

Abstract
Recent identification of an enhancer element, RDINK4/ARF (RD), in the prominent INK4/ARF locus provides a novel mechanism to simultaneously regulate the transcription of p15INK4B (p15), p14ARF, and p16INK4A (p16) tumor suppressor genes. While genetic inactivation of p15, p14ARF, and p16 in human tumors has been extensively studied, little is known about genetic alterations of RD and its impact on p15, p14ARF, and p16 in human cancer. The purpose of this study was to investigate the potential existence of genetic alterations of RD in human cancer cells. DNAs extracted from 17 different cancer cell lines and 31 primary pheochromocytoma tumors were analyzed for deletion and mutation of RD using qPCR and direct DNA sequencing. We found that RD was deleted in human cancer cell lines and pheochromocytoma tumors at frequencies of 41.2% (7/17) and 13.0% (4/31), respectively. While some of these RD deletion events occurred along with deletions of the entire INK4/ARF locus, other RD deletion events were independent of genetic alterations in p15, p14ARF, and p16. Furthermore, the status of RD was poorly associated with the expression of p15, p14ARF, and p16 in tested cancer cell lines and tumors. This study demonstrates for the first time that deletion of the RD enhancer is a prevalent event in human cancer cells. Its implication in carcinogenesis remains to be further explored.
INTRODUCTION
The INK4/ARF (CDKN2A) locus plays a central role in tumor suppression as reflected by the fact that this locus is inactivated in nearly 50% of all human cancers [1, 2]. This locus of ∼50 kb encodes three distinct tumor suppressors; namely, P16INK4A (P16), P15INK4B (P15), and P14ARF (Figure 1). P16 and P15 specifically inhibit cyclin D-dependent kinase 4/6 (CDK4/6)-mediated phosphorylation of pRB (the retinoblastoma susceptible gene product) and the subsequent E2F-mediated transactivation of genes required for entry into the S phase; P14ARF negatively modulates the E3 ubiquitin ligase activity of MDM2, thus activating P53-mediated apoptosis or cell cycle arrest. While the p15, p14ARF, and p16 genes are driven by their respective individual promoters [2], accumulating evidence indicates that there are locus control region mechanisms able to modulate these three genes simultaneously [1]. One such mechanism involves a DNA element located upstream of the p15 gene (Figure 1) [3, 4]. This regulatory element, RDINK4/ARF (hereafter, RD), is conserved among mammalian INK4/ARF loci and exhibits a global impact on enhancing the concomitant expression of p15, p14ARF, and p16. Moreover, CDC6, an essential DNA replication modulator frequently over-expressed in a number of human cancers [1], is able to bind to RD and recruit histone deacetylase 1 (HDAC1) and histone deacetylase 2 (HDAC2) to the INK4/ARF locus, resulting in heterochromatinization and transcriptional silencing [3]. Further studies have demonstrated that such CDC6-mediated repression of the INK4/ARF locus involves physical interactions between CDC6 and Bmi1, a Polycomb (PcG) protein that functions to generate and recognize histone modifications in genomic domains with clusters of genes. Upon senescence, JMJD3 (KDM6B, a histone demethylase) and MLL (a histone methytrasferase) are recruited to the INK4/ARF locus, provoking the dissociation of PcG proteins from RD and the transcription of p16 [5]. The RD/CDC6 interaction provides a novel mechanism that is important in cell cycle control and carcinogenesis since its perturbation by RD alterations and/or increased expression of CDC6 may down-regulate both the Rb and P53 pathways. While genetic alterations of p16 (as well as p15 and p14ARF) in human cancer cells and tumors have been extensively investigated [1, 2], little is known about changes in the genetic status of RD in human cancer. In this study, we investigated the genetic status of RD and its correlation with the expression of p16 (as well as p15 and p14ARF) in 17 human cancer cell lines and a cohort of 31 human pheochromocytoma primary tumors.
Figure 1.
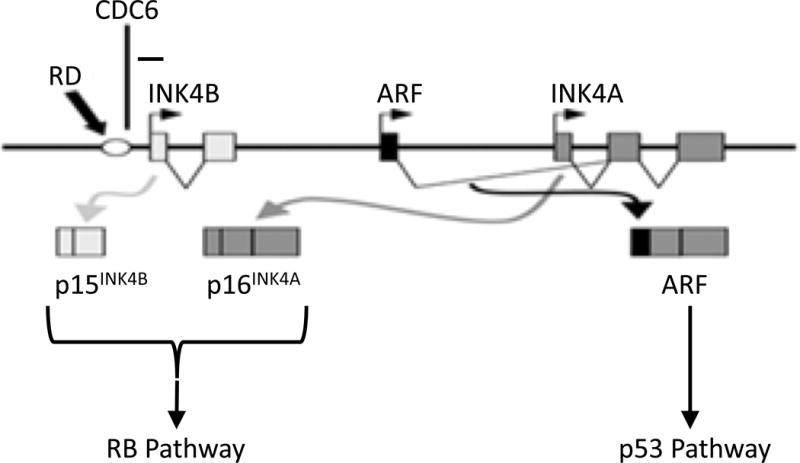
The schematic structure of the INK4/ARF locus. Promoters for p15, p14ARF, and p16 genes are indicated by horizontal arrows. Boxes represent exons, and the empty circle represents the regulatory element RD. The minus sign indicates negative regulation. This figure was adapted from Ref. [4].
MATERIALS AND METHODS
Cell Culture
All cell lines used in this study are listed in Table 1. TE-1177, SCC-83-01-82, and SCC-83-01-82CA were kindly provided by Dr. S.M. D’Ambrosio of The Ohio State University [6]; UM-SCC-22A was obtained from the University of Michigan. These four cell lines were cultured in Advanced D-MEM/F-12 medium (Life Technologies/Invitrogen, Carlsbad, CA) containing 5% fetal bovine serum (FBS; Invitrogen) in a 90% relative humidity incubator at 37°C supplied with 5% CO2 [6]. The other cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA) and cultured according to the provider’s recommendations.
Table 1.
The Genetic Status of the INK4/ARF Locus and mRNA Expression of p15, p14ARF, p16, and CDC6 in Human Cancer Cell Lines
| Cell line | Source | Genetic Status(a) | mRNA Expression(b) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| RD | p15 | p14ARF | p16 | p15 | p14ARF | p16 | CDC6 | ||||
|
|
|||||||||||
| TE-1177 | Oral (tonsil) | Normal epithelia | +/+ | +/+ | +/+ | +/+ | Control | Control | Control | Control | |
| SCC-83-01-82 | Oral | Premalignant epithelia | +/+ | +/+ | +/+ | +/+ | Low | High | Normal | High | |
| SCC-83-01-82CA | Oral | Squamous cell carcinoma | +/+ | +/+ | +/+ | +/+ | Low | High | Low | Normal | |
| UM-SCC-22A | Oral (hypopharynx) | Squamous cell carcinoma | −/− | −/− | −/− | −/− | NE | NE | NE | Normal | |
| CAL-27 | Oral (tongue) | Squamous cell carcinoma | +/+ | M | +/+ | +/+ | NE | High | High | High | |
| SCC-15 | Oral (tongue) | Squamous cell carcinoma | +/− | +/+ | +/+ | +/+ | Normal | High | Low | High | |
| SCC-4 | Oral (tongue) | Squamous cell carcinoma | +/− | +/+ | +/+ | +/+ | Low | High | Normal | Normal | |
| SCC-9 | Oral (tongue) | Squamous cell carcinoma | −/− | −/− | −/− | +/+ | NE | NE | NE | High | |
| SCC-25 | Oral (tongue) | Squamous cell carcinoma | +/− | +/+ | +/+ | −/− | Low | Normal | NE | Normal | |
| HeLa | Cervical | Adenocarcinoma | +/+ | +/+ | +/+ | +/+ | Low | High | High | Normal | |
| C-33A | Cervical | Carcinoma | +/+ | +/+ | +/+ | +/+ | Low | High | High | High | |
| SiHa | Cervical | Squamous cell carcinoma | +/+ | +/+ | +/+ | +/+ | Low | High | High | High | |
| MIA PaCa-2 | Pancreatic | Carcinoma | −/− | −/− | −/− | −/− | NE | NE | NE | High | |
| PANC-1 | Pancreatic | Epithelioid carcinoma | −/− | −/− | −/− | −/− | NE | NE | NE | High | |
| PC3 | Prostate | Adenocarcinoma | +/+ | +/+ | +/+ | M | High | High | High | High | |
| DU145 | Prostate | Carcinoma | +/+ | +/+ | R88L | D74Y | Normal | High | High | High | |
| COLO320 | Colorectal | Adenocarcinoma | +/+ | +/+ | +/+ | +/+ | Low | High | Normal | High | |
| SW480 | Colorectal | Adenocarcinoma | +/+ | +/+ | +/+ | +/+ | Low | High | Normal | High | |
The genetic status of a target sequence: +/+, wild type; +/−, hemizygous deletion; −/−, homozygous deletion; M, methylation.
The mRNA expression levels of p15, p14ARF, p16, and CDC6 were quantitatively determined using TaqMan assays, in which TE-1177 was the control cell line, and HPRT1 was the reference gene for normalization. Results were interpreted as follows: increase of two or more than two-fold, high; decrease of two or more than two-fold, low; increase or decrease by less than two-fold, normal; NE, not expressed.
Detecting Genetic Alterations of RD in Cancer Cells
Deletion, methylation, and point mutations of p16, p15, and p14ARF were evaluated as previously reported with primers and probes listed in Table 2 [7–12]. The genetic status of RD was investigated as follows.
Table 2.
PCR Primers and Probes
| DNA Fragment Analysis | ||
|
| ||
| RD | F | 5′-CAATTGCCCTCCTGTTAAGAC-3′ |
| R | 5′-CCCACTTTGTCAGGTATCTTATTT-3′ | |
| FPR1(a) | F | 5′-CTCACCACAGTGCTTGCTGC-3′ |
| R | 5′-CTTACCTGGACTGTTCACCAG-3′ | |
| p16-1α | F | 5′-CGGCTGCGGAGAGGGGGAGAC-3′ |
| R | 5′-CAGCGCCCGCACCTCCTCTA-3′ | |
| p14-1β | F | 5′-GGAGGCGGCGAGAACAT-3′ |
| R | 5′-GGGCCTTTCCTACCTGGTCTT-3′ | |
| p16-2a | F | 5′-GGGCTCTACACAAGCTTCCTT-3′ |
| R | 5′-AGCCAGGTCCACGGGCAGGGTACA-3′ | |
| p16-2b | F | 5′-GGGAGGGCTTCCTGGACAC-3′ |
| R | 5′-TTTGGAAGCTCTCAGGGTACA-3′ | |
| p16-3 | F | 5′-GCCTGTTTTCTTTCTGCCCTCTG-3′ |
| R | 5′-CGAAAGCGGGGTGGGTTGT-3′ | |
| p15-1 | F | 5′-GCGTCTGGGGGCTGCGGAATG-3′ |
| R | 5′-CCTCGCCAACGTAGACTCCTGTA-3′ | |
| p15-2 | F | 5′-CCCGGCCGGCATCTCCCATAC-3′ |
| R | 5′-GTTGTGGGCTGGGGAACC-3′ | |
|
| ||
| DNA Deletion Analysis (Multiplex PCR) | ||
|
| ||
| RD | F | 5′-TTATGCAGTTCCTCACCAAAGTTTT-3′ |
| R | 5′-GGATAGGAAGAGAACCGCAAGTT-3′ | |
| P | 5′-FAM-AGGCAACAAATCCA-TAMRA-3′ | |
| p16 | F | 5′-GGCTCTACACAAGCTTCCTTTCC-3′ |
| R | 5′-TCATGACCTGCCAGAGAGAACA-3′ | |
| P | 5′-FAM-CCCCCACCCTGGCTCTGACCA-TAMRA-3′ | |
| p14ARF | F | 5′-GCGCATTCGGCACTTGTT-3′ |
| R | 5′-CTGGGCTAGAGACGAATTATCTGTT-3′ | |
| P | 5′-FAM-TGTTTGGTGTGATTTCGT-MGB-3′ | |
| p15 | F | 5′-GGCCGGAGGTGTGCATT-3′ |
| R | 5′-CTCTTTAGGATTTTTGCTGGGTAAA-3′ | |
| P | 5′-FAM-CACGCGTAAAACAG-MGB-3′ | |
| ACTB(b) | F | 5′-CGTAGCACAGCTTCTCCTTAATGTC-3′ |
| R | 5′-AGCGCGGCTACAGCTTCA-3′ | |
| P | 5′-TET-ATTTCCCGCTCGGCCGTGGT-TAMRA-3′ | |
|
| ||
| DNA Methylation Analysis (Methylation-Specific PCR) | ||
|
| ||
| p15 | CpG Wiz p15 amplification kit (Chemicon, S7802) | |
| p16 | CpG Wiz p16 amplification kit (Chemicon, S7800) | |
| p14ARF | F | 5′-TTTTTGGTGTTAAAGGGTGGTGTAGT-3′ (unmethylated) |
| R | 5′-CACAAAAACCCTCACTCAC AACAA-3′ (unmethylated) | |
| F | 5′-GTGTTAAAGGGCGGCGTAGC-3′ (methylated) | |
| R | 5′-AAAACCCTC ACTCGCGACGA-3′ (methylated) | |
|
| ||
| Gene Expression Analysis (TaqMan Assays) | ||
|
| ||
| HPRT1(c) | Hs99999909_m1 | |
| CDC6 | Hs00154374_m1 | |
| p14ARF | Hs00924091_m1 | |
| p15 | Hs00394703_m1 | |
| p16 | Hs00233365_m1 | |
ACTB, β-actin; FPR1, N-formyl peptide receptor gene [20]
F, forward primer; R, reverse primer; P, probe
DNA Fragment Analysis control
Deletion Analysis control
Gene Expression Analysis control
RD Point Mutations
The RD fragment was amplified by PCR using the following primers: 5′-CAA TTG CCC TCC TGT TAA GAC-3′ (forward) and 5′-CCC ACT TTG TCA GGT ATC TTA TTT-3′ (reverse), and analyzed by electrophoresis (Figure 1A). After purification, both strands of RD fragments were sequenced to identify potential mutations.
As shown in Figure 2A, RD was homozygously deleted in UM-SCC-22A and PANC-1. Thus, UM-SCC-22A (RD−/−) together with TE-1177 (normal oral epithelial cell line, RD+/+) were used in the qPCR-based genomic deletion assay.
Figure 2.
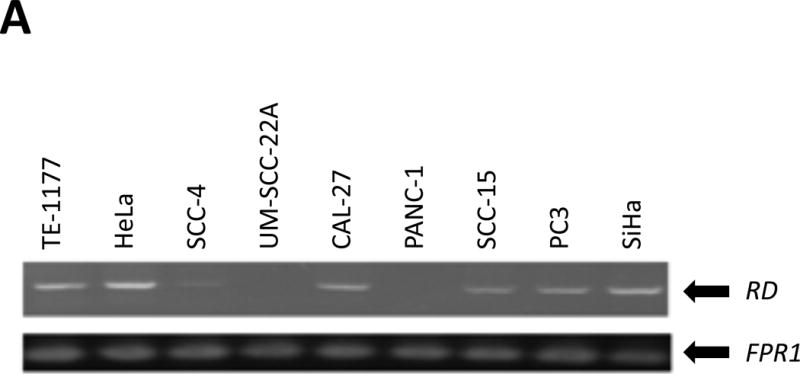
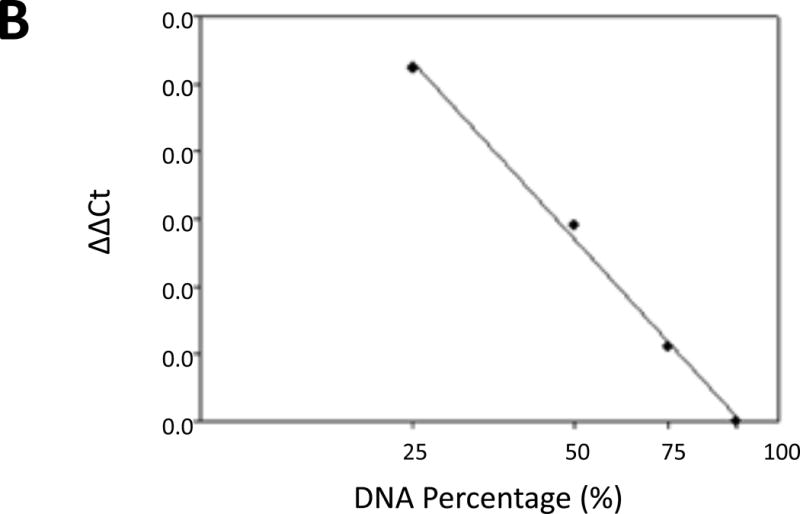
qPCR assays to detect genetic alterations of RD. (A) Amplification of RD from selected cell lines. FPR1 (N-formyl peptide receptor) was used as an endogenous control [20]. The PCR products of RD and FPR1 are 406 and 500 bp, respectively. (B) The calibration curve for qPCR-based analyses of RD deletion. Multiplex qPCR for RD and ACTB was performed with a series of mixtures of genomic DNAs from TE-1177 (RD+/+) and UM-SCC-22A (RD−/−) cells at various ratios. The resulting Cq values were normalized as described in Materials and Methods Section. The ΔΔCq values were plotted against the relative ratio of normal RD in the mixtures (in exponential form) to get a calibration curve with a linear correlation coefficient of −0.996.
RD Genomic Deletion
Homozygous and hemizygous deletions of RD were determined using multiplex qPCR on a Cepheid Smart Cycler as previously described [13]. The primers and probes for RD and β-actin (ACTB, endogenous control) are listed in Table 2. All experiments were performed in triplicate.
Samples containing mixed genomic DNAs from TE-1177 (RD+/+) and UM-SCC-22A (RD−/−) at various ratios (100:0, 75:25, 50:50, 25:75, 0:100) were analyzed and the resulting quantification cycle value (Cq) values were normalized using the following equation [14]:
The ΔΔCq values were then plotted against known ratios of RD (in exponential form) to get a calibration line with a correlation coefficient of −0.996 (Figure 2B), indicating that the dosage of RD in a sample can be accurately measured using this technique [15]. Subsequently, the gene dosage of RD in different cancer cell lines and tumors was determined and the results were interpreted as follows [15]: dosage <25%, homozygous deletion (RD−/−); dosage between 25% and 75%, hemizygous deletion (RD+/−); and dosage >75%, RD wild type (RD+/+).
RD methylation status
While methylation is a primary mechanism in transcriptionally inactivating genes, we analyzed the RD sequence with two widely used programs, CpG Island Searcher [16] and EMBOSS CpGPlot [17], and no potential CpG island was identified. This is not unexpected since the RD sequence is 63.8% A/T-rich. Hence, RD methylation was not further investigated in this study.
Quantitatively Determining the Expression of p15, p16, p14ARF, and CDC6 in Cancer Cells
Extraction of total RNA and first-strand cDNA synthesis were performed as previously described [6]. Quantitative RT-qPCR reactions were carried out using a StepOne Real-Time PCR System (Applied Biosystems) and pre-validated TaqMan gene expression assays as listed in Table 2. Human hypoxanthine phosphoribosyltransferase (HPRT1) was used as an endogenous control for gene expression. Each gene (p16, p15, p14ARF, CDC6, and HPRT1) was amplified separately, and all experiments were performed in triplicate.
The relative gene expression level of a target was determined using a comparative Cq method, in which data from cancer cells were normalized to that from TE-1177 [6]:
A two-fold increased (≥2) or decreased (≤0.5) value was considered significant for mRNA over-expression or down-regulation, respectively.
Western Blot
Cell pellets were washed three times in ice-cold PBS and then lysed with RIPA buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM PMSF, and 1 μg/mL of leupeptin; Cell Signaling Technologies; Danvers, MA] on ice for 20 min. Subsequently, cell lysates were clarified by centrifugation at 4°C, 13,000 rpm for 5 min and the concentration of total proteins in the supernatants was determined using the BCA assay (Thermo Fisher Scientific/Pierce, Rockford, IL). The samples were subjected to SDS-PAGE (10% or 15%) at 50 μg of total proteins per lane and then transferred to nitrocellulose membrane and immunoblotted with the following antibodies: mouse anti-β actin monoclonal antibody (sc-56459; Santa Cruz Biotechnology; Santa Cruz, CA), rabbit anti-P14ARF polyclonal antibody (sc-8340; Santa Cruz Biotechnology), rabbit anti-P15 polyclonal antibody (sc-613; Santa Cruz Biotechnology), mouse anti-P16 monoclonal antibody (sc-1661; Santa Cruz Biotechnology); rabbit anti-CDC6 monoclonal antibody (C42F7; Cell Signaling Technologies). The concentration of each antibody was used as suggested by the suppliers. Immunodetection was performed with the Enhanced Chemiluminescence Kit for Western blotting (GE Healthcare Biosciences, Piscataway, NJ).
Analyzing Pheochromocytoma Tumors
Pheochromocytomas are adrenal gland tumors that have been shown to have molecular defects in cell cycle associated genes, including p16 [13]. A cohort of 31 human pheochromocytoma tumors and seven histologically normal specimens were obtained from The Ohio State University Medical Center under the guidance of the Institutional Review Board as previously reported [13]. Genomic DNA was extracted from paraffin-embedded specimens using a ChargeSwitch gDNA Kit (Life Technologies/Invitrogen) and subjected to assays to detect the genetic status of RD and p16 [13]. Protein expression of P16 was evaluated by immunohistochemistry (IHC)-based tissue microarrays using a primary antibody against human P16 protein (16P04; Cell Marque; Rockland, CA).
RESULTS AND DISCUSSION
The Genetic Status of the INK4/ARF Locus in Human Cancer Cells
We first extracted genomic DNAs from 18 human cell lines (one normal oral epithelial cell line, 17 neoplastic cell lines) and investigated potential genetic abnormalities of RD including homozygous/hemizygous deletions and point mutations. In parallel, genetic statuses of p15, p14ARF, and p16 in these cell lines were evaluated as previously described [7–12]. Table 1 summarizes the genetic statuses of RD, p15, p14ARF, and p16 in these cell lines. While four cell lines, SCC-9, UM-SCC-22A, PANC-1, and MIA PaCa-2, harbored homozygous deletions of RD, hemizygous deletions of RD were found in SCC-15, SCC-4, and SCC-25 cells. Interestingly, no point mutations of RD were identified in any of these 17 tumor cell lines, demonstrating that hemizygous (3/17) and homozygous (4/17) deletions are the major mechanisms to inactivate RD at the DNA level. Of the four RD−/− cell lines, UM-SCC-22A, PANC-1, and MIA PaCa-2 also harbored homozygous deletions of p15, p14ARF, and p16, indicating that the entire INK4/ARF locus was deleted in these cell lines. In contrast, SCC-9 harbored homozygous deletions of RD, p15, and p14ARF, but retained intact p16, suggesting that the deletion event in SCC-9 occurs at the upstream of the p16 gene. Out of the three RD+/− cell lines, SCC-4 had intact p15, p14ARF, and p16 genes, indicative of RD-specific genetic alterations in this cell line; SCC-15 and SCC-25 had methylated p16 and homozygous deletion of p16, respectively, whereas their p15 and p14ARF genes remained intact, suggesting that there are two independent inactivating events targeting RD and p16, respectively, in SCC-15 and SCC-25.
In addition, genetic alterations of p15, p14ARF or p16 were observed in some RD+/+ cell lines. For example, CAL-27 and PC3 (both RD+/+) had promoter hypermethylation in p15 and p16, respectively, whereas DU145 had a point mutation (G → T) in exon 2 of p16, which leads to mutated P16 (Asp74Tyr) and P14ARF (Arg88Leu) proteins [18]. These findings support the notion that molecular alterations of RD and p16 (as well as p15 and p14ARF) represent independent mechanisms of regulation.
Expression of p15, p14ARF, and p16 in Human Cancer Cells
We then determined the expression levels of p15, p14ARF, and p16 in these cell lines to evaluate how the genetic status of RD influences the transcription of these INK4/ARF genes (Figure 3). Since binding of CDC6 directly down-regulates the enhancer activity of RD, we also assessed the expression level of CDC6 in these cell lines. As expected, no expression of p15, p14ARF, and p16 was detected in cell lines with homozygous deletions of the entire INK4/ARF locus (UM-SCC-22A, PANC-1, and MIA PaCa-2). Similar results were observed with SCC-9, which has homozygous deletions of RD, p15, and p14ARF but intact p16, suggesting that the transcription of p16 in this cell line is silenced through yet-to-be identified mechanisms. As for the other 13 cell lines, the overall expression patterns of p15, p14ARF, and p16 are different from each other. The p16 gene was over-expressed in six cell lines (HeLa, SiHa, C-33A, COLO320, CAL-27, and DU145) and normally expressed in five cell lines (PC3, SW480, SCC-4, SCC-83-01-82, and SCC-83-01-82CA). The expression of p16 was nominally detected in SCC-15 and SCC-25 since the p16 gene was either methylated (SCC-15) or homozygously deleted (SCC-25). Notably, even though methylation of the p16 promoter was identified in PC3 cells [18], the expression of p16 remained comparable to that in TE-1177. Overall, these results indicate that unless the p16 gene is homozygously deleted or highly silenced by methylation, it tends to be over-expressed or normally expressed at the mRNA level in most of cancerous or precancerous cancer cell lines. These findings are consistent with previous studies showing that p16 (wild type or with point mutations) was over-expressed prevalently in human tumors, such as prostate, lung, brain tumors, and oral squamous cell carcinomas [1, 19]. Similarly, p14ARF was over-expressed in all tested cell lines except UM-SCC-22A, PANC-1, MIA PaCa-2, SCC-9 (with homozygous deletions of the entire INK4/ARF locus) and SCC-25. In SCC-25, p14ARF was expressed at a level comparable with that in TE-1177. Interestingly, both p16 and p14ARF were over-expressed in DU145, a cell line harboring mutated P16 and P14ARF proteins. In contrast, while a moderately increased expression of p15 was found in COLO320 cells, the expression levels of p15 were significantly reduced or unchanged (in comparison with TE-1177) in nine and three cell lines, respectively, indicating that the expression of p15 is down-regulated in the majority of cancerous or precancerous cell lines. While CDC6 was expressed in HeLa, SCC-83-01-82CA, SW480, and SCC-25 at a level comparable to that in TE-1177, its over-expression was observed in the remaining 13 cell lines.
Figure 3.
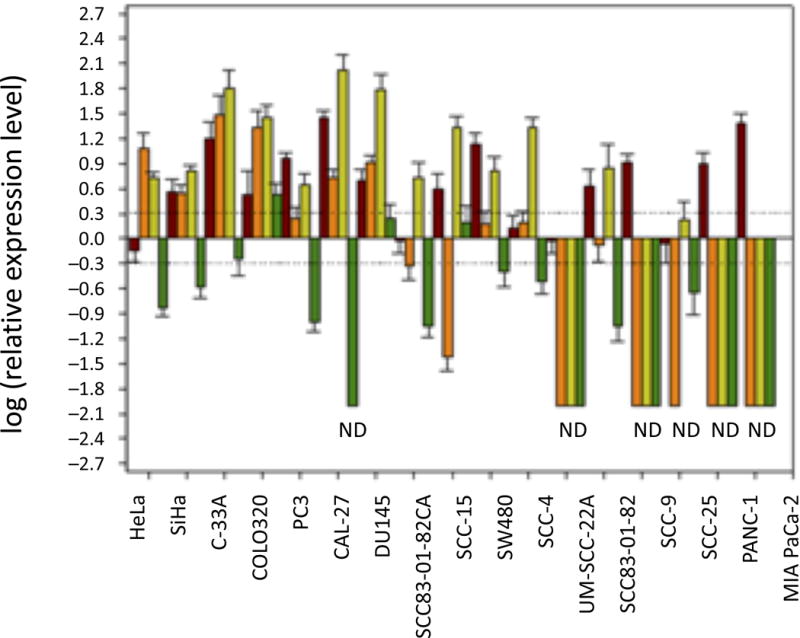
Quantitative determination of CDC6, p16, p14ARF, and p15 mRNA expression in human cell lines. Brown, CDC6; orange, p16; lime green, p14ARF; dark green, p15. HPRT1 was used as an endogenous control, and TE-1177 cells were used to represent the reference baseline state for normalization [6]. Quantitative RT-PCR-based assays were conducted in triplicate. Values (y-axis) represent fold-changes (in the log form) calibrated to the expression levels of indicated genes in TE-1177. Error bars represent standard deviations. Dashed lines represent twofold increases (upper) and decreases (lower) in gene expression. ND, not detected.
As for each individual cell line, the expression of p15, p14ARF, and p16 appears to be differential. For example, in SCC-4 cells (RD+/−), p15, p14ARF, and p16 were down-regulated, over-expressed, and normally expressed, respectively. In comparison, SCC-15 (RD+/−) had normally expressed, over-expressed, and lowly-expressed p15, p14ARF, and p16, respectively. Similar observations were obtained among RD+/+ cell lines. For example, p15, p14ARF, and p16 were down-regulated, over-expressed, and normally expressed, respectively, in SCC-83-01-82 cells.
We continued to evaluate the expression of CDC6, P16, P14ARF, and P15 at the protein level in these cell lines using Western blot analyses. As shown in Figure 4, CDC6 was highly expressed in all tested cell lines relative to the TE-1177 control cell line. P14ARF was highly expressed in CAL-27 (RD+/+), SCC-4 (RD+/−), SCC-15 (RD+/−), SCC-83-01-82, SCC-83-01-82CA (RD+/+), but not detected in SCC-9 (RD−/−) and UM-SCC-22A (RD−/−), in which the p14ARF gene was homozygously deleted. P16 was highly expressed in CAL-27, SCC-83-01-82, and SCC-83-01-82CA, all of which retain intact p16 gene. In contrast, P16 protein was not detected in SCC-4, SCC-9, SCC-15, and UM-SCC-22A. While the p16 gene is homozygously deleted in UM-SCC-22A, it is transcriptionally silenced in SCC-15 (through methylation) and SCC-9 (through unknown mechanisms). In SCC-4, p16 was expressed at the mRNA level, but not at the protein level, indicating that post-transcriptional mechanisms are involved in the regulation of P16 [1]. As for p15, we failed to detect P15 protein in all tested cell lines (data not shown). Such inability to detect P15 protein in our Western blot assay could be attributed to the following reasons: (1) the expression of P15 in these cell lines is below the sensitivity of our Western immunoblot assay and (2) P15 protein was degraded following cell lysis and sample preparation. Overall, these Western blot results are consistent with our previous observations with the gene expression: P14ARF and P16 are differentially expressed and the genetic status of individual genes, not the status of RD, appears to be associated with their expression.
Figure 4.
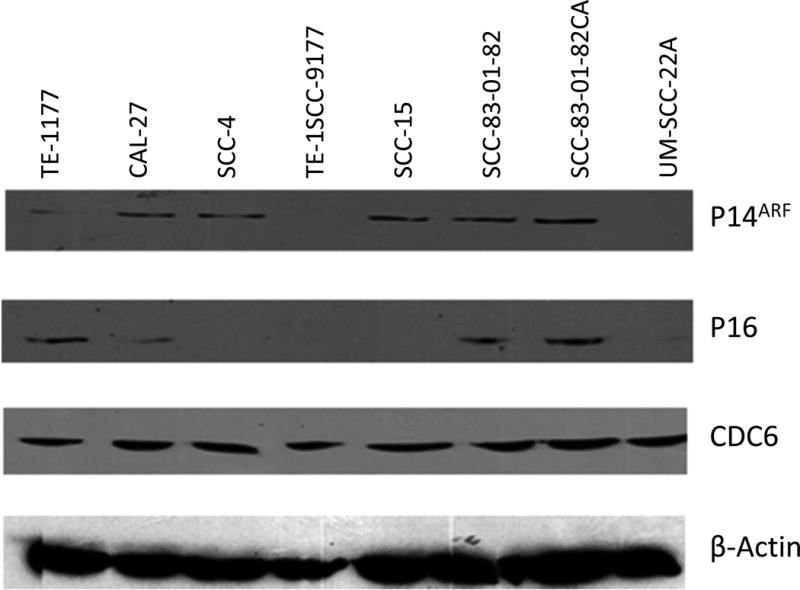
Western Blot Analyses. Soluble proteins (50 mg) from selected cell lines were subjected to SDS–PAGE analysis, and after electro-transfer to nitrocellulose membrane, the proteins were probed using primary antibodies against β-actin (sc-56459; Santa Cruz Biotechnology), CDC6 (C42F7; Cell Signaling Technologies), P16 (sc-1661; Santa Cruz Biotechnology), and P14ARF (sc-8340; Santa Cruz Biotechnology). Immunodetection was performed with the Enhanced Chemiluminescence Kit for Western blotting (GE Healthcare Bioscience). TE-1177 was the reference cell line. β-actin was the internal reference protein.
Genetic Alterations of RD in Human Pheochromocytoma Tumors
We further analyzed the genetic status of RD in a cohort of human pheochromocytoma tumors, of which the genetic status of p16 and the expression level of P16 protein were determined previously [13]. As listed in Table 3, homozygous and hemizygous deletions of RD were identified in one and three tumors, respectively, while no “normal” specimens harbored RD alterations. The overall frequency of RD alterations in this cohort of pheochromocytomas is 13.0% (4/31). In comparison, deletions (hemizygous and homozygous), point mutation, and aberrant CpG methylation of p16 were found in 9, 1, and 11 tumors with an overall frequency of 67.7% (21/31). In the group of four pheochromocytomas with RD alterations, one tumor (T21) harbored homozygous deletions of both RD and p16, and another tumor (T5) had hemizygous deletions of both RD and p16, indicating that alterations of RD and p16 in these two tumors may result from a single molecular event in the INK4/ARF locus. The remaining pheochromocytomas with RD alterations, T1 and T18L, harbored hemizygously deleted RD; however, the status of p16 in these two tumors were different: p16 was methylated in T18L, but remained intact in T1. Hence, these results suggest that RD alteration is independent of, or at least not always concomitant with p16 alteration in pheochromocytoma tumors.
Table 3.
Genetic Analyses of RD in Human Pheochromocytoma Tumor Specimens
| Specimen(a,b) | Expression of P16(b) | p16 (c) | RD | Specimen(a,b) | Expression of P16(b) | p16(c) | RD |
|---|---|---|---|---|---|---|---|
| T1 | Low | +/+ | +/− | T18R | Negative | −/− | +/+ |
| T2 | Negative | +/+ | +/+ | T18L | Low | M | +/− |
| T3 | Low | +/+ | +/+ | T19 | Negative | M | +/+ |
| T4 | Negative | +/− | +/+ | T20 | Negative | +/− | +/+ |
| T5 | Low | +/− | +/− | T21 | Low | −/− | −/− |
| T6 | Negative | +/+ | +/+ | T22 | Negative | +/+ | +/+ |
| T7 | Low | +/− | +/+ | T23 | Negative | +/+ | +/+ |
| T8 | Low | +/+ | +/+ | T24R | Low | +/+ | +/+ |
| T9R | Low | +/+ | +/+ | T24L | Low | M | +/+ |
| T9L | Negative | M | +/+ | T25 | Low | M | +/+ |
| T10 | Low | M | +/+ | T26 | Low | M | +/+ |
| T11 | Low | −/− | +/+ | T27 | Low | M | +/+ |
| T12 | Negative | +/− | +/+ | C3 | High | +/+ | +/+ |
| T13 | Negative | +/− | +/+ | C4 | Low | +/+ | +/+ |
| T14 | Low | M | +/+ | C9R | High | +/+ | +/+ |
| T15 | Negative | M | +/+ | C9L | High | +/+ | +/+ |
| T16R | Low | +/+ | +/+ | C22 | High | +/+ | +/+ |
| T16L | High | A20P | +/+ | C25 | High | +/+ | +/+ |
| T17 | Negative | M | +/+ | C27 | Low | +/+ | +/+ |
T, tumor specimen; C, normal specimen [13]; R, right-sided tumor; L, left-sided tumor
Determined by immunohistochemistry (IHC)-based tissue microarray [13] Based on the percentage of positive nuclear P16 staining by IHC, the samples were categorized as P16-negative (complete absence of nuclear immunostaining), P16-low (1–25% of positive nuclei) or P16-high (>25% of positive nuclei).
The genetic status of a target sequence: +/+, wild-type; +/−, hemizygous deletion; −/−, homozygous deletion; M, methylation.
It is also worthwhile to note that P16 protein was expressed at low levels or even undetected in all but one pheochromocytomas (30/31, 96.8%), which is different from our previous finding that p16 was over-expressed at the mRNA level in the majority of 17 tested neoplastic cell lines. Such inconsistency could be attributed to the following reasons: (1) the transcriptional levels of p16 mRNA and protein expression levels of P16 are not necessarily stoichiometrically coupled, since a number of confounding cellular processes can mediate the stabilities (expression) of p16 mRNA and protein in cancer cells [1] and (2) the microenvironment surrounding a tumor tissue in vivo is different from that defined in vitro for cultured cells. Importantly, the only tumor (T16L) exhibiting highly expressed P16 had an intact RD, but a point mutation in p16 leading to a partial loss of P16 function [13]. Since no genetic alteration of RD was identified in the majority of low-expressing P16 tumors, there does not appear to be a strong association between the RD status and the expression of P16 in pheochromocytoma tumors.
Here, we present our studies on the prevalence of RD alterations in human cancer cells. In a panel of 17 human cell lines derived from different types of cancers, up to 41.2% (7/17) demonstrated homozygous (4/17) or hemizygous (3/17) RD deletions. We also showed that in a cohort of 31 human pheochromocytoma tumors, three tumors harbored hemizygous deletions of RD and one tumor had homozygous deletions of RD. These results reveal that deletion of RD occurs in human cancer cells and tumor tissues with a considerable incidence (11/48, 22.9%). As described earlier, some RD-altering events identified in this study were global and involved the entire INK4/ARF locus, while other RD-altering events were localized specifically to the RD region. Moreover, some cancer cell lines and tumors had intact RD, but harbored genetic alterations (i.e., deletion, methylation, and point mutations) in one or more of the p15, p14ARF, and p16 genes. These results support a model in which some RD alterations are independent of genetic alterations of the p15, p14ARF, and p16 tumor suppressor genes. Notably, point mutations (not detected) and methylation (not investigated) are not supported as significant mediators of RD functional activities. However, hemizygous and homozygous deletions of RD were identified both in diverse human cancer cell lines and pheochromocytoma tumor tissues, but not in non-malignant cells in vitro or non-tumor human tissue samples. These findings suggesting that deletion is the primary mechanism to functionally inactivate RD activities at the genomic level. In contrast, a spectrum of genetic alterations of p15, p14ARF, and p16 were present in these cell lines and tumor specimens, including not only deletion, but also methylation and point mutations. Specifically, both methylation and deletion are major mechanisms to inactive p16 in pheochromocytomas as evidenced by their incidences of 29.0% (10/31) and 35.5% (11/31), respectively. This difference in “inactivating” mechanism between RD and three INK4/ARF genes could be partially ascribed to the A/T-rich nature of RD, which is distinct from the G/C-rich nature of p15, p14ARF, and p16 genes [1, 2]. To our knowledge, this is the first report on genetic alterations of RD in human cancer cells. However, it is important to note that how genetic alterations of RD contribute to the regulation of tumor suppressors P15, P14ARF, and P16 as well as carcinogenesis remains to be further investigated. The small sample size of either the in vitro studies (n = 17) or clinical pheochromocytoma samples (n = 31) impairs our effort to definitively correlate the status of RD with the expression of p15, p14ARF, and p16. Furthermore, RD-mediated regulation of p15, p14ARF, and p16 directly or indirectly involves a number of regulatory proteins, including CDC6, Bmi1, HDAC1, and HDAC2. While CDC6 is highly expressed in most of tested cancer cell lines regardless of the status of RD, the expression (and the functioning) of Bmi1 as well as other factors may further contribute to the differential expression of p15, p14ARF, and p16 in these cell lines. From this perspective, it is of significance to investigate the status of RD, its regulatory factors (including CDC6, Bmi1, HDAC1, and HDAC2), and its downstream effectors (p15, p14ARF, and p16) in a large cohort of patient-matched normal and tumor specimens in regard to the potentials of RD in mediating both pRb and P53 tumor suppressive pathways. Indeed, such a study in a cohort of human squamous cell carcinomas of the head and neck is ongoing in our laboratory.
Acknowledgments
This work was supported by a research grant from NIH, R01 CA69472 (J.L.), R21 DE016361 (TJK, CMW), R01 CA127368 (TJK, CMW).
References
- 1.Li J, Poi MJ, Tsai M-D. Regulatory mechanisms of tumor suppressor p16INK4a and their relevance to cancer. Biochemistry. 2011;50:5566–5582. doi: 10.1021/bi200642e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez S, Serrano M. A new mechanism of inactivation of the INK4/ARF locus. Cell Cycle. 2006;5:1382–1384. doi: 10.4161/cc.5.13.2901. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez S, Klatt P, Delgado S, et al. Oncogenic activity of Cdc6 through repression the INK4/ARF locus. Nature. 2006;440:702–706. doi: 10.1038/nature04585. [DOI] [PubMed] [Google Scholar]
- 5.Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, Djabali M. Polycomb mediated epigenetic silencing and replication timing at the INK4a/ARF locus during senescence. PLoS ONE. 2009;4:e5622. doi: 10.1371/journal.pone.0005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Knobloch TJ, Kresty LA, et al. Gankyrin, an epithelial biomarker, is frequently over-expressed in human oral cancers. Anticancer Res. 2011;31:2683–2692. [PubMed] [Google Scholar]
- 7.Poi MJ, Yen T, Li J, et al. Somatic INK4a-ARF locus mutations: A significant mechanism of gene inactivation in squamous cell carcinomas of the head and neck. Mol Carcinogenesis. 2001;30:26–36. doi: 10.1002/1098-2744(200101)30:1<26::aid-mc1010>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 8.Smith EM, Rubenstein LM, Hoffman H, Haugen TH, Turek LP. Human papollomavirus, p16 and p53 expression associated with survival of head and neck cancer. Infect Agent Cancer. 2010;5:4–10. doi: 10.1186/1750-9378-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanellou P, Zaravinos A, Zioga M, et al. Genomic instability, mutations and expression analysis of the tumor suppressor p14ARF, p15INK4b, p16INK4a and p53 in actinic keratosis. Cancer Lett. 2008;264:145–161. doi: 10.1016/j.canlet.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 10.Smeds J, Berggren P, Ma X, Xu Z, Hemminki K, Kumar R. Genetic status of cell cycle regulators in squamous cell carcinoma of the esophagus: The CDKN2A (p16INK4a and p14ARF) and p53 genes are major targets for inactivation. Carcinogenesis. 2002;23:645–655. doi: 10.1093/carcin/23.4.645. [DOI] [PubMed] [Google Scholar]
- 11.Esteller M, Tortola S, Toyota M, et al. Hypermethylation-associated inactivation of p14ARF is independent of p16INK4a methylation and p53 mutational status. Cancer Res. 2000;60:129–133. [PubMed] [Google Scholar]
- 12.Kresty LA, Mallery SR, Knobloch TJ, et al. Frequent alterations of p16INK4a and p14ARF in oral proliferative verrucous leukoplakia. Cancer Epidemiol Biomarkers Prev. 2008;17:3179–3187. doi: 10.1158/1055-9965.EPI-08-0574. [DOI] [PubMed] [Google Scholar]
- 13.Muscarella P, Bloomston M, Brewer AR, et al. Expression of p16INK4A/Cdkn2a gene is prevalently down-regulated in human pheochromocytoma tumor specimens. Gene Expr. 2008;14:207–216. doi: 10.3727/105221608786883825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Weghorst CM, Tsutsumi M, et al. Frequent p16INK4A/CDKN2A alterations in chemically induced Syrian golden hamster pancreatic tumors. Carcinogenesis. 2004;25:263–268. doi: 10.1093/carcin/bgh007. [DOI] [PubMed] [Google Scholar]
- 16.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 18.Jarrard DF, Bova GS, Ewing CM, et al. Deletional, mutational, and methylation analyses of CDKN2 (p16/MTS1) in primary and metastatic prostate cancer. Genes Chromosomes Cancer. 1997;19:90–96. [PubMed] [Google Scholar]
- 19.Lang JC, Borchers J, Danahey D, et al. Mutational status of overexpressed p16 in head and neck cancer: Evidence for germline mutation of p16/p14ARF. Int J Oncol. 2002;21:401–408. doi: 10.3892/ijo.21.2.401. [DOI] [PubMed] [Google Scholar]
- 20.Murphy PM, Tiffany HL, McDermott D, Ahuja SK. Sequence and organization of the human N-formyl peptide receptor-encoding gene. Gene. 1993;133:285–290. doi: 10.1016/0378-1119(93)90653-k. [DOI] [PubMed] [Google Scholar]