

Abstract
We evaluated whether small molecule correctors could rescue four nucleotide-binding domain 1 (NBD1) mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (A455E, S492F, ΔI507, and R560T). We first transfected Cos-7 cells (green monkey kidney cells) with A455E, S492F, ΔI507, or R560T and created HEK-293 (human embryonic kidney cells) cell lines stably expressing these CFTR mutations. The mutants showed lowered protein expression, instability at physiological temperature, and rapid degradation. After treatment with correctors CFFT-002, CFFT-003, C3, C4, and/or C18, the combination of C18+C4 showed the most correction and resulted in increased CFTR residing in the plasma membrane. We found a profound decrease in binding of CFTR to histone deacetylases (HDAC) 6 and 7 and heat shock proteins (Hsps) 27 and 40. Silencing Hsp27 or 40 rescued the mutants, but no additional amount of CFTR was rescued when both proteins were knocked down simultaneously. Thus, CFTR mutations in NBD1 can be rescued by a combination of correctors, and the treatment alters the interaction between mutated CFTR and the endoplasmic reticulum machinery.
Keywords: correctors, cystic fibrosis, mutagenesis, protein misfolding, proteostasis network
Introduction
Cystic fibrosis (CF) is a Mendelian hereditary disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. The mutated CFTR gene translates into a misfolded protein with reduced or no activity.[1] CFTR is a 1,480-amino acid channel that transports chloride across the plasma membrane in the cells of the sweat glands, the pancreas, and the gastrointestinal, reproductive, and respiratory systems.[2] As a member of the ATP-binding cassette (ABC) protein family, CFTR possesses two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs), but it is the only ABC protein that has a regulatory domain.[3] The most common mutation in CFTR is a deletion of the phenylalanine at position 508 (ΔF508) within NBD1. Worldwide, ΔF508 is present in ∼80% of CF patients.[4,5] The wild-type (wt)-CFTR creates a fully glycosylated protein (∼180 kDa, C band), present at the cell surface. In contrast, ΔF508-CFTR forms an incomplete glycosylated protein (∼ 150 kDa, B band), which is held in the endoplasmic reticulum (ER) and degraded by proteasomes.[6]
Clinically, the most common CF manifestations occur within the respiratory tract. CF patients are constantly subjected to a vicious cycle of mucus obstruction, inflammation, and infection that progressively damages the airway tissue, leading to pulmonary remodeling and end-stage lung disease. CF patients also present with pancreatic insufficiency, obstructive azoospermia in men, and reduced fertility in women. A salty taste in the sweat is the first feature observed in CF newborns, as the reabsorption of chloride by CFTR in their sweat glands is compromised, increasing the elimination of electrolytes in the sweat.[7] Given the aforementioned symptoms, a high treatment burden is necessarily associated with overcoming CF’s multisystem symptoms and complications, all of which limits patients’ quality of life. Throughout the past decades, implementation of early diagnosis and pursuance of more effective treatments have lengthened the life expectancy of CF patients.[8] Nevertheless, there is still no therapeutic approach for fully correcting the misfolding, and thus, cellular misprocessing, in mutant CFTR.
Numerous mutations (over 2000) have been reported in the CFTR1 database [Cystic Fibrosis Mutation Database at http://www.genet.sickkids.on.ca ], with the clinical features stored in the CFTR2 database [Clinical and Functional Translation of CFTR at http://www.cftr2.org]. ΔF508 has been well studied, as it is the most prevalent mutant causing CF.[3, 4] However, there are other mutations, including A455E, S492F, ΔI507, and R560T. According the CFTR2 database, A455E, ΔI507, and R560T are among the 23 most common mutations. S492F is less common. Patients with A455E and S492F are likely to have sufficient pancreatic activity, whereas ΔI507 and R560T, when combined with another CF-causing mutation, result in pancreatic insufficiency, indicating more severe disease [clinical and functional translation of CFTR]. All four of these mutations are located at sites that are highly evolutionarily conserved in NBD1: A455E is in the F1-type ATP-binding core subdomain, S492F is situated in the Q loop, ΔI507 and ΔF508 are in the ABC α-domain, and R560T is located between the LSGGQ and Walker B motifs. The ΔF508 mutation has a profound effect on CFTR, decreasing the thermal stability of NBD1 and precluding correct interactions between NBD1 and intracellular loop 4 (ICL4).[2, 3] The impact of the other NBD1 mutations on CFTR is still poorly established, and a better understanding should suggest ways to rescue CFTR and thus benefit the patients bearing these mutations.
The knowledge obtained since the discovery of the primary defect in CFTR has been translated from experimental research into clinical application for CF patients. Two chemical compounds found by high-throughput screening have led to new perspectives in CF treatment: lumacaftor and ivacaftor. Lumacaftor (also known as VX-809) is a corrector that improves the conformational stability of ΔF508-CFTR, resulting in increased processing and trafficking of the mature C band to the cell surface.[9] In contrast, ivacaftor (also called VX-770) is a potentiator that might activate conductance in cases of defective CFTRs in which the protein is already expressed at the cell surface.[10] Both drugs have been used individually in clinical trials for ΔF508-homozygous patients, but they showed only limited benefit.[11, 12] Later, another clinical study tested the combination of lumacaftor/ivacaftor as a treatment for homozygous and heterozygous patients carrying the ΔF508 mutation. The drug combination was safe, and it reduced chloride concentration in the sweat and improved lung function in ΔF508-homozygous CF patients.[13] The US Food and Drug Administration (FDA) has approved the use of ivacaftor (Kalydeco) for 10 CF mutations with defective conductance (G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, G1349D, and R117H). Furthermore, the FDA recently approved the use of the combination lumacaftor/ivacaftor (Orkambi) in CF patients who are ΔF508-homozygous. The goal of personalized medicine is to find appropriate treatments for all CF patients, but the impact of correctors on other (non-ΔF508) mutations is still uncertain.
In the present study, we analyzed the effect of correctors on four CFTR constructs bearing mutations in NBD1 (A455E, S492F, ΔI507, and R560T) and explored whether correctors could modulate the interaction between CFTR and ER-associated degradation (ERAD) components, and thus rescue the trafficking of CFTR to the plasma membrane.
Results and Discussion
CFTR expression and thermal sensitivity
In order to begin to understand how CFTR constructs bearing mutations in NBD1 affect CFTR, we transiently expressed CFTR constructs bearing mutations in NBD1 (A455E, S492F, ΔI507, ΔF508, and R560T) in Cos-7 cells (green monkey kidney cells) under identical conditions (Figure 1). As expected, the C band of ΔF508 was barely detected when compared to that of wt-CFTR. S492F, ΔI507, and R560T showed similar CFTR expression levels to ΔF508. Otherwise, A455E expressed more CFTR than did the other four CFTR constructs bearing mutations in NBD1, but the expression levels were still lower than for wt-CFTR.
Figure 1.

Expression of CFTR. Cos-7 cells were transfected with 4 μg of wt-CFTR, A455E, S492F, ΔI507, ΔF508, or R560T for 48 h. A) Immunoblots and protein expression quantification of B) immature B band and C) mature C band in wt-CFTR and CFTR constructs bearing mutations in NBD1 (ANOVA, n = 3). Data were normalized to wt-CFTR; vs. wt: **P < 0.01; vs. A455E: ##P < 0.01. Ezrin (Ez) was used as a loading control.
As observed for ΔF508, incomplete folding in the other NBD1 mutations could be responsible for the reduced protein levels.[5, 14] To evaluate whether there were similarities between each of the four CFTR constructs bearing mutations in NBD1 and ΔF508, we incubated the cells at a lower temperature (27°C) than mammalian physiological temperature (37°C). Both the immature B and mature C bands of all CFTR constructs bearing mutations in NBD1 increased significantly when the cells were grown at reduced temperature (Figure 2). Interestingly, the increases in the B band (3.5- to 4.5-fold) and C band (2.5- to fourfold) followed the same pattern for all mutants, with the B band increasing to a greater extent than did the C band.
Figure 2.

Thermal sensitivity of CFTR constructs bearing mutations in NBD1. Cos-7 cells were transfected with 4 μg of A455E, S492F, ΔI507, or R560T for 48 h. Cells were incubated at 27°C or 37°C to evaluate the temperature correction of the mutations. Immunoblots and densitometry quantification of CFTR (bands B and C) in A) A455E, B) S492F, C) ΔI507, and D) R560T (Student’s t test, n = 3). Data were normalized to 37°C; vs. 37°C: **P < 0.01, ***P < 0.001. Ezrin (Ez) was used as a loading control.
Degradation pathway
The mutant ΔF508 is well known to be a misfolded protein that is partially glycosylated, arrested in the ER, and quickly degraded by proteasomes.[6] The mutant A455E was found to be degraded mainly by proteasomes but also by aggresomes.[15] To determine how the mutations S492F, ΔI507, and R560T were degraded, we treated cells with MG-132, a nonspecific proteasome inhibitor; tubacin, an HDAC6 inhibitor that transports misfolding proteins to the aggresome; and bafilomycin A1, a vacuolar-type H+-ATPase inhibitor that inhibits lysosomal degradation.[16] The greatest effect was observed with MG-132. Figure 3 shows that the amount of S492F, ΔI507, and R560T steady-state protein increased dramatically (three- to sevenfold) when cells were treated with MG-132. Treatment of the cells with tubacin showed that the mutants were degraded by aggresomes (1.5- to 2.5-fold increase) but to a lesser extent than by proteasomes (Figure 4A–C). In contrast, bafilomycin A1 had a very little effect on either S492F or ΔI507 (< 1.5-fold increase) and produced no change in R560T (Figure 4D–F).
Figure 3.

Proteasomal degradation pathway. Cos-7 cells transfected with 4 μg of S492F, ΔI507, or R560T were treated for 16 h with incremental doses of MG-132, a nonspecific proteasome inhibitor. Immunoblots and protein quantification of CFTR in A) S492F, B) ΔI507, and C) R560T (ANOVA, n = 3). Data were normalized to 0 μm (control); vs. 0 μm: *P < 0.05, **P < 0.01, ***P < 0.001. Ezrin (Ez) was used as a loading control.
Figure 4.
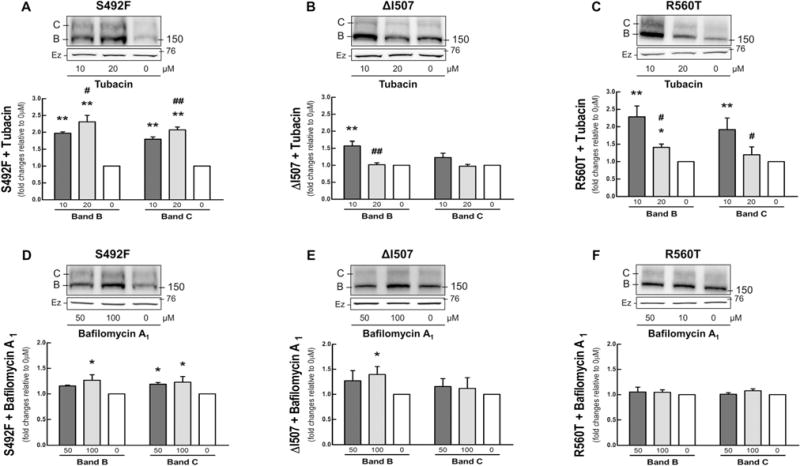
Aggresomal and lysosomal degradation pathways. Cos-7 cells transfected with 4 μg of S492F, ΔI507, or R560T were treated for 16 h with increasing doses of tubacin, an aggresome inhibitor, and bafilomycin A1, a vacuolar-type H+-ATPase lysosome inhibitor, respectively. Immunoblots and densitometry quantification of CFTR in A, D) S492F, B), E) ΔI507, and C, F) R560T (ANOVA, n = 3–4). Data were normalized to 0 μm (control); vs. 0 μm: *P < 0.05, **P < 0.01; vs. 10 μm: #P < 0.05, ##P < 0.01. Ezrin (Ez) was used as a loading control.
Small molecule correctors rescue CFTR expression in CFTR constructs bearing NBD1mutations
Many correctors have been investigated as a prospective therapeutic approach to retrieve the processing and trafficking activities in the ΔF508 mutant. However, the effect of these molecules on other NBD1 mutations is not well established. To assess the effect of these correctors on A455E, S492F, ΔI507, and R560T, we tested Cystic Fibrosis Foundation Therapeutics (CFFT) compounds 002 and 003; C3 and C18, discovered by Vertex Pharmaceuticals;[17] and C4, discovered by the Verkman laboratory.[18]
Mutations A455E (Figure S1 in the Supporting Information), S492F (Figure S2), ΔI507 (Figure S3), and R560T (Figure S4) all responded to treatment with either CFFT-002 or -003 when they were applied individually. All four mutations showed an increased amount of protein in both the immature B band (1.5- to 2.5-fold) and mature C band (1.5- to 2.5-fold), depending on the dose and corrector used. Greater variation was observed in the response to the remaining correctors.
For mutant A455E (Figure S1), C3 and C4 were previously tested and showed a small effect.[15] C18, applied by itself, increased the mature C band by two- to 2.5-fold. The combination of correctors C3+C4 and C18+C3 increased the expression of both the immature B and mature C bands (1.5- to twofold) but to a lesser extent than did C18 alone. On the other hand, the combination of C18+C4 (Figure 5A) showed a synergistic effect and increased the amount of CFTR rescued (two-to threefold).
Figure 5.

The combination of correctors C18 and C4 rescues CFTR in CFTR constructs bearing mutations in NBD1. Cos-7 cells transfected with 4 μg of A455E, S492F, ΔI507, or R560T were treated for 16 h with incremental doses of C18+C4. Immunoblots and protein quantification of CFTR in A) A455E, B) S492F, C) ΔI507, and D) R560T (ANOVA, n = 4). Data were normalized to 0 μm (control); vs. 0 μm: *P < 0.05, **P < 0.01; vs. 10 μm: #P < 0.05. Ezrin (Ez) was used as a loading control.
For mutant S492F (Figure S2), C3, C4, or C18 applied alone had a lesser effect than did CFFT-002 or -003 (1.5- to twofold). C3 and C18 showed less of an effect when applied together than individually (< 1.5-fold for the combination). In contrast, the combination of C3+C4 increased the correction (2.5- to threefold). The greatest effect was observed when C18 and C4 were administrated together (2.5- to 3.5-fold; Figure 5B).
For mutant ΔI507 (Figure S3), C3 and C18 applied individually or in combination had little or no effect (<1.5-fold). C4 applied alone increased both the immature B and mature C band (1.5- to twofold). The combination of C3+C4 did not show any synergy, increasing the immature B and mature C bands to an extent similar to that caused by C4 alone (1.5- to twofold). The maximum effect was obtained with the combination of C4+C18, yielding ∼1.7- to 2.3-fold increases in the immature B band and 2.3- to threefold increases in the mature C band (Figure 5C).
In the mutant R560T (Figure S4), C3 and C18 applied individually or in combination had little effect (<1.5-fold), similar to observations for ΔI507. C4 applied alone increased the amount of both the immature B and mature C bands (two- to 2.5-fold). The combination of C3+C4 was not synergistic, giving approximately the same response as C4 alone (two- to 2.5-fold). In contrast, the combination of C4+C18 showed synergism, increasing both the B and C bands two- to threefold (Figure 5D).
The combination of C4 and C18 had the most pronounced effect in rescuing CFTR. Thus, this combination was used to perform the next experiments, designed to help us understand how CFTR bearing the mutations A455E, S492F, ΔI507, and R560T were rescued.
Correctors enhance CFTR stability and direct the protein to the plasma membrane
The mature C band of wt-CFTR is described as a relatively stable protein with a long half-life. In contrast, the immature B band of ΔF508 is less stable, with a short half-life.[19] In this study, we used the rate of disappearance of A455E, S492F, ΔI507, and R560T in cells treated with cycloheximide (to stop the protein translation) as an indication of protein stability and degradation. As shown in Figures 6 and 7, the mutants expressed significantly more immature B band than mature C band. After cycloheximide administration, little of the C band was observed, and what could be detected disappeared rapidly during the time course of cycloheximide treatment. The immature B band of the mutants was still detectable after 8 h of cycloheximide treatment. As discussed above, the amount of CFTR protein in the CFTR constructs bearing mutations in NBD1 increased after treatment with a combination of C18 and C4. For S492F and R560T, the combination of these two correctors increased the amount of C band present after 8 h. Thus, the correctors were effective in stabilizing the mature C band. Interestingly, there appeared to be a rapid decline in the C band during the first 1–2 h, followed by a slower decline. On the other hand, A455E and ΔI507 showed a significantly reduced decline in both immature B and mature C bands after treatment with the correctors. All four mutations showed some amount of immature B band at 8 h, which was probably arrested in the ER and perhaps amenable to rescue by an additional therapeutic approach.
Figure 6.
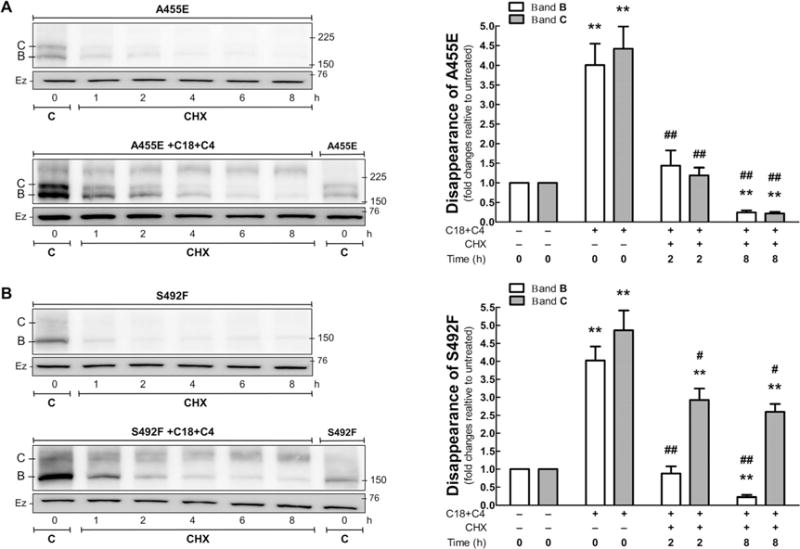
Disappearance of CFTR in the A455E and S492F mutants untreated or treated with C18 + C4. HEK-293 cell lines stably expressing A455E and S492F were treated with C18 and C4 for 16 h. Subsequently, protein translation was stopped by cycloheximide application (CHX, 25 μg mL−1), and the cells were harvested at different time points. Immunoblots and protein quantification of CFTR in A) A455E and B) S492F (ANOVA, n = 4). Data were normalized to control (mutant with no treatment); vs. 0 h, without C18 + C4: **P < 0.01; vs. 0 h with C18 + C4: #P < 0.05, ##P < 0.01. Bars represent the densitometry quantification of immature band B (□) and mature band C (■). Ezrin (Ez) was used as a loading control. C: control.
Figure 7.

Disappearance of CFTR from ΔI507 and R560T mutations untreated or treated with C18 + C4. HEK-293 cell lines stably expressing ΔI507 and R560T were treated with C18 and C4 for 16 h. Subsequently, protein translation was stopped by cycloheximide administration (CHX, 25 μg mL−1), and the cells were harvested at different time points. Immunoblots and protein quantification of CFTR in A) ΔI507 and B) R560T (ANOVA, n = 4). Data were normalized to the control (mutants with no treatment) values; vs. 0 h without C18 + C4: *P < 0.05, **P < 0.01; vs. 0 h with C18 + C4: ##P < 0.01. Bars represent the densitometry quantification of immature band B (□) and mature band C (■). Ezrin (Ez) was used as a loading control. C: control.
To evaluate whether the treatment with correctors restores trafficking of the C band to the plasma membrane, we biotinylated the proteins at the cell surface (Figure 8A and B). The combination of C18+C4 was able to increase the surface expression of A455E (3.3-fold), S492F (2.8-fold), ΔF508 (5.1-fold), and R560T (5.7-fold). Although the correctors increased the CFTR amount in the ΔI507 mutant, the treatment was not able to restore trafficking of the C band to the plasma membrane. As the maximum amount of C band delivered to the cell surface was obtained for R560T after corrector therapy, we tested whether the protein was stable (Figure 8C). Treatment of R560T with C18+C4 resulted in an increase in cell-surface residence time, with expression of the mature C band remaining stable for 6 h after protein synthesis was blocked.
Figure 8.

Correctors address CFTR mutations in NBD1 to the cell surface. HEK-293 cell lines stably expressing A455E, S492F, ΔI507, ΔF508, and R560T were treated with C18 and C4 for 16 h. Immunoblots show the effect of C18 and C4 on CFTR A) in the total protein lysate and B) biotinylation of the cell surface. Band C at the cell surface was quantified in untreated and treated mutations (Student’s t test, n = 3). Data were normalized to control (untreated mutants); vs. untreated: **P < 0.01. C) The stability of band C in the R560T mutant treated with C18 and C4 was assessed by cycloheximide application (CHX, 25 μg mL−1), followed by biotinylation at the cell surface (ANOVA, n = 3). Data were normalized to R560Twith no cycloheximide. Ezrin (Ez) was used as a loading control. C: control.
A proteostasis network is engaged in the rescue of CFTR by correctors
Protein misfolding and aggregation are widely involved in several human diseases. The cellular response to preserve homeostasis is highly regulated by stress signaling pathways, molecular chaperones, and transport and clearance machineries that serve as a protein homeostasis (proteostasis) network.[20] The interaction of mutant ΔF508 with proteins from ERAD has been studied intensively, and therapeutic strategies that target these proteins might rescue ΔF508-CFTR.[21, 22] To explore whether correctors rescue CFTR by reducing the interaction with ERAD proteins, we performed co-immunoprecipitation (co-IP) experiments with five chaperones/co-chaperones: the heat shock proteins (Hsps) 27, 40, 70, and 90, and the activator of 90 kDa Hsp ATPase homologue 1 (AHSA-1). Hsp27 (HspB1) is a member of the small Hsp family that targets ΔF508 for degradation by connecting CFTR with the small ubiquitin-like modifier 2 (SUMO)-2.[23] Hsp40 works as a co-chaperone for Hsp70.[24] Hsp70 and Hsp90 are core ER chaperones that signal the degradation of ΔF508 by the 26S proteasome,[25] and AHSA-1 is the co-chaperone for Hsp90.[26] In addition to studying these chaperones, we also determined the interaction of CFTR constructs bearing mutations in NBD1 with the vasolin-containing protein (VCP, also termed p97) and histone deacetylase 6 (HDAC6), which translocate mutant proteins to proteasomes[27] and aggresomes,[28] respectively. Yet, inhibition of HDAC7[29] or VCP[30] has been shown to rescue CFTR from the misprocessing caused by ΔF508.
The combination of correctors C18+C4 had little or no effect on the steady-state levels of the ERAD proteins tested although we observed a profound effect on their binding to CFTR (Figures 9–12). The binding of HDAC6 and 7, as well as of Hsp27 and 40, was reduced sharply in the A455E, S492F (Figure 9 and 10), ΔI507, and R560T (Figures 11 and 12) mutants after treatment with C18 and C4.
Figure 9.

Correctors reduce the interaction of CFTR in A445E and S492F mutants with proteostasis components. HEK-293 cell lines stably expressing A455E or S492F were treated with C18 and C4 (10 μm of each) for 16 h. CFTR was immunopreciptated with the anti-CFTR antibody M3A7. Protein samples from A) total lysate (TL) and B) co-immunoprecipitation (co-IP) were subjected to immunoblotting and incubated with various primary antibodies as detailed in the Experimental Section (n = 4).
Figure 12.

Correctors reduce the interaction of CFTR in ΔI507 and R560T mutants with proteostasis components, as shown by densitometry quantification of the immunoblots shown in Figure 10. Data are from total lysate (TL), co-immunoprecipitation (co-IP), and co-IP adjusted for CFTR amount of the level of A) VCP, B) HDAC6, C) HDAC7, D) AHSA1, E) Hsp90, F) Hsp70, G) Hsp40, or H) Hsp27 (Student’s t test, n = 4). Data were normalized to the control (mutant with no treatment); vs. untreated: *P < 0.05, **P < 0.01. Please note: there was a 25–40% reduction in the pull-down of HDAC6 and 7 and of Hsp40 and 27 after treatment. However, when the increase in CFTR was taken into account, there was a large reduction in the binding of the proteins to CFTR.
Figure 10.

Correctors reduce the interaction of CFTR in A445E and S492F mutants with proteostasis components, as shown by densitometry quantification of the immunoblots shown in Figure 9. Data are from total lysate (TL), co-immunoprecipitation (co-IP), and co-IP adjusted for CFTR amount of the level of A) VCP, B) HDAC6, C) HDAC7, D) AHSA1, E) Hsp90, F) Hsp70, G) Hsp40, or H) Hsp27 (Student’s t test, n = 4). Data were normalized to the control (mutant with no treatment); vs. untreated: *P < 0.05, **P < 0.01. Please note: there was a 20–30% reduction in the pull-down of HDAC6 and 7 and of Hsp40 and 27 after treatment. However, when the increase in CFTR was taken into account, there was a large reduction in the binding of the proteins to CFTR.
Figure 11.
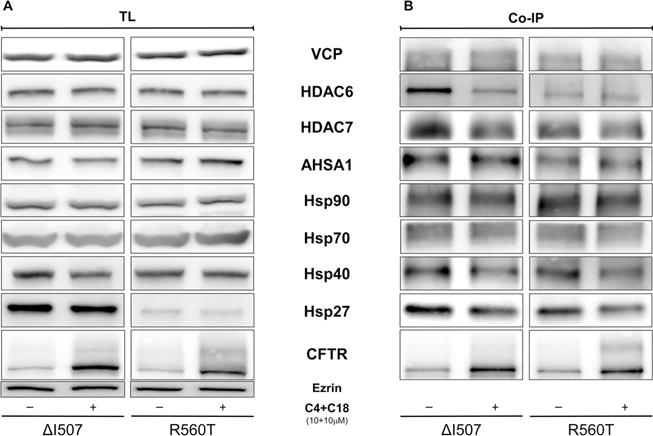
Correctors reduce the interaction of CFTR in the ΔI507 and R560T mutants with proteostasis components. HEK-293 cell lines stably expressing ΔI507 and R560T were treated with C18 and C4 (10 μm of each) for 16 h. CFTR was immunoprecipitated with the anti-CFTR antibody M3A7. Protein samples from A) total lysate (TL) and B) co-immunopreciptation (co-IP) were subjects to immunoblotting and incubated with various primary antibodies as described in the Experimental Section (n = 4).
To confirm the findings from these co-IP studies, we used the proteins most affected by the treatment with correctors: Hsp27 and 40. We tested whether small interfering (si)RNA-mediated silencing of the proteins individually or together would rescue CFTR in CFTR constructs bearing mutations in NBD1 (Figure 13). We observed that knocking down either Hsp27 or 40 increased the expression of both the immature B and mature C bands in all four mutations. However, no additional amount of CFTR was rescued when both chaperones were knocked down together.
Figure 13.

Effect of Hsp40 and Hsp27 on CFTR in CFTR constructs bearing mutations in NBD1. HEK-293 cell lines stably expressing A455E, S492F, ΔI507, or R560T were transfected with siRNAs against Hsp40 (10 nm) and Hsp27 (10 nm) for 72 h. Immunoblots and protein quantification of CFTR in A) A455E, B) S492F, C) ΔI507, and D) R560T (ANOVA, n = 3–4). Data were normalized to the corresponding untreated mutation (□); vs. untreated: *P <0.05, **P <0.01. Ezrin (Ez) was used as a loading control.
In the present study, we explored the nature of four CFTR mutants in NBD1 (A455E, S492F, ΔI507, and R560T) and showed that small molecule correctors can rescue their protein expression. Like ΔF508, the mutants S492F, ΔI507, and R560T are associated with severe CF and lead to disruption of CFTR processing, which is characteristic of class II mutations. The ER machinery arrests CFTR, precluding mature glycosylation of the protein. On the other hand, A455E expresses more of the immature B and mature C bands than the other CFTR constructs bearing mutations in NBD1, but it still has less of both than does wt-CFTR. The amino acid substitution in A455E causes inefficient protein maturation, but part of the C band traffics to the cell surface. Thus, A455E is a class V mutant, causing a milder CF phenotype.
Soon after the discovery that CF is caused by a loss of CFTR function, ΔF508 was shown to be a temperature-sensitive mutant. The incubation of cells at reduced temperature partially reverses the misprocessing.[31] Similar to ΔF508, A455E, S492F, ΔI507, and R560T are also rescued by low-temperature incubation. The lower temperature increases the folding efficiency and corrects the NBD1 instability present in these mutants.[5, 14] Furthermore, the exposure of S492F, ΔI507, and R560T to inhibitors has shown that the mutants share a degradation pathway with ΔF508[6] and A455E.[15] The ER machinery recognizes the CFTR instability created by NBD1 mutations, and a large fraction of the protein is sent for degradation by the proteasomes. Aggresomes degrade by autophagy the portion of the misfolded CFTR that cannot be destroyed by the proteasome. Although the A455E, S492F, ΔI507, and R560T mutants are located in different subdomains within NBD1, they exhibit the same classical features of misfolded proteins: they express less protein than wt-CFTR, are unstable at human physiological temperature, and are degraded mainly by proteasomes.
Many of the newly proposed treatments for CF have focused on overcoming the cellular defect that causes the disease. Toward this goal, the processing, channel activity, and protein stability must be rectified to produce therapeutic levels of the CFTR. The multiple defects in CFTR might suggest a combination of therapeutic approaches for complete rescue of the mutant protein.[32] In clinical studies with ΔF508-homozygous patients, the combination of lumacaftor and ivacaftor showed improvement in lung function,[13] but treatment with lumacaftor or ivacaftor individually achieved only limited benefits.[11, 12] Here, we tested several correctors for A455E, S492F, ΔI507, and R560T. The most effective rescue of CFTR expression was obtained with the combination of C18 and C4 (Scheme 1). C18, a class I corrector, is hypothesized to restore the NBD1-ICL4 interaction. C4, a class II corrector, is supposed to stabilize NBD2 and its interfaces.[33] Interestingly, the same treatment (C18 + C4) showed different amounts of C band rescued to the cell surface, depending on the particular mutation tested. ΔI507 exhibited increased amounts of C band after treatment with correctors, but it was not trafficked to the plasma membrane. Therefore, each mutation apparently has a different impact on the overall structure of the domain, with a more drastic effect evidenced for ΔI507, making it harder to correct. Intriguingly, C18 and C3 (Scheme 1), which are correctors from the same class, rescued the same or lesser amounts of CFTR when applied together compared to individually. We speculate that these compounds might compete for the same mechanism of action and thus show a deleterious effect when they are applied together.
Scheme 1.
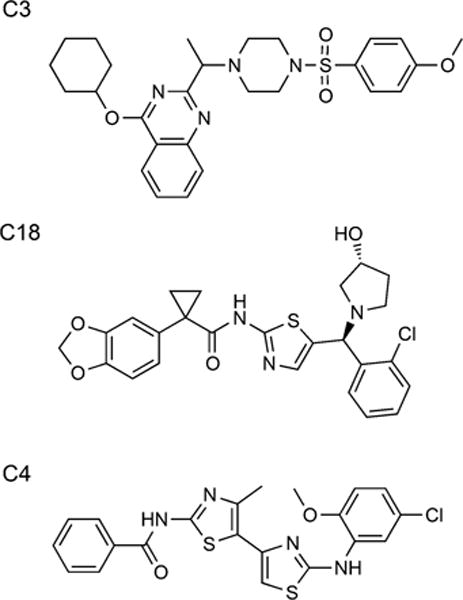
Corrector chemical structures. Corrector C3 and C18 are class I correctors, whereas C4 is a class II corrector.[33] Structures taken from CF Therapeutics Compound Library at http://cftrfolding.org/CFFTReagents.htm.
Misfolded proteins cause diseases as a result of the inability of their altered peptide sequences to interact properly with components of the proteostasis network. Chronic expression of misfolded proteins leads to a sustained activation of the Hsp response.[20] The CFTR mutant ΔF508 fails to acquire a mature folded conformation, and the proteostasis network arrests the processing of ΔF508 at the ER.[21, 22] Mutations A455E, S492F, ΔI507, and R560T interact with a proteostasis network similar to that observed for ΔF508, a finding that might explain the similarity in their degradation pathways. In our hands, treatment with C18 and C4 resulted in a remarkable decrease in the binding of Hsp27 and 40, as well as HDAC6 and 7, to the CFTR constructs bearing mutations in NBD1. HDAC6 translocates mutated proteins to be degraded by aggresomes,[28] whereas Hsp27 and 40 and HDAC7 are correlated with proteasomal degradation.[23, 24, 29] Thus, correctors affect the interaction between CFTR and the proteostasis network.
There are at least two events that must occur for CFTR to be rescued by correctors. Firstly, the correctors must act as a pharmacological chaperone, aiding the folding process.[34, 35] Subsequently, treatment with correctors alters the interaction between CFTR and proteostasis components (CFTR interactome), thus allowing the newly synthesized CFTR to reach the plasma membrane.[20, 21] Here, we have shown that administration of correctors reduces the binding of HDAC6 and 7 and Hsp27 and 40 to CFTR. In order to corroborate our findings from the co-IP studies, we performed siRNA-mediated silencing of Hsp27 and 40, the most affected proteins after corrector treatment. The levels of steady-state protein for mutants A455E, S492F, ΔI507, and R560T all increased after the individual knockdown of Hsp27 or 40. The combined knockdown of Hsp27 and 40 did not result in a synergistic effect in rescuing CFTR when compared to the effect observed when the chaperones were knocked down individually. Therefore, these chaperones might share the same pathway for rescuing the NBD1 mutations. The impact of Hsp27 and 40 on CFTR also differs for each mutation, as different amounts of CFTR were rescued after the chaperones were knocked down. For rescuing CFTR, protein folding has to be restored, and our data indicate that this folding affects the CFTR interactome.
Once the CFTR reaches the plasma membrane, the protein must be stable, or it will be removed by peripheral quality control mechanisms.[36] Low-temperature incubation of ΔF508 restored trafficking of CFTR to the cell surface, but it still showed a reduced half-life, resulting from both increased endocytosis and reduced recycling.[37, 38] After treatment with correctors, the S492F and R560T mutants had a longer half-life (> 8 h), with increased residence time at the cell surface. Mutations A455E and ΔI507 showed a lower rate of disappearance but a shorter half-life than did S492F or R560T. A455E showed an increased amount of C band at the surface after administration of correctors; thus, an additional compound that promotes stability could improve its half-life. ΔI507 did not reach the cell surface after treatment with correctors. For ΔI507, a third compound that rescues trafficking is still required, or another combination of drugs should be tested.
At first glance, mutations A455E, S492F, ΔI507, ΔF508 and R560T appeared to exhibit similar features, although they are located in different sites in NBD1. A better understanding of each mutant reveals that they each have a different impact on NBD1 structure and its interactions with the other CFTR domains. Despite our finding that the best correction occurred with the combination of C18 and C4, CF is still a paradigmatic monogenic disease with incredible genetic and protein variance. Therefore, individualized therapeutic strategies will be needed to treat patients with different CFTR mutations.
Conclusion
In conclusion, our data show that for mutants in NBD1, a combination of correctors has a greater effect if the correctors are derived from different classes than if the compounds are applied individually. Treatment affected the binding of CFTR to proteostasis components, allowing the folded protein to be delivered to the cell surface. Finally, the results indicate that patients bearing A455E, S492F, or R560T mutations might benefit from the combination of C4 and C18. However, more studies are required to provide adequate rescue of ΔI507.
Experimental Section
Cell culture
The African green monkey fibroblast-like cell line, Cos-7 (catalogue CRL-1651, American Type Culture Collection [ATCC]) was maintained in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies) high glucose (1 × glucose), supplemented with 10% fetal bovine serum (FBS, Invitrogen), penicillin (100 U mL−1), and streptomycin (100 μg mL−1) at 37°C in humidified incubator in 5% CO2. To evaluate thermal sensitivity, cells were maintained at 27°C for 24 h before being harvested. Flp-In human embryonic kidney (HEK)-293 cells (catalog CRL-1573, Life Technologies), cultured in DMEM containing 10% FBS with penicillin (100 U mL−1), streptomycin (100 μg mL−1), and Zeocin (100 μg mL−1) at 37°C, were used to generate the stably transfected A455E, S492F, ΔI507, and R560T mutant cell lines.
Transfection and gene silencing
The wt-CFTR, A455E, S492F, ΔI507, ΔF508, and R560T clones in the PBI-CMV2 vector were transfected into Cos-7 cells. Transfections were performed using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. Forty-eight hours after transfection, the cells were harvested and used for experiments. siRNA-mediated silencing was carried out against Hsp40 (10 nM, catalogue sc-35599, Santa Cruz Biotechnology) and Hsp27 (10 nM, catalogue 6526, Cell Signaling Technology) in HEK-293 cell lines for 72 h. Transfections were performed using INTERFERin (Polyplus Transfection), according to the manufacturer’s instructions.
Generation of stably transfected HEK-293 cell lines
The NBD1-CFTR mutant (pEGFP-A455E, S492F, ΔI507, and R560T) clones were subcloned into the pcDNA5-FRT expression vector to generate stably transfected cells. Flp-In HEK-293 cells were transfected with pcDNA5/FRT carrying CFTR pEGFP-A455E, S492F, ΔI507, or R560T, according to the manufacturer’s protocol (Life Technologies). After transfection, the medium was changed to DMEM with 10% FBS, penicillin (100 U mL−1), streptomycin (100 μg mL−1), and hygromycin (100 μg mL−1). The hygromycin-resistant foci were isolated, expanded, and analyzed for the expression of CFTR by immunoblotting.
Treatment
The degradation pathway was evaluated by exposing the S492F, ΔI507, and R560T mutants to a proteasome, aggresome, or lysosome inhibitor (MG-132, tubacin, and bafilomycin A1, respectively) for 16 h. Small molecule correctors were applied for 16 h to analyze the effect of rescuing CFTR. N-((1S,4S)-4-methylcyclohexyl)-5-(4-morpholinophenyl)-4-(oxetan-3-yloxy)pyrimidin-2-amine(CFFT-002), and 5-(2-cyclopropylbenzo[d]oxazol-5-yl)-N-((1S,4S)-4-methyl-cyclohexyl)-4-(oxetan-3-yloxy)pyrimidin-2-amine (CFFT-003) were kindly provided by Martin Mense, and 4-cyclohexyloxy-2-{1-[4-(4-methoxy-benzensulfonyl)-piperazin-1-yl]ethyl}quinazoline (C3), N-[2-(5-chloro-2-methoxy-phenylamino)-4′-methyl-[4,5′]bithiazolyl-2′-yl]-benzamide (C4), and 1-(benzo[d][1,3]dioxol-5-yl)-N-(5-((S)-(2-chlorophenyl)((R)-3-hydroxypyrrolidin-1-yl)methyl)thiazol-2-yl)cyclopropanecarboxamide (C18) were obtained from the CFFT panel library [CFTR Folding Consortium]. These last three compounds were used alone or in combinations of two. The stability of the CFTR protein was analyzed by cycloheximide treatment (25 μg mL−1, Sigma-Aldrich). Cells were harvested 1, 2, 4, 6, or 8 h after protein translation was blocked by cycloheximide.
Biotinylation of CFTR at the cell surface
The plasma membrane proteins of HEK-293 cells stably expressing pEGFP-A455E, S492F, ΔI507, ΔF508, or R560T were submitted to EZ-Link sulfo-NHS-SS-biotin (Thermo Scientific) for 30 min at 4°C. The cells were then washed gently three times with glycine-quenching buffer (200 mM glycine and 25 mM Tris·HCl, pH 8.0, in DPBS plus Ca2+ and Mg2+) and solubilized in lysis buffer (50 mM Tris·HCl, pH 7.4, with 150 mM NaCl, 1% Nonidet P-40, and protease inhibitors). The lysates were rotated for 30 min at 4°C, then centrifuged at 14000g for 20 min at 4°C. The total amount of cellular protein was determined using Protein Assay Dye Reagent (Bio-Rad). The cell surface proteins were isolated from the total lysate (2000 μg) by incubation with NeutrAvidin Plus UltraLink Resin (Thermo Scientific) for 45 min at 4°C (25 μg of protein per 1 μL of beads). After a brief centrifugation, the supernatant was discarded, and the beads were washed five times with lysis buffer. The bound proteins were eluted with 2× Laemmli sample buffer with 5% β-mercaptoethanol. The eluted proteins were then subjected to SDS-PAGE and immunoblotting as described below.
Immunoblotting
The cells were harvested and solubilized in lysis buffer (described above), supplemented with protease inhibitor cocktail (catalogue 78429, Thermo Scientific). The cell lysates were centrifuged at 14000g for 20 min at 4°C to pellet the insoluble material. The supernatants (50 μg of proteins) were subjected to 10% SDS-PAGE and immunoblotting, followed by enhanced chemiluminescence (SuperSignal West Dura Extended Duration Substrate, catalogue 34075, Thermo Scientific). The chemiluminescent signal on the polyvinylidene difluoride (PVDF) membrane (Bio-Rad) was directly captured by a FujiFilm LAS-4000 Plus system with a cooled CCD camera. CFTR protein was detected with monoclonal anti-human CFTR (217; 1:1,000) antibody (provided by Dr. John Riordan, University of North Carolina). Ezrin, used as a loading control, was detected with monoclonal antibody (1:10,000; Santa Cruz Biotechnology). Mouse monoclonal antibodies directed against VCP (1:500), HDAC7 (1:1000), AHSA1 (1:1000), Hsp90 (1:1000), Hsp70 (1:1000), Hsp40 (1:1000), Hsp27 (1:500) and a rabbit polyclonal antibody specific for HDAC6 (1:500) were used. Image Gauge version 3.2 software (Fuji Film) was used to quantify the bands.
Immunopreciptation
The proteins were extracted, and protein concentration was measured as described above. The protein lysates (2 000 μg) were then rotated with 80 μL of protein A/G-agarose beads (Santa Cruz Biotechnology) and 5 μg of anti-CFTR antibody (M3A7, Millipore) for 4 h at 4°C. A/G beads were washed four times with lysis buffer supplemented with protease inhibitor. Sample buffer (2×) with 5% β-mercaptoethanol was added 1:1 with the A/G beads. The protein samples were used for immunoblotting as described above.
Statistical assays
Statistical comparisons were performed using one-way ANOVA, followed by Tukey’s test or an unpaired Student’s t-test (Prism 5.0, GraphPad Software). All data are present as means±SD. Treatment experiments (proteasome inhibitor and small molecule correctors) used the two most expressive doses in rescuing CFTR to perform the statistical assay. All measurements were taken from at least in three independent experiments, and values were considered significant at P < 0.05.
Supplementary Material
Acknowledgments
This work was funded by the U.S. Cystic Fibrosis Foundation and the National Council for Scientific and Technological Development (CNPq) Brazil (to M.L.-P.). The authors acknowledge Dr. Deborah McClellan for editing the manuscript. The authors also thank Assistant Professor Tasuku Ueno (University of Tokyo) for help with the corrector chemical structures in Scheme 1.
Footnotes
Supporting information and ORCID(s) from the author(s) for this article are available on the WWW under http://dx.doi.org/10.1002/cbic.201500620.
Author contributions: M.L-P., W.B.G., and L.C. conceived and designed the experiments; M.L-P, IS., and D.R. performed the experiments; M.L-P, M.M.M., W.B.G., and LC. analyzed the data; M.L.-P, W.B.G., and LC wrote the paper.
The authors declare no conflict of interests.
References
- 1.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dowart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, Emtage S. EMBO J. 2004;23:282–293. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Dokholyan NV, Riordan JR. Proc Natl Acad Sci USA. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski J, Masica DL, Karchin R, Millen L, Thomas PJ, Patrinos GP, Corey M, Lewis MH, Rommens JM, Castellani C, Penland CM, Cutting GR. Nat Genet. 2013;45:1160–1167. doi: 10.1038/ng.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopes-Pacheco M, Boinot C, Sabirzhanova I, Morales MM, Guggino WB, Cebotaru L. J Biol Chem. 2015;290:25636–25645. doi: 10.1074/jbc.M115.671925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 7.Lyczak JB, Cannon CL, Pier GB. Clin Microbiol Rev. 2002;15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen-Cymberknoh M, Shoseyov D, Kerem E. Am J Respir Crit Care Med. 2011;183:1463–1471. doi: 10.1164/rccm.201009-1478CI. [DOI] [PubMed] [Google Scholar]
- 9.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Straley JH, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzel RA, Ashlock M, Negulescu PA. Proc Natl Acad Sci USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumgam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, Boeck KDe, Donaldson SH, Dorkin HL, Dunitz JM, Durie PR, Jain M, Leonard A, McCoy KS, Moss RB, Pilewski JM, Rosenbluth DB, Rubenstein RC, Schechter MS, Botfield M, Ordoñez CL, Spencer-Green GT, Vernillet L, Wisseh S, Yen K, Konstan MW. Thorax. 2012;67:12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordoñez CL, Geller DE, VX 08-770-104 Study Group Chest. 2012;142:718–724. doi: 10.1378/chest.11-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D, VX-809–102 Study Group Lancet Respir Med. 2014;2:527–538. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- 14.Hoelen H, Kleizen B, Schmidt A, Richardson J, Charitou P, Thomas PJ, Braakman I. PLoS One. 2010;5:e15458. doi: 10.1371/journal.pone.0015458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cebotaru L, Rapino D, Cebotaru V, Guggino WB. PLoS One. 2014;9:e85183. doi: 10.1371/journal.pone.0085183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabirzhanova I, Lopes-Pacheco M, Rapino D, Grover R, Handa JT, Guggino WB, Cebotaru L. J Biol Chem. 2015;290:19743–19755. doi: 10.1074/jbc.M115.647685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goor FVan, Straley KS, Cao D, González J, Hadida S, Hazlewood A, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Grootenhuis PD, Negulescu P. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 18.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, Verkman AS. J Clin Invest. 2005;115:2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, Gristein S. J Biol Chem. 1993;268:21592–21598. [PubMed] [Google Scholar]
- 20.Roth DM, Hutt DM, Bouchecareih M, Wang N, Seeley T, Dekkers JF, Beekman JM, Garza D, Drew L, Masliah E, Morimoto RI, Balch WE. PLoS Biol. 2014;12:e1001998. doi: 10.1371/journal.pbio.1001998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balch WE, Roth DM, Hutt DM. Cold Spring Harbor Perspect Biol. 2011;3:a004499. doi: 10.1101/cshperspect.a004499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amaral MD. J Mol Neurosci. 2004;23:41–48. doi: 10.1385/JMN:23:1-2:041. [DOI] [PubMed] [Google Scholar]
- 23.Ahner A, Gong X, Schmidt BZ, Peters KW, Thibodeau PH, Lukacs GL, Frizzell RA. Mol Biol Cell. 2013;24:74–84. doi: 10.1091/mbc.E12-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grove DE, Fan CY, Ren HY, Dyr DM. Mol Biol Cell. 2011;22:301–314. doi: 10.1091/mbc.E10-09-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Janich S, Cohn JA, Wilson JM. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Soroka J, Buchner J. Biochem Biophys Acta. 2012;1823:624–635. doi: 10.1016/j.bbamcr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 27.Boyault C, Gliquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Müller CW, Khochbin S. EMBO J. 2006;25:3357–3366. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 29.Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, Thomas PJ, Matsumura Y, Skach WR, Gentzsch M, Riordan JR, Sorscher EJ, Okiyoneda T, Yates JR, 3rd, Lukacs GL, Fizzell RA, Manning G, Gottesfeld JM, Balch WE. Nat Chem Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vij N, Fang S, Zeitlin PL. J Biol Chem. 2006;281:17369–17378. doi: 10.1074/jbc.M600509200. [DOI] [PubMed] [Google Scholar]
- 31.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 32.Farinha CM, Matos P. FEBS J. 2016;283:246–264. doi: 10.1111/febs.13531. [DOI] [PubMed] [Google Scholar]
- 33.Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, Hegedus T, Beekman JM, Lukacs GL. Nat Chem Biol. 2013;9:444–454. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Loo TW, Bartlett MC, Clarke DM. J Biol Chem. 2007;282:33247–33251. doi: 10.1074/jbc.C700175200. [DOI] [PubMed] [Google Scholar]
- 35.Sinha C, Zhang W, Moon CS, Actis M, Yarlagadda S, Arora K, Woodroofe K, Clancy JP, Lin S, Ziady AG, Frizzell R, Fujii N, Naren AP. ChemBioChem. 2015;16:2017–2022. doi: 10.1002/cbic.201500123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okiyoneda T, Barrière H, Bagdány M, Rabeh WM, Du K, Young JC, Lukacs GL. Science. 2010;329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma M, Pampinella F, Nemes C, Benharouga M, So J, Du K, Papsin KG, Zerangue N, Stenmark H, Lukacs GL. J Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swiatecka-Urban A, Brown A, Moreau-Marquis S, Renuka J, Coutermarsh B, Barnaby R, Karlson KH, Flotte TR, Fukuda M, Langford GM, Stanton BA. J Biol Chem. 2005;280:36762–36772. doi: 10.1074/jbc.M508944200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.