

Abstract
Fragile X syndrome (FXS) is the most common cause of heritable intellectual disability and autism and affects ~1 in 4000 males and 1 in 8000 females. The discovery of effective treatments for FXS has been hampered by the lack of effective animal models and phenotypic readouts for drug screening. FXS ensues from the epigenetic silencing or loss-of-function mutation of the fragile X mental retardation 1 (FMR1) gene, which encodes an RNA binding protein that associates with and represses the translation of target mRNAs. We previously found that the activation of LIM kinase 1 (LIMK1) downstream of augmented synthesis of bone morphogenetic protein (BMP) type 2 receptor (BMPR2) promotes aberrant synaptic development in mouse and Drosophila models of FXS and that these molecular and cellular markers were correlated in patients with FXS. We report that larval locomotion is augmented in a Drosophila FXS model. Genetic or pharmacological intervention on the BMPR2-LIMK pathway ameliorated the synaptic abnormality and locomotion phenotypes of FXS larvae, as well as hyperactivity in an FXS mouse model. Our study demonstrates that (i) the BMPR2-LIMK pathway is a promising therapeutic target for FXS and (ii) the locomotion phenotype of FXS larvae is a quantitative functional readout for the neuromorphological phenotype associated with FXS and is amenable to the screening novel FXS therapeutics.
INTRODUCTION
Fragile X syndrome (FXS) is linked to other neurologic and psychiatric disorders and is the most prevalent monogenetic cause of autism (1, 2). It is caused by an expansion of a triplet (CGG) more than 200 repeats in the 5′ untranslated region of fragile X mental retardation 1 (FMR1), which silences its expression (1, 3). The fragile X mental retardation protein (FMRP) binds various mRNA targets and itself inhibits mRNA stability and function, including translation (3). FXS patients exhibit abnormal dendritic spine density and morphology in the central nervous system (CNS), which leads to cognitive impairment, anxiety, hyperactivity, and autistic behavior (1–3). It has been proposed that, during brain development, FMRP plays a role in dendritic spine development by repressing the translation of molecules critical for cytoskeleton remodeling and receptor signaling.
The search for an animal model of FXS has led to the generation and characterization of Drosophila and rodent FMR1 mutants. It has been reported that Drosophila third-instar larvae with a mutation of the Drosophila ortholog of FMR1 (dFMR1) have an increased number of branches and synaptic boutons at neuromuscular junctions (NMJs) (4). We found that the branching and bouton abnormalities were reduced by decreasing the gene dosage of the Drosophila ortholog of bone morphogenetic protein receptor type 2 (BMPR2) gene, wishful thinking (Wit), suggesting that excess production of BMPR2 and of its downstream signaling plays a role in aberrant synaptic growth (5).
Homozygous deletion of FMR1 (FMR1-KO) in rodents produces a dendritic spine abnormality that resembles the phenotype seen in FXS patients (6, 7). Furthermore, FMR1-KO mice and rats exhibit cognitive and behavioral traits, some of which are consistent with those of the FXS patients, such as hyperactivity as well as learning and memory defects (8). Hyperactivity is one of the hallmark characteristics of human FXS. FMR1-KO mice also show abnormal locomotor activity and behavioral hyperactivity (8). Therefore, locomotor hyperactivity has been used as a measurement to assess drug effect on FXS in preclinical studies. An open-field test (OFT) is one of the most commonly used mouse behavioral tests to observe locomotor activity and hyperactivity as well as anxiety. Unfortunately, the behavioral and cognitive traits of the rodent models of FXS are highly variable depending on age, gender, and strain of the animals (8). These are major limitations for the development of therapies for FXS. In addition, the behavioral tests using rodents require cost and time. Thus, there continues to be a pressing need for rapid, quantitative, sensitive, and time- and cost-effective animal assays to screen drugs for treatment of FXS, possibly using an invertebrate FXS model.
Our work that implicates BMPR2 in FXS suggests that BMPR2 may be a novel target for controlling FMRP-dependent translational regulation in FXS development (5). Both the FMR1-KO mouse and human FXS patients exhibit an increased abundance of BMPR2 protein in CNS neurons (5). In addition, increased BMPR2 results in activation of LIM kinase 1 (LIMK1), which interacts with the cytoplasmic domain of BMPR2 and promotes actin remodeling and dendritic spine abnormalities (5). Reduction of the BMPR2 gene dosage as well as pharmacological inhibition of LIMK1 ameliorate the aberrant spine development in the FMR1-KO mouse (5), but there have been no reported characterizations of LIMK1 inhibition on cognitive and behavioral traits of FXS model animals.
Behavioral manifestations in the Drosophila FXS model have been reported (9, 10) and include abnormal crawling and locomotion of third-instar larvae. Here, we developed quantitative behavioral assays that showed that reduction of Wit gene dosage in dFMR1 mutant larvae reverts the locomotion phenotype and that oral administration of LIMK antagonists and a protein synthesis inhibitor restores normal crawling velocity and reduces NMJ bouton numbers. We also confirmed that administration of a LIMK antagonist in the mouse FXS model rescues the rodent behavioral abnormalities. Thus, this study demonstrates that (i) the locomotion phenotype in dFMR1 mutant larvae serves as a readout of NMJ bouton phenotype; (ii) the larval crawling assay system that we developed can be used for the genetic or chemical screening of therapeutic molecules for FXS as well as other synapse formation abnormalities; and (iii) targeting the LIMK1 pathway, which is conserved from Drosophila to human, is a potential therapeutic strategy for FXS.
RESULTS
Correlation between larval locomotion activity and synaptic bouton number
A loss-of-expression mutation of dFMR1 (dFMR1Δ113) causes a significant increase in synaptic bouton formation in the larval NMJ (4, 5). In contrast, loss-of-expression mutants of the Drosophila ortholog of BMPR2 Wit exhibit a reduced number of synaptic boutons (11, 12). When one allele of Wit is mutated in dFMR1 mutants (dFMR1Δ113,WitA12/+), the bouton number is decreased by ~20% (5), suggesting that the overgrowth of boutons in dFMR1Δ113/+ mutants is mediated in part by augmented Wit and its downstream signaling pathway. This finding is consistent with results obtained in the mouse FXS model and in human FXS patients. We first hypothesized that an overgrowth of synaptic boutons at the NMJ alters the crawling activities of larvae. We tested the crawling behavior of 10 larvae on an agarose plate by video recording for 1 min (Fig. 1A) and visually measuring their velocity (Fig. 1B). The velocity of dFMR1Δ113/+ mutants was 62% faster than that of wild-type larvae (Fig. 1B, right), suggesting that an increased number of boutons correlates with the augmented crawling velocity. Similar to dFMR1Δ113/+, other dFMR1 heterozygous mutants, such as dFMR1Δ50/+ (13, 14) and dFMR13/+ (14), also exhibited increased NMJ bouton number and augmented crawling velocity compared to wild-type larvae (Fig. 1C), suggesting that these phenotypes are the result of dFMR1 mutation. Conversely, the velocity of WitA12/+ mutants was 40% slower than that in wild-type larvae (Fig. 1B, right), indicating that the decreased number of boutons correlates with a decreased crawling velocity. The velocity of dFMR1Δ113/+ larvae with a Wit mutation (dFMR1Δ113,WitA12/+ double mutant) was similar to that of wild-type larvae (Fig. 1B, right), paralleling the reduction in bouton number (5). These results suggest that morphological changes in the neuromuscular synapses in dFMR1Δ113/+ larvae can alter the speed at which they crawl and indicate that the increased velocity in dFMR1Δ113/+ mutants is rescued by a concurrent mutation in Wit. The results also indicate that the aberrant synaptic morphology and the crawling activity of dFMR1Δ113/+ and other dFMR1 mutants are, at least in part, caused by increased Wit abundance and downstream Wit signaling (such as through the LIMK1 pathway) in presynaptic neurons, in agreement with our observations in the CNS neurons of the mouse FXS model (5).
Fig. 1. Increased larval locomotion activity of the Drosophila FXS model.
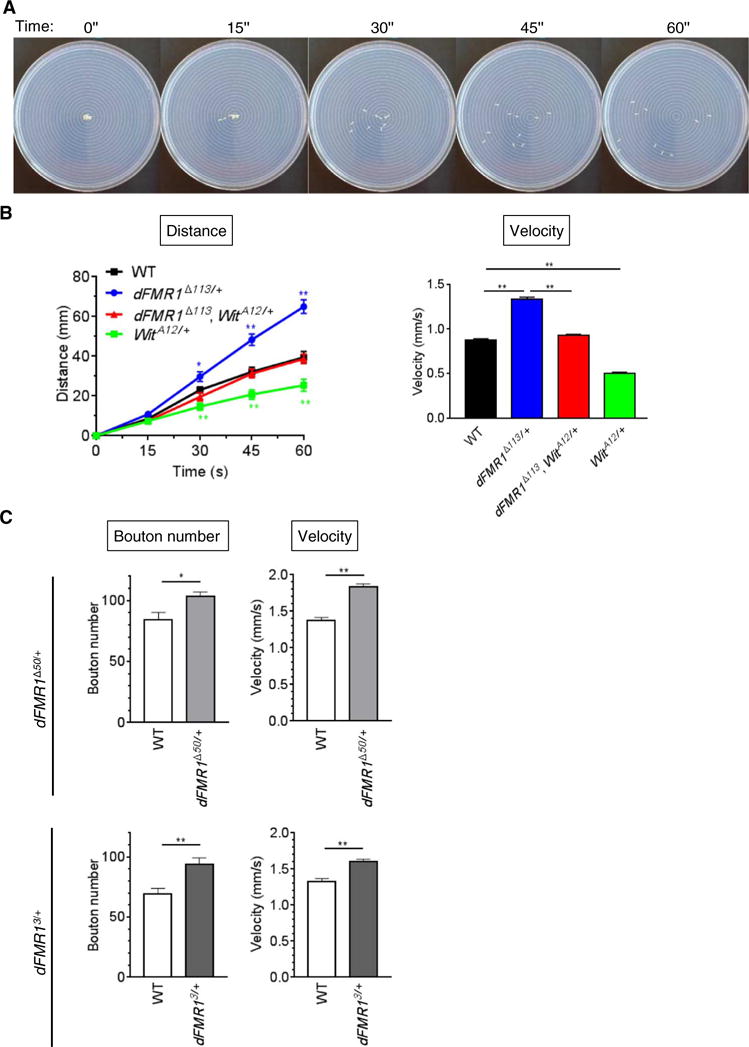
(A) Representative images of the larval crawling assay performed on an agarose plate. (B) The larval crawling assay of wild-type (WT), dFMR1Δ113/+, WitA12/+, and dFMR1Δ113, WitA12/+ larvae (n = 10 each) was performed by video-recording the crawling behavior for 1 min, followed by measuring the distance that the larvae traveled every 15 s (bottom left). The velocity was calculated on the basis of the plot shown in (A) (bottom right). Data are means ± SEM. **P < 0.01, by analysis of variance (ANOVA) with post hoc Tukey’s test. (C) The NMJ bouton number at muscle 6/7 in segment A3 and the larval crawling velocity of WT, dFMR1Δ50/+, and dFMR13/+ third-instar larvae were assessed. Data are means ± SEM of 19 to 20 independent images. The crawling velocity of 10 larvae was analyzed simultaneously and repeated four times. Data are means ± SEM. *P < 0.05 and **P < 0.01, by t test.
Reversing bouton number and locomotion phenotype in dFMR1D113/+ larvae by oral administration of a LIMK1 inhibitor
Our previous study correlated augmented BMPR2 protein amount in FXS animal models and patients with activation of LIMK1, phosphorylation of cofilin, and actin remodeling (5). To investigate whether the LIMK1-cofilin axis is also activated in dFMR1Δ113/+ larvae, we performed immunoblot analysis of phosphorylated Twinstar (P-Tsr), the Drosophila ortholog of cofilin, relative to total Tsr (t-Tsr). The relative basal amount of P-Tsr in dFMR1Δ113/+ mutants was 26% greater than that in wild-type larvae (Fig. 2A, mock), indicative of augmented Drosophila LIMK (dLIMK) activity in dFMR1Δ113/+ mutants. This finding provided us the opportunity to test whether oral delivery of the LIMK inhibitor LIMKi-3 (LIMK-i) to dFMR1Δ113/+ larvae could inhibit dLIMK activity. Increasing concentrations of LIMK-i (5, 10, or 50 μM) were administered orally to dFMR1Δ113/+ or wild-type third-instar larvae, followed by assessment of the relative P-Tsr amount (Fig. 2A). LIMK-i dose-dependently decreased the relative amount of P-Tsr ultimately to a value similar to mock-treated wild-type larvae (Fig. 2A), indicating that augmented dLIMK activity in dFMR1Δ113/+ was suppressed by oral delivery of LIMK-i. Unlike dFMR1Δ113/+ larvae, the relative amount of P-Tsr in wild-type larvae that received LIMK-i was not affected upon visual assessment of the blots (Fig. 2A), indicating little to no effect on the physiological dLIMK activity.
Fig. 2. Inhibition of LIMK1 restores the number of boutons as well as the crawling behavior in dFMR1D113/+ larvae.
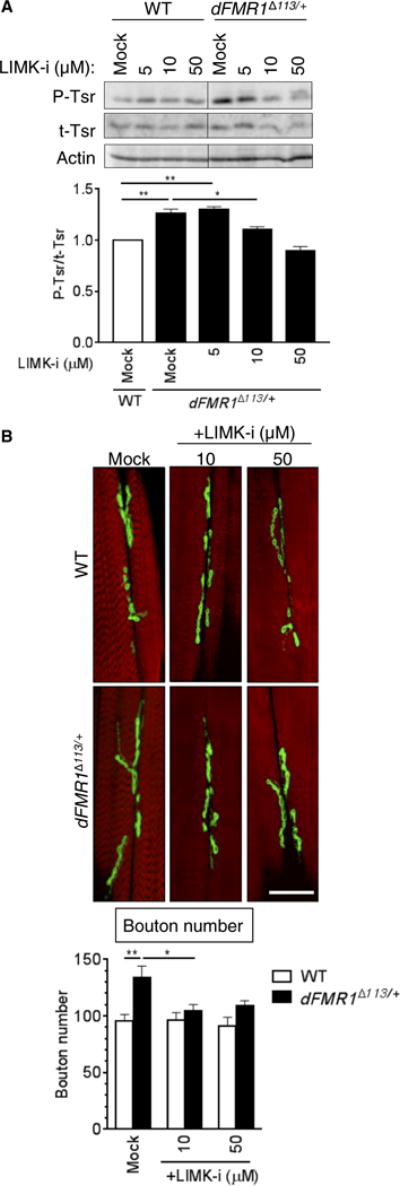
(A) Third-instar WT or dFMR1Δ113/+ larvae were fed with food containing different doses of LIMK-i as indicated, and total larval lysates were subjected to immunoblot analysis of P-Tsr, t-Tsr, and actin (loading control). The immunoblot result (top) is shown as the relative P-Tsr/t-Tsr ratio, when the P-Tsr/t-Tsr ratio of mock-treated WT larvae is 1 (bottom). Data are means ± SEM of five independent immunoblots. *P < 0.05 and **P < 0.01, by ANOVA with post hoc Tukey’s test. (B) WT or dFMR1Δ113/+ mutant third-instar larvae were treated with increasing doses of LIMK-i, as indicated, and boutons and muscle were stained with Dlg1 antibody (green) and Alexa Fluor 568–conjugated phalloidin (red), respectively. Representative confocal microscopy images of muscle 6/7 in segment A3 are presented (top). Scale bar, 25 μm. Data are means ± SEM of 9 to 16 independent images (bottom). *P < 0.05 and **P < 0.01, by ANOVA with post hoc Tukey’s test.
To examine whether the inhibition of LIMK1 leads to changes in the number of NMJ boutons in dFMR1Δ113/+ larvae, we fed dFMR1Δ113/+ or wild-type larvae increasing concentrations of LIMK-i and analyzed the number of mature synaptic boutons by immunofluorescence staining of the postsynaptic marker discs large 1 (Dlg1) (Fig. 2B, green) and phalloidin (Fig. 2B, red) on M6/M7 muscles in the A3 segment of third-instar larvae. Consistent with previously reported data (4, 5), mock-treated dFMR1Δ113/+ mutants had ~40% more boutons than did mock-treated wild-type larvae (Fig. 2B, graph), and even the lowest concentration(10μM) of LIMK-i used decreased the average number of boutons to a similar number observed in the mock-treated wild-type larvae (96) (Fig. 2B, graph). This result indicates that administration of LIMK-i can reverse the abnormal NMJ bouton phenotype in dFMR1Δ113/+ larvae similarly to the result of the gene dosage reduction of Wit (5).
Development of an algorithm to measure the crawling distance of larvae
The change in the NMJ bouton number mirrored by the locomotion behavior in dFMR1Δ113/+ larvae prompted us to use the crawling assay for a screening of drugs to treat conditions associated with abnormal synapse formation, such as FXS and autism (15). To develop a high-throughput drug screen with locomotion velocity as functional readout of NMJ bouton phenotype, we devised a semiautomated analysis of the larval crawling path and distance traveled based on an algorithm we named “LarvaTrack” (see Materials and Methods and text S1). Larva-Track can trace the path of as many as 10 larvae simultaneously (Fig. 3A) while measuring their crawling distance (Fig. 3B), which allows auto mated calculation of the average velocity (Fig. 3C). Using LarvaTrack to analyze the locomotion videos, we found that the mock-treated dFMR1Δ113/+ mutants traveled ~30% farther than mock-treated wild-type larvae in 60 s (Fig. 3B). The distance traveled by dFMR1Δ113/+ mutants was reduced by treatment with LIMK-i to a value similar to or lower than the distance traveled by mock-treated wild-type larvae (Fig. 3B). The velocity of mock-treated dFMR1Δ113/+ mutants (1.5 ± 0.03 mm/s) was ~20% faster than that of mock-treated wild-type larvae (Fig. 3C). LIMK-i treatment reduced the velocity of dFMR1Δ113/+ larvae, which, to a measure, is similar to that of mock-treated wild-type larvae (Fig. 3C). LIMK-i treatment also decreased the velocity of wild-type larvae to 0.89 ± 0.06 mm/s (Fig. 3C). Furthermore, unlike mock-treated larvae, which crawled from the center to the periphery in a linear fashion, LIMK-i treatment (100 μM) induced frequent turning and circling behavior in both wild-type and dFMR1Δ113/+ larvae (Fig. 3D), which suggests that high doses of LIMK-i cause neurotoxicity. To further confirm the effects of LIMK-i in dFMR1Δ113/+ larvae, we administered LIMK-i to heterozygous (dFMR13/+) and transheterozygous (dFMR1Δ113/3) lines of dFMR1 mutants, as well as to a transgenic line in which small inhibitory RNA (RNAi) against dFMR1 is expressed in neurons (dFMR1RNAi), followed by assessment of the NMJ bouton phenotype and the crawling velocity. Consistent with dFMR1Δ113/+ larvae, dFMR13/+, dFMR1Δ113/3, and dFMR1RNAi larvae all had greater numbers of boutons and faster velocity compared to wild-type larvae, and inhibition of LIMK activity ameliorated both parameters to amounts similar to wild type (Fig. 3E). Thus, the semiautomated assessment of crawling velocity by LarvaTrack is a sensitive and quantitative assay for monitoring changes in synaptic bouton phenotype in the Drosophila FXS model, indicating that this method might be usable as a screening strategy to discover and assess the toxicity of new FXS therapies.
Fig. 3. Development of an algorithm that measures the crawling distance of larvae.
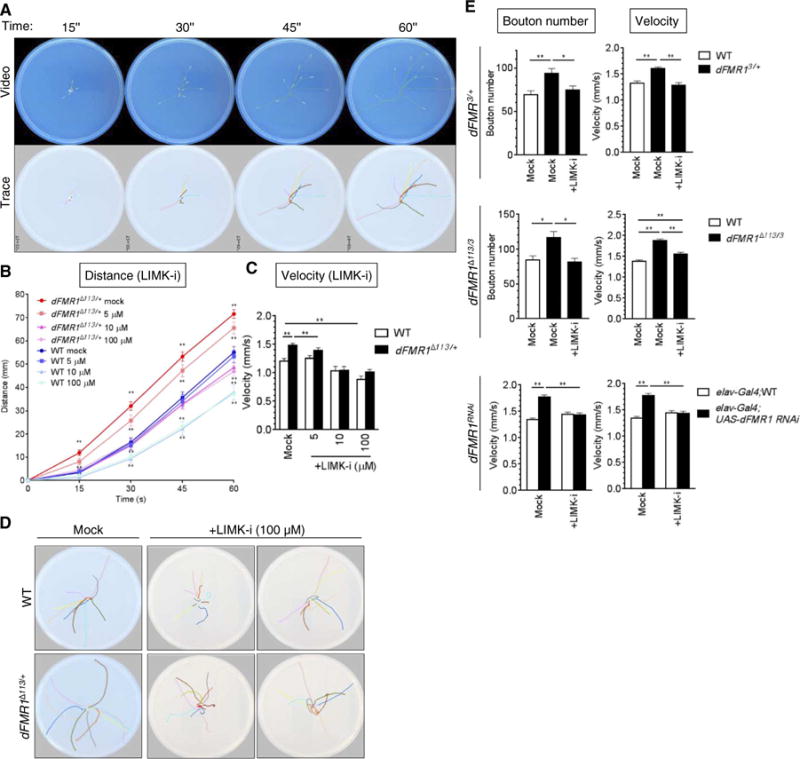
(A) Representative video images and path analysis data of WT larvae crawling assay. The snapshots of video imaged every 15 s (top) were used to trace a travel path for each individual larva by the LarvaTrack algorithm and shown with lines of different colors (bottom). (B) dFMR1Δ113/+ or WT third-instar larvae were treated with LIMK-i (5, 10, or 100 μM) and subjected to the crawling assay. Ten larvae were analyzed simultaneously, and the assay was repeated four times. The average travel distance was quantitated by LarvaTrack. Data are means ± SEM. (C) The velocity was calculated on the basis of the plot shown in (B). Data are means ± SEM. **P < 0.01, by ANOVA with post hoc Tukey’s test. (D) Crawling paths of larvae treated with dimethyl sulfoxide (DMSO) (mock) or LIMK-i (100 μM). Each color indicates a crawling path of an individual larva (dFMR1Δ113/+) for 1 min. (E) The NMJ bouton number (left) and the velocity (right) were examined in the third-instar larvae of WT, dFMR13/+ (top) or dFMR1Δ113/3 (middle) or of elav-Gal4 or elav-Gal4;UAS-dFMR1 RNAi (dFMR1RNAi) (bottom), treated with DMSO (mock) or LIMK-i (5 μM top, middle; 10 μM bottom). Data are means ± SEM of at least 18 independent images of muscle 6/7 in segment A3 (left). The crawling from 10 larvae was analyzed simultaneously, and the assay was repeated five times. The velocity was calculated by the LarvaTrack. Data are means ± SEM. *P < 0.05 and **P < 0.01, by two-way ANOVA with post hoc Tukey’s test.
Application of dFMR1 mutant larval crawling assay to screen drugs for FXS and other disease associated with aberrant dendritic spine phenotype
To test whether the larval crawling velocity assay can be used to screen drugs that ameliorate the abnormal synaptic bouton phenotype in the FXS model, several small molecules were examined. It was previously reported that FMRP bound to its target mRNAs inhibits translation in association with polyribosomes and that derepression of FMRP target mRNAs induced by the ablation of FMRP can be reversed by puromycin, an inhibitor of protein synthesis (16). Administration of puromycin ameliorates the long-term olfactory memory in dFMR1 mutant Drosophila (17). Thus, we tested whether puromycin might reverse the locomotion phenotype in dFMR1 mutant larvae. Various concentrations of puromycin (0.1, 0.5, or 1 mM) or vehicle (H2O, “mock”) were administered to third-instar larvae, followed by the locomotion assay (Fig. 4A) and analysis of NMJ boutons (Fig. 4B). Similar to the result of LIMK-i (Fig. 3B), the velocity of mock-treated dFMR1Δ113/+ mutants was ~20% faster than that of mock-treated wild-type larvae (Fig. 4A). The two lower doses (0.1 and 0.5 mM) of puromycin reduced the velocity of dFMR1Δ113/+ larvae to that comparable to mock-treated wild-type larvae (Fig. 4A). At the higher dose (1 mM), the velocity of both wild-type and dFMR1Δ113/+ mutants was severely affected (Fig. 4A). Along with the decrease in locomotion, puromycin caused a dose-dependent decrease in the number of NMJ boutons in dFMR1 mutants (Fig. 4B). We also tested the effect of two allosteric, highly selective antagonists of LIMK1 and LIMK2 (LIMK1/2), TH251 [LIMK2; median inhibitory concentration (IC50), 0.003 μM] and TH255 (LIMK2; IC50, 0.039 μM) (18), as well as an inactive analog of TH251 and TH255, called TH263 (Fig. 4C and fig. S1), in dFMR1Δ113/+ or wild-type larvae. Different concentrations (1, 10, or 50 μM) of these compounds were administered through the food, followed by assessment of crawling velocity by LarvaTrack and measurement of the NMJ bouton number (Fig. 4C). Similar to the results of LIMK-i (Figs. 2B and 3C), both active LIMK antagonists (TH251 and TH255) reduced the crawling velocity in dFMR1Δ113/+ in a dose-dependent manner (Fig. 4C and fig. S1). The reduction of crawling velocity was accompanied by a reduced bouton number (Fig. 4C). When the inactive compound (TH263) was administered, neither the locomotion phenotype nor the bouton number of dFMR1Δ113/+ mutants was affected (Fig. 4C), demonstrating the selectivity of the effect of LIMK inhibitors. Neither TH251 nor TH255 exhibited an effect on the crawling velocity or the bouton number in wild-type larvae (Fig. 4C). These results demonstrate that changes in larval locomotion can reflect changes in NMJ bouton number and that the dFMR1 larvae crawling assay can serve as a screening strategy for compounds that ameliorate synaptic abnormalities. They also support a therapeutic potential of LIMK1 antagonists for FXS.
Fig. 4. Administration of a translational inhibitor or a LIMK antagonist rescues the bouton and the crawling phenotype in dFMR1 mutants.
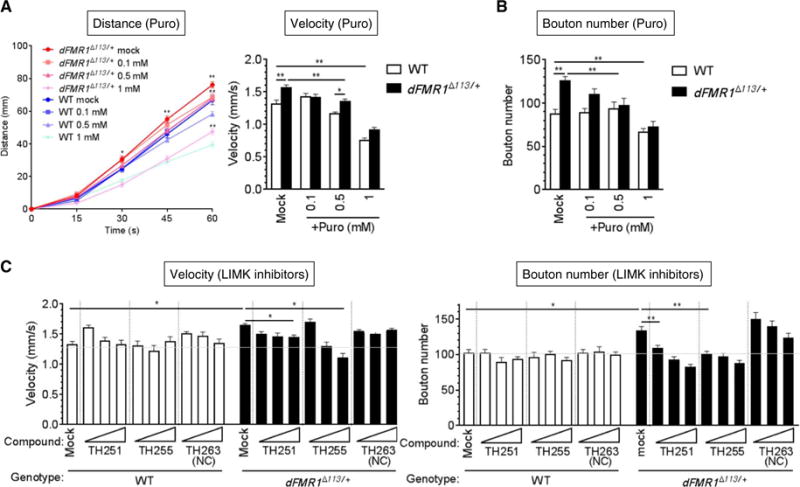
(A) dFMR1Δ113/+ or WT third-instar larvae were treated with puromycin (Puro) (0.1, 0.5, or 1 mM) and subjected to the crawling assay. Ten larvae were analyzed simultaneously. The assay was repeated four times. The velocity was calculated, and the average travel distance was quantitated by LarvaTrack. Data are means ± SEM.*P < 0.05 and **P < 0.01, by ANOVA with post hoc Tukey’s test. (B) WT or dFMR1Δ113/+ mutant third-instar larvae were treated with increasing doses of puromycin (Puro) as indicated, and bouton numbers were counted. Data are means ± SEM of at least 12 independent images of muscle 6/7 in segment A3. **P < 0.01, by ANOVA with post hoc Tukey’s test. (C) The velocity (left) and the NMJ bouton number (right) were examined in dFMR1Δ113/+ or WT third-instar larvae treated with a LIMK antagonist (TH251 or TH255) (1, 5, or 50 μM) or an inactive analog (TH263). The crawling from 10 larvae was analyzed simultaneously, and the assay was repeated four times. The velocity was calculated by LarvaTrack. Data are means ± SEM. The average number of boutons was calculated from at least 15 independent images of muscle 6/7 in segment A3.*P < 0.05 and **P < 0.01, by two-way ANOVA with post hoc Dunnett’s test. NC, negative control.
Reversing behavioral deficits in the mouse FXS model by administration of LIMK inhibitor
To support the therapeutic potential of LIMK1 antagonists for FXS in a mammalian model, we performed behavioral studies in FXS model mice. Our previously reported results show that when FMR1 homozygous null (FMR1-KO) mice were treated with LIMK-i postnatally at postnatal day 1 (P1) and P4, both spine density and the fraction of immature spines in the dentate gyrus (5) and CA3 (fig. S2) were reversed to those observed in vehicle-treated wild-type mice. To examine whether the postnatal inhibition of LIMK1 could ameliorate the FMR1-KO cognitive and behavioral deficits, we administered LIMK-i or vehicle (DMSO) to male FMR1-KO mice at P1, P4, and 3 months of age, and the mice were subjected to a “repeated open-field test” (rOFT) at 3 to 3.5 months of age. rOFT not only provides a simple assessment of spontaneous locomotion activity and exploratory behavior in a new environment but also monitors habituation to a spatial environment and memory for that environment throughout the test (19). The test mice were given four 5-min sessions in a clear acrylic OFT chamber twice per day over two consecutive days (sessions #1 and #2 on day 1 and sessions #3 and #4 on day 2). On day 16, the mice were retested in the OFT chamber for two more sessions (sessions #5 and #6) to examine their spatial memory. The total number of spontaneous movements, including horizontal (fine or ambulatory movement; total activities) and vertical (rearing) movements, was individually counted by a Flex-Field/Open-Field Photobeam Activity System, and average values of multiple mice are presented (Fig. 5A). The total activities of mock-treated FMR1-KO mice reached a plateau on day 2 at session #3 (Fig. 5A, red line), in contrast to mock-treated wild-type mice, which reached a plateau a day earlier at session #2 (Fig. 5A, black line). This result indicates that the FMR1-KO mice require a longer period to familiarize with a novel environment compared to control mice. This observation has been previously reported as an indication of memory deficit (20, 21). When FMR1-KO mice were treated with LIMK-i (Fig. 5A, blue line), their learning curve became similar to that of mock-treated wild-type mice (Fig. 5A, black), indicating that the LIMK-i treatment improves memory in FMR1-KO mice. To measure the behavior of animals placed in an OFT chamber without previous acclimation, we averaged the total activities at day 1 (sessions #1 and #2) and day 16 (sessions #5 and #6). This analysis revealed that the mock-treated FMR1-KO mice exhibited a significantly higher number of total activities compared to mock-treated wild-type mice (Fig. 5B, left), indicating that FMR1-KO mice present a hyperactive behavior in a new environment, as reported previously (22–24). Consistently, the mock-treated FMR1-KO mice exhibited 2.1-fold higher frequency of rearing behavior compared to mock-treated wild-type littermates (Fig. 5B, right). Compared to mock-treated FMR1-KO mice, LIMK-i–treated FMR1-KO mice exhibited a reduced number of total activities similar to that in mock-treated wild-type littermates (Fig. 5B, left), suggesting that LIMK-i treatment may be effective in treating hyperactivity and anxiety in FMR1-KO mice. No significant difference in total activity or rearing behavior was observed in LIMK-i–treated wild-type mice compared to mock-treated wild-type mice (Fig. 5B). Together, these data suggest that LIMK-i treatment of FMR1-KO mice improves memory and reduces hyperactivity, two common characteristics of FXS patients (8). Thus, the results obtained in the Drosophila and mouse models of FXS converge in supporting the role of LIMK-i in reversing part of the FXS phenotype. They also support an association between dysregulation of the LIMK1-cofilin pathway and the pathogenesis of FXS. Inhibition of the LIMK1-cofilin or the BMP signaling pathways is, therefore, a potentially promising therapy for the behavioral and cognitive deficits of FXS. Finally, these results validate the semiautomated assessment of the crawling velocity by LarvaTrack as a sensitive and quantitative assay for indirectly monitoring changes in synaptic bouton phenotype in the Drosophila FXS model, which could serve as a novel screening strategy for the discovery of FXS therapies.
Fig. 5. Administration of LIMK-i ameliorates neuromorphological and behavioral abnormalities in mouse model of FXS.
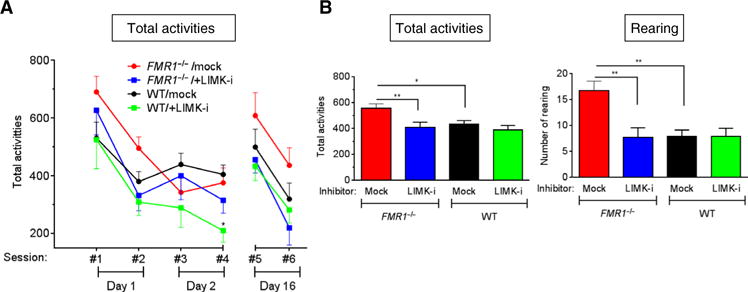
(A and B) The FMR1-KO mice or WT littermates received vehicle (DMSO) alone (mock) or LIMK-i and were subjected to the rOFT at 3 to 3.5 months of age. The number of total activities (A) was counted during six sessions spanning 16 days (#1 and #2 on day 1, #3 and #4 on day 2, and #5 and #6 on day 16). (B) The total activities (left) and rearing behavior (right) during the first two and the last two sessions (sessions #1, #2, #5, and #6) were computed, and the averages per session are indicated. Data are means ± SEM.*P<0.05and **P<0.01, by ANOVA with posthoc Tukey’s test. n=9micefortheFMR1−/−/mock group. n = 11 for FMR1−/−/+LIMK-i. n = 10 for WT/mock. n = 7 for WT/+LIMK-i.
DISCUSSION
Presynaptic Wit as a critical receptor for the development and function of neuromuscular synapses
In Drosophila, glass bottom boat (Gbb), which is produced by the post-synaptic muscle, binds to the presynaptic receptor Wit and plays a critical role in modulating neuromuscular synaptic growth, stability, and function. Upon Gbb binding, Wit forms a heteromeric receptor complex with Thickveins (Tkv) and Saxophone (Sax), which then phospho-rylate Mothers against decapentaplegic (MAD), a Drosophila homolog of Smad1/5/8 (25). It has been reported that loss of Spartin, a Drosophila homolog of SPG20 that promotes endocytotic degradation of Wit and represses the BMP-Wit signaling pathway, results in an increment of neuromuscular synapses (26). The result with the dFMR1Δ113/+ mutant is consistent with the Spartin study and confirms the effect of increased Gbb-Wit signal on abnormal synapse development (26). Loss-of-expression mutants of Spartin develop age-dependent and progressive neuronal defects resembling hereditary spastic paraplegia (HSP) (26). Because frameshift mutations in the SPG20 gene cause a form of HSP known as Troyer syndrome (Online Mendelian Inheritance in Man no. 275900) (27), these results underscore the significance of a presynaptic BMP signal finely tuned by multiple regulatory molecules, including SPG20 and FMRP, for proper motor neuron development and function. Beyond the domain of the NMJ, multiple studies reinforce the notion that the correct intensity and spatiotemporal dynamics of the BMP signaling pathway are critical for axon regeneration upon neuronal and glial injury responses after CNS injury (28). Furthermore, BMP signaling specifies large and fast-transmitting synapses in the auditory system in a process that largely shares homologies with retrograde BMP signaling in Drosophila neuromuscular synapses (29). In line with these findings, our results propose an essential role for the FMRP-BMPR2 axis in the development of the neuropathology of patients with FXS.
Limitations of behavioral assays in mice
The major obstacle against the development of drugs for neurodevelopmental and neurodegenerative diseases is the lack of proper animal models that recapitulate the range of intellectual disability and/or cognitive dysfunction found in human patients. The existing, inadequate models also lack quantitative and reproducible assays to examine cognitive phenotypes. The mouse model of FXS exhibits cognitive and behavioral phenotypes that are both consistent and inconsistent with the symptoms of patients with FXS. For example, a frequent FXS trait is a high-anxiety behavior, whereas the FMR1-KO mice exhibit lower anxiety–like behaviors in the “light-dark compartment” test (30, 31). Furthermore, the result of the OFT is confusing because the FMR1-KO mice tend to spend a longer period in the center. The number of crossings and their velocity are both augmented compared to control mice because of their hyperactivity, but this behavior is interpreted as lower anxiety–like (24, 30, 32, 33). The results of the “elevated plus maze (EPM)” test, which is frequently used to investigate anxiety-like behaviors in FXS mice, exhibit both a decrease (34) and an increase (35) in anxiety. Furthermore, the tests show great variability and, sometimes, opposing outcomes in behavior depending on the genetic background of the mice, for example, FVB versus C57BL/6J (8). Considering the variability and lack of reproducibility of behavioral test results in FMR1-KO mice, as well as the concerns of animal welfare and the cost of husbandry, there is a strong need for an animal model and phenotypic readout to screen for FXS drugs.
Advantages of the Drosophila FXS model
There are multiple advantages of the Drosophila FXS model over the rodent models. Flies are invertebrates, which are inexpensive and easily cared for. They have a shorter life span and produce numerous externally laid embryos than rodent models. Their genome is small, minimally redundant, and easy to genetically manipulate in a tissue-specific manner (36, 37). It is easy to orally administer drugs to larvae by adding compounds to the Drosophila medium Formula 4–24 (Carolina Biological Supply Company). Previously, small molecules had to be delivered through conventional fly food that requires boiling followed by the addition of propionic acid, which disables the effect of heat- or acid-sensitive molecules. The use of Formula 4–24, which can be dissolved in water at room temperature and does not require exposure to high temperature nor addition of acid, expands the range of molecules that can be delivered to larvae without loss of activity.
Drosophila has contributed extensively to the discovery and validation of drug targets, as well as to the mechanistic understanding of their genetic cause (4, 14). In the context of FXS studies, it has been reported that dFMR1 adult mutant flies exhibit defects in learning/memory assays, such as Pavlovian olfactory association (17) and courtship conditioning (38). These behavioral abnormalities can be restored by various compounds known to target different FMRP targets, including protein synthesis inhibitors, such as puromycin and cycloheximide (17), the metabotropic glutamate receptor 5 antagonist MPEP (38), γ-aminobutyric acid agonists (39, 40), phosphodiesterase-4 inhibitor (41), and glycogensynthasekinase3inhibitor (38,42).Our study demonstrates that several dFMR1 mutant larvae exhibit an abnormally high number of NMJ synaptic boutons that correlate with their locomotion abnormality. Both are reversed by LIMK-i treatment, similarly to the effect of this drug in the FMR1-KO mouse. Thus, we propose the crawling assay in dFMR1 mutant Dro-sophila larvae as a rapid, quantitative, and reproducible preclinical screening strategy for potential FXS therapies that is alternative to behavioral tests using dFMR1 adult mutant flies or vertebrate FXS models. To facilitate the transition to a high-throughput screen of FXS drugs, the current assay will benefit from an improvement in the number of larvae that can be simultaneously assessed and in the robustness of the phenotype of dFMR1Δ113/+ mutants.
Larval crawling assay as a functional readout for the change in NMJ boutons
Larval locomotion abnormalities are described in Drosophila models of CNS diseases, such as Alzheimer’s (43, 44) and Huntington’s (45, 46). It has been reported that a different strain of dFMR1 mutant Drosophila larvae (dFMR14) (47) exhibits frequent turnings compared to wild-type larvae (44, 48). We observed that various dFMR1 mutants, including dFMR1Δ113/+, dFMR1Δ50/+, dFMR13/+, and dFMR1Δ113/3, as well as the dFMR1-RNAi line, crawled from the center to the periphery in a linear manner with an enhanced velocity compared to wild-type larvae. We speculate that this discrepancy might be due to the different nature of the mutations. For example, dFMR1Δ113 harbors a deletion of the first three exons of the dFMR1 gene, including exon 3 that contains the translation initiation methionine (4). Consequently, the dFMR1Δ113 allele results in a loss of dFMRP (4). On the contrary, the dFMR14 allele has a replacement of amino acid 289 with a stop codon; hence, it expresses a partial dFMRP missing the C terminus (47). Furthermore, in the process of creating the dFMR14 mutant, a Gal4-binding site was inserted into the first intron between exons 1 and 2 of the dFMR1 gene to overexpress a truncated dFMRP upon coexpression of Gal4 transcription factor (47). These differences might explain the distinct larval crawling behavior of the dFMR1Δ113 and dFMR14 mutants. The homozygous dFMR1Δ50 (13, 14) and homozygous dFMR13 mutants (14) are viable and develop into adulthood similarly to the homozygous dFMR14 mutant (47, 48). The homozygous dFMR1Δ50andhomozygousdFMR13 mutants exhibited frequent turns in the locomotion assay (fig. S3) similar to the previous study of the homozygous dFMR14 mutant (48). The velocity of dFMR13 and dFMR1Δ50 homozygous mutants was slower than wild-type, presumably because of the frequent changes of direction, unlike the transheterozygous dFMR1Δ113/3 mutant, which crawled in a linear fashion with an augmented velocity. We speculate that the turning phenotype observed in the homozygous mutants is due to complete loss of dFMRP, affecting the CNS neurons. FMRP activity has already been shown to be important for CNS neuron development and function in Drosophila (4, 49–51).
Performing the locomotion assay with larvae instead of adults is beneficial as they present an accessible, anatomically simple, and well-described peripheral nervous system (for example, NMJ boutons), which allows the molecular and biochemical assessment of the mechanism underlying the locomotion dysfunction and the therapeutic effects of drugs (52). For chemical screens of known pathways or targets, the NMJ synapses of larvae that exhibit an altered crawling phenotype should be subjected to synaptic bouton phenotype analysis as well as biochemical investigation to rapidly validate the “on-target” and eliminate the “off-target” effects of the candidate molecules. Sinadinos et al. (53) previously proposed a Drosophila larvae locomotion assay as a way to screen drugs for neurodegenerative diseases, such as Alzheimer’s disease. They subjected Drosophila larvae expressing the human three-repeat tau gene in motor neurons to crawling assays, such as a five-lane assay and a four-plate open-field assay, video-recorded the locomotion with an Ikegami digital video camera and a 5-mm digital video camera lens, and analyzed locomotion using EthoVision 3.0 software (53). In comparison, the advantage of our strategy is that the assay does not require specialized equipment, but a common video recording device, such as an iPhone camera, and an algorithm that is accessible and free to the scientific community. Furthermore, our system can simultaneously track and assess the crawling activities of multiple larvae through the open-access algorithm LarvaTrack, which we developed to trace and measure larval crawling activity. We have successfully simultaneously assessed up to 15 larvae using a 15-cm agarose plate. The method can be easily adapted to a larger number of larvae by using a larger agarose plate to avoid larvae to cross paths during crawling. Thus, our semiautomated assay of locomotion behavior can allow the higher-throughput assay that is essential for the screen of candidate molecules. In conclusion, activation of the FMRP–BMPR2 axis plays a role in synaptic abnormalities in both mouse and Drosophila models of FXS. The larval crawling assay is an easy, fast, and well-suited medium-throughput screen for genetic or chemical modulators of locomotion dysfunction in the Drosophila FXS model, which can be further evaluated in cognitive and behavioral tests using mammalian FXS models.
MATERIALS AND METHODS
Chemicals and antibodies
LIMK-i (Tocris Bioscience) and puromycin (Clontech) were dissolved in DMSO (final stock concentration, 100 mM) and water (final stock concentration, 100 mM), respectively, and administered to mice or Drosophila. LIMK allosteric antagonists (TH251 and TH255) were synthesized as previously reported (18). The synthesis of the inactive control compound (TH263) is described in text S2. All the compounds (TH251, TH255, and TH263) were dissolved in DMSO (final stock concentration, 10 mM) and administered to Drosophila. We used antibodies for phosphorylated cofilin (SC-12912-R, Santa Cruz Biotechnology), Drosophila Twinstar (a gift from T. Uemura) (54), actin (AC-15, Thermo Scientific), and Dlg1 (4F3, Developmental Studies Hybridoma Bank). Alexa Fluor 488–conjugated secondary antibody against mouse immunoglobulin G (IgG) and Alexa Fluor 568–conjugated phalloidin (A12380, Life Technologies) were also used.
Drosophila lines
The mutant lines carrying the WitA12, dFMR1Δ113M, dFMR1Δ50M, and UAS-dFMR1 RNAi (Bloomington Drosophila Stock Center ID #35200) alleles were obtained from the Bloomington Drosophila Stock Center (http://flybase.org/reports/FBal0131035.html). The dFMR13 and elav[C155]-Gal4 lines were obtained from T. A. Jongens and Y. N. Jan, respectively. To obtain the WitA12/+, dFMR1Δ113/+, and dFMR1Δ113,WitA12/+, dFMR1Δ50/+, and dFMR13/+ third-instar larvae, the mutant flies were crossed to the w1118 line. To obtain dFMR1Δ113/3 third-instar larvae, dFMR1Δ113/TM6B, Tb and dFmr13/TM6C, Tb, Sb flies were crossed. The wild-type Drosophila melanogaster w1118 line was used as a control. Drosophila strains were cultured at 25°C in 50 to 70% humidity in a 12 hour/12 hour light/dark cycle, on cornmeal-sucrose-yeast medium supplemented with the mold inhibitor methylparaben and autoclaved.
To express UAS-dFMR1 RNAi in the neuron, UAS-dFMR1 RNAi female flies were crossed with elav-Gal4 male flies, kept at 16°C until the first and second larval stages, and incubated at 29°C for 1 day. As a control, w1118 female flies were crossed with elav-Gal4 male flies and kept as the RNAi line. The third-instar female larvae were used for analysis.
Oral administration of drugs to Drosophila larvae
Distilled water (20 ml) at room temperature was added to two measuring cups of the instant Drosophila medium Formula 4–24 (Carolina Biological Supply Company) into the culture bottle. LIMK-i in DMSO or puromycin in water was placed on the formula and mixed well, and the formula was brought to the final concentration by addition of 10 ml of water at room temperature. Ten female and 5 male flies were put in the bottle with LIMK-i formula to set up mating. After 5 to 7 days, third-instar larvae were subjected to the crawling assay and the immunoblot analysis.
Immunoblot analysis
Third-instar larvae were lysed in lysis buffer (tris-buffered saline containing 1% NP-40, 1 μM NaF, and 1 mM EDTA) and incubated for 1 hour at 4°C. Lysed samples were separated by SDS–polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane (Millipore), immunoblotted with antibodies, and visualized using the LI-COR imaging system.
Larva immunofluorescence and image quantitation
Wandering late third-instar larvae were dissected for NMJ phenotype analysis. Briefly, the tissue was fixed with 8% formaldehyde in phosphate-buffered saline (PBS) for 10 min, washed with PBT (PBS containing 0.1% Triton X-100), and blocked with 5% normal donkey serum in PBT for 1 hour at room temperate. The larvae were incubated with Dlg1 antibody overnight at 4°C, followed by incubation with Alexa Fluor 488–conjugated secondary antibody against mouse IgG and Alexa Fluor 568–conjugated phalloidin. Confocal images were acquired using a point scanning confocal microscope (Leica). Images for quantification of NMJ bouton number on muscles 6 and 7 at segment 3 were produced by projection of z-sections using the ImageJ software blindly.
Larval crawling assay
Larval crawling abilities were examined as reported previously (55). Briefly, third-instar larvae (n = 10) were placed on the center of a 15-cm petri dish containing 2% agarose, and their crawling behavior was digitally recorded for 1 min using an iPhone 4S (Apple) in QuickTime movie (MOV) format. The videos were analyzed either manually or computationally. For manual analysis of crawling, the relative crawling distance every 15 s was quantitated by counting the number of concentric lines crossed, which was converted to the definitive distance (in millimeters). For the computational analysis, the relative crawling distance of each individual larva was quantitated on the basis of the crawling path traced by the LarvaTrack algorithm (https://github.com/plredmond/ larva-tracker), as described below. The position of each larva was recorded on most frames, and the cumulative distance (in millimeters) was computed for four 15-s intervals. The velocity was calculated by determining the average speed from 15 to 45 s. Beginning and end of the path were excluded because the larvae did not move the first 10 s and some of them reached the edge of the petri dish by the last 10 s.
LarvaTrack algorithm
LarvaTrack takes a video satisfying the following constraints. A time-lapse video was taken by a motionless camera at two frames per second (fps) and encoded via wall-clock time for playback at 30 fps. The total length of the video was 4 s, or 120 frames, representing 60-s wall-clock time. The camera was positioned motionless for the duration of the video. A 15-cm petri dish filled the narrow dimension of the frame. A coin (in this case, a penny measuring 19.05 mm), was present in the frame for scale. The background observable through the petri dish was a solid blue color distinct from the color of the larva.
We constructed a deterministic multiple-point object tracker in the following way. We detected larva locations (“detected location”) in each video frame with the OpenCV blob detector. We updated those larva locations (“flowed location”) in each frame with data from the following frame using the OpenCV optical-flow algorithm. Finally, for each successive pair of frames, we assigned some flowed locations from the earlier frame to some detected locations in the later frame. For this, we applied a variation of the Gale-Shapley stable matching algorithm (www.jstor.org/stable/2312726). The resulting digitized larval paths were used to compute average velocity and distance traveled over four 15-s intervals. For additional resources and code, see the Supplementary Materials (text S1 and fig. S4) and the online repository (https://github.com/ plredmond/larva-tracker).
FXS mouse model
B6.129P2-FMR1tm1Cgr/J mice were maintained under standard conditions. To obtain FMR1 knockout littermates, FMR1 heterozygous female and hemizygous male mice were mated, and wild-type and knockout (FMR1−/−) male mice were used. Genotyping was performed following the Jackson Laboratory protocol.
Mouse behavior analyses
LIMK-i or DMSO was administered to B6.129P2-FMR1tm1Cgr/J and littermate wild-type mice by intracerebroventricular injection at P1 and P4 (20 μg per pup) and by intraperitoneal injection twice at about 3 months of age (80 mg/kg) and again after 2 weeks. Two weeks after the final injection, the mice were examined by the rOFT at the Behavioral Core Facility of the Gladstone Institute (19). Briefly, mice were transferred to the testing room 60 min before testing to acclimate them to the testing conditions under normal lighting. For the rOFT, mice were given four 5-min sessions in a clear acrylic OFT chamber (41 × 41 × 30 cm) twice per day, once in the morning and once in the afternoon over two consecutive days (sessions #1 and #2 on the first day and sessions #3 and #4 on the second day). After these sessions, the mice were retested after 14 days in the OFT chamber for two more 5-min sessions separated by 3 to 4 hours (sessions #5 and #6). Spontaneous locomotion activity at each session was measured in an OFT chamber with two 16 × 16 photobeam arrays that detect horizontal (fine or ambulatory movement) and vertical (rearing) movements, which is also known as a Flex-Field/Open-Field Photobeam Activity System (San Diego Instruments). Fine movements were defined as having broken less than three photobeams. Ambulatory movements were defined as having broken more than three photobeams. Ambulatory movements were interpreted as walking, whereas fine movements accounted for grooming or perhaps stretching. Total activities were measured by calculating ambulatory and fine movements. Locomotion and exploratory activities were monitored by measuring the total activity and the rearing movement throughout the six sessions, respectively. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved Institutional Animal Care and Use Committee protocols (AN108100) of University of California, San Francisco (UCSF). The protocol was approved by the Committee on the Ethics of Animal Experiments of UCSF and Gladstone Institute.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism 7.0 package and reviewed by M. Nojima at the University of Tokyo. Statistical test and significance are denoted in the figure legends. Statistical significance is denoted as described within the figure legends.
Supplementary Material
Acknowledgments
We thank M. Gill and the Gladstone Institute Behavioral Core Facility staff members for mouse behavioral analysis; T. A. Jongens (University of Pennsylvania) for dFmr13/TM6C, Tb, Sb fly stock; Y. N. Jan (UCSF) for elav[C155]-Gal4 fly stock; T. Uemura (Kyoto University) for the antibody against Twinstar; and H. Li (Airware) and J. A. Trono (Saint Michael’s College) for advice in writing the algorithm. We also want to thank M. Nojima for the statistical analysis and S. Liu and S. Manghise for the technical support.
Funding: R.K. is a recipient of a fellowship from Japan Society for the Promotion of Science. This work was supported by grants from the NIH (HL093154 and HL108317) to A.H. and the Program for Breakthrough Biomedical Research to R.K. S.K. is grateful for support by the Structural Genomics Consortium, a registered charity (no. 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada through Ontario Genomics Institute, Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation (FAPESP), Takeda, the Centre of Excellence Macromolecular Complexes at Frankfurt University, and the Wellcome Trust.
Footnotes
Author contributions: R.K., P.L.R., S.R., P.G., T.H., S.K., T.B.K., and G. L. designed and performed the experiments and interpreted the data. R.K., P.L.R., T.B.K., S.R., T.H., S.K., and G.L. edited the manuscript. A.H. conceived the project, designed the experiments, interpreted the data, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
www.sciencesignaling.org/cgi/content/full/10/477/eaai8133/DC1
REFERENCES AND NOTES
- 1.Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16:595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Saldarriaga W, Tassone F, González-Teshima LY, Forero-Forero JV, Ayala-Zapata S, Hagerman R. Fragile X syndrome. Colomb Med. 2014;45:190–198. [PMC free article] [PubMed] [Google Scholar]
- 3.Darnell JC, Klann E. The translation of translational control by FMRP: Therapeutic targets for FXS. Nat Neurosci. 2013;16:1530–1536. doi: 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang YQ, Bailey AM, Matthies HJG, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 5.Kashima R, Roy S, Ascano M, Martinez-Cerdeno V, Ariza-Torres J, Kim S, Louie L, Lu Y, Leyton P, Bloch KD, Kornberg TB, Hagerman PJ, Hagerman R, Lagna G, Hata A. Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci Signal. 2016;9:ra58. doi: 10.1126/scisignal.aaf6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc Natl Acad Sci USA. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Till SM, Asiminas A, Jackson AD, Katsanevaki D, Barnes SA, Osterweil EK, Bear MF, Chattarji S, Wood ER, Wyllie DJA, Kind PC. Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Hum Mol Genet. 2015;24:5977–5984. doi: 10.1093/hmg/ddv299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santos AR, Kanellopoulos AK, Bagni C. Learning and behavioral deficits associated with the absence of the fragile X mental retardation protein: What a fly and mouse model can teach us. Learn Mem. 2014;21:543–555. doi: 10.1101/lm.035956.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tauber JM, Vanlandingham PA, Zhang B. Elevated levels of the vesicular monoamine transporter and a novel repetitive behavior in the Drosophila model of fragile X syndrome. PLOS ONE. 2011;6:e27100. doi: 10.1371/journal.pone.0027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Fang Z, Jud C, Vansteensel MJ, Kaasik K, Lee CC, Albrecht U, Tamanini F, Meijer JH, Oostra BA, Nelson DL. Fragile X-related proteins regulate mammalian circadian behavioral rhythms. Am J Hum Genet. 2008;83:43–52. doi: 10.1016/j.ajhg.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhães TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in. Drosophila Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- 12.Marqués G, Bao H, Haerry TE, Shimell MJ, Duchek P, Zhang B, O’Connor MB. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- 13.Bozzetti MP, Specchia V, Cattenoz PB, Laneve P, Geusa A, Sahin HB, Di Tommaso S, Friscini A, Massari S, Diebold C, Giangrande A. The Drosophila fragile X mental retardation protein participates in the piRNA pathway. J Cell Sci. 2015;128:2070–2084. doi: 10.1242/jcs.161810. [DOI] [PubMed] [Google Scholar]
- 14.Pan L, Zhang YQ, Woodruff E, Broadie K. The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol. 2004;14:1863–1870. doi: 10.1016/j.cub.2004.09.085. [DOI] [PubMed] [Google Scholar]
- 15.Doll CA, Broadie K. Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front Cell Neurosci. 2014;8:30. doi: 10.3389/fncel.2014.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stefani G, Fraser CE, Darnell JC, Darnell RB. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci. 2004;24:7272–7276. doi: 10.1523/JNEUROSCI.2306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11:1143–1145. doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodwin NC, Cianchetta G, Burgoon HA, Healy J, Mabon R, Strobel ED, Allen J, Wang S, Hamman BD, Rawlins DB. Discovery of a type III inhibitor of LIM kinase 2 that binds in a DFG-out conformation. ACS Med Chem Lett. 2015;6:53–57. doi: 10.1021/ml500242y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci USA. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventura R, Pascucci T, Catania MV, Musumeci SA, Puglisi-Allegra S. Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: Involvement of dopamine in the medial prefrontal cortex. Behav Pharmacol. 2004;15:433–442. doi: 10.1097/00008877-200409000-00018. [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76:325–337. doi: 10.1016/j.neuron.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu ZH, Chuang DM, Smith CB. Lithium ameliorates phenotypic deficits in a mouse model of fragile X syndrome. Int J Neuropsychopharmacol. 2011;14:618–630. doi: 10.1017/S1461145710000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74:49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olmos-Serrano JL, Corbin JG, Burns MP. The GABAA receptor agonist THIP ameliorates specific behavioral deficits in the mouse model of fragile X syndrome. Dev Neurosci. 2011;33:395–403. doi: 10.1159/000332884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman SH, Dani N, Rushton E, Broadie K. Fragile X mental retardation protein regulates trans-synaptic signaling in Drosophila. Dis Model Mech. 2013;6:1400–1413. doi: 10.1242/dmm.012229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nahm M, Lee MJ, Parkinson W, Lee M, Kim H, Kim YJ, Kim S, Cho YS, Min BM, Bae YC, Broadie K, Lee S. Spartin regulates synaptic growth and neuronal survival by inhibiting BMP-mediated microtubule stabilization. Neuron. 2013;77:680–695. doi: 10.1016/j.neuron.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel H, Cross H, Proukakis C, Hershberger R, Bork P, Ciccarelli FD, Patton MA, McKusick VA, Crosby AH. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- 28.Zhong J, Zou H. BMP signaling in axon regeneration. Curr Opin Neurobiol. 2014;27:127–134. doi: 10.1016/j.conb.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao L, Michalski N, Kronander E, Gjoni E, Genoud C, Knott G, Schneggenburger R. BMP signaling specifies the development of a large and fast CNS synapse. Nat Neurosci. 2013;16:856–864. doi: 10.1038/nn.3414. [DOI] [PubMed] [Google Scholar]
- 30.Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL. (Over) correction of FMR1 deficiency with YAC transgenics: Behavioral and physical features. Hum Mol Genet. 2000;9:1145–1159. doi: 10.1093/hmg/9.8.1145. [DOI] [PubMed] [Google Scholar]
- 31.Veeraragavan S, Graham D, Bui N, Yuva-Paylor LA, Wess J, Paylor R. Genetic reduction of muscarinic M4 receptor modulates analgesic response and acoustic startle response in a mouse model of fragile X syndrome (FXS) Behav Brain Res. 2012;228:1–8. doi: 10.1016/j.bbr.2011.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- 33.Restivo L, Ferrari F, Passino E, Sgobio C, Bock J, Oostra BA, Bagni C, Ammassari-Teule M. Enriched environment promotes behavioral and morphological recovery in a mouse model for the fragile X syndrome. Proc Natl Acad Sci USA. 2005;102:11557–11562. doi: 10.1073/pnas.0504984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol. 2010;79:632–646. doi: 10.1016/j.bcp.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bilousova TV, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, Ethell IM. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009;46:94–102. doi: 10.1136/jmg.2008.061796. [DOI] [PubMed] [Google Scholar]
- 36.Mohr SE, Smith JA, Shamu CE, Neumüller RA, Perrimon N. RNAi screening comes of age: Improved techniques and complementary approaches. Nat Rev Mol Cell Biol. 2014;15:591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirth F. Drosophila melanogaster in the study of human neurodegeneration. CNS Neurol Disord Drug Targets. 2010;9:504–523. doi: 10.2174/187152710791556104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McBride SMJ, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, McDonald TV, Jongens TA. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 39.Chang S, Bray SM, Li Z, Zarnescu DC, He C, Jin P, Warren ST. Identification of small molecules rescuing fragile X syndrome phenotypes in Drosophila. Nat Chem Biol. 2008;4:256–263. doi: 10.1038/nchembio.78. [DOI] [PubMed] [Google Scholar]
- 40.Idrissi A El, Boukarrou L, Dokin C, Brown WT. Taurine improves congestive functions in a mouse model of fragile X syndrome. Adv Exp Med Biol. 2009;643:191–198. doi: 10.1007/978-0-387-75681-3_19. [DOI] [PubMed] [Google Scholar]
- 41.Kanellopoulos AK, Semelidou O, Kotini AG, Anezaki M, Skoulakis EMC. Learning and memory deficits consequent to reduction of the fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J Neurosci. 2012;32:13111–13124. doi: 10.1523/JNEUROSCI.1347-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLOS ONE. 2010;5:e9706. doi: 10.1371/journal.pone.0009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Folwell J, Cowan CM, Ubhi KK, Shiabh H, Newman TA, Shepherd D, Mudher A. Aβ exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer’s disease. Exp Neurol. 2010;223:401–409. doi: 10.1016/j.expneurol.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 44.Günther MN, Nettesheim G, Shubeita GT. Quantifying and predicting Drosophila larvae crawling phenotypes. Sci Rep. 2016;6:27972. doi: 10.1038/srep27972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinadinos C, Burbidge-King T, Soh D, Thompson LM, Marsh JL, Wyttenbach A, Mudher AK. Live axonal transport disruption by mutant huntingtin fragments in Drosophila motor neuron axons. Neurobiol Dis. 2009;34:389–395. doi: 10.1016/j.nbd.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 47.Lee A, Li W, Xu K, Bogert BA, Su K, Gao FB. Control of dendritic development by the Drosophila fragile X-related gene involves the small GTPase Rac1. Development. 2003;130:5543–5552. doi: 10.1242/dev.00792. [DOI] [PubMed] [Google Scholar]
- 48.Xu K, Bogert BA, Li W, Su K, Lee A, Gao FB. The fragile X-related gene affects the crawling behavior of Drosophila larvae by regulating the mRNA level of the DEG/ENaC protein pickpocket1. Curr Biol. 2004;14:1025–1034. doi: 10.1016/j.cub.2004.05.055. [DOI] [PubMed] [Google Scholar]
- 49.Michel CI, Kraft R, Restifo LL. Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J Neurosci. 2004;24:5798–5809. doi: 10.1523/JNEUROSCI.1102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dockendorff TC, Su HS, McBride SMJ, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–984. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 51.Morales J, Hiesinger PR, Schroeder AJ, Kume K, Verstreken P, Jackson FR, Nelson DL, Hassan BA. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron. 2002;34:961–972. doi: 10.1016/s0896-6273(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 52.Gramates LS, Budnik V. Assembly and maturation of the Drosophila larval neuromuscular junction. Int Rev Neurobiol. 1999;43:93–117. doi: 10.1016/s0074-7742(08)60542-5. [DOI] [PubMed] [Google Scholar]
- 53.Sinadinos C, Cowan CM, Wyttenbach A, Mudher A. Increased throughput assays of locomotor dysfunction in Drosophila larvae. J Neurosci Methods. 2012;203:325–334. doi: 10.1016/j.jneumeth.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 54.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 55.Nichols CD, Becnel J, Pandey UB. Methods to assay Drosophila behavior. J Vis Exp. 2012;2012:e3795. doi: 10.3791/3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuen HK, Princen J, Illingworth J, Kittler J. Comparative-study of hough transform methods for circle finding. Image Vis Comput. 1990;8:71–77. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.