

Abstract
Background
Structural chromosomal rearrangements that lead to expressed fusion genes are a hallmark of acute lymphoblastic leukemia (ALL). In this study, we performed transcriptome sequencing of 134 primary ALL patient samples to comprehensively detect fusion transcripts.
Methods
We combined fusion gene detection with genome-wide DNA methylation analysis, gene expression profiling, and targeted sequencing to determine molecular signatures of emerging ALL subtypes.
Results
We identified 64 unique fusion events distributed among 80 individual patients, of which over 50% have not previously been reported in ALL. Although the majority of the fusion genes were found only in a single patient, we identified several recurrent fusion gene families defined by promiscuous fusion gene partners, such as ETV6, RUNX1, PAX5, and ZNF384, or recurrent fusion genes, such as DUX4-IGH. Our data show that patients harboring these fusion genes displayed characteristic genome-wide DNA methylation and gene expression signatures in addition to distinct patterns in single nucleotide variants and recurrent copy number alterations.
Conclusion
Our study delineates the fusion gene landscape in pediatric ALL, including both known and novel fusion genes, and highlights fusion gene families with shared molecular etiologies, which may provide additional information for prognosis and therapeutic options in the future.
Electronic supplementary material
The online version of this article (doi:10.1186/s13045-017-0515-y) contains supplementary material, which is available to authorized users.
Keywords: Pediatric acute lymphoblastic leukemia, RNA sequencing, Fusion genes, BCP-ALL, T-ALL, Translocation
Background
Chromosomal rearrangements giving rise to fusion genes play a central role in the malignant transformation of many cancers, including acute lymphoblastic leukemia (ALL) [1]. Recurrent large-scale structural rearrangements that result in expressed fusion transcripts are a hallmark of ALL and are included in the predictors of clinical outcome of individual patients that form the basis for treatment stratification [2]. The well-known subgroups of pediatric ALL based on expressed fusion genes include BCR-ABL1, ETV6-RUNX1, TCF3-PBX1, and 11q23/MLL rearrangements, such as KMT2A-AFF1, KMT2A-MLLT3, KMT2A-MLLT1, and KMT2A-MLLT10. In the Nordic countries, these fusion genes are routinely screened for at ALL diagnosis using fluorescent in situ hybridization (FISH) or polymerase chain reaction (PCR)-based assays [3].
In addition to recurrent rearrangements, ALL cells typically harbor other chromosomal aberrations that are detectable by routine cytogenetic screening and are non-recurrent or have not yet been associated with expressed fusion genes. Moreover, additional copy-neutral or cryptic translocations or inversions, which are not detectable by clinical routine methods, may occur in ALL cells. Therefore, there are potentially many fusion genes that have so far remained undetected in ALL. Identifying new fusion genes is important as they can serve as novel therapeutic targets and provide prognostic information [4]. Recent developments in transcriptome sequencing have enabled precise and sensitive detection of fusion genes in T cell acute lymphoblastic leukemia (T-ALL) [5, 6] and in B cell precursor acute lymphoblastic leukemia (BCP-ALL) [4, 7–13]. The most significant discoveries are the characterization of in-frame fusion genes involving DUX4, ZNF384, and MEF2D, which each define a new molecular subgroup of pediatric ALL with a distinct gene expression profile [12, 14, 15]. However, there is large heterogeneity regarding the cytogenetic subtypes screened and type of fusion genes that have been reported in these studies.
In the current study, we performed systematic analysis of expressed fusion genes by transcriptome sequencing in BCP-ALL and T-ALL cells collected at diagnosis from 134 patients with pediatric ALL. We surveyed the fusion gene landscape in 74 BCP-ALL patients with well-characterized recurrent subtypes, 42 BCP-ALL patients with karyotypes denoted “other” or “normal” that lack a defined cytogenetic subtype at diagnosis (BCP-ALL “other”), and 18 T-ALL patients. In total, we identified and validated 64 unique fusion events in 80 of the patients of which several have not been previously observed in pediatric ALL. We also identified distinct DNA methylation and gene expression profiles associated with recurrent fusion genes.
Methods
Patient and control samples
Bone marrow aspirates or peripheral blood samples were collected at diagnosis from 134 pediatric ALL patients. ALL diagnosis was established by analysis of leukemic cells with respect to morphology, immunophenotype, and cytogenetics (Table 1). The samples included in the study were of B cell precursor (BCP-ALL; n = 116) or of T cell immunophenotype (T-ALL; n = 18). Lymphocytes were isolated from the samples by Ficoll-isopaque centrifugation, and the proportion of leukemic blasts was determined by light microscopy as previously described [16]. The samples selected for analysis contained at least 80% leukemic blasts (average 91%). High-quality RNA was extracted with the AllPrep DNA/RNA Mini Kit (QIAGEN). No systematic differences in blast count or RNA quality was observed between the bone marrow aspirates and peripheral blood samples. The majority of patients were treated according to Nordic Society of Pediatric Hematology and Oncology (NOPHO) protocols except for patients with t(9;22)BCR-ABL1 who were treated with the EsPh-ALL protocol and children below 1 year who were treated according to Interfant protocols [3, 17, 18] (Additional file 1: Table S1). RNA was extracted from normal CD19+ B cells (n = 5) and CD3+ T cells (n = 5) isolated from peripheral blood mononuclear cells from five healthy Swedish blood donors as described previously [19]. Additional details about the RNA samples, characteristics of the leukemic cells, and treatment protocols can be found in Additional file 2. The guardians of the patients provided written or oral consent to the study. The study was approved by the regional ethics board in Uppsala, Sweden.
Table 1.
Summary of ALL samples included in the study
| Immuno-phenotype | Cytogenetic abnormality | Fusion gene | N | Median WBC at diagnosis, × 10^9/L (range) | Median age at diagnosis, years (range) |
|---|---|---|---|---|---|
| T-ALL | Various | 18 | 173.5 (1–588) | 11.9 (1.9–16.8) | |
| BCP-ALL | HeH | – | 42 | 9.6 (0.8–124) | 3.6 (1.0–17.74 |
| t(12;21) | ETV6-RUNX1 | 18 | 6.5 (1.1–95) | 4.9 (2.2–12.7) | |
| 11q23/MLL | MLL-r | 7 | 193 (1.8–744) | 0.6 (0.5–1.7) | |
| t(9;22) | BCR-ABL1 | 6 | 88.3 (14.1–180) | 12.1 (9.3–13.5) | |
| dic(9;20) | – | 1 | 41.8 (41.8–41.8) | 4.4 (4.4–4.4) | |
| Various (BCP-ALL “other”)a | – | 42 | 7.9 (0–213) | 8.8 (1.4–17.7) | |
| Total | 134 |
aBCP-ALL samples negative for targeted assays for known ALL cytogenetic aberrations with either non-recurrent aberrations, normal karyotypes, or no results available from cytogenetic analysis
WBC white blood cell count at diagnosis, MLL-r rearrangements involving the KMT2A (MLL) gene
Library preparation and transcriptome sequencing
Strand-specific RNA sequencing libraries were constructed from rRNA-depleted RNA using the ScriptSeq V2 Kit according to the manufacturer’s instructions (Epicentre). The libraries were sequenced using a Hiseq2000/2500 instrument (Illumina), 50 bp paired-end, with the exception of one sample (ALL_707) that was sequenced on a MiSeq instrument, 83 bp paired-end. Raw sequence reads were trimmed using Cutadapt 1.2.1 [20] and mapped to the human 1000 Genomes build 37 (GRCh37) using Tophat 2 (2.0.4) [21]. Quality control of RNA sequencing data was performed with RNA-SeQC [22] (Additional file 1: Table S2). Detailed information of RNA sequencing and computational analysis is provided in Additional file 2.
Fusion gene detection
Fusion genes were detected using a two-pronged approach. First, FusionCatcher v0.99.4a beta was used to screen for novel and known fusion genes in an unbiased manner (de novo search) [23]. Next, we screened a set of predefined established fusion genes in pediatric ALL (targeted search) by counting uniquely aligned reads supporting the fusion gene (Additional file 3: Fig. S1, Additional file 1: Table S4). To reduce false positives, a filtering process of the fusion genes detected by FusionCatcher was applied. These measures consisted of removing fusion genes that met any of the following criteria: blacklisted fusion genes based on reference data provided by FusionCatcher (Additional file 1: Table S3), fusion genes with fusion-supporting reads that map to multiple genomic locations indicative of sequence homology (common mapping reads), fusion genes where the 5′ or the 3′ fusion gene partner mapped to many other genes (highly promiscuous gene) (Additional file 1: Table S3), and fusion genes with less than three unique sequencing reads that support a detected chimeric transcript. Genes previously described to be involved in ALL-related fusion events were retained throughout all filtering steps. All fusion genes were experimentally validated by Sanger sequencing (Additional file 1: Table S5, Additional file 3: Fig. S1). Additional information about the fusion gene detection, the filtering procedure, and the validation is provided in Additional file 2.
Gene expression profiling
For quantification of gene expression, counts of aligned reads were summarized using featureCounts [24] and normalized using variance stabilizing transformation (R package DESeq2) [25]. Gene expression levels in fragments per kilobase per million mapped reads (FPKM) were determined using Cufflinks version 2.2.0 [26], and Cuffdiff was subsequently used to detect differentially expressed genes [27]. Analysis of differential gene expression was performed by pair-wise comparison of the expression levels between each of the following subgroups: DUX4-IGH (n = 8), ZNF384 rearrangements (n = 6), t(12;21)ETV6-RUNX1 (n = 18), t(9;22)BCR-ABL1 (n = 6), 11q23/MLL (n = 7), high hyperdiploid (HeH) (n = 42), and normal CD19+ B cells (n = 5). For determining differentially expressed genes in the groups with DUX4-IGH and ZNF384 rearrangements, only pair-wise comparisons with a false discovery rate corrected p value less than 0.1 and twofold difference in mean expression detected between three or more compared groups were regarded as differentially expressed. Lowly expressed genes with average FPKM < 5 were excluded from the analysis.
DNA methylation analysis
Previously published genome-wide DNA methylation data for ~450,000 CpG sites (450 k) was available for 130 out of the 134 patients included in the present study (GSE49031) [19]. Differentially methylated CpG sites (DMCs) were determined as previously described [19]. Briefly, DMCs were determined in the DUX4-IGH- and ZNF384-rearranged subgroups using remission bone marrow, CD19+ B cells, and CD34+ progenitor cells as the reference. A minimal cut-off value of 0.2 was applied for the mean absolute difference in DNA methylation (∆β) to highlight CpG sites with large differences between groups. The R package “CopyNumber450kCancer” was used to detect copy number alterations with the 450 k data [28].
Detection of somatic variants
Data from previously performed mutational analysis was available for 75 of the patients included in the present study [29]. Briefly, the exons of 872 cancer genes were captured using a HaloPlex Target Enrichment panel and sequenced to high depth (> 500×). DNA from matched remission samples and from healthy Swedish blood donors were used as controls. Single nucleotide variants (SNVs) were called using Freebayes and putative SNVs were filtered and annotated as previously described [29, 30].
Rapid amplification of cDNA ends
Rapid amplification of cDNA ends (RACE) was performed using the SMARTer RACE 5′/3′ Kit (Clontech) with 250 ng total RNA as input. The primers used for 3′ RACE of DUX4 and 5′ RACE of ZNF384 can be found in Additional file 1: Table S5. Sequencing libraries were prepared from the RACE fragments using the MicroPlex Library Preparation Kit v2 kit (Diagenode). Libraries were pooled and sequenced on a MiSeq instrument, PE150 bp read length using V2 chemistry. The reads were aligned to GRCh37 using Tophat 2 (2.0.4) [21] and STAR [31], and data were visualized in IGV [32].
Data availability
RNA sequencing data are available for academic purposes by contacting the corresponding author, as the patient/parent consent does not cover depositing data that can be used for large-scale determination of germline variants.
Results
Transcriptome sequencing and detection of fusion genes
To identify fusion genes in pediatric ALL, we sequenced the transcriptomes of 134 ALL samples, including 116 BCP-ALL and 18 T-ALL patients (Table 1; Additional file 1: Table S1). RNA sequencing yielded between 19 and 120 million (average 46 million) paired-end reads per sample. We detected 2136 candidate fusion transcripts with the FusionCatcher software across all the samples prior to filtering [23]. On average, we discovered 31 candidate fusion genes per sample (range 2–158). To reduce the number of potential false-positive fusion transcripts, we performed stringent filtering of the candidate fusion transcripts, including filtering of fusion genes called in the normal B and T cell to enrich for cancer-specific fusion genes in the ALL samples. This filtering procedure rendered a set of 197 unique candidate fusion genes identified in 97 of the ALL patient samples (Additional file 3: Fig. S1B). Next, we validated candidate fusion genes by visual examination of the aligned sequencing reads that supported a fusion junction in the RNA sequencing data. Of these candidates, 104 were selected for further validation by PCR followed by Sanger sequencing, where 61 of the fusion genes were experimentally validated (Additional file 1: Table S6). In addition, we performed targeted screening of 22 well-established ALL fusion genes whereby additional fusion genes were detected, including DUX4-IGH (n = 8 patients), TAF15-ZNF384 (n = 1), and STIL-TAL1 (n = 1) (Additional file 1: Table S4). Thus, after filtering and experimental validation, we detected a total of 64 unique fusion events, corresponding to 136 fusion genes in 80 of the patients included in the study (Fig. 1).
Fig. 1.

Distribution of validated fusion transcripts among 134 ALL patients. a Number of fusion genes detected per sample in BCP-ALL and T-ALL patients. The vertical axis shows the number of fusion genes identified per sample ranging from zero (no detectable fusion gene) up to four co-expressed fusion genes. b Violin plots showing the number of fusion genes per patient in each ALL subtype. No fusion genes were identified in the single dic(9;20) patient, and thus, this sample is excluded from the plot. The vertical axis shows the number of fusion genes identified per sample. Each dot represents one patient. c Chromosomal location of the 64 unique fusion events. The 5′ fusion gene partners are plotted in the left panel, and the 3′ fusion gene partners are plotted in the right panel together with their respective chromosomal locations. The thickness of the connecting lines reflects the recurrence of the fusion gene. Blue lines represent the canonical fusion genes associated with the t(12;21), t(9;22), or 11q23/MLL subtypes. Green lines represent recurrent non-canonical fusion genes and dashed gray lines represent fusion genes identified in a single patient. d Number of fusion genes per ALL subtype. The bars indicate the number of patients in which a given fusion gene was observed by subtype. The panel to the right of the plot shows the number of patients with any fusion gene out of all of patients belonging to the given subtype. The canonical fusion genes are highlighted in blue. Novel fusion genes discovered in this study are highlighted in red. + = in-frame fusion events. * = intra-chromosomal fusion events
Characteristics of the fusion genes in pediatric ALL
A fusion gene was detected in 74 of the 116 BCP-ALL patients, whereas a fusion gene was detected in only six out of the 18 T-ALL patients analyzed. No difference in fusion gene calls was observed between samples originating from the bone marrow or peripheral blood. Among the BCP-ALL patients with a detectable fusion gene, the frequency of fusion genes varied from one to four co-occurring fusion genes, although in the majority of the BCP-ALL patients (38/74), only one fusion gene was detected per sample (Fig. 1a). In the six T-ALL patients with a detectable fusion gene, only a single fusion gene was observed per sample.
In the BCP-ALL subtypes t(12;21)ETV6-RUNX1, t(9;22)BCR-ABL1, and 11q23/MLL, a fusion gene was detected by RNA-sequencing in all but one t(12;21) patient (ALL_504) and patients with the HeH subtype had the lowest frequency of fusion genes (Fig. 1b). In addition to fusion genes that are characteristic for ALL, we identified several other fusion genes in our patient cohort (Fig. 1c, d). The largest proportion of unique fusion genes was detected in the BCP-ALL “other” subgroup where a fusion gene was detected in 33 of the 42 patients, of which 16 patients expressed more than one.
Forty of the 64 unique fusion genes occurred between two genes on different chromosomes, while 24 fusion genes were caused by presumptive intra-chromosomal rearrangements, of which 18 (75%) of the involved genes are located over 1 Mbp distance from each other (Fig. 1c, d, Additional file 1: Table S6). Thirty-six fusion genes had an open reading frame, and in-frame fusion genes were more common in t(12;21), t(9;22), or the 11q23/MLL than in the ALL patients with HeH, where only two out of 12 fusion genes were in-frame. Most fusion genes (43/64) were only detected in a single patient and 36 (56%) have not been previously described in ALL.
The most common fusion gene was the well-known ETV6-RUNX1 and its reciprocal RUNX1-ETV6 in the t(12;21) BCP-ALL subtype (Fig. 1d). We also detected eight fusion genes, including six novel in-frame fusion genes that were expressed concurrently with ETV6-RUNX1. Contrary to the t(12;21) subtype, we only identified a single in-frame fusion gene (TNKS-ATL3) in addition to the canonical BCR-ABL1/ABL1-BCR in the t(9;22) subtype. In the 11q23/MLL subtype, no in-frame fusion genes were detected besides the canonical KMT2A-AFF1/AFF1-KMT2A and KMT2A-MLLT3. Among the BCP-ALL “other” patients, DUX4-IGH was detected in eight out of 42 patients and was thus the most common fusion gene in this subgroup, followed by recurrent fusion events involving ZNF384 or PAX5.
Genomic breakpoints
Information from karyotyping performed at diagnosis provided support for genomic breakpoints giving rise to the fusion genes. In addition to the canonical fusion genes in t(12;21), t(9;22), and 11q23/MLL, we found evidence for genomic breakpoints for 12 fusion genes in the karyotype data. Supporting evidence in the karyotype data was primarily observed as translocations between the chromosomes where the fusion genes are located (Additional file 1: Table S7). Furthermore, we used array-based copy number analysis (CNA) to detect evidence for chromosomal rearrangements within or in close proximity of the genes involved in a fusion event. Deletions or amplifications in the genomic regions of genes involved in a fusion event were identified for an additional 11 fusion genes (Additional file 1: Table S7, Additional file 3: Fig. S2). Thus, in total, we detected the presumable genomic breakpoint that gave rise to 23 out of the 57 unique non-canonical fusion genes.
Recurrent fusion genes
Of the 64 fusion genes, 21 were recurrent in BCP-ALL (Fig. 2a). Patients with t(12;21)ETV6-RUNX1 (n = 18), t(9;22)BCR-ABL (n = 6), and 11q23/MLL (n = 7), all harbored translocations resulting in fusion genes that had been verified either by FISH or RT-PCR and served as positive controls for fusion gene detection by RNA sequencing. We detected the characteristic subtype-defining fusion transcripts, including their reciprocal fusion genes, in 29/31 (94%) of the patients in this positive control group (Additional file 1: Table S1). The expected canonical fusion gene was not detected in two patients. In both cases (ALL_504 t(12;21) and ALL_16 11q23/MLL), the libraries were among those with the lowest sequence depth in the study (32 and 35 million read-pairs, respectively) (Additional file 1: Table S1). The targeted approach to identify fusion-supporting reads for ETV6-RUNX1 in ALL_504 revealed only two reads, while no fusion supporting reads in ALL_16 were detected for 11q23/MLL-related fusion partners, although this does not exclude the presence of an unknown or rare fusion partner that was not included in our target approach. It is highly likely that low sequencing depth in these two cases contributed to the false negative result.
Fig. 2.
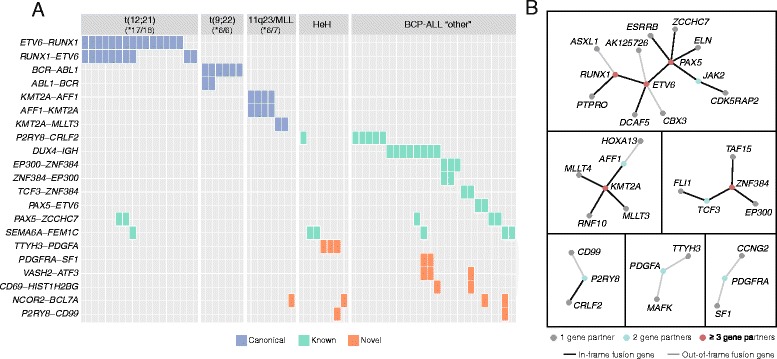
Recurrent fusion gene families in ALL. a Twenty-one fusion genes were recurrent in BCP-ALL patients. The fusion genes are plotted along the vertical axis, and the patients are plotted along the horizontal axis by the subtype given at ALL diagnosis. The numbers in the upper panels indicate the number of patients where the expected canonical fusion gene was detected by RNA sequencing in each subtype. The canonical subtype-associated fusion genes are marked in blue, the previously reported fusion genes in ALL are indicated in green, and the novel fusion genes discovered here are indicated in red. b Frequently translocated genes giving rise to expressed fusion genes formed six independent node groups. The nodes representing genes that fused with two different partners are shown with green dots, and genes that can have three or more fusion partners are shown with red dots. In-frame fusion genes are highlighted with a black line
Balanced translocations, which can result in expression of reciprocal fusion genes, occur in the t(12;21)ETV6-RUNX1, t(9;22)BCR-ABL1, and 11q23/MLL subtypes. In agreement with this, we found co-expression of the reciprocal fusion gene in several patients belonging to these three BCP-ALL subtypes (Fig. 2a). In the 11q23/MLL subgroup, we found co-expression of KMT2A-AFF1 and AFF1-KMT2A in all four patients with t(4;11), but no evidence for co-expression of the reciprocal fusion gene in the two patients with KMT2A-MLLT3. We also identified reciprocal BCR-ABL1 and ABL1-BCR in two out of the six t(9;22) patients. In the t(12;21) subtype, we detected co-expressed ETV6-RUNX1 and reciprocal RUNX1-ETV6 in eight out of 18 t(12;21) patients. Moreover, in three out of the seven patients with ETV6-RUNX1, but without the reciprocal RUNX1-ETV6, we identified co-expression of two other previously unreported in-frame fusion genes, namely DCAF5-ETV6 (ALL_386) and RUNX1-PTPRO (ALL_9 and ALL_678), which appear to have arisen from cryptic unbalanced translocation of the truncated ETV6 or RUNX1 gene to another chromosomal region (Additional file 1: Table S1). Interestingly, the PTPRO gene is located on chromosome 12 approximately 3.6 Mbp downstream of ETV6. The DCAF5 gene is located on chromosome 14, and the karyotype of ALL_386 revealed a complex translocation involving chromosomes 3, 12, and 14.
The DUX4-IGH fusion gene (n = 8) and ZNF384 rearrangements involving EP300 (n = 3) or TCF3 (n = 2) were the most frequently occurring fusion genes in the BCP-ALL “other” group in addition to P2RY8-CRLF2. Together with PAX5-ETV6 (n = 2), DUX4-IGH and ZNF384 rearrangements were observed exclusively in BCP-ALL “other” patients. The novel PDGFRA-SF1 (n = 2), VASH2-ATF3 (n = 3), and CD69-HIST1H2BG (n = 2) were also found exclusively in BCP-ALL “other” and co-expressed with aforementioned fusion genes such as DUX4-IGH or ZNF384-TCF3. Interestingly, TTHY3-PDGFA was found exclusively in three patients with HeH.
Fusion gene nodes
In addition to the recurrent fusion genes described above, we identified promiscuous genes that were fused with several gene partners (Fig. 2b). Of the 89 unique genes involved in fusion events, 11 genes had more than one fusion partner and constitute six independent nodes. These genes include the well-known ALL genes ETV6, RUNX1, KMT2A, and PAX5 that form fusion genes with up to five different partners; the emerging BCP-ALL subgroup defined by ZNF384 rearrangements; and the new P2RY8, PDGFA, and PDGFRA nodes.
ETV6 and PAX5 were the most frequently translocated genes and formed the largest network of gene connections often resulting in in-frame fusion genes. Although fusion genes involving ETV6 and RUNX1 were predominantly detected in the t(12;21) subgroup, CBX3-ETV6, RUNX1-ASXL1, and ETV6-AK125726 were detected in patients with other subtypes. These three out-of-frame fusion genes have been previously described in these patients based on a t(12;21)-like DNA methylation signature [8]. This is contrary to the general pattern in the t(12;21) subgroup, where additional in-frame fusion genes with ETV6 or RUNX1 are expected. For example, DCAF5-ETV6 and RUNX1-PTPRO are both in-frame.
DNA methylation and transcriptional signatures
Next, to obtain a view of the molecular variation associated with recurrent fusion genes in our dataset, we performed unsupervised clustering analysis using array-based genome-wide DNA methylation and gene expression data from RNA sequencing (Fig. 3, Additional file 3: Fig. S3). T(12;21)-like DNA methylation patterns have previously been described for three of the patients included in this study (ALL_11, ALL_106, and ALL_495) as mentioned above. These patients were found to harbor fusion genes involving either ETV6 or RUNX1, but not the canonical ETV6-RUNX1 fusion gene [8]. In agreement with these findings, these three patients clustered together with the t(12;21)ETV6-RUNX1 patients in DNA methylation and gene expression data (Fig. 3a, b, e, f). Notably, the patient with a CBX3-ETV6 fusion gene that was diagnosed as HeH and verified by CNA clustered together with the t(12;21) rather than with the HeH subgroup. Furthermore, three t(12;21) patients (ALL_386, ALL_9, and ALL_678) harboring unbalanced t(12;21)-translocations with DCAF5-ETV6 or RUNX1-PTPRO, and six patients in which only one of the reciprocal fusion genes was detected, clustered with the t(12;21) based on both DNA methylation and gene expression data. The two patients with PAX5-ETV6 did not cluster with the t(12;21) patients, most likely due to the downstream effect of altered PAX5, rather than ETV6 in these patients.
Fig. 3.
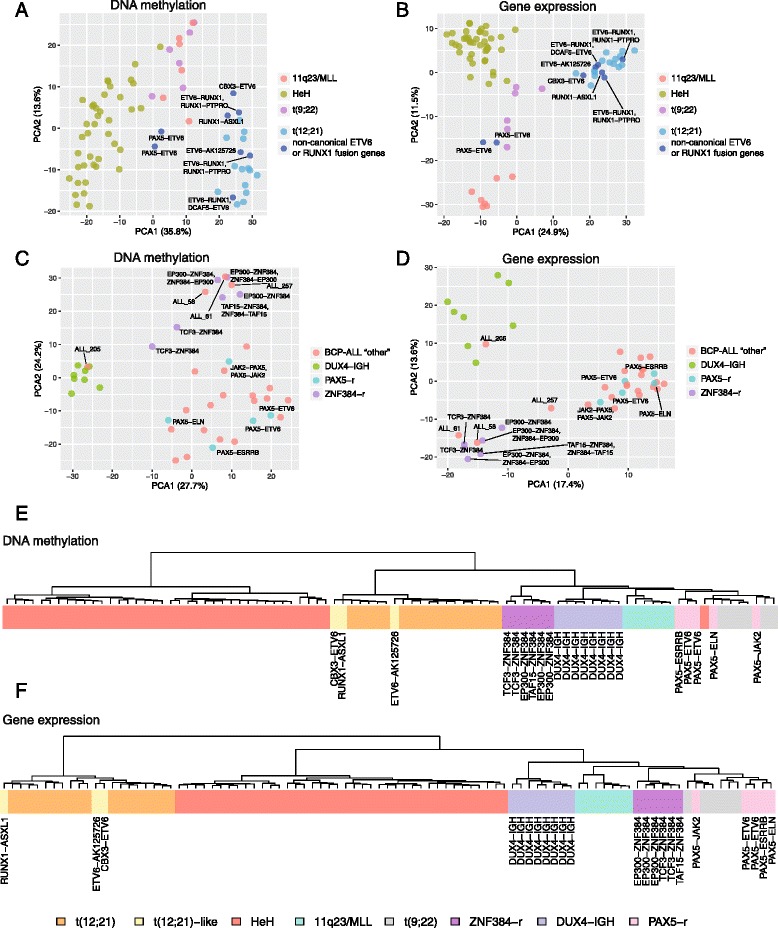
Clustering analysis using genome-wide DNA methylation and gene expression patterns in BCP-ALL patients with expressed fusion genes. a, b Principal component analysis of patients harboring non-canonical fusion genes involving ETV6 or RUNX1 together with patient samples with an established cytogenetic subtype (t(12;21)ETV6-RUNX1, t(9;22)BCR-ABL1, 11q23/MLL, and HeH) using a DNA methylation and b gene expression data. The fraction of the variance explained by principal component 1 (PC1) and principal component 2 (PC2) are shown on the horizontal and vertical axes, respectively. c, d Principal component analysis of BCP-ALL “other” patients using c DNA methylation and d gene expression data. Patients with DUX4-IGH rearrangements, ZNF384 rearrangements (ZNF384-r), and PAX5 rearrangements (PAX5-r), together with BCP-ALL “other” patients harboring other fusion genes or no detectable fusion gene, are shown in the figures. Patients that cluster together with IGH-DUX4- or ZNF384-rearranged patients, although with no characterizing fusion gene, are highlighted in the figure. The fraction of the variance explained by principal component 1 (PC1) and principal component 2 (PC2) are shown on the horizontal and vertical axes, respectively. e, f Unsupervised hierarchical clustering using e DNA methylation and f gene expression data. Patients belonging to the t(12;21)ETV6-RUNX1, t(9;22)BCR-ABL1, 11q23/MLL, and HeH subtypes are shown together with patients harboring t(12;21)-like fusion genes, DUX4-IGH, ZNF384, and PAX5 rearrangements. For ease of interpretation, only one of the paired reciprocal fusion genes detected in a sample is shown in panels e and f. The 1000 most variably methylated CpG sites or expressed genes were used in all panels
We also examined the DNA methylation and gene expression patterns for recurrent fusion gene families characterized by DUX4-IGH, ZNF384, or PAX5 rearrangements in the BCP-ALL “other” group. Three major clusters defined by the recurrent fusion gene families emerged (Fig. 3c, d). One additional patient (ALL_205) clustered together with the eight DUX4-IGH patients, although DUX4-IGH was not detected by RNA sequencing. DUX4-rearranged cases have previously been associated with a distinct over-expression of the DUX4 gene [14]. In agreement with this, ALL_205 displayed similarly high levels of DUX4 expression as the DUX4-IGH-positive cases at much higher levels compared to other BCP-ALL subgroups and controls (Additional file 3: Fig. S4A). 3′ RACE confirmed the presence of a DUX4-IGH fusion gene in ALL_205; however, ~470 bp from chromosome 8q24.21 (chr8:130,691,944-130,692,413) was identified as inserted between the DUX4 and IGH genes in the fusion transcript; thus, it was not initially detected by our targeted screening approach (Additional file 3: Fig. S5A).
The six patients with ZNF384 rearrangements clustered together in both the DNA methylation and gene expression data (Fig. 3c–f). In the ZNF384 cluster, we observed three additional patients (ALL_58, ALL_61, and ALL_257) with no evidence for a ZNF384 rearrangement despite targeted screening (Fig. 3c, d). Unlike the DUX4-IGH group, patients with ZNF384 rearrangements lacked differential expression of the ZNF384 gene and its associated fusion partners (Additional file 3: Fig. S4B). 5′ RACE was performed to amplify the fusion transcripts without prior knowledge of the ZNF384 fusion partner in these three patients. A novel fusion between the ATP5C1 gene on chromosome 10 (5′ fusion partner) and ZNF384 (3′ fusion partner) was detected in ALL_257 (Additional file 3: Fig. S5B). The RACE experiments were inconclusive for ALL_58 and ALL_61, and additional experiments will be needed to identify which, if any, ZNF384 fusion gene is present in these patients.
Consistent with the previously reported Philadelphia-like signature associated with PAX5-JAK2, patient ALL_539 clustered together with the t(9;22)BCR-ABL1 patients (Fig. 3e, f) [4, 33]. The remaining patients with other PAX5 fusion genes clustered together.
Differential DNA methylation and gene expression
To date, differential DNA methylation has not been comprehensively studied in the DUX4-IGH- and ZNF384-rearranged subgroups. We therefore highlight DMCs in combination with differentially expressed genes in patients with the DUX4-IGH and ZNF384 rearrangements compared to patients with well-established ALL subtypes and normal CD19+ B cells.
We detected 2740 and 3516 DMCs specific to the DUX4-IGH- and ZNF384-rearranged subgroups, respectively (Additional file 1: Table S8–S9). DUX4-IGH was characterized by widespread hypomethylation compared with normal B cells and the other ALL subtypes, whereas the group with ZNF384 rearrangements was hypermethylated (Fig. 4, Additional file 1: Table S10). The DMCs were distributed across 245 and 192 genes unique to the DUX4-IGH and ZNF384-rearranged groups, respectively (Additional file 1: Table S10). While no enrichment to known pathways was observed, an enrichment of genes regulated by the transcription factor E2F1 was found in the DUX4-IGH subgroup (p = 8.2 × 10− 5). E2F1 is a member of the E2F family of transcription factors and acts as a potent transcriptional activator and master regulator of cell cycle progression.
Fig. 4.
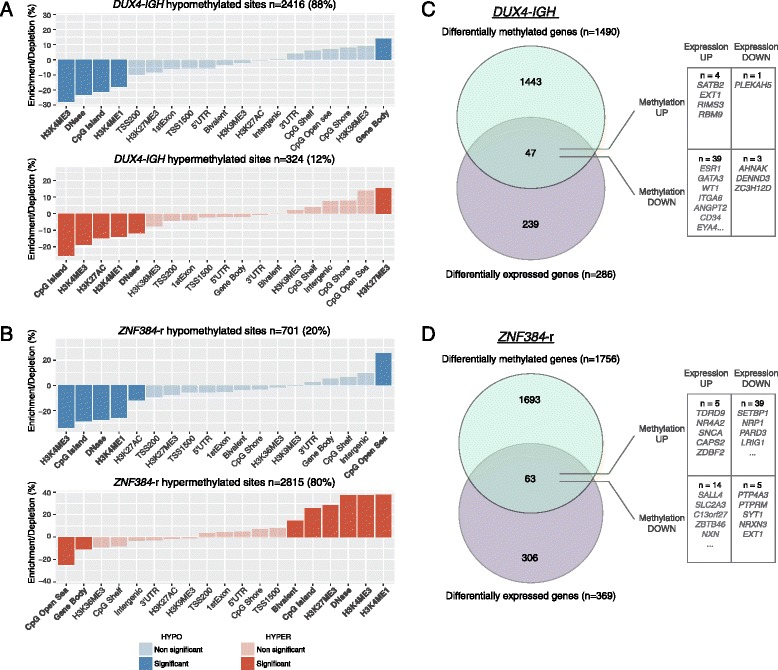
Functional genomic distribution of differentially methylated CpG sites (DMCs) in patients with DUX4-IGH and ZNF384 rearrangements. a, b The enrichment and depletion of hypomethylated DMCs in blue and hypermethylated DMCs in red were determined in relation to CpG island context (island, shore, shelf, open sea), gene regions, and chromatin states in groups with a DUX4-IGH and b ZNF384 rearrangements. The bars show the difference in proportion of the CpG sites annotated to each functionally annotated region between the 450 k array and the DMCs. The colored bars represent the annotations to which the DMCs significantly differ compared with the distribution of probes on the 450 k array (Bonferroni-corrected two-sided Fisher’s exact P value < 0.01 and absolute difference in proportion > 10). c, d Overlap between differentially methylated genes (green) and differentially expressed genes (purple) in patients with c DUX4-IGH and d ZNF384 rearrangements. The overlapping genes with hypermethylated and hypomethylated DMCs and up- or downregulated gene expression are listed in the right panel
To provide additional insights into possible functional implications of the subtype-specific DMCs, we examined their distribution across functional genomic regions defined by chromatin marks and DNaseI hypersensitive sites and in relation to CpG islands and gene-centric regions. The majority of the DMCs (88%) in DUX4-IGH were hypomethylated, enriched to gene bodies, and depleted in regions with open chromatin and high CG density (Fig. 4a). In contrast, the predominantly hypermethylated DMCs (80%) in the ZNF384-rearranged group were strongly enriched in CpG islands marked by bivalent chromatin marks (H3K4me3 and H3K27me3) and in open chromatin regions (Fig. 4b).
In order to investigate whether differential methylation is associated with gene expression, differential gene expression analysis was performed in the DUX4-IGH- and ZNF384-rearranged groups (Additional file 1: Table S11–S12). Approximately 3% of the genes with DMCs overlapped with differentially expressed genes corresponding to 47 and 63 overlapping genes in the DUX4-IGH- and ZNF384-rearranged groups, respectively (Fig. 4c, d, Additional file 1: Table S13). Most of the overlapping genes in the DUX4-IGH group showed an inverse correlation between methylation and gene expression (85%), with the majority of genes upregulated in DUX4-IGH compared to the other subtypes. Several of the overlapping genes (n = 9) were directly regulated or associated with the hypomethylated ESR1 gene such as GATA3, WT1, and ITGA6. Alterations involving ESR1 has mainly been described in breast cancer [34]. However, a study performed in non-hyperdiploid multiple myeloma proposed that ESR1 contributes to cell cycle dysregulation, thus affecting the transcription of several downstream genes including E2F1 [35]. Similarly, in the ZNF384-rearranged group, the majority of the genes showed an inverse correlation between methylation and gene expression (84%); however, the differentially expressed and methylated genes were downregulated compared to the other ALL subgroups. These include genes involved in hematological system development such as SETBP1 and NRP1 and putative tumor suppressor genes such as PARD3 and LRIG1 [36–38]. A notable exception is the overexpression of SALL4 in the ZNF384-rearranged group, which has been described as an oncogene in leukemia [39, 40].
Genetic alterations
To further characterize the subgroups with DUX4-IGH and ZNF384 rearrangements, we utilized previously published targeted exome sequencing data including 872 cancer genes [29] and array-based copy number data derived from the HumanMethylation 450 k arrays from the same patients [19]. Targeted sequencing data was available for five out of the nine DUX4-IGH positive cases. Non-synonymous somatic mutations in the mutation hotspot p.G12 of the NRAS gene were found in all five DUX4-IGH-positive samples that were analyzed by targeted sequencing (Table 2). We screened for NRAS mutations in the RNA-sequencing reads confirming the presence of the five aforementioned NRAS mutations, but no additional NRAS mutations were detected in the remaining four patients. We detected ERG deletions, which is a common alteration in this subgroup [14, 15], in seven out of the nine DUX4-IGH patients (Additional file 3: Fig. S6). Interestingly, we also detected shared non-synonymous PTPN11 mutations and chr7q deletions in the two patients with TCF3-ZNF384 (Table 2). These mutations were confirmed in the RNA-sequencing data, and no additional PTPN11 mutations were detected in the other patients with ZNF384 rearrangements. Furthermore, Hirabayashi et al. reported that two out of six patients with TCF3-ZNF384 harbored PTPN11 mutations detected by whole exome sequencing or RNA sequencing; however, no additional copy number analysis was performed [13]. Together, these findings, albeit in a small sample set, suggest a common pattern related with the TCF3-ZNF384 fusion gene.
Table 2.
Genetic alterations in patients with DUX4-IGH and ZNF384 rearrangements
| Fusion gene | Sample | Mutated ALL driver genesa | Copy number alterations |
|---|---|---|---|
| DUX4-IGH fusion gene | |||
| DUX4-IGH | ALL_176 | na | ERG deletion |
| DUX4-IGH | ALL_218 | na | Inconclusive |
| DUX4-IGH | ALL_312 | NRAS (p.G12D) | ERG deletion |
| DUX4-IGH | ALL_371 | NRAS (p.G12D) | ERG deletion |
| DUX4-IGH | ALL_390 | na | ERG deletion |
| DUX4-IGH | ALL_392 | na | ERG deletion |
| DUX4-IGH | ALL_501 | NRAS (p.G12S), KMT2D (p.H3883fs) | ERG deletion |
| DUX4-IGH | ALL_546 | NRAS (p.G12S) | ERG deletion |
| DUX4-chr8q24.21-IGH | ALL_205 | NRAS (p.G12D) | Inconclusive |
| ZNF384-rearranged fusion genes | |||
| TCF3-ZNF384 | ALL_604 | PTPN11 (p.E76Q) | chr7q deletion |
| TCF3-ZNF384 | ALL_622 | PTPN11 (p.T73I) | chr7q deletion |
| TAF15-ZNF384, ZNF384-TAF15 | ALL_8 | NRAS (p.Q61H) | – |
| EP300-ZNF384, ZNF384-EP300 | ALL_52 | na | – |
| EP300-ZNF384, ZNF384-EP300 | ALL_613 | na | – |
| EP300-ZNF384 | ALL_693 | na | – |
| ATP5C1-ZNF384 | ALL_257 | CREBBP (p.R1408C) | – |
aKnown ALL driver genes are reported in this table
na no targeted sequencing data available
Clinical features
Recent studies have shown that DUX4 rearrangements are associated with a favorable prognosis of pediatric ALL, whereas ZNF384 rearrangements appear to be associated with an intermediate outcome. To assess the prognostic impact of recurrent fusion genes in our cohort, we determined the event-free survival (EFS) in the BCP-ALL subgroups (Additional file 3: Fig. S7). One relapse was observed in the nine patients with DUX4-IGH, which confirms previous reports of a generally favorable outcome associated with DUX4 rearrangements [9, 14, 15]. Inferior prognosis has been associated with TCF3-ZNF384, while favorable prognosis has been associated with the EP300-ZNF384 fusion gene [13]. In the present study, no relapses were observed in patients with EP300-ZNF384. Relapses were observed in patients with all of the other fusion gene partners (TCF3-ZNF384 (ALL_622), TAF15-ZNF384 (ALL_8), and ATP5C1-ZNF384 (ALL_257)).
Discussion
In our study, we screened the whole transcriptome of 134 pediatric ALL patients by RNA sequencing to comprehensively identify expressed fusion genes. Our results highlight recurrent fusion gene families, comprising recurrent fusion genes and promiscuous genes that form fusion genes with several partners in the heterogeneous BCP-ALL “other” group, and add to the repertoire of recurrent fusion events in pediatric ALL.
Accurate identification of fusion genes requires a balance between increasing sensitivity in combination with reduction of false positives by filtering and validation experiments. Even after computational filtering, we performed PCR and Sanger sequencing to validate each unique fusion gene in comparison with CD19+ B cells and CD3+ T cells from healthy blood donors to ascertain that all identified putative fusion gene was expressed exclusively in ALL cells. This approach excluded 2075 of the putative fusion genes called by FusionCatcher [23], and thus, we identified an average of 1.8 fusion genes per sample validated with high confidence in BCP-ALL and 1.0 fusion genes per sample in T-ALL.
The most significant recent findings in terms of fusion genes in pediatric ALL are the fusion genes involving DUX4, ZNF384, and MEF2D [9, 10, 14]. We detected DUX4-IGH in 21% of our BCP-ALL “other” patients and various ZNF384 rearrangements in 17% of our BCP-ALL “other” patients. The children in these groups were older at diagnosis (median 9.3 years in DUX4-IGH and 10.7 in ZNF384-rearranged vs 4.5 years in BCP-ALL) and exhibited similar clinical outcome as in previous studies [9, 14, 15, 41–43]. Because of these consistent findings, DUX4-IGH should be considered as a potential favorable clinical marker in BCP-ALL and evaluated together with other prognostic factors in larger ALL cohorts.
To date, four studies have screened for somatic mutations in DUX4-IGH- and ZNF384-rearranged cases [12–15]. NRAS mutations are present in approximately 20% of BCP-ALL samples [29] and are enriched in the HeH subtype [44]. NRAS mutations have been reported in 13–26% of DUX4-rearranged cases, yet we found them in > 50% of cases. Our results, although based on a small number of samples, indicate that the frequency of NRAS mutation may in fact be higher in this subgroup. Both of the TCF3-ZNF384 patients in the present study harbored somatic PTPN11 mutations. In the few ZNF384-rearranged cases described in the literature, PTPN11 mutations have only been observed in ZNF384-rearranged cases when they have the TCF3 partner gene [12, 13]. These observations, however, are based on a small number of samples, and larger cohorts should be investigated to determine the actual mutational frequencies and if the mutations have any prognostic impact.
MEF2D fusion genes have recently been reported to comprise a new biological subtype of BCP-ALL with an overall frequency of 4–7% but are predominantly found in young adults [9, 10, 45, 46]. We did not identify any MEF2D fusion genes, despite performing additional targeted screening (Additional file 1: Table S4). Notably, Lilljebjörn et al. detected one patient with a MEF2D fusion gene in an independent cohort of 195 Swedish pediatric BCP-ALL patients [14]. The younger average age of the patients in Lilljebjörn et al. and the present study (7.1 and 5 years, respectively) most likely explains why so few MEF2D fusion genes were detected in the two Nordic cohorts.
Fusion genes can affect cellular functions through several means including formation of constitutively active chimeric proteins or by creating dominant negative proteins that inhibit normal protein function, which in turn could lead to the malignant transformation into cancer cells [47]. Fifty-six percent of the fusion genes detected in the current study were in-frame and the subtype-defining fusion genes are typically in-frame (ETV6-RUNX1, BCR-ABL1, and KMT2A rearrangements; TCF3-PBX1, DUX4-IGH, and ZNF384 rearrangements). Even reciprocal fusion genes arising from balanced breakpoints, when expressed, are typically in-frame. A notable exception to this pattern is the t(12;21)-like fusion genes consisting of RUNX1-ASXL1, CBX3-ETV6, and ETV6-AK125726, which were all out-of-frame. Despite this, the patients harboring t(12;21)-like out-of-frame fusion genes, display highly similar DNA methylation patterns as the t(12;21)-positive cases, as we have previously described [8] and similar gene expression patterns as showed in the present study and by others [14]. T(12;21)-like fusion genes also infer similar favorable clinical outcomes as ETV6-RUNX1, which raises the interesting point whether these fusion genes could be used for treatment stratification purposes. We also identified fusion genes in 11/42 HeH patients, which is more common than previously observed [14]. With the exception of ARHGEF6–ORC4 and P2RY8-CRLF2, all the other fusion genes detected in HeH were out-of-frame and most were intra-chromosomal.
Conclusion
Expressed fusion genes are a recognized form of driver mutations in ALL, and the insights gained from our study will help to shape our understanding of ALL pathogenesis and the heterogeneity between patients. Moreover, studies like these offer the possibility to identify targetable fusion genes as has been done for tyrosine kinase-related fusion genes [11, 48, 49]. Considering the decrease in sequencing costs coupled with recent advances in deep-sequencing technology, unbiased and detailed screening of fusion genes is now feasible. The next challenge will be to identify the downstream targets and cellular processes that are affected by fusion genes, which in turn may lead to refined stratification of patients for existing therapies and the discovery of new therapies for ALL.
Additional files
Supplementary Tables S1-S13. (XLSX 1165 kb)
Additional materials and methods. (DOCX 29 kb)
Supplementary Figures S1-S7. (PDF 2992 kb)
Acknowledgements
We thank Anna-Karin Lannergård, Christina Leek, Lili Milani, Elin Övernäs, and Anders Lundmark for the technical assistance. We especially thank our colleagues from NOPHO and the ALL patients who contributed samples to this study. RNA sequencing was performed by the SNP&SEQ Technology Platform, which is part of Science for Life Laboratory and the National Genomics Infrastructure at Uppsala University, supported by the Swedish Research Council (VR-RFI) and the Knut and Alice Wallenberg Foundation. Computational analysis was performed using resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science.
Funding
This work was supported by grants from the Swedish Foundation for Strategic Research (RBc08-008 ACS and GL), the Swedish Cancer Society (130440; 160711 ACS), the Swedish Childhood Cancer Foundation (11098 ACS), the Swedish Research Council for Science and Technology (C0524801; 2016-03691_3 ACS), and the Erik, Karin, and Gösta Selanders Stiftelse (JN).
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available as the patient/parent consent does not cover depositing data that can be used for large-scale determination of germline variants but are available from the corresponding author on reasonable request.
Abbreviations
- ALL
Acute lymphoblastic leukemia
- BCP-ALL
B cell precursor acute lymphoblastic leukemia
- CNA
Copy number analysis
- DMC
Differentially methylated CpG site
- EFS
Event-free survival
- FPKM
Fragments per kilobase per million mapped reads
- HeH
High hyperdiploid
- NOPHO
Nordic Society of Pediatric Hematology and Oncology
- RACE
Rapid amplification of cDNA ends
- SNVs
Single nucleotide variants
- T-ALL
T cell acute lymphoblastic leukemia
Authors’ contributions
ACS, GL, and JN conceived the study. YMZ, JD, SNi, and JN analyzed the data. YMZ, JD, ACS, and JN wrote the paper. AR performed the lab work. CML and ECB provided the Haloplex data. YMZ, SNi, and SNy performed the validation experiments. EF provided the karyotype data. JA, LC, EF, MH, and GL provided the ALL patient material and clinical data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the regional ethics board in Uppsala, Sweden, and the patients and/or guardians provided written or oral consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13045-017-0515-y) contains supplementary material, which is available to authorized users.
Contributor Information
Yanara Marincevic-Zuniga, Email: yanara.marincevic_zuniga@medsci.uu.se.
Johan Dahlberg, Email: johan.dahlberg@medsci.uu.se.
Sara Nilsson, Email: saranilsson9258@gmail.com.
Amanda Raine, Email: amanda.raine@medsci.uu.se.
Sara Nystedt, Email: sara.nystedt@medsci.uu.se.
Carl Mårten Lindqvist, Email: carl_marten.lindqvist@medsci.uu.se.
Eva C. Berglund, Email: eva.berglund@medsci.uu.se
Jonas Abrahamsson, Email: vobjab@gmail.com.
Lucia Cavelier, Email: lucia.cavelier@igp.uu.se.
Erik Forestier, Email: erik.forestier@medbio.umu.se.
Mats Heyman, Email: mats.heyman@ki.se.
Gudmar Lönnerholm, Email: gudmar.lonnerholm@kbh.uu.se.
Jessica Nordlund, Phone: +46 70 425 08 06, Email: jessica.nordlund@medsci.uu.se.
Ann-Christine Syvänen, Email: ann-christine.syvanen@medsci.uu.se.
References
- 1.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 2.Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26:123–135. doi: 10.1016/j.blre.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S, Taskinen M. Nordic Society of Paediatric H, Oncology. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia. 2010;24:345–354. doi: 10.1038/leu.2009.251. [DOI] [PubMed] [Google Scholar]
- 4.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, Mentens N, Chiaretti S, Durinck K, Uyttebroeck A, et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013;9:e1003997. doi: 10.1371/journal.pgen.1003997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gianfelici V, Chiaretti S, Demeyer S, Di Giacomo F, Messina M, La Starza R, Peragine N, Paoloni F, Geerdens E, Pierini V, et al. RNA sequencing unravels the genetics of refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic and therapeutic implications. Haematologica. 2016;101:941–950. doi: 10.3324/haematol.2015.139410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lilljebjorn H, Agerstam H, Orsmark-Pietras C, Rissler M, Ehrencrona H, Nilsson L, Richter J, Fioretos T. RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia. 2014;28:977–979. doi: 10.1038/leu.2013.324. [DOI] [PubMed] [Google Scholar]
- 8.Nordlund J, Backlin CL, Zachariadis V, Cavelier L, Dahlberg J, Ofverholm I, Barbany G, Nordgren A, Overnas E, Abrahamsson J, et al. DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin Epigenetics. 2015;7:11. doi: 10.1186/s13148-014-0039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, Imoto N, Kohsaka S, Kunita A, Doi K, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet. 2016;48:569–574. doi: 10.1038/ng.3535. [DOI] [PubMed] [Google Scholar]
- 10.Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, Jiang L, Li JF, Wang MJ, Dai YJ, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine. 2016;8:173–183. doi: 10.1016/j.ebiom.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imamura T, Kiyokawa N, Kato M, Imai C, Okamoto Y, Yano M, Ohki K, Yamashita Y, Kodama Y, Saito A, et al. Characterization of pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia with kinase fusions in Japan. Blood Cancer J. 2016;6:e419. doi: 10.1038/bcj.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian M, Zhang H, Kham SK, Liu S, Jiang C, Zhao X, Lu Y, Goodings C, Lin TN, Zhang R, et al. Whole-transcriptome sequencing identifies a distinct subtype of acute lymphoblastic leukemia with predominant genomic abnormalities of EP300 and CREBBP. Genome Res. 2017;27:185–195. doi: 10.1101/gr.209163.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirabayashi S, Ohki K, Nakabayashi K, Ichikawa H, Momozawa Y, Okamura K, Yaguchi A, Terada K, Saito Y, Yoshimi A, et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica. 2017;102:118–129. doi: 10.3324/haematol.2016.151035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lilljebjorn H, Henningsson R, Hyrenius-Wittsten A, Olsson L, Orsmark-Pietras C, von Palffy S, Askmyr M, Rissler M, Schrappe M, Cario G, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun. 2016;7:11790. doi: 10.1038/ncomms11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, Wu G, Li Y, Wei L, Iacobucci I, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet. 2016;48:1481–1489. doi: 10.1038/ng.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milani L, Lundmark A, Kiialainen A, Nordlund J, Flaegstad T, Forestier E, Heyman M, Jonmundsson G, Kanerva J, Schmiegelow K, et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood. 2010;115:1214–1225. doi: 10.1182/blood-2009-04-214668. [DOI] [PubMed] [Google Scholar]
- 17.Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, Hovi L, LeBlanc T, Szczepanski T, Ferster A, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370:240–250. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 18.Biondi A, Schrappe M, De Lorenzo P, Castor A, Lucchini G, Gandemer V, Pieters R, Stary J, Escherich G, Campbell M, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol. 2012;13:936–945. doi: 10.1016/S1470-2045(12)70377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nordlund J, Backlin CL, Wahlberg P, Busche S, Berglund EC, Eloranta ML, Flaegstad T, Forestier E, Frost BM, Harila-Saari A, et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013;14:r105. doi: 10.1186/gb-2013-14-9-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011. doi:10.14806/ej.17.1.200.
- 21.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nicorici D, Satalan M, Edgren H, Kangaspeska S, Murumagi A, Kallioniemi O, Virtanen S, Kilkku O. FusionCatcher—a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv. 2014. doi:10.1101/011650.
- 24.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 25.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marzouka NA, Nordlund J, Backlin CL, Lonnerholm G, Syvanen AC, Carlsson AJ. CopyNumber450kCancer: baseline correction for accurate copy number calling from the 450k methylation array. Bioinformatics. 2016;32:1080–1082. doi: 10.1093/bioinformatics/btv652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindqvist CM, Lundmark A, Nordlund J, Freyhult E, Ekman D, Carlsson Almlof J, Raine A, Overnas E, Abrahamsson J, Frost BM, et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget. 2016;7:64071–64088. doi: 10.18632/oncotarget.11773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindqvist CM, Nordlund J, Ekman D, Johansson A, Moghadam BT, Raine A, Overnas E, Dahlberg J, Wahlberg P, Henriksson N, et al. The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Hum Mutat. 2015;36:118–128. doi: 10.1002/humu.22719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nebral K, Denk D, Attarbaschi A, Konig M, Mann G, Haas OA, Strehl S. Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia. 2009;23:134–143. doi: 10.1038/leu.2008.306. [DOI] [PubMed] [Google Scholar]
- 34.Bailey SD, Desai K, Kron KJ, Mazrooei P, Sinnott-Armstrong NA, Treloar AE, Dowar M, Thu KL, Cescon DW, Silvester J, et al. Noncoding somatic and inherited single-nucleotide variants converge to promote ESR1 expression in breast cancer. Nat Genet. 2016;48:1260–1266. doi: 10.1038/ng.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Yan Z, Fulciniti M, Li Y, Gkotzamanidou M, Amin SB, Shah PK, Zhang Y, Munshi NC, Li C. Transcription factor-pathway coexpression analysis reveals cooperation between SP1 and ESR1 on dysregulating cell cycle arrest in non-hyperdiploid multiple myeloma. Leukemia. 2014;28:894–903. doi: 10.1038/leu.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonastre E, Verdura S, Zondervan I, Facchinetti F, Lantuejoul S, Chiara MD, Rodrigo JP, Carretero J, Condom E, Vidal A, et al. PARD3 inactivation in lung squamous cell carcinomas impairs STAT3 and promotes malignant invasion. Cancer Res. 2015;75:1287–1297. doi: 10.1158/0008-5472.CAN-14-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kou C, Zhou T, Han X, Zhuang H, Qian H. LRIG1, a 3p tumor suppressor, represses EGFR signaling and is a novel epigenetic silenced gene in colorectal cancer. Biochem Biophys Res Commun. 2015;464:519–525. doi: 10.1016/j.bbrc.2015.06.173. [DOI] [PubMed] [Google Scholar]
- 38.Hedman H, Nilsson J, Guo D, Henriksson R. Is LRIG1 a tumour suppressor gene at chromosome 3p14.3? Acta Oncol. 2002;41:352–354. doi: 10.1080/028418602760169398. [DOI] [PubMed] [Google Scholar]
- 39.Wang F, Zhao W, Kong N, Cui W, Chai L. The next new target in leukemia: the embryonic stem cell gene SALL4. Mol Cell Oncol. 2014;1:e969169. doi: 10.4161/23723548.2014.969169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y, Cui W, Yang J, Qu J, Di C, Amin HM, Lai R, Ritz J, Krause DS, Chai L. SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice. Blood. 2006;108:2726–2735. doi: 10.1182/blood-2006-02-001594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martini A, La Starza R, Janssen H, Bilhou-Nabera C, Corveleyn A, Somers R, Aventin A, Foa R, Hagemeijer A, Mecucci C, Marynen P. Recurrent rearrangement of the Ewing’s sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res. 2002;62:5408–5412. [PubMed] [Google Scholar]
- 42.Nyquist KB, Thorsen J, Zeller B, Haaland A, Troen G, Heim S, Micci F. Identification of the TAF15-ZNF384 fusion gene in two new cases of acute lymphoblastic leukemia with a t(12;17)(p13;q12) Cancer Genet. 2011;204:147–152. doi: 10.1016/j.cancergen.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Gocho Y, Kiyokawa N, Ichikawa H, Nakabayashi K, Osumi T, Ishibashi T, Ueno H, Terada K, Oboki K, Sakamoto H, et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia. 2015;29:2445–2448. doi: 10.1038/leu.2015.111. [DOI] [PubMed] [Google Scholar]
- 44.Paulsson K, Horvat A, Strombeck B, Nilsson F, Heldrup J, Behrendtz M, Forestier E, Andersson A, Fioretos T, Johansson B. Mutations of FLT3, NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2008;47:26–33. doi: 10.1002/gcc.20502. [DOI] [PubMed] [Google Scholar]
- 45.Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, McCastlain K, Reshmi SC, Payne-Turner D, Iacobucci I, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun. 2016;7:13331. doi: 10.1038/ncomms13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki K, Okuno Y, Kawashima N, Muramatsu H, Okuno T, Wang X, Kataoka S, Sekiya Y, Hamada M, Murakami N, et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J Clin Oncol. 2016;34:3451–3459. doi: 10.1200/JCO.2016.66.5547. [DOI] [PubMed] [Google Scholar]
- 47.Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15:371–381. doi: 10.1038/nrc3947. [DOI] [PubMed] [Google Scholar]
- 48.Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood. 2015;125:3977–3987. doi: 10.1182/blood-2015-02-580043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwab C, Ryan SL, Chilton L, Elliott A, Murray J, Richardson S, Wragg C, Moppett J, Cummins M, Tunstall O, et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): genetic profile and clinical implications. Blood. 2016;127:2214–2218. doi: 10.1182/blood-2015-09-670166. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables S1-S13. (XLSX 1165 kb)
Additional materials and methods. (DOCX 29 kb)
Supplementary Figures S1-S7. (PDF 2992 kb)
Data Availability Statement
RNA sequencing data are available for academic purposes by contacting the corresponding author, as the patient/parent consent does not cover depositing data that can be used for large-scale determination of germline variants.
The datasets generated and/or analyzed during the current study are not publicly available as the patient/parent consent does not cover depositing data that can be used for large-scale determination of germline variants but are available from the corresponding author on reasonable request.