 1.Bos, J. D. & Meinardi, M. M. H. M. (2000) Exp. Dermatol. 9, 165-169. [DOI] [PubMed] [Google Scholar]
- 2.Williams, I. & Kupper, T. (1996) Life Sci. 58, 1485-1507. [DOI] [PubMed] [Google Scholar]
- 3.Kupper, T. (1990) J. Clin. Invest. 86, 1783-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Draize, J., Woodward, G. & Calvery, H. (1944) J. Pharmacol. Exp. Ther. 82, 377-390. [Google Scholar]
- 5.Bauerova, K., Matusova, D. & Kassai, Z. (2001) Eur. J. Drug Metab. Pharmacokinet. 26, 85-94. [DOI] [PubMed] [Google Scholar]
- 6.Asbill, C. S., El-Kattan, A. F. & Michniak, B. (2000) Crit. Rev. Ther. Drug Carrier Syst. 17, 621-658. [PubMed] [Google Scholar]
- 7.Kanikkannan, N., Kandimalla, K., Lamba, S. S. & Singh, M. (2000) Curr. Med. Chem. 7, 593-608. [DOI] [PubMed] [Google Scholar]
- 8.Karande, P., Jain, A. & Mitragotri, S. (2004) Nat. Biotechnol. 22, 192-197. [DOI] [PubMed] [Google Scholar]
- 9.Kligman, A. & Christophel, E. (1963) Arch. Dermatol. 88, 702-705. [DOI] [PubMed] [Google Scholar]
- 10.Simon, G. & Maibach, H. (2000) Skin Pharmacol. Physiol. 13, 229-234. [DOI] [PubMed] [Google Scholar]
- 11.Larsen, C. G., Larsen, F. G. & Thestrup-Pederson, K. (1988) Acta Dermatol. Venerol. 68, 474-479. [PubMed] [Google Scholar]
- 12.Morejohn, L. C. & Pratley, J. N. (1979) Cell Tissue Res. 198, 349-362. [DOI] [PubMed] [Google Scholar]
- 13.Sznitowska, M., Janicki, S., Williams, A., Lau, S. & Stolyhwo, A. (2003) J. Pharmacol. Sci. 92, 173-179. [DOI] [PubMed] [Google Scholar]
- 14.Byler, M. & Susi, H. (1986) Biopolymers 25, 469-487. [DOI] [PubMed] [Google Scholar]
- 15.Krimm, S. & Bandekar, J. (1986) Adv. Protein Chem. 38, 181-364. [DOI] [PubMed] [Google Scholar]
- 16.Viswanadhan, V., Ghose, A., Revankar, G. & Robins, R. (1988) J. Chem. Inf. Comput. Sci. 29, 163-172. [Google Scholar]
- 17.Hansch, C. & Leo, A. (1979) Substituent Constants for Correlation Analysis in Chemistry and Biology (Wiley, New York). [DOI] [PubMed]
- 18.Bodor, N. & Buchwald, P. (1997) J. Phys. Chem. B 101, 3404-3412. [Google Scholar]
- 19.Moriguchi, I., Hirono, S., Liu, Q., Nakagome, I. & Matshusita, Y. (1992) Chem. Pharm. Bull. 40, 127-130. [Google Scholar]
- 20.Moriguchi, I., Hirono, S., Nakagome, I. & Hirano, H. (1994) Chem. Pharm. Bull. 42, 976-978. [Google Scholar]
- 21.van Krevelen, D. (1990) Properties of Polymers: Their Correlation with Chemical Structure, Their Numerical Estimation and Prediction from Additive Group Contributions (Elsevier, Amsterdam).
- 22.Hoy, K. (1970) J. Paint Technol. 42, 76-78. [Google Scholar]
- 23.Karande, P. & Mitragotri, S. (2002) Pharm. Res. 19, 655-660. [DOI] [PubMed] [Google Scholar]
- 24.Senak, L., Moore, D. & Mendelsohn, R. (1992) J. Phys. Chem. 96, 2749-2754. [Google Scholar]
- 25.Kluge, T., Olejnik, J., Smilowitz, L. & Rothschild, K. (1998) Biochemistry 37, 10279-10285. [DOI] [PubMed] [Google Scholar]
- 26.Ghafourian, T., Zandasrar, P., Hamishekar, H. & Nokhodchi, A. (2004) J. Controlled Rel. 99, 113-125. [DOI] [PubMed] [Google Scholar]
- 27.Williams, A. C. & Barry, B. W. (2004) Adv. Drug Deliv. Rev. 56, 603-618. [DOI] [PubMed] [Google Scholar]
- 28.Welss, T., Basketter, D. & Schroder, K. (2004) Toxicol. In Vitro 18, 231-243. [DOI] [PubMed] [Google Scholar]
- 29.Holden, C. (1988) Science 242, 185-186. [DOI] [PubMed] [Google Scholar]
- 30.Eun, H. & Suh, D. (2000) J. Dermatol. Sci. 24, 77-91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.