

Abstract
Mitochondrial complex II (CII) is an emerging target for numerous human diseases. Sixteen analogues of the CII inhibitor natural product atpenin A5 were prepared to evaluate the structure-activity relationship of the C-5 pyridine side chain. The side chain ketone moiety was determined to be pharmacophoric, engendering a bioactive conformation. One analogue (16c) displayed CII IC50 = 64 nM, retained selectivity for CII over mitochondrial complex I (>156-fold) and possessed a ligand-lipophilicity efficiency of 5.62, desirable metrics for a lead compound. This derivative and other highly potent complex II inhibitors possess potent and selective anti-proliferative activity in multiple human prostate cancer cell lines under both normoxia and hypoxia, acting to inhibit mitochondrial electron transport.
Graphical abstract
1. Introduction
Mitochondrial respiratory complex II (CII) also known as succinate dehydrogenase (SDH) or succinate-coenzyme Q reductase (SQR) is a 124 kDa protein complex located in the inner membrane of mitochondria.9[1] CII forms part of the electron transport chain (ETC) as well as being implicated in succinate signaling and substantial reactive oxygen species (ROS) generation.[2] The protein connects the tricarboxylic acid cycle (TCA) and the ETC, while lacking any contribution to maintaining the proton gradient across the mitochondrial inner membrane.[3]
Despite the association of some aggressive forms of cancer with CII mutation (probably linked to hypoxia-inducible factor (HIF) activation resulting from inhibition of HIF prolyl-hydroxylases by succinate accumulation),[4] far greater evidence supports an anti-proliferative role for CII inhibition. Five facets of CII inhibition, beyond the generation of ROS,[5] have significant promise for the development of selective small molecule chemotherapeutics. i) CII is rarely mutated within cancers and thus presents a unique invariant target to address cancer chemotherapy. Mutations that are observed are only associated in infrequent and nonaggressive neoplasias such as pheochromocytomas.[6] In most common cancers such as prostate cancer, only one in a million patients carry mutation in CII.[3] ii) Inhibition of the TCA cycle impairs glutaminolysis, a major source of energy for tumor cells. iii) Along with complex I, complex II (also known as fumarate reductase) is a key member of the NADH-fumarate reductase system which is important for maintaining mitochondrial energy production in tumor microenvironments under hypoxic conditions.[7] iv) CII inhibition leads to prolonged activation of autophagy and cancer cell death.[8] As cancer cells are known to develop resistance to apoptosis,[9] activation of autophagy could be a viable strategy to combat drug resistance.v) Promisingly, low potency CII inhibitors are selectively cytotoxic to cancer cells, albeit with weak effect, while conveying minimal, to no toxicity, to non-malignant cells.[10]
Several CII inhibitors are known, but all suffer from low potency. 3-Bromopyruvate (1, 3BP, Figure 1) was the first identified CII inhibitor, however no IC50 has been reported. Malonate (2) possesses an IC50 = 40 μM.[11] The vitamin E analogue α -Tocopheryl succinate (α-TOS, 3) has a CII IC50 = 42 μM,[12] inducing apoptosis in cancer cells by ROS generation. Mitochondrially targeted vitamin E succinate (MitoVES, 4) has an IC50 = 70 μM.[13] However, MitoVES was found to be 20-50 times more effective in inducing apoptosis in cancer cells than α-TOS.[14] This was attributed to the introduction of a cationic triphenylphosphonium (TPP) group which acts to target the compound to the mitochondria. Inhibition of CII was selective to cancer cells with MitoVES possessing an IC50 of 0.5-3 μM for apoptosis induction in cancer cells and approximately 20-60 μM for non-malignant cells.[14] Thenoyltrifluoroacetone (TFFA, 5), is widely used as a control compound in CII assay kits with an IC50 = 30 μM.[15] The clinical vasodilator and CII inhibitor diazoxide 6 (IC50 = 32 μM),[16] is known to regulate ROS production inducing specific cancer cell death.[17]
Figure 1.
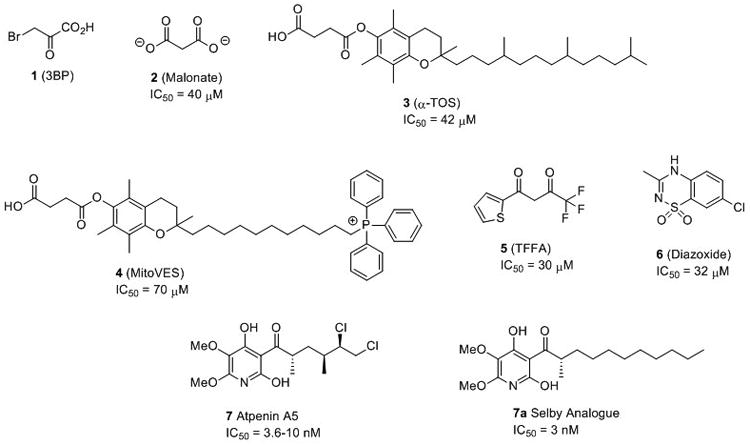
Structures of known complex II inhibitors.
A potent CII inhibitor would be expected to elicit greater anticancer efficacy and would be an excellent candidate for development as a chemotherapeutic agent and as a probe compound for further study of the role of mitochondria in cancer and other diseases. For example, accumulation of the TCA cycle metabolite succinate is an important pathologic event in tissue ischemia, and this is thought to occur via reverse operation of CII.[18] As such, it has been shown that inhibiting CII with low potency compounds,[18-19] as well as Atpenin A5,[20] is capable of protecting the heart against ischemic reperfusion injury, thereby ascribing CII as a potential target for drug development in stroke. While CII mutations contribute to the pathophysiology of a number of diseases,[21] the pharmacological inhibition of CII by a number of compounds, including clinically approved drugs, has not been linked to increased susceptibility to these diseases.[22]
The natural product atpenin A5 (AA5, 7, Figure 1) is a potent and specific CII inhibitor with an IC50 = 3.6-10 nM.[20, 23] Inhibition of CII by AA5 results in production of ROS.[24] AA5 and several naturally occurring analogues demonstrate antineoplastic activity in DU-145 prostate cancer cells.[25] However, atpenin B, a close analogue of AA5, was found to have limited anticancer effect in in vivo studies attributed to poor absorption, distribution, metabolism and excretion (ADME) properties.[26] AA5 derivative 7a was prepared by Selby et. al. as part of an antifungal library and gives bovine CII IC50 = 3 nM.[27] However, the compound retains a stereocenter on the side chain alpha to the ketone. This moiety is present in all of the most active CII inhibitors described in the Selby library. Excision of the methyl resulted in significant amelioration of CII inhibition, suggesting a pharmacophoric role and complicating the generation of synthetic derivatives.
The inhibitory potency for CII and the observed in vitro anticancer effects make AA5 an excellent compound for further drug discovery efforts. However, its suitability as a chemotherapeutic is hindered by low abundance, complex structure, 50-fold reduction in in vivo activity and, until this study, incomplete assignment of its pharmacophore.
2. Results and Discussion
2.1 Chemistry
The known crystal structure of E.coli CII with AA5,[28] and porcine CII (PDB ID: 3AEE) indicates a role for the highly substituted pyridine ring in active site recognition and hence in our first series of designed analogues this moiety remained unchanged. The hydrocarbon side chain of AA5 contains three stereocenters that hinder large scale preparation, similarly AA5 derivative 7a (Figure 1), while simplified, still contained a stereocenter alpha to the side chain ketone. We sought to simplify the side chain through the generation of unfunctionalized and non-chiral hydrocarbon chain derivatives.
Several hydrocarbon chain derivatives of AA5 have been synthesized to evaluate antifungal activity.[27] However, only one has shown enhanced activity. Three total synthesis of the AA5 scaffold have been reported in the literature from the groups of Omura,[29] Selby,[27] and Carreira[30] as well as one report of racemic atpenin B by Quéguiner et. al.[31] Our synthetic strategy to access AA5 derivatives for structure-activity relationship (SAR) study is based on elements from all four routes.
Commercially available 2,3-dimethoxypyridine (8) was lithiated with nbutyl lithium and exposed to trimethylborate, followed by oxidation with peracetic acid to provide 2,3-dimethyoxy-4-hydroxypyridine (9) in moderate yield (Scheme 1). Regioselective addition of bromine was achieved at the 5-position by exposure to N-bromosuccinimide and purification by column chromatography to afford 5-bromo-2,3-dimethoxypyridin-4-ol (10) as the major product. Subsequent protection as the methoxymethyl ether (11) was achieved in excellent yield. Rearrangement of the bromine of pyridine 11 from the 5-position to the 6-position was accomplished by a ‘halogen dance’ reaction upon exposure to LDA and catalytic bromine, providing 6-bromo-pyridine (12) in excellent yield. Metal-halogen exchange with nbutyl lithium, critically with just one minute of stirring at -78 °C, followed by trapping with trimethyl borate[32] and subsequent oxidation with peracetic acid provided 5,6-dimethoxy-4-(methoxymethoxy)pyridin-2-ol (13). Protection of the hydroxy functionality of 13 as the methoxymethyl ether afforded the critical intermediate 2,3-dimethoxy-4,6-bis(methoxymethoxy)pyridine (14) in excellent yield. Subsequent exposure of pyridine (14) to nbutyl lithium and a suitably functionalized aldehyde provided the respective alcohol which was oxidized to the ketone with DMP to afford a series of methoxymethyl ether protected atpenin A5 derivatives of type 15. Deprotection by exposure to TFA resulted in a variety of hydrocarbon chain atpenin A5 derivatives (16a-k) in moderate to good yield.
Scheme 1.
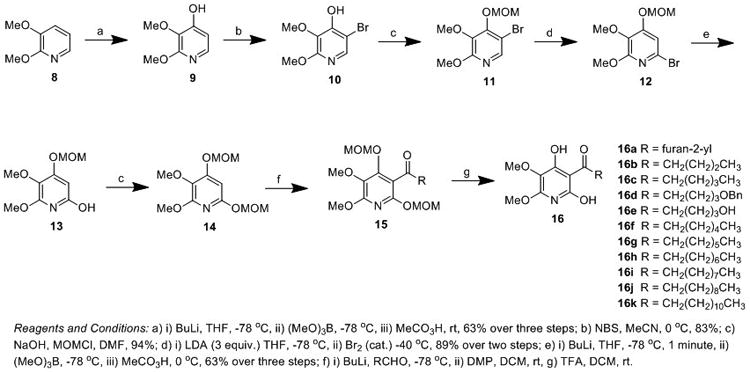
Synthesis of hydrocarbon side chain derivatives of atpenin A5.
To determine the effect of the oxidation state of the first position of the hydrocarbon chain derivatives on CII inhibition, triol (18) (Scheme 2) was isolated from the methoxymethyl ether intermediate (17) by omitting the oxidation step from the developed synthetic route.
Scheme 2.
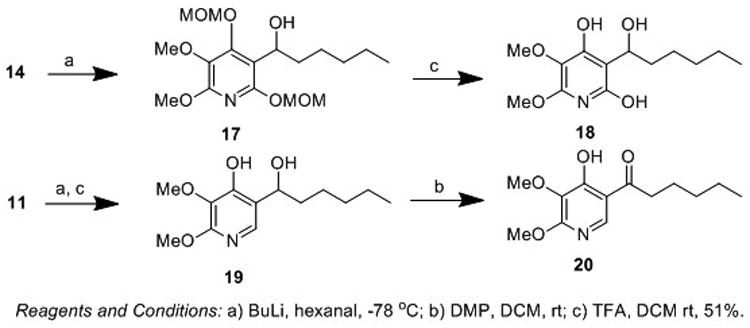
Synthesis of oxidation state derivatives of atpenin A5.
Given the structural similarity between AA5 and the natural CII substrate ubiquinone, we hypothesized that the 2,3-dimethoxy and 4-hydroxy substituents on the AA5 pyridine ring would be essential for CII binding site recognition. However, as the 6-hydroxy substituent of AA5 is not present in ubiquinone we sought to determine the effects of removing this group. To this end we synthesized the 6-deshydroxy derivative (20) by direct addition of a suitably functionalized aldehyde to bromide (11), lacking the 6-hydroxy functionality (Scheme 2). This route provided diol intermediate (19) in good yield which was selectively oxidized with DMP to provide the desired 1-(4-hydroxy-5,6-dimethoxypyridin-3-yl)hexan-1-one (20) in moderate yield.
During the course of our investigation into the synthesis of AA5 derivatives we partially adopted the route of Quéguiner et. al. to atpenin B.[31] Protection of alcohol (9) as the N,N-diisopropyl carbamate (21) followed by addition of Br2 provided access to bromide (22) in excellent yield (Scheme 3). Subsequent transmetallation and trapping with hexanal provided alcohol (23) in good overall yield. However, deprotection of the N,N-diisopropyl carbamate by exposure to methanolic potassium hydroxide failed to yield the desired compound, with the respective methyl ether derivative (24) obtained in 50% yield.
Scheme 3.
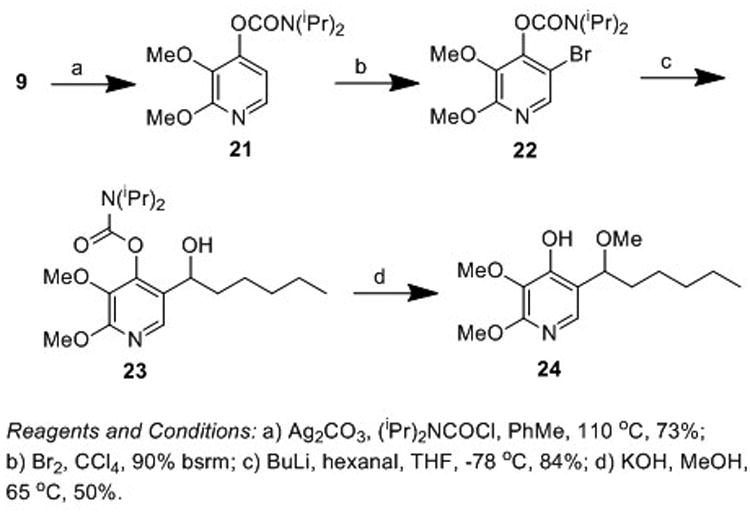
Synthesis of a methyl ether derivative of atpenin A5.
2.2 Complex II inhibition assay
Complex II activity was measured spectrophotometrically according to a literature method using isolated rat heart mitrochondria, with suitable modifications to ensure rapid isolation.[11] The control compound AA5 was determined to have an IC50 = 3.3 nM, in accordance with the 3.6-10 nM reported in the literature,[20, 23] the clinically approved agent Diazoxide has a literature IC50 = 32 μM and is included in Table 1 for comparative purposes of CII inhibitors of non-AA5 structure.[16] With the sole exception of the furan-2-yl moiety, all of the synthesized side chain derivatives displayed rat heart CII inhibition activity ranging from 3.3 nM to 3.7 μM. Compounds 16j and 16k display CII inhibition activity of IC50 8.6 and 3.3 nM respectively, two of the most potent inhibitors ever described. To compare CII species differences directly, two compounds were synthesized that were included in a recently described antifungal library (16b & 16f),[27] which demonstrate bovine CII IC50 = 96 nM and 20 nM respectively. In rat heart CII, activity is substantially attenuated with IC50 = 346 nM and 282 nM respectively. Therefore, suggesting that compound 16k described herein, may represent the most potent CII inhibitor yet described.
Table 1.
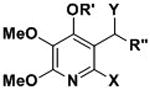
Complex II inhibition activity, ClogP, Mw, PSA and Lipophilic efficiency of AA5 derivatives. IC50 values are mean of n = 4 experiments ± S.D.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R′ | X | Y | R″ | Mw | ClogPa | PSAb | LLEc | CII IC50 (nM)d |
| 16a | H | OH | O | 2-furan | 265.22 | -0.02 | 102.02 | n/a | 0%e |
| 16b | H | OH | O | butyl | 255.27 | 1.09 | 88.88 | 5.37 | 345.5 ± 7.6 |
| 16c | H | OH | O | pentyl | 269.29 | 1.53 | 88.88 | 5.62 | 64± 4.3 |
| 16d | H | OH | O | 5-OBn butyl | 361.39 | 2.02 | 98.11 | 3.41 | 280.8 ± 88.7 |
| 16e | H | OH | O | 5-hydroxybutyl | 271.27 | -0.35 | 109.11 | 6.90 | 3730.2 ± 1021.1 |
| 16f | H | OH | O | hexyl | 283.32 | 1.98 | 88.88 | 4.57 | 282.9 ± 135.5 |
| 16g | H | OH | O | heptyl | 297.35 | 2.42 | 88.88 | 4.89 | 49.1 ± 22.1 |
| 16h | H | OH | O | octyl | 311.37 | 2.87 | 88.88 | 4.93 | 15.7 ± 3.5 |
| 16i | H | OH | O | nonyl | 325.40 | 3.31 | 88.88 | 4.69 | 9.9 ± 1.9 |
| 16j | H | OH | O | decyl | 339.43 | 3.75 | 88.88 | 4.32 | 8.6 ± 2.9 |
| 16k | H | OH | O | dodecyl | 367.48 | 4.64 | 88.88 | 3.76 | 3.3 ± 2.4 |
| 17 | MOM | OMOM | OH | pentyl | 359.42 | 2.95 | 88.5 | n/a | 0% |
| 18 | H | OH | OH | pentyl | 271.31 | 0.97 | 92.04 | n/a | 0% |
| 19 | H | H | OH | pentyl | 255.31 | 2.24 | 71.81 | n/a | 0% |
| 20 | H | H | O | pentyl | 253.29 | 2.97 | 68.65 | n/a | 0% |
| 24 | H | H | OMe | pentyl | 269.34 | 2.88 | 60.81 | n/a | 0% |
| Atpenin A5 | n/a | n/a | n/a | n/a | 366.24 | 2.64 | 88.88 | 5.36 | 3.3± 2.0 |
| Diazoxide | n/a | n/a | n/a | n/a | 230.67 | 1.0 | 58.53 | 1.85 | 32,000[6] |
Calculated by MarvinSketch 5.10.3
Polar Surface Area (pH = 7.4), calculated by MarvinSketch 5.10.3
Ligand-Lipophilicity Efficiency (LLE = pIC50-ClogP)
Mean value of four experiments
0% inhibition of CII at 100 nM concentration
Addition of an aromatic furnan-2-yl moiety (16a) to the 5-position of the AA5 pyridine ring abrogates CII inhibition activity. Potentially due to the presence of the extended conjugated system. Shortening the side chain to a simple butyl chain provided a compound with an IC50 of 345 nM and a 1.5-fold lower ClogP (16b). Interestingly, this derivative when assayed in bovine CII provided an IC50 = 96 nM.[27] Retaining the length of the natural AA5 hydrocarbon chain but with no substituents, resulted in compound 16c which displays an IC50 of 64 nM, lower lipophilicity, but identical PSA to 16b. The ligand-lipophilicity efficiency (LLE) of 16c, a measure of potency and lipophilicity, was calculated to be 5.62, the optimum value obtained across all of the derivatives herein. A LLE greater than 5, combined with a lipophilic ClogP is often considered desirable for a lead or clinical compound.[33]
Retaining the length of the natural product side chain but replacing the terminal methyl with a benzyl ether (16d) reduced activity (IC50 = 281 nM) compared with AA5. Removal of the benzyl protecting group to provide alcohol (16e) further decreased activity providing a CII IC50 of 3.7 μM, but still provided a compound of 10-fold greater activity than diazoxide. This observation suggests the alcohol moiety of 16e is not engaging in hydrogen bonding within the complex II protein given its lower potency compared with the identical side chain length derivative 16c. The importance of lipophilicity in the design of CII inhibitors is also supported by this data.
Addition of a methyl group to the pentyl side chain of 16c provided hexyl derivative 16f which displayed equipotent activity with the benzyl ether derivative (16d) with an IC50 of 283 nM, further suggesting that the oxygen of 16d and 16e is not involved in intermolecular interactions with CII. Again, a difference in activity between species of CII is observed, when derivative 16f is assayed in bovine CII an IC50 = 20 nM is obtained.[27] This suggests that the CII inhibitors detailed herein may possess much greater activity when assayed in bovine CII.
Homologation from hexyl through to dodecyl resulted in a progressive increase in CII inhibition resulting in the identification of three compounds with highly potent CII inhibition activity; 16i which displays IC50 = 9.9 nM, 16j with IC50 = 8.6 nM and 16k with IC50 = 3.3 nM. Homologation was ceased at the dodecyl chain (16k) as this compound represents comparable molecular weight and PSA to the AA5 natural product, albeit with increased ClogP. In general, increasing lipophilicity increases potency, a common phenomenon in medicinal chemistry.Compound 16f however, does not fit this trend when going from pentyl (16c) to heptyl (16g) wherein the activity of hexyl side chain derivate 16f displays attenuated CII inhibition activity.
A similar trend is observed in the MitoVES (4, Figure 1) series of compounds wherein shortening the alkyl chain results in progressive loss of CII inhibition activity.[14] Penetration of the mitochondrial inner membrane to access the CII target is necessary for inhibitory activity, with lipophilicity a key determinant in this process along with cationic charge.[34] To achieve greater delivery to the mitochondria we envision the appendage of a mitochondrial targeting triphenylphosphonium (TPP) group. However, an optimum hydrocarbon linker length between the active CII inhibitor ‘warhead’ and the TPP group has been shown to be essential to activity.[14]
Computational analysis of the crystal structure of the CII ubiquinone binding site with bound AA5 predicts a pharmacophoric role for the highly substituted pyridine moiety but no role for the 5-position side chain carbonyl in intermolecular binding.[28] We next sought to generate analogues of our AA5 derivatives about the ketone moiety to confirm this model. The intermediate hexanol derivative 17 displays 0% inhibition of CII at 100 nM concentration, suggesting that both pyridine alcohols are required for intermolecular binding to the CII quinone site, or alternatively, the additional steric bulk of the MOM groups precludes access of the compound to the CII binding site. Interestingly, alcohol 18, an oxidation state analogue of the active CII inhibitor 16c displayed no CII inhibition activity. Excision of the pyridine 6-position alcohol (19) also resulted in an inactive compound. Oxidation of alcohol 19 to the respective carbonyl (20) and thus a direct bioisostere of 16c wherein the pyridine 6-positon alcohol is excised, displayed no activity at 100 nM concentration. Methyl ether 24, like its alcohol counterpart, showed no activity to inhibit CII at 100 nM. Together these observations suggest a pharmacophoric role for the carbonyl of the side chain in the design of AA5 derivatives. While the possibility that the inactivity of these derivatives may indicate a failure of the compounds to penetrate into the mitochondria inner membrane, analysis of their physicochemical properties disputes this. PSA values of 18 and 24 (71.81 and 60.81) calculated at pH 7.4 are significantly lower than active inhibitor 16c and the AA5 natural product (both 88.88). ClogP values, a direct measure of lipophilicity, of 18 and 24 (2.24 and 2.88) are higher than the active and structurally similar inhibitor 16c (1.53) and in the case of compound 24, higher than the AA5 scaffold (2.64) itself, although shape, size and charge requirements also play a role in mitochondrial penetration.
2.3 Molecular modeling
Through detailed SAR studies we have shown that the side chain ketone of AA5 is critical for CII inhibition activity and thus constitutes part of the pharmacophore of this class of inhibitor. Quantitative structure-activity relationship prediction was employed to understand the SAR of our compound series. The known crystal structure of AA5 bound in the ubiquinone binding site of porcine heart mitochondria complex II (PDB ID: 3AEE) provided the basis of our analysis. The AA5 binding site was defined and the ligand replaced with compounds 16c, 18 or 24 using SeeSAR 5.5 (BioSolveIT GmbH). Binding poses for each ligand were generated and scoring performed by HYDE.[35]
Active CII inhibitor 16c is predicted to form hydrogen bond interactions between the pyridine 4-OH of 16c to the amine of the TrpB173 amino acid residue, between the 3-OMe and the hydroxy moiety of TyrD91 and between the pyridine 6-OH and the hydroxy moiety of MetC39 (Figure 2A). This is in agreement with the observed inactivity of the 6-deshydroxy or protected analogues 17, 19, 20 and 24. These predicted active site interactions match those obtained from the crystal structure of AA5 with porcine heart mitochondria CII (PDB ID: 3AAE), when 16c is overlaid with AA5 (Figure 2B).
Figure 2.

Modelling of selected AA5 derivatives in the ubiquinone binding site of porcine heart mitochondria complex II (PDB ID: 3AEE). A) Hypothetical binding interactions of CII inhibitor 16c. B) Overlay of AA5 (7) (turquoise) and 16c (gold) in active site of porcine heart mitochondria complex II. C) Hypothetical binding interactions of inactive compound 18. D) Hypothetical binding interactions of inactive compound 24 (gold), overlayed with AA5 (7) (turquoise). Red;oxygen, blue; nitrogen, white; hydrogen, gold; carbon. Green dotted lines represent hydrogen bond (greater opacity represents stronger bond).
QSAR prediction of the inactive alcohol derivative 18 shows a significant change of binding pose, which is sufficient to prevent formation of, or reduce the strength of, hydrogen bond interactions between the pyridine 3-OMe and TyrD91 and between the pyridine 6-OH and MetC39. The 6-OH hydrogen is predicted instead to partake in intramolecular hydrogen bonding to the side chain alcohol, forming a thermodynamically stable six-membered ring (Figure 2C). The methyl ether derivative 24, lacking the pyridine 6-OH, undergoes a significant change in binding pose compared with AA5 (Figure 2D) resulting in only hydrogen bond interactions involving the pyridine 4-OH moiety and thus loss of CII inhibition activity.
The QSAR ligand-protein interaction predictions show excellent correlation with the determined SAR. The molecular modelling studies and SAR data reveal a pharmacophoric role for the side chain ketone moiety, not as might be expected by participating in hydrogen bonding to CII binding site residues, but by conferring a bioactive conformation to active inhibitors. Indeed, in all three compound interaction models (16c, 18 and 24, Figure 2) and in the original crystal structure of AA5 bound with porcine heart mitochondria CII, the ketone oxygen is not predicted to be involved in intermolecular hydrogen bonding. Excision of the carbonyl moiety leads to conformational changes that reduce distances between hydrogen bond donors and receptors and leads to suboptimal bonding angles.
2.4 Cytotoxicity assay
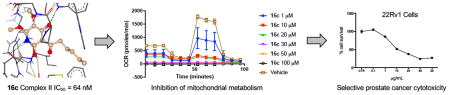
Derivative 16c was taken forward for cytotoxicity evaluation. This derivative was chosen for further study due to its structural resemblance to natural AA5, same PSA, yet lower lipophilicity engendering greater solubility, structural simplicity and favorable LLE value (5.62) combined with CII inhibition potency (IC50 = 64 nM, 17.23 × 10-3 μg/mL).Three human prostate cancer cell lines and HEK293 cells were exposed to compound 16c and cytotoxicity measured by MTS assay after 48 hours (Figure 3).
Figure 3.

Complex II inhibitors are potent anti-proliferative agents. A) Dose-response curve of effect of complex II inhibitor 16c on percentage cell viability of DU-145 prostate cancer cells. B) Dose-response curve of effect of complex II inhibitor 16c on percentage cell viability of PC3 prostate cancer cells. C) Dose-response curve of effect of complex II inhibitor 16c on percentage cell viability of 22Rv1 prostate cancer cells. D) Dose-response curve of effect of complex II inhibitor 16c on percentage cell viability of low tumorigenic HEK293 cells. E) Dose-response curve of the effect of structurally similar but inactive complex II inhibitor 24 on percentage cell viability of DU-145 prostate cancer cells. F) Dose-response curve of the effect of clinical chemotherapeutic enzalutamide on percentage cell viability of 22Rv1 prostate cancer cells after 72 hour incubation. Cell viability measured after 48-hour incubation of compound unless otherwise noted. Values are the mean ± S.D. of triplicate experiments.
Treatment of DU-145 prostate cancer cells with CII inhibitor 16c provided a significant inhibitory effect on cell growth in a dose-dependent manner, resulting in an IC50 of 13 μg/mL (Figure 3A). When inhibitor 16c was exposed to PC3 prostate cancer cells a much lower inhibitory effect was observed, resulting in an IC50 of 55 μg/mL (Figure 3B). In 22Rv1 prostate cancer cells, inhibitor 16c demonstrated an IC50 of 11 μg/mL (Figure 3C). Selectivity to cancerous cells over low tumorigenic cells, was confirmed by the use of human embryonic kidney cells (HEK293) where 16c demonstrated an IC50 of 71 μg/mL, an approximate 7-fold selectivity for DU-145 and 22Rv1 prostate cancer cells over this transformed cell line (Figure 3D). The inactive control compound 24 displayed no growth inhibition effect upon treatment of DU-145 cells up to 30 μg/mL (Figure 3E), implicating CII inhibition as a primary mechanism of action to inhibit prostate cancer cell growth. The clinically available prostate cancer drug enzalutamide displayed an IC50 of 29 μg/mL after 72 hour treatment in 22Rv1 prostate cancer cells (Figure 3F), a cell line known to be resistant to this chemotherapeutic.[36] CII inhibitor 16c displayed an IC50 of 11 μg/mL after just 48 hours. This observed cytotoxicity, along with that of CII inhibitor 16k (Figure 4), suggests that CII inhibition may be a novel target for the treatment of advanced drug-resistant prostate cancer.
Figure 4.

Complex II inhibitors with greater potency convey greater cytotoxicity. A) Dose-response curve of the effect of complex II inhibitor 16k on percentage cell viability of 22Rv1 prostate cancer cells. B) Dose-response curve of effect of complex II inhibitor 16k on the percentage cell viability of low tumorigenic HEK293 cells. Cell viability measured after 48-hour incubation of compound. Values are the mean ± S.D. of triplicate experiments.
Further, when the more potent CII inhibitor 16k (IC50 = 3.3 nM, 1.21 × 10-3 μg/mL) was exposed to 22Rv1 prostate cancer cells, an even greater reduction of cell viability was observed with an IC50 of just 2 μg/mL (Figure 4A). This shows that CII inhibition potency is directly proportional to anti-proliferative effect. However, maximal cytotoxicity is reached at 10 μg/mL, beyond which a plateau is observed. Selectivity over the low tumorigenic HEK293 cell line was again observed with 16k providing an IC50 of 28 μg/mL, conferring 14-fold selectivity (Figure 4B). A summary of the IC50 values for compounds 16c and 16k across prostate cancer cell lines and HEK293 cells compared with enzalutamide positive control is depicted in table 2.
Table 2.
Activity of CII inhibitors 16c and 16k, the inactive control 24 and the clinical chemotherapeutic enzalutamide across a panel of human prostate cancer cells and low tumorigenic human endothelial kidney cells. IC50 values are mean of n = 3 experiments ± S.D.
| Compound | DU-145 IC50 (μg/mL) | PC3 IC50 (μg/mL) | 22Rv1 IC50 (μg/mL) | HEK293 IC50 (μg/mL) |
|---|---|---|---|---|
| 16c | 13 ± 1.6 | 55 ± 9.4 | 11 ± 1 | 71 ± 5.2 |
| 16k | N.D.a | N.D. | 2 ± 0.5 | 28 ± 2 |
| 24 | >30b | N.D. | ND | N.D. |
| Enzalutamide | N.D. | N.D. | 29 ± 2.4 | N.D. |
Not Determined
No cell death at 30 μg/mL concentration.
To determine if our synthetic CII inhibitors retain the selectivity of the parent compound for CII over other constituent members of the electron transport chain we assayed 16c and 16K for mitochondrial complex I (CI) inhibition activity. Both compounds were found to be inactive up to 10 μM leading to a selectivity ratio for CII:CI of >156-fold for 16c and >3030-fold for 16k (Table 3).This data highlights these compounds as promising chemical probes for further interrogation of the NADH-fumarate reductase system, specifically targeting complex II.
Table 3.
Selectivity of compounds 16c, 16k and AA5 for inhibition of mitochondrial complex II over complex I.
| Compound | Complex II IC50 (nM) | Complex I IC50 (nM) | Selectivity Ratio (CII:CI) |
|---|---|---|---|
| 16c | 64 ± 4.3 | >10,000 | >156 |
| 16k | 3.3 ± 2.4 | >10,000 | >3030 |
| 7 (AA5) | 3.3 ± 2.0 | >10,000 | >3030 |
A recent report identified AA5 as an active hit in a screen for the identification of compounds that target cells in dormant tumor spheroid regions.[37] Cancer cells in poorly vascularized tumor regions react to diminished nutrients by stopping cell cycle progression and becoming dormant. Hence those cells in hypoxic regions of the tumor are more likely to be resistant to chemotherapeutics. Utilizing a microfluidic culture system,[38] we exposed PC3 prostate cancer cells under hypoxic conditions to the clinical chemotherapeutic etoposide, CII inhibitor 16c and inactive control 18 (Figure 5). As expected, the inactive compound 18 demonstrated no cytotoxic effect up to 10 μM concentration within experimental error, further supporting the antineoplastic role of the CII inhibition mechanism. Complex II inhibitor 16c provided 12% cell death at 1 μM concentration and 20% cell death at 10 μM concentration. Etoposide is known to have poor cytotoxicity in hypoxic environments,[39] displaying only 7% cell death at 10 μM concentration in hypoxic assay. Our designed CII inhibitor (16c), at 10-fold lower concentration, provides superior cytotoxicity in PC3 cells under hypoxia than etoposide.
Figure 5.

Cytotoxic effect of the clinical chemotherapeutic etoposide, complex II inhibitor 16c and inactive control 18 in PC3 prostate cancer cells under hypoxia. Cell death was measured with Annexin V and Sytox Green. Values are the mean ± S.D. for triplicate experiments. A one-way ANOVA analysis was used to compare statistical difference between etoposide and 16c at 1 μM concentrations, p = 0.0068.
We next sought to determine the effect of complex II inhibition by our developed inhibitors on mitochondrial metabolic processes. The effect of 16c on electron transport parameters of 22Rv1 cells after 48 hour incubation was studied using a Seahorse XF Extracellular Flux Analyzer. The oxygen consumption rate of 22Rv1 cells is substantially reduced in a dose-dependent manner upon exposure to 16c (Figure 3A). CII inhibitor 16c significantly inhibited mitochondrial function in 22Rv1 prostate cancer cells in a dose-dependent manner, including reducing basal respiration (Figure 3B), inhibition of ATP production (Figure 3C) and reduction of maximal respiration (Figure 3D) compared with no treatment control.
Conclusion
We disclose the design, synthesis and evaluation of ten highly potent CII inhibitors. Analysis of physicochemical properties indicated compound 16c as the most ‘drug-like’ molecule for further study, while the potency of 16c is reduced (CII IC50 = 64 nM) over 16k (CII IC50 = 3.3 nM) the ligand-lipophilicity efficiency of 16c is the most optimal of the CII inhibitors synthesized and represents far greater potency than existing inhibitors while retaining ‘drug-like’ properties and selectivity for mitochondrial complex II over mitochondrial complex I. Significant anti-proliferative activity was demonstrated by 16c in DU-145 and 22Rv1 human prostate cancer cell lines. This effect was shown to be selective with a 7-fold greater anti-proliferative activity over low tumorigenic human embryonic kidney cells. Both compounds 16c and 16k showed superior anti-proliferative activity to the clinically approved chemotherapeutic enzalutamide. Further, this effect is retained under hypoxic conditions. Inhibitor 16c significantly inhibited mitochondrial electron transport parameters, reducing oxygen consumption rate, basal respiration, ATP production and maximal respiration.
In summary, for the first time, truly potent CII inhibitors with nanomolar activity are shown to elicit significant and selective anti-proliferative activity. CII inhibitors 16c, 16j and 16k represent valuable molecular tools to study the effect of CII in cancer and other diseases.
4. Experimental
4.1 Chemistry
General
All reactions were carried out in oven- or flame-dried glassware under positive nitrogen pressure unless otherwise noted. Reaction progress was monitored by thin-layer chromatography (TLC) carried out on silica gel plates (2.5 cm × 7.5 cm, 200 μm thick, 60 F254) and visualized by using UV (254 nm) or by potassium permanganate and/or phosphomolybdic acid solution as indictor. Flash column chromatography was performed with silica gel (40-63 μm, 60 Å) or on a Teledyne Isco (CombiFlash Rf 200 UV/Vis). Commercial grade solvents and reagents were purchased from Fisher Scientific (Houston, TX) or Sigma Aldrich (Milwaukee, WI) and were used without further purification except as indicated. Anhydrous solvents were purchased from Across Organics and stored under and atmosphere of dry nitrogen over molecular sieves.
[1]H, 13C, COSY, HMQC and DEPT NMR spectra were recorded in the indicated solvent on a Bruker 400 MHz Avance III HD spectrometer at 400 and 100 MHz for 1H and 13C respectively with TMS as an internal standard. Multiplicities are indicated by s (single), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), br (broad). Chemical shifts (δ) are reported in parts per million (ppm), and coupling constants (J), in hertz. High-resolution mass spectrometry was performed on a LC/MS IT-TOF (Shimadzu) using an ESI source conducted at the University of Texas at Arlington, Shimadzu Center for Advanced Analytical Chemistry. High-pressure liquid chromatography was performed on a Gilson HPLC system with 321 pumps and 155 UV/Vis detector using trilution software v2.1 with an ACE Equivalence 3 (C18, 3 μM, 4.6 × 150 mm) column. All samples were determined to possess >95% purity.
2,3-dimethoxypyridin-4-ol (9)
[29] To a solution of 2,3-dimethoxypyridine (0.30 g, 2.16 mmol) in THF (15 mL) at -78 °C was added nBuLi (1.3 mL, of a 2.5 M solution in hexanes, 3.24 mmol). The mixture was stirred for 1 h at 0 °C in an ice-water bath. The reaction mixture was cooled to -78 °C, B(OMe)3 (0.62 mL, 5.4 mmol) was added and the mixture stirred at -78 °C for 2 h. A solution of peracetic acid (32 wt % in dilute acetic acid, 1.02 mL, 4.32 mmol) was added and the mixture was slowly warmed to room temperature over one hour. To the reaction solution was added an aqueous solution of sodium bisulfite. The solution was extracted with ethyl acetate, washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and purification by flash column chromatography (DCM:Et2O 94:6) afforded the title compound as a colorless oil (211 mg, 63%): 1H NMR (400 MHz, CDCl3) δ 3.71 (s, 3H), 3.90 (s, 3H), 6.48(d, 1H, J = 5.7 Hz), 7.61 (d, 1H, J = 5.7 Hz), 7.99 (s, 1H); 13CNMR (100 MHz, CDCl3) δ 53.30, 60.20, 107.30, 130.40, 140.70, 156.60, 157.80.
5-bromo-2,3-dimethoxypyridin-4-ol (10)
To a solution of 2,3-dimethoxypryidin-4-ol (0.106 g, 0.68 mmol) in MeCN (5 mL) at 0 °C was added NBS (0.134g, 0.75 mmmol). The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction mixture was extracted with ethyl acetate, the organic layer washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and purification by flash column chromatography (Hexane:EtOAc 20:1) afforded the title compound as a yellow powder (0.132 g, 83%): 1H NMR (400 MHz, CDCL3) δ 3.91 (s, 3H), 3.99 (s, 3H), 6.77 (s, 1H),7.90 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 53.76, 60.99, 100.88, 130.63, 141.98, 152.60, 156.59; HRMS (ESI) m/z calcd for C7H8BrNO3 (M+Na+): 255.9580, found 255.9574.
5-bromo-2,3-dimethoxy-4-(methoxymethoxy)pyridine (11)
To a solution of 5-bromo-2,3-dimethoxypyridin-4-ol (10) (3.68 g, 15.7 mmol) in DMF (50 mL) at 0 °C was added NaH (0.943 g, 23.6 mmol of a 60% dispersion in mineral oils). The mixture was stirred for 10 minutes and methoxymethyl chloride (1.79 ml, 23.6 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred for 1 h. The reaction mixture was extracted with ethyl acetate, the organic layer washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and purification by flash column chromatography (Hexane:EtOAc 20:1) afforded the title compound as a yellow powder (4.11g, 94%): 1H NMR (400 MHz, CDCl3) δ 3.57(s, 3H), 3.82 (s, 3H), 3.94 (s, 3H), 5.33 (s, 2H), 7.90 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 53.88, 57.68, 60.51, 98.57, 107.24, 136.29, 142.25, 153.16, 158.56; HRMS (ESI) m/z calcd for C9H12NBrO4 (M+Na+) 299.9842 found 299.9835.
6-bromo-2,3-dimethoxy-4-(methoxymethoxy)pyridine (12)
Lithium diisopropylamide (LDA) was prepared by addition of nBuLi (1.41 mL, 1.6 M in hexanes, 2.26 mmol) to diisopropylamine (0.317 mL, 2.26 mmol) in THF (10 mL) at -78 °C and warmed up to 0 °C for 30 min with stirring. LDA solution was cooled to -78 °C and a solution of 5-bromo-2,3-dimethoxy-4-(methoxymethoxy)pyridine (11) (0.21 g, 0.76 mmol) in THF (10 mL) was added. The solution was warmed to -40 °C in an acetonitrile-dry ice bath and 2 uL of bromine was added. The reaction mixture was stirred for 1 h at -40 °C and then cooled to -78 °C. An excess of EtOH (4 mL) was added and the mixture was warmed up to 23 °C, which was treated with a saturated aqueous solution of NH4Cl (5 mL) and then extracted with ethyl acetate. The organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and purification by flash column chromatography (Hexane:EtOAc 10:1) afforded the title compound as a yellow powder (0.187 g with 89%): 1H NMR (400 MHz, CDCl3) δ 3.39 (s, 3H), 3.72 (s, 3H), 3.86 (s, 3H), 5.14 (s, 2H), 6.82 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 54.06, 56.30, 60.36, 94.42, 108.95, 131.10, 131.69, 158.5, 157.43; HRMS (ESI) m/z calcd for C9H12NBrO4 (M+Na+) 299.9842 found 299.9839.
5,6-dimethoxy-4-(methoxymethoxy)pyridin-2-ol (13)
To a solution of 6-bromo-2,3-dimethoxy-4-(methoxymethoxy)pyridine (12) (3.79 g, 13.6 mmol) in THF (120 mL) at -78 °C was quickly added BuLi (18.7 mL, of a 1.6 M solution in hexanes, 40.9 mmol). The mixture was stirred for 1 minute and B(MeO)3 (4.89 mL, 40.9 mmol) was added with stirring for 2 h at -78 °C. A solution of peracetic acid (32 wt % in dilute acetic acid, 12.9 mL, 54.5 mmol) was then added and the reaction mixture allowed to warm to 0 °C under stirring for 2 h. An aqueous solution of sodium hydrogenosulfite was added dropwise, and the mixture extracted with ethyl acetate. The organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and purification by flash column chromatography (Hexane:EtOAc 10:1) afforded the title compound as a yellow powder (1.86g, 63%): 1H NMR (400 MHz, CDCl3) δ 3.41(s, 3H), 3.69 (s, 3H), 3.86 (s, 3H), 5.16 (s, 2H), 6.04 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 54.90, 56.45, 60.99, 89.02, 94.36, 126.31, 155.69, 158.34, 160.32; HRMS (ESI) m/z calcd for C9H13NO5 (M+Na+) 238.0686 found 238.0678
2,3-dimethoxy-4,6-bis(methoxymethoxy)pyridine (14)
To a solution of 5,6-dimethoxy-4-(methoxymethoxy)pyridin-2-ol (13) (1.805 g, 8.38 mmol) in DMF (10 mL) at 0 °C was added NaH (0.537 g, 13.4 mmol, of a 60% dispersion in mineral oils). After 10 minutes, methoxymethyl chloride (0.96 mL, 12.6 mmol) was added. The reaction mixture was allowed to warm to room temperature and stirred for 1h. The reaction mixture was extracted with ethyl acetate, the organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and purification by flash column chromatography (Hexane:EtOAc 10:1) afforded the title compound as a yellow powder (2.05 g, 94%): 1H NMR (400 MHz, CDCl3) δ 3.48 (s, 3H), 3.49 (s, 3H), 3.77 (s, 3H), 3.92 (s, 3H), 5.22 (s, 2H), 5.41 (s, 2H), 6.21 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 53.67, 56.47, 56.96, 60.86, 88.99, 91.95, 94.54, 127.50, 156.02, 156.46, 159.21; HRMS (ESI) m/z calcd for C11H17NO6 (M+Na+) 282.0948 found 282.0940.
(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)(furan-2-yl)methanone (16a). General method for the synthesis of compounds 16a-16k
To a solution of 2,3-dimethoxy-4,6-bis(methoxymethoxy)pyridine (14) (0.0290 g, 0.11 mmol) in THF (5 mL) at -78 °C was added nBuLi (0.09 mL, 2.5 M, 0.22 mmol) and the reaction mixture stirred for 1 h. Furan-2-carbaldedhye (10.6 mg, 0.11 mmol) was added and the mixture stirred for 1 h. The reaction was quenched by addition of EtOH and extracted with EtOAC, the organic washings were washed with brine and dried over Na2SO4, the solvent was removed in vacuo and the crude product used for the next step without further purification. To a solution of the crude material in CH2Cl2 (5 mL) at room temperature was added Dess-Martin Periodinane (0.071 g, 0.17mmol), and the reaction mixture stirred for 30 minutes. The reaction was quenched by addition of saturated sodium thiosulfate and extracted with EtOAc, the organic washings were washed with brine and dried over Na2SO4, the solvent was removed in vacuo and the crude product used for the next step without further purification. To a solution of the crude material in CH2Cl2 (2 mL) at 0 °C was added TFA (0.5 mL) and the reaction mixture stirred for 30 minutes. The solvent was removed in vacuo and the residue purified by flash column chromatography (DCM:MeOH 97:3) to afford the title compound as a colorless oil (0.014 g, 48%): 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 4.11 (s, 3H), 6.57 (1H, s), 7.68 (2H, m, 2× CH); 13C NMR (100 MHz, CDCl3) δ 57.31, 61.54, 111.97, 120.97, 122.16, 146.87, 151.46, 156.19, 160.53, 182.98.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)pentan-1-one (16b)
A colorless oil (0.015 g, 53%): 1H NMR (400 MHz, CDCl3) δ 0.96 (3H, t, J = 7.0 Hz), 1.33 (m, 2H), 1.66 (m, 2H), 3.05 (2H, t, J = 7.0 Hz), 3.79 (s, 3H), 4.16 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 13.94, 22.51, 26.32, 42.25, 61.51, 101.04, 121.05, 155.56, 162.07, 205.93; HRMS (ESI) m/z calcd for C12H17NO5 (M+Na+) 278.0999 found 278.1005.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)hexan-1-one (16c)
A colorless oil (0.037 g, 51%): 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, t, J = 7.0 Hz), 1.30 (m, 4H), 1.65 (m, 2H), 3.04 (2H, t, J = 7.0 Hz), 3.80 (s, 3H), 4.16 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 14.0, 22.4, 24.07, 31.82, 42.50, 57.78, 60.61, 101.55, 121.06, 155.08, 161.82, 206.60; HRMS (ESI) m/z calcd for C13H19NO5 (M+Na+) 292.1161 found 292.1155.
5-(benzyloxy)-1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)pentan-1-one (16d)
A colorless oil (0.098 g, 58%): 1H NMR (400 MHz, CDCl3) δ 1.77 (m, 4H), 1.25 (m, 18 H), 3.09 (2H, t, J = 6.3 Hz), 3.51 (2H, t, J = 6.3 Hz), 3.78 (s, 3H), 4.15 (s, 3H), 4.50 (s, 2H), 7.32 (m, 5 H); 13C NMR (100 MHz, CDCl3) δ 20.94, 29.37, 42.26, 57.94, 61.49, 70.16, 72.92, 100.67, 121.49, 127.49, 127.60, 128.34, 138.55, 155.94, 161.82, 205.74; HRMS (ESI) m/z calcd for C19H23NO6 (M+Na+) 384.1423 found 384.1417.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)-5-hydroxypentan-1-one (16e)
A colorless oil (0.045 g, 56%): 1H NMR (400 MHz, CD3OD) δ 1.60 (m, 2H), 1.73 (m, 2 H), 3.10 (2H, t, J = 7.4 Hz), 3.59 (2H, t, J = 7.4 Hz), 3.71 (s, 3H), 4.99 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 21.86, 33.26, 43.84, 61.19, 62.75, 101.06, 124.70, 160.08, 162.86, 207.51; HRMS (ESI) m/z calcd for C12H17NO6 (M+Na+) 294.0948 found 294.0954.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)heptan-1-one (16f)
A colorless oil (0.025 g, 52%): 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, t, J = 7 Hz), 1.31 (m, 6H), 1.66 (m, 2H), 3.03 (2H, t, J = 7 Hz), 3.79 (s, 3H), 4.16 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 13.94, 22.51, 26.32, 42.25, 57.63, 61.51, 101.04, 121.05, 155.56, 162.07, 205.93; HRMS (ESI) m/z calcd for C14H21NO5 (M+Na+) 306.1312 found 306.1308.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)octan-1-one (16g)
A colorless oil (0.017 g, 51%): 1H NMR (400 MHz, CDCl3) δ 0.88 (3H, t, J = 7.0 Hz), 1.31 (m, 8H), 1.64 (m, 2H), 3.06 (2H, t, J = 7.0 Hz), 3.79 (s, 3H), 4.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 14.08, 22.64, 24.21, 29.17, 29.39, 31.75, 42.56, 57.63, 61.50, 100.99, 121.25, 155.95, 161.82, 206.33; HRMS (ESI) m/z calcd for C15H23NO5 (M+Na+) 320.1468 found 320.1463.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)nonan-1-one (16h)
A colorless oil (0.019 g, 55%): 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, t, J = 7.0 Hz), 1.29 (m, 10 H), 1.69 (m, 2H), 3.10 (2H, t, J = 7.0 Hz), 3.82 (s, 3H), 4.21 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 14.10, 22.67, 24.15, 29.19, 29.34, 29.44, 31.85, 42.77, 58.33, 61.68, 100.48, 121.96, 154.84, 161.67, 206.41; HRMS (ESI) calcd for C16H25NO5 (M+Na+) 334.1625 found 334.1626.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)decan-1-one (16i)
A colorless oil (0.018 g, 52%): 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 7.0 Hz), 1.26 (m, 12 H), 1.65 (m, 2H), 3.06 (2H, t, J.0 = 7 Hz), 3.78 (s, 3H), 4.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 14.10, 22.65, 24.19, 29.29, 29.42, 29.51, 31.87, 42.56, 57.63, 61.52, 100.05, 121.10, 155.19, 161.65, 206.76; HRMS (ESI) m/z calcd for C17H27NO5 (M+Na+) 348.1781 found 348.1778.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)undecan-1-one (16j)
A colorless oil (0.028 g, 55%): 1H NMR (400 MHz, CDCl3) δ 0.87 (3H, t, J = 7.0 Hz), 1.26 (m, 14 H), 1.65 (m, 2H), 3.04 (2H, t, J = 7.0 Hz), 3.78 (s, 3H), 4.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 14.09, 22.66, 24.20, 29.32, 29.43, 29.51, 29.55, 29.59, 31.88, 42.57, 57.25, 61.50, 100.69, 121.26, 155.67, 160.99, 206.08; HRMS (ESI) m/z calcd for C18H29NO5 (M+Na+) 362.1938 found 362.1937.
1-(2,4-dihydroxy-5,6-dimethoxypyridin-3-yl)tridecan-1-one (16k)
A colorless oil (0.035 g, 56%): 1H NMR (400 MHz, CDCl3) δ 0.89 (3H, t, J = 7.0 Hz), 1.25 (m, 18 H), 1.68 (m, 2H), 3.07 (2H, t, J = 7.0 Hz), 3.79 (s, 3H), 4.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 14.10, 22.67, 24.20, 29.34, 29.42, 29.51, 29.55, 29.64, 29.67, 31.90, 42.55, 57.6361.51, 101.07, 121.25, 155.19, 161.80, 206.42; HRMS (ESI) m/z calcd for C18H29NO5 (M+Na+) 390.2251 found 390.2256.
1-(5,6-dimethoxy-2,4-bis(methoxymethoxy)pyridin-3-yl)hexan-1-ol (17)
To a solution of 2,3-dimethoxy-4,6-bis(methoxymethoxy)pyridine (14) (0.030 g, 0.116 mmol) in THF (3 mL) at -78 °C was added nBuLi (0.19 mL, of a 2.5 M solution in hexanes, 0.35 mmol), which was stirred for 1 h. Hexanal (0.074 mL, 0.58 mmol) was added and the reaction stirred for 1 h. The reaction was quenched with ethanol and extracted with ethyl acetate, the organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and the residue purified by column chromatography (Hexanes:EtOAc 4:1) to afford the title compound as a colorless oil (33.3 mg, 80%): 1H NMR (400 MHz, CDCl3) δ 0.89 (t, 3H, J = 7.0 Hz), 1.30 (m, 4H), 1.52 (m, 2H), 1.74 (m, 1H),1.91 (m, 1H), 3.22 (s, 1H), 3.53 (s, 3H), 3.56 (s, 3H), 3.75 (s, 3H), 3.93 (s, 3H), 4.95 (t, 1H, J = 6.7 Hz), 5.29 (d, 1H, J = 5.0 Hz), 5.32 (d, 1H, J = 5.0 Hz), 5.49 (d, 1H, J = 5.0 Hz), 5.60 (d, 1H, J = 5.0 Hz); 13C NMR (100 MHz, CDCl3) δ 13.9, 22.2, 25.9, 31.8, 37.5, 53.6, 57.3, 57.8, 60.6, 67.3, 91.8, 99.2, 112.0, 129.0, 153.0, 154.9, 156.8.
(rac)-3-(1-hydroxyhexyl)-5,6-dimethoxypyridine-2,4-diol (18)
To a solution of 2,3-dimethoxy-4,6-bis(methoxymethoxy)pyridine (14) (0.020 g, 0.077 mmol) in THF (5 mL) at -78 °C was added nBuLi (0.068 mL, of a 2.5 M solution in hexanes, 0.17 mmol), which was stirred for 1 h. Hexanal (0.010 mL, 0.077 mmol) was added and the reaction stirred for 1 h. The reaction was quenched with ethanol and extracted with ethyl acetate, the organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and the crude product used for the next step without further purification. To a solution of the crude material in CH2Cl2 (4 mL) at 0 °C was added TFA (1 mL) and the reaction stirred for 30 minutes. The solvent was removed in vacuo and the residue purified by flash column chromatography (DCM:MeOH 97:3) to afford the title compound as a racemic mixture as a colorless oil (0.061 g, 63%): 1H NMR (400 MHz, CDCl3) δ 0.87 (m, 4H), 1.29 (m, 6H), 3.81 (m, 4H), 3.93 (s, 3H), 5.55 (d, 1H, J = 7.0 Hz), 7.71 (br. s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.89, 22.63, 25.17, 31.79, 36.94, 72.83, 80.03, 157.16, 163.58; HRMS (ESI) m/z calcd for C13H21NO5 (M+H-H2O+) 254.1387 found 254.1366.
(rac)-5-(1-hydroxyhexyl)-2,3-dimethoxypyridin-4-ol (19)
To a solution of 5-bromo-2,3-dimethoxy-4-(methoxymethoxy)pyridine (11) (0.032 g, 0.115 mmol) in THF (5 mL) at -78 °C was added nBuLi (0.10 mL, of a 2.5 M solution in hexanes, 0.25 mmol), which was stirred for one minute. Hexanal (0.012 g, 0.15 mmol) was added and stirring continued for 1 h. The reaction was quenched by ethanol and extracted with ethyl acetate, the organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and the residue purified by flash column chromatography (Hexane:EtOAc 4:1) to afford the title compound as a colorless oil (25 mg, 85%): 1H NMR (400 MHz, CDCl3) δ 0.89 (m, 2H), 1.30 (m, 6H), 1.73 (q, J = 5.5 Hz), 3.90 (s, 3H), 4.27 (s, 3H), 4.86 (br. t, 1H, J = 5.5 Hz), 7.76 (s, 1H), 10.59 (br. s, 2H); 13C NMR (100 MHz, CDCl3) δ 13.90, 22.45, 25.0, 31.37, 37.0, 58.58, 61.40, 69.54, 114.39, 117.28, 124.97, 130.53, 132.05, 155.08, 162.0.
1-(4-hydroxy-5,6-dimethoxypyridin-3-yl)hexan-1-one (20)
To a solution of 5-(1-hydroxyhexyl)-2,3-dimethoxypyridin-4-ol (19) (28 mg, 0.11 mmol) in CH2Cl2 (5 mL) at room temperature was added Dess-Martin Periodinane (0.073 g, 0.17 mmol) and the reaction stirred for 30 minutes. The reaction was quenched by addition of saturated sodium thiosulfate and extracted with ethyl acetate, the organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and the crude product used for the next step without further purification. To a solution of the crude material in CH2Cl2 (4 mL) at 0 °C was added TFA (1 mL) and the reaction stirred for 30 minutes. The solvent was removed in vacuo and the residue purified by flash column chromatography (DCM:MeOH 97:3) to afford the title compound as a colorless oil (0.015 g, 51%): 1H NMR (400 MHz, CDCl3) δ 0.91 (3H, t, J = 7.0 Hz), 1.37 (m, 6H), 1.75 (m, 2H), 2.93 (2H, t, J = 7.0 Hz), 3.89 (s, 3H), 4.05 (s, 3H), 8.44 (s, 1H), 12.63 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.88, 22.43, 24.44, 31.41, 38.19, 54.40, 60.57, 114.55, 130.525, 145.07, 160.81, 161.39, 206.02; HRMS (ESI) m/z calcd for C13H19NO4 (M+Na+) 276.1206 found 276.1207.
2,3-dimethoxypyridin-4-yl diisopropylcarbamate (21)
[31] To a solution of 2,3-dimethoxypyridin-4-ol (9) (0.30 g, 1.93mmol) in toluene (10 mL) was added silver carbonate (0.80 g, 2.9 mmol) and diisopropylcarbamyl chloride (0.484 g, 2.9 mmol) and the reaction mixture was refluxed for 3 h. The suspension was allowed to cool and passed through celite® washing with MeOH, removal of the solvent in vacuo and purification by chromatography (DCM:Et2O 96:4) provided the title compound as a colorless oil with yield (398 mg, 73%): 1H NMR (400 MHz, CDCl3) δ 1.26 (d, 12H, J = 6.8 Hz), 3.81 (s, 3H), 3.95 (s, 3H), 3.99 (m, 2H), 6.68 (d, 1H, J = 5.6 Hz), 7.77 (d, 1H, J = 5.6 Hz); 13CNMR (100 MHz, CDCl3) δ 20.2, 21.0, 46.6, 53.5, 60, 112.7, 135.7 140.4, 150.8, 152.1, 158.8.
5-bromo-2,3-dimethoxypyridin-4-yl diisopropylcarbamate (22)
[31] To a solution of 2,3-dimethoxypyridin-4-yl diisopropylcarbamate (21) (0.60 g, 2.12 mmol) in CCl4 (20 mL) was added bromine (0.273 mL, 5.31 mmol) and the reaction mixture stirred at room temperature for 48 h, which was shielded from light with aluminum foil. To the reaction mixture was added a solution of saturated aqueous NaHCO3 (5 mL) and Na2S2O3 (5 mL) and extracted with ethyl acetate. The organic phase was washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and the residue purified by column chromatography (DCM:Et2O 98:2) to afford the title compound as a yellow solid (383 mg, 50%): 1H NMR (400 MHz, CDCl3) δ 1.29 and 1.34 (2d, 12H, J = 6.8 Hz), 3.86 (s, 3H), 3.97 (s, 3H), 4.01-4.04 (m, 2 H), 7.99 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 19.9, 20.8, 46.6, 46.9, 53.4, 58.9, 107.5, 137.1, 141.1, 148.3, 150.5, 157.5.
5-(1-hydroxyhexyl)-2,3-dimethoxypyridin-4-yl diisopropylcarbamate (23)
To a solution of 5-bromo-2,3-dimethoxypyridin-4-yl diisopropylcarbamate (22) (0.12 g, 0.33 mmol) in THF (3 mL) at -78 °C was added nBuLi (0.45 mL, of a 2.5 M solution in hexanes, 1.1 mmol) which was stirred for 1 minute. Hexanal (0.24 mL, 1.66 mmol) was added and the reaction mixture stirred for 1 h. The reaction was quenched by addition of ethanol and extracted with ethyl acetate, the organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacuo and the residue purified by column chromatography (Hexane:EtOAc 4:1) to afforded the title compound as a colorless oil (104.7 mg, 83%): 1H NMR (400 MHz, CDCl3) δ 0.86 (t, 3H, J = 7.0 Hz), 1.1-1.5 (m, 18H), 1.70-2.00 (m, 2H), 3.82 (s, 3H), 4.00 (s, 3H), 3.98-4.12 (m, 2H), 4.69 (t, 1H, J=6.7 Hz), 7.97 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.1, 20.4, 21.3, 22.6, 25.7, 31.6, 36.7, 47, 47.1, 53.8, 60.1, 60.3, 128.1, 135.6, 138.2, 148.9, 153.1, 158.
2,3-dimethoxy-5-(1-methoxyhexyl)pyridin-4-ol (24)
5-(1-hydroxyhexyl)-2,3-dimethoxypyridin-4-yl diisopropylcarbamate (23) (0.090 g, 0.23 mmol) was refluxed in KOH solution (5 M, in methanol, 10 mL) for 20 h. The solvent was removed in vacuo and diluted acetic acid added. The reaction mixture was neutralized by addition of saturated aqueous NaHCO3, extracted with ethyl acetate. The organic washings were washed with brine and dried over sodium sulfate, the solvent was removed in vacco and purification by column chromatography afforded the title compound as a colorless oil (50%): 1H NMR (400 MHz, CDCl3) δ 0.85 (t, 3H, J = 7.0 Hz), 1.26 (m, 6H), 1.68-1.83 (m, 2H), 3.34 (s, 3H), 3.90 (s, 3H), 3.98 (s, 3H) 4.28 (t, 1H, J =6.7 Hz), 7.55 (s, 1H), 7.68 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9, 22.2, 25, 31.5, 35.7, 53.6, 57.1, 60.7, 80.7, 117.9, 130.4, 139.8, 154.8, 157.1; HRMS (ESI) m/z calcd for C14H23NO4 (M+Na+) 292.1525 found 292.1517.
Biology
Complex II inhibition Assay
Mitochondria were isolated from fresh rat hearts by differential centrifugation in sucrose-based buffer as previously described.[11] Complex II enzymatic activity was determined spectrophotometrically as the rate of succinate-driven, co-enzyme Q2-linked reduction of dichlorophenolindophenol (DCPIP). Mitochondria or sub-mitochondrial particles were incubated in phosphate buffer (pH 7.4) containing 40 μM DCPIP, 1 mM KCN, 10 μM rotenone, and 50 μM co-enzyme Q2. The rate of reduction of DCPIP to DCPIPH2 was followed at 600 nM (ε = 21 mM-1 cm-1).[40] The reaction was initiated by addition of succinate (10 mM), and varying amounts of inhibitors were used to determine an IC50 value. At the end of each run thenoyltrifluoroacetone (1 mM) was added and the residual TTFA-insensitive rate subtracted.
Complex I inhibition Assay
Complex I (NADH ubiquinone-oxidoreductase) was measured spectrophotometrically (340 nm) in frozen/thawed mouse cardiac mitochondria, as the rotenone-sensitive oxidation of NADH (40 uM) in the presence of co-enzyme Q1 (10 uM), in phosphate buffer at pH 7.2, according to literature procedures.[41]
Cytotoxicity Assays
DU-145, PC3, 22RV1 and HEK293 cell lines were purchased from ATCC and cultured according to the suppliers recommended protocol. To determine the cell growth inhibition ability of the synthesized compounds, cells were seeded at a density of 0.1 × 106 cells/mL in 96 well plates containing 100 μL cell suspension per well. Stock solutions of the synthesized compounds were prepared in DMSO. Cells were treated at the indicated concentrations of test compounds, limiting the final DMSO concentration to less than 1%. After incubation at 37°C, 5% CO2 for 48 hr, 20 μL of (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) reagent (CellTiter 96® AQueous One Solution Reagent) was added to each well and incubated at the above mentioned conditions for 3-4 hr. Plates were read at OD 490 nm on a plate reader and the viability of cells were plotted as percentage of controls.
Human PC3 prostate cancer cells were cultured in a microfluidic chip for 16 hours and the chip placed in a hypoxia chamber and pre-conditioned at <1% oxygen before treating the cells with various concentrations of synthesized compounds. Apoptosis was assayed using Annexin V and Sytox Green dye.[42]
Assessment of Metabolic Parameters
Changes in oxygen consumption rate were measured using a XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA). 22Rv1 cells were plated in XF24-well plates (40,000 cells/well) and allowed to adapt overnight. Treatments were performed with 16c at concentrations of 1, 10, 20, 30, 50 and 100 μM and incubated for 48 hours. Mito Stress test was performed using oligomycin (10 μM), FCCP (5 μM) and Rotenone/Antimycin A (5 μM) based on the manufacturer's recommended protocol.
Supplementary Material
Figure 6.

Complex II inhibitor 16c blocks mitochondrial respiration and function in 22Rv1 prostate cancer cells. A) Oxygen consumption rate (OCR), B) basal respiration, C) ATP production and D) maximal respiration are reduced in a dose-dependent manner. Error bars represent mean ± S.D. n = 3.
Acknowledgments
This work was funded by Texas Tech University Health Sciences Center School of Pharmacy. B.H. thanks the Kingdom of Saudi Arabia Cultural Mission to the U. S. A. for a graduate studentship. Work in the lab of PSB is funded by a grant from the US National Institutes of Health (R01-HL071158).
References
- 1.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Cell. 2005;121:1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 2.Ernster L, Dallner G. Biochim Biophys Acta. 1995;1271:195–204. doi: 10.1016/0925-4439(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 3.Kluckova K, Bezawork-Geleta A, Rohlena J, Dong L, Neuzil J. Biochim Biophys Acta. 2013;1827:552–564. doi: 10.1016/j.bbabio.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 5.Kluckova K, Sticha M, Cerny J, Mracek T, Dong L, Drahota Z, Gottlieb E, Neuzil J, Rohlena J. Cell Death Dis. 2015;6:e1749. doi: 10.1038/cddis.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peczkowska M, Cascon A, Prejbisz A, Kubaszek A, Cwikla BJ, Furmanek M, Erlic Z, Eng C, Januszewicz A, Neumann HP. Nat Clin Pract Endocrinol Metab. 2008;4:111–115. doi: 10.1038/ncpendmet0726. [DOI] [PubMed] [Google Scholar]
- 7.Tomitsuka E, Kita K, Esumi H. J Biochem. 2012;152:171–183. doi: 10.1093/jb/mvs041. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. J Cell Sci. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- 9.Fulda S. Int J Cancer. 2009;124:511–515. doi: 10.1002/ijc.24064. [DOI] [PubMed] [Google Scholar]
- 10.Neuzil J, Dyason JC, Freeman R, Dong LF, Prochazka L, Wang XF, Scheffler I, Ralph SJ. J Bioenerg Biomembr. 2007;39:65–72. doi: 10.1007/s10863-006-9060-z. [DOI] [PubMed] [Google Scholar]
- 11.Wojtovich AP, Brookes PS. Biochim Biophys Acta. 2008;1777:882–889. doi: 10.1016/j.bbabio.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gruber J, Staniek K, Krewenka C, Moldzio R, Patel A, Bohmdorfer S, Rosenau T, Gille L. Bioorg Med Chem. 2014;22:684–691. doi: 10.1016/j.bmc.2013.12.036. [DOI] [PubMed] [Google Scholar]
- 13.Neuzil J, Dong LF, Rohlena J, Truksa J, Ralph SJ. Mitochondrion. 2013;13:199–208. doi: 10.1016/j.mito.2012.07.112. [DOI] [PubMed] [Google Scholar]
- 14.Dong LF, Jameson VJ, Tilly D, Cerny J, Mahdavian E, Marin-Hernandez A, Hernandez-Esquivel L, Rodriguez-Enriquez S, Stursa J, Witting PK, Stantic B, Rohlena J, Truksa J, Kluckova K, Dyason JC, Ledvina M, Salvatore BA, Moreno-Sanchez R, Coster MJ, Ralph SJ, Smith RA, Neuzil J. J Biol Chem. 2011;286:3717–3728. doi: 10.1074/jbc.M110.186643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nadanaciva S, Bernal A, Aggeler R, Capaldi R, Will Y. Toxicol in Vitro. 2007;21:902–911. doi: 10.1016/j.tiv.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 16.Dzeja PP, Bast P, Ozcan C, Valverde A, Holmuhamedov EL, Van Wylen DG, Terzic A. Am J Physiol Heart Circ Physiol. 2003;284:H1048–1056. doi: 10.1152/ajpheart.00847.2002. [DOI] [PubMed] [Google Scholar]
- 17.Ralph SJ, Moreno-Sanchez R, Neuzil J, Rodriguez-Enriquez S. Pharm Res. 2011;28:2695–2730. doi: 10.1007/s11095-011-0566-7. [DOI] [PubMed] [Google Scholar]
- 18.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valls-Lacalle L, Barba I, Miro-Casas E, Alburquerque-Bejar JJ, Ruiz-Meana M, Fuertes-Agudo M, Rodriguez-Sinovas A, Garcia-Dorado D. Cardiovasc Res. 2016;109:374–384. doi: 10.1093/cvr/cvv279. [DOI] [PubMed] [Google Scholar]
- 20.Wojtovich AP, Brookes PS. Basic Res Cardiol. 2009;104:121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoekstra AS, Bayley JP. Biochim Biophys Acta. 2013;1827:543–551. doi: 10.1016/j.bbabio.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Touati G, Poggi-Travert F, Ogier de Baulny H, Rahier J, Brunelle F, Nihoul-Fekete C, Czernichow P, Saudubray JM. Eur J Pediatr. 1998;157:628–633. doi: 10.1007/s004310050900. [DOI] [PubMed] [Google Scholar]
- 23.Miyadera H, Shiomi K, Ui H, Yamaguchi Y, Masuma R, Tomoda H, Miyoshi H, Osanai A, Kita K, Omura S. Proc Nat Acad Sci U S A. 2003;100:473–477. doi: 10.1073/pnas.0237315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siebels I, Drose S. Biochim Biophys Acta. 2013;1827:1156–1164. doi: 10.1016/j.bbabio.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Kawada M, Momose I, Someno T, Tsujiuchi G, Ikeda D. J Antibiot. 2009;62:243–246. doi: 10.1038/ja.2009.20. [DOI] [PubMed] [Google Scholar]
- 26.Kawada M, Inoue H, Ohba S, Masuda T, Momose I, Ikeda D. Int J Cancer. 2010;126:810–818. doi: 10.1002/ijc.24915. [DOI] [PubMed] [Google Scholar]
- 27.Selby TP, Hughes KA, Rauh JJ, Hanna WS. Bioorg Med Chem Lett. 2010;20:1665–1668. doi: 10.1016/j.bmcl.2010.01.066. [DOI] [PubMed] [Google Scholar]
- 28.Horsefield R, Yankovskaya V, Sexton G, Whittingham W, Shiomi K, Omura S, Byrne B, Cecchini G, Iwata S. J Biol Chem. 2006;281:7309–7316. doi: 10.1074/jbc.M508173200. [DOI] [PubMed] [Google Scholar]
- 29.Ohtawa M, Ogihara S, Sugiyama K, Shiomi K, Harigaya Y, Nagamitsu T, Omura S. J Antibiot. 2009;62:289–294. doi: 10.1038/ja.2009.29. [DOI] [PubMed] [Google Scholar]
- 30.Krautwald S, Nilewski C, Mori M, Shiomi K, Omura S, Carreira EM. Angew Chem. 2016;55:4049–4053. doi: 10.1002/anie.201511672. [DOI] [PubMed] [Google Scholar]
- 31.Trecourt F, Mallet M, Mongin O, Queguiner G. J Org Chem. 1994;59:6173–6178. [Google Scholar]
- 32.Trippier PC, McGuigan C, Balzarini J. Antivir Chem Chemother. 2010;20:249–257. doi: 10.3851/IMP1632. [DOI] [PubMed] [Google Scholar]
- 33.Leeson PD, Springthorpe B. Nat Rev Drug Discov. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 34.Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Chem Biol. 2008;15:375–382. doi: 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Reulecke I, Lange G, Albrecht J, Klein R, Rarey M. ChemMedChem. 2008;3:885–897. doi: 10.1002/cmdc.200700319. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Cancer Res. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wenzel C, Riefke B, Grundemann S, Krebs A, Christian S, Prinz F, Osterland M, Golfier S, Rase S, Ansari N, Esner M, Bickle M, Pampaloni F, Mattheyer C, Stelzer EH, Parczyk K, Prechtl S, Steigemann P. Exp Cell Res. 2014;323:131–143. doi: 10.1016/j.yexcr.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 38.Germain T, Ansari M, Pappas D. Anal Chim Acta. 2016;936:179–184. doi: 10.1016/j.aca.2016.07.040. [DOI] [PubMed] [Google Scholar]
- 39.Maxwell PJ, Gallagher R, Seaton A, Wilson C, Scullin P, Pettigrew J, Stratford IJ, Williams KJ, Johnston PG, Waugh DJ. Oncogene. 2007;26:7333–7345. doi: 10.1038/sj.onc.1210536. [DOI] [PubMed] [Google Scholar]
- 40.Ragan CI, Wilson MT, Darley-Usmar VM, Lowe PN. Sub-fractionation of mitochondria and isolation of the roteins of oxidative phosphorylation Chapter 4 In: Mitochondria, a Practical Approach. IRL Press; Oxford, UK: 1988. [Google Scholar]
- 41.Kirby DM, Thurburn DR, Turnbull DM. Methods Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- 42.Khanal G, Hiemstra S, Pappas D. Analyst. 2014;139:3274–3280. doi: 10.1039/c3an02324a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.