

SUMMARY
Transporters shuttle molecules across cell membranes by alternating among distinct conformational states. Fundamental questions remain about how transporters transition between states and how such structural rearrangements regulate substrate translocation. Here we capture the translocation process by crystallography and unguided molecular dynamics simulations, providing an atomic-level description of alternating access transport. Simulations of a SWEET-family transporter initiated from an outward-open, glucose-bound structure reported here spontaneously adopt occluded and inward-open conformations. Strikingly, these conformations match crystal structures, including our inward-open structure. Mutagenesis experiments further validate simulation predictions. Our results reveal that state transitions are driven by favorable interactions formed upon closure of extracellular and intracellular “gates” and by an unfavorable transmembrane helix configuration when both gates are closed. This mechanism leads to tight allosteric coupling between gates, preventing them from opening simultaneously. Interestingly, the substrate appears to take a “free ride” across the membrane without causing major structural rearrangements in the transporter.
Keywords: molecular dynamics (MD) simulation, membrane protein, transporter, sugar transporter, SemiSWEET, structure
Graphical abstract
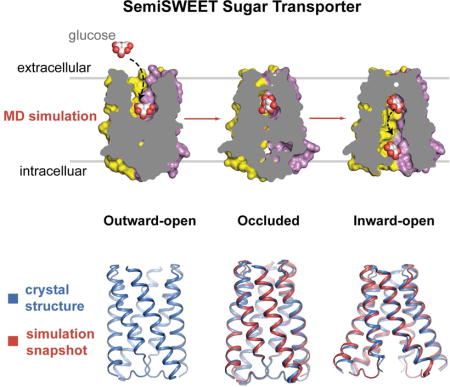
INTRODUCTION
Membrane transporters—which control the flow of a wide variety of molecules into and out of the cell—are generally thought to operate by a common “alternating access” model, proposed over 50 years ago (Jardetzky, 1966; Mitchell, 1957). According to this model, a transporter undergoes structural transitions that alternately expose a substrate-binding site to either side of the membrane. Crystallography has lent support to this model by capturing snapshots of transporters in multiple distinct conformations, including outward-open and inward-open conformations in which the substrate-binding site is accessible to the extracellular and intracellular regions, respectively (Drew and Boudker, 2016; Forrest et al., 2011; Locher, 2016; Palmgren and Nissen, 2011; Yan, 2015). Fundamental questions about the transport mechanism have proven difficult to tackle, however, despite extensive investigation (Forrest et al., 2011; LeVine et al., 2016; Quistgaard et al., 2016). What drives the large-scale conformational changes of the transporter? What are the intermediate conformations between the outward-open and inward-open state, and what functional role do they play, if any? How exactly are structural rearrangements of transporters linked to substrate movement across the membrane?
Molecular dynamics (MD) simulations reveal protein motions at an atomic level and thus appear particularly well suited to answer these questions, in conjunction with crystallography and other experimental techniques (Adelman et al., 2016; Dror et al., 2012; Faraldo-Gomez and Forrest, 2011; Fukuda et al., 2015; Lee et al., 2016; Li et al., 2015; Watanabe et al., 2010). Capturing critical events such as the translocation of a substrate through a transporter or the transition of a transporter between its outward-open and inward-open conformation, however, has proven challenging in simulation (Li et al., 2015; Shaikh et al., 2013). This reflects not only the relative scarcity of experimental structures of transporters but also the large size of many transporters and the intensive computational requirements of MD simulations on the relatively long timescales at which transport occurs. Several elegant studies have provided important insights into these events through atomic-level simulations by relying on biased or guided simulation techniques (Cheng and Bahar, 2014; Gu et al., 2009; Liu et al., 2015; Moradi et al., 2015; Moradi and Tajkhorshid, 2013; Shimamura et al., 2010), but the artificial forces generally used to accelerate transitions in such simulations might give rise to unphysical transition pathways, complicating the mechanistic interpretation of these simulations.
To overcome this barrier, we focused on a “minimal” alternating access transporter, the sugar transporter SemiSWEET. SemiSWEET is among the smallest characterized transporters (<20 kDa), has a simple geometry (a symmetric dimer of three-helix bundles), and is thought to transport a single substrate rather than engaging in coupled transport (Xu et al., 2014; Xuan et al., 2013). The structure of SemiSWEET, a SWEET family transporter found in bacteria, mimics the core of the eukaryotic SWEET transporter (Tao et al., 2015; Xuan et al., 2013). Members of the widespread SWEET family of sugar transporters play critical roles in many important biological processes in plants, including phloem loading, nectar secretion, pollen development, seed filling, and host-pathogen interactions, and are important regulators of metabolic homeostasis in animals (Chen et al., 2015; Chen et al., 2010; Wright, 2013).
Recent crystal structures have captured several distinct conformations of SemiSWEET (Lee et al., 2015; Wang et al., 2014; Xu et al., 2014). These conformations were captured in orthologous proteins from different species, however, and structural deviations caused by sequence divergence thus complicate their interpretation (Figure S1). A full set of conformations in a single SemiSWEET isoform is needed to ascertain the precise conformational changes that constitute the transport cycle. Furthermore, none of the existing structures for any SWEET family transporter has had a known substrate bound, and thus the basis for substrate recognition in this family has been unclear. And, of course, the fundamental mechanistic questions that remain unanswered for other transporters apply to SemiSWEET as well.
We report crystal structures of both outward-open and inward-open conformations of L. biflexa SemiSWEET (LbSemiSWEET), with the substrate glucose bound in the former. Together with the previously reported structure of an occluded conformation, in which the substrate-binding pocket is open to neither the inside nor the outside of the cell (Xu et al., 2014), experimental structures of all three conformations are now available for a single SemiSWEET ortholog. To the best of our knowledge, such a complete set of structures was not previously available for any uniporter or SWEET family transporter.
Using the glucose-bound SemiSWEET structure as a starting point, we performed unguided atomic-level MD simulations of the glucose translocation process, capturing the spontaneous transition of the transporter from its outward-open to its inward-open conformation. We also performed simulations of glucose-free SemiSWEET and captured a similar spontaneous conformational transition in the absence of substrate. Both sets of simulations spontaneously reached the experimentally observed occluded and inward-open conformations—even though these experimental structures were not used to inform the simulations. These results, further validated through mutagenesis experiments, allow us to describe an alternating access transport mechanism in atomic detail. In particular, they enable us to describe the sequence of conformational changes that gives rise to substrate translocation, the forces that drive these conformational changes, and the nature of the allosteric coupling that ensures that the substrate-binding pocket is not simultaneously accessible from both sides of the membrane.
RESULTS
Determination of a substrate-bound, outward-open crystal structure
To determine the structure of LbSemiSWEET in an outward-open state, we designed a mutation predicted to favor this conformational state. In the previously reported occluded-state crystal structure (Xu et al., 2014), a network of interactions between the two protomers near the extracellular and intracellular surfaces seal the substrate-binding cavity from solvent (Figure S2A). We reasoned that mutations that disrupt interactions on the extracellular surface might favor an outward-open conformation. Indeed, a mutant in which the extracellular residue Asp57 was replaced by alanine (D57A) crystallized in an outward-open conformation, yielding a crystal structure with a resolution of 2.8 Å (Figure 1A and table S1).
Figure 1. Crystal structure of LbSemiSWEET captured in an outward-open conformation.
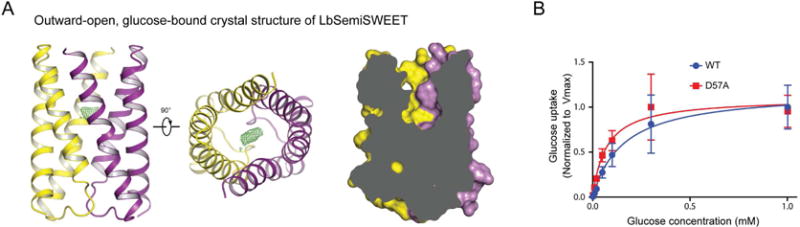
(A) LbSemiSWEETD57A captured in an outward-open conformation. The green mesh shows the Fo–Fc omit map of D57A contoured to 3σ. The disc-shaped density likely corresponds to glucose (left and middle). Slab views of LbSemiSWEET in an outward-open conformation (right). Two protomers are shown in yellow and purple, respectively. (B) Cell-based radioactive glucose uptake by wild-type LbSemiSWEET (blue trace) compared to the D57A mutant (red trace). See also Figure S1, S2 and Table S1.
Notably, the D57A mutant exhibited more than double the glucose affinity of the wild type (apparent Km of ∼70 μM for D57A and ∼160 μM for the wild type; Figure 1B) and the growth of these crystals depended on the presence of glucose. After all protein atoms were modeled for the outward-open D57A LbSemiSWEET, we observed a strong electron density in the substrate-binding pocket that fits the transporter substrate, glucose (Figure 1A). We were thus able to place glucose into the density, and the resulting pose proved compatible with the local protein environment. This substrate-bound structure of a SWEET-family transporter provides insight into the structural basis for substrate recognition in these transporters (discussed below). It also provides a starting point for simulations of substrate translocation.
Simulations capture alternating access transport in action
We initiated all-atom MD simulations of LbSemiSWEET from the glucose-bound, outward-open crystal structure (20 simulations totaling 66.8 μs). We performed additional simulations beginning from the same crystal structure but with glucose removed (20 simulations totaling 57.8 μs). In all simulations, we placed the transporter in a hydrated lipid bilayer and reverted residue 57 to its wild-type identity (Asp). The complete set of simulation conditions and durations is listed in table S2.
In several simulations (four with glucose and four without), the transporter transitioned spontaneously from its outward-open conformation to an inward-open conformation, in which the binding cavity was accessible to solvent from the intracellular side (Figure 2A). The magnitude of this conformational change was substantial, with individual helices displacing up to 5 Å from their starting position. Interestingly, the simulations that transitioned to the inward-open conformation first visited a metastable, occluded conformation, in which the binding pocket was inaccessible to solvent from either side of the membrane. The simulations that did not transition to an inward-open conformation remained in the outward-open conformation, apart from three that ended in the occluded conformation.
Figure 2. Conformational states and substrate translocation in LbSemiSWEET revealed by unguided simulations.
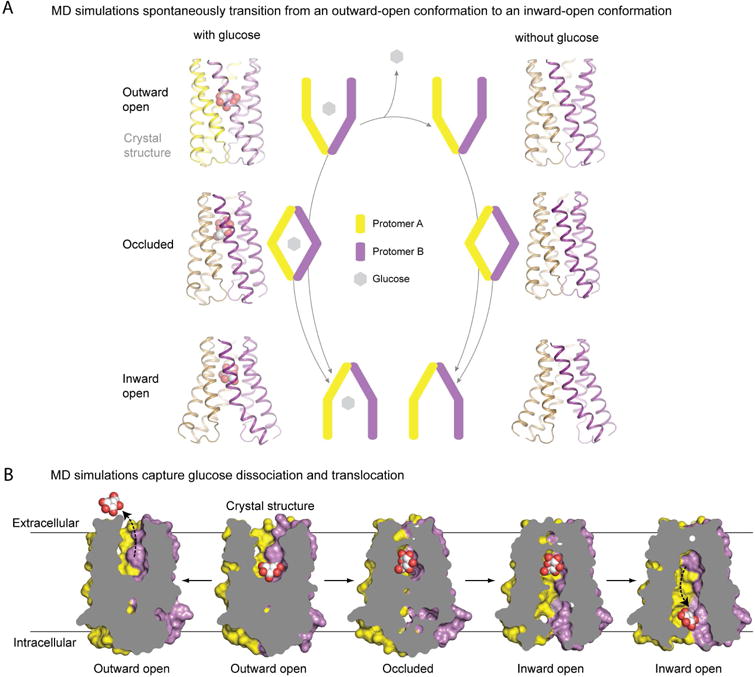
(A) Schematic of major conformational states in the SemiSWEET transport cycle (center). Arrows indicate the conformational transitions observed during MD simulations. The two protomers are colored yellow and purple, respectively. Corresponding simulation snapshots are shown to the left and right. (B) Slab representations of the major conformational states observed in simulations initiated from the outward-open, glucose-bound crystal structure. The transition to the left depicts glucose dissociation into the bulk from Simulation 3. The transition to the right shows the position of glucose in occluded and inward-open states before escaping the binding pocket in Simulation 9. See also Table S2.
Interestingly, our simulations indicate that the conformational states visited in simulation with glucose bound are essentially identical to those without any substrate present (Figure 6). In addition, the sequence of events involved in the transition from the outward-open to inward-open conformation appears to be the same in the presence and absence of glucose (Figure S4).
Figure 6. Comparison of simulations in the presence and absence of substrate.
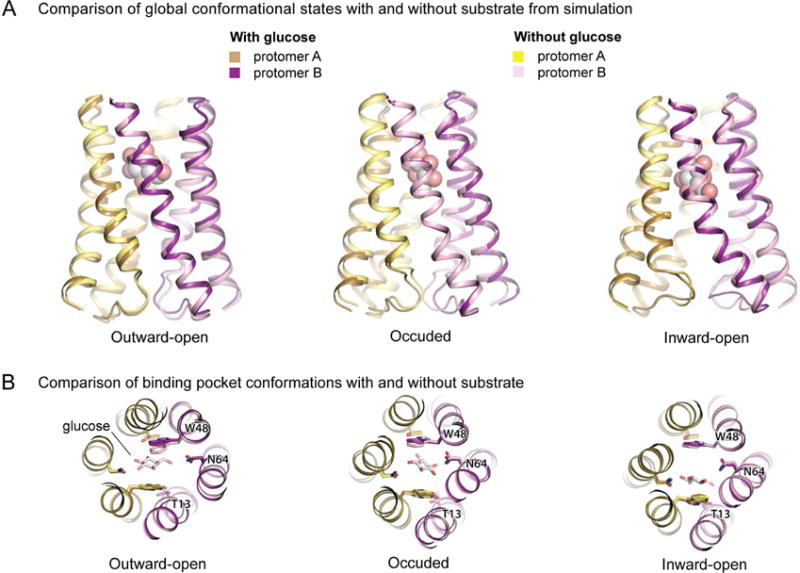
Snapshots are taken from simulation frames closest in protein backbone Cα RMSD to the ensemble average structure for each conformational state (see Methods). (A) Overlays of snapshots taken from simulations with and without glucose reveal that essentially the same conformational states are adopted under each condition. (B) Overlays of simulation snapshots of the substrate-binding pocket in each major conformational state reveal similar binding pocket conformations in the presence and absence of glucose. See also Figure S6 and Table S2.
Our simulations also captured the substrate translocation process (Figure 2B). In six of the simulations initiated from the glucose-bound outward-open structure, glucose dissociated from the transporter into the extracellular solvent while the transporter remained in the outward-open conformation. In one of these simulations, glucose later re-associated with the transporter, which was still in an outward-open conformation. In one of the glucose-bound simulations in which the transporter transitioned to an inward-open conformation, glucose subsequently unbound from the substrate-binding site and translocated to the intracellular surface of the transporter.
Crystallographic validation of simulated conformational states
To validate our simulations, we sought to solve the structure of LbSemiSWEET in an inward-open conformation. We reasoned that mutations that disrupt interactions on the intracellular side might favor an inward-open conformation. We designed a mutant in which the intracellular residue Gln20 was replaced by alanine (Q20A). This construct crystallized in an inward-open conformation (albeit in a domain-swapped configuration; see Methods; Figure S2), yielding a crystal structure with a resolution of 2.8 Å (Figure 3A), which closely matches a previously published inward-open structure of E. coli SemiSWEET (Lee et al., 2015).
Figure 3. Crystallographic validation of simulations.
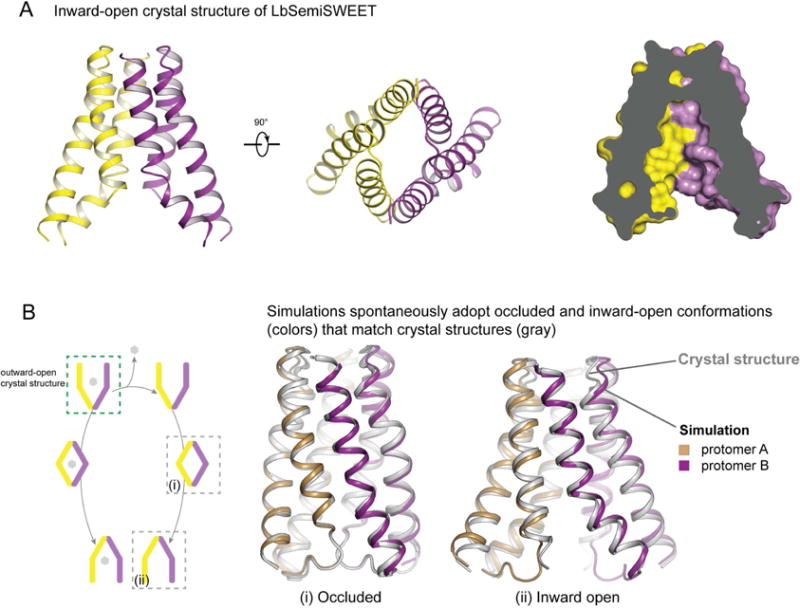
(A) Ribbon representation of LbSemiSWEETQ20A in an inward-open conformation (left) and slab views of LbSemiSWEET in an inward-open conformation. One protomer is shown in yellow and the other is in purple. (B) Comparison of simulated and crystallographic conformational states. Simulations started from the outward-open crystal structure, with and without glucose bound, transition spontaneously to occluded and inward-open conformational states (dark colors) that closely match crystal structures (silver). Boxes in B (left) indicate conformational states captured crystallographically, and simulation overlays (right) compare conformational states visited in simulations to crystal structures. See also Figure S2, S3 and Table S1.
Strikingly, simulations initiated from the outward-open crystal structure transitioned to a conformation that matched our inward-open structure very closely (Figure 3B). Indeed, the deviations of the simulations from the inward-open crystal structure after this transition were comparable to the deviations of a simulation initiated from an inward-open crystal structure (Figure S3A). The observed deviations most likely represent thermal fluctuations.
Additionally, the occluded intermediate conformation visited during simulations of the outward-open-to-inward-open conformational transition very closely matched the previously published occluded-state crystal structure of LbSemiSWEET (Figure 3B). This suggests that the occluded conformation is in fact an on-pathway intermediate visited as part of the transport process. Additional simulations initiated from the occluded-state crystal structure, both with and without glucose added to the substrate-binding pocket, also transitioned spontaneously to the same inward-open conformational state seen in simulations initiated from the outward-open conformation.
None of the simulations initiated from the outward-open or occluded states incorporated any prior knowledge of the crystallographically observed inward-open conformation, so the fact that they reached it spontaneously provides an indication of their predictive power. The same is true for simulations initiated from the outward-open state that reached the crystallographically observed occluded state without incorporating any prior knowledge of the occluded state structure. We further validated our simulations through a number of mutagenesis experiments, discussed below.
Mechanism for conformational change in alternating access transport
The most notable conformational changes in the transition from an outward-open to an inward-open conformation are the closing of an extracellular gate and the opening of an intracellular gate. These two gates appear to control the accessibility of the substrate-binding pocket to the extracellular and intracellular solvent, respectively. Both gates are formed by residues at the interface between the two symmetric protomers that make up SemiSWEET. The extracellular gate consists of residues Tyr51 of the second transmembrane helix (TM2) and Ile60 of TM3, immediately above the substrate-binding pocket, and Arg55 and Asp57 near the extracellular surface (Figure 4A). The intracellular gate consists of residues Phe17 on TM1 and Phe41 on TM2, immediately beneath the substrate-binding pocket, and Met37 and Tyr38 on TM2 near the intracellular surface (Figure 4A).
Figure 4. Key molecular events during the outward-open to inward-open transition.
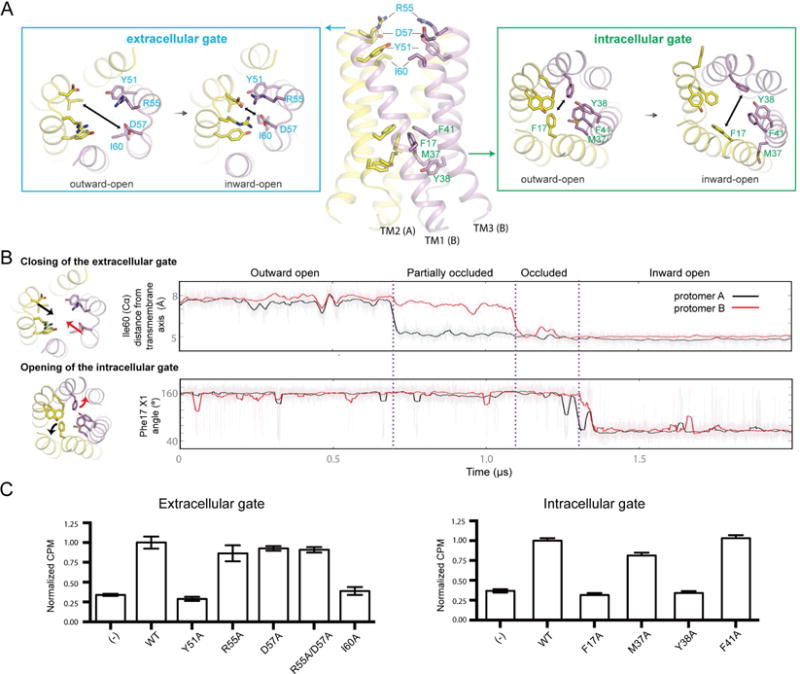
(A) During state transitions, changes to the extracellular gate (top-down view) and the intracellular gate (bottom-up view) are shown in the boxed areas. Two-headed arrows indicate direction of helical movement observed during simulation. (B) Sequential order of events during the outward-open to inward-open transition (Simulation 4). The conformational transition begins when TM3 from each protomer displaces inward, as measured by the distance of the I60 Cα atom to the central axis of the helical bundle (top panel). Inward displacement of TM3 destabilizes packing between TM1 and TM3 in the region around Pro19 (Figure S5). Inward displacement of TM3 on the second protomer occurs within ∼0.4 μs of the first protomer displacing inward, forming a fully occluded state. Transition to the inward-open state is favored by the formation of backbone hydrogen bonds along TM1, which correlates with formation of the kinked conformation of TM1 (Figure S5). These collective motions favor the rotation of Phe17 to move away from the central pore and pack against TM3 (bottom panel). (C) Glucose (14C) uptake activities in E. coli are shown for LbSemiSWEET with alanine point mutations in extracellular (left) and intracellular (right) gates compared to wild type and control (empty vector) (±s.d., n=3). See also Figure S4, S5, S7 and Table S2.
We found that each outward-open to inward-open transition began with closure of the extracellular gate (Figure 4B). In particular, TM3 tilted such that its extracellular end moved inward towards the central axis of the transporter, accompanied by a smaller inward motion of the extracellular end of TM2. This movement—which usually took place either simultaneously or within a few hundred nanoseconds in both protomers—resulted in tight packing between the extracellular gate residues Ile60 on TM3 and Tyr51 on TM2 of the opposite protomer (Figure 4A). As a result, the substrate-binding pocket became sealed from the extracellular or intracellular solution. Upon inward displacement of TM3, cross-protomer ionic contacts also formed between Asp57 and Arg55, although these contacts appeared less stable, breaking and reforming intermittently. Notably, the transporter persisted in this state with both extracellular and intracellular gates closed for up to several hundred nanoseconds (Figure S4).
The outward-open to inward-open transition concluded with the intracellular gate opening. In particular, the intracellular end of TM1 kinked at Pro19, with Phe17 flipping to point away from the central translocation path, packing instead against TM3 (Figure 4, A and B). Again, this movement usually took place in the two protomers simultaneously or almost simultaneously (within a few tens of nanoseconds). These motions were accompanied by outward displacement of TM2, which caused Met37, Tyr38, and Phe41 to shift away from the central substrate translocation pore, exposing the binding pocket to solvent.
Closure of the extracellular and intracellular gates, which is tightly linked to larger motions of the helices, appears to be driven by formation of favorable contacts between the gate residues. Hydrophobic and electrostatic interactions stabilize the closed state of the extracellular gate, and hydrophobic and aromatic interactions stabilize the closed state of the intracellular gate (Figure 4A). These contacts also appear to be responsible for the cooperativity of conformational changes across the two protomers: formation of the most favorable set of contacts requires that both sides of a gate be in the closed conformation, so once one side closes, the other is more likely to adopt a closed conformation, and once one side opens, the other is more likely to open.
But what drives the gates to open? And why does the intracellular gate generally open only after the extracellular gate closes? Such negative cooperativity between the two gates is critical, because it prevents the “forbidden state” in which both gates are open, resulting in a continuously conducting pore (King and Wilson, 1990). The intracellular gate is apparently driven to open because TM1 energetically favors a conformation in which it is kinked at Pro19, allowing the formation of backbone hydrogen bonds between Thr13 and Phe17 and between Thr14 and Leu18. In simulations of an isolated TM1, the helix quickly adopts a kinked conformation matching that observed in the inward open state, with the inner gate residue Phe17 favoring the rotamer it adopts in the inward open state (Figure S5). The kinking of TM1 is apparently prevented in the outward-open state, however, by favorable packing interactions between TM1 and TM3 (Figure 5). Upon extracellular gate closure, TM3 tilts such that its extracellular end moves inwards, causing TM1 to move slightly outwards. These helical motions substantially reduce the number of contacts between TM1 and TM3 (Figure S5), freeing TM1 to kink and thus allowing the intracellular gate to open.
Figure 5. Model for state transitions in an alternating access transporter, SemiSWEET.
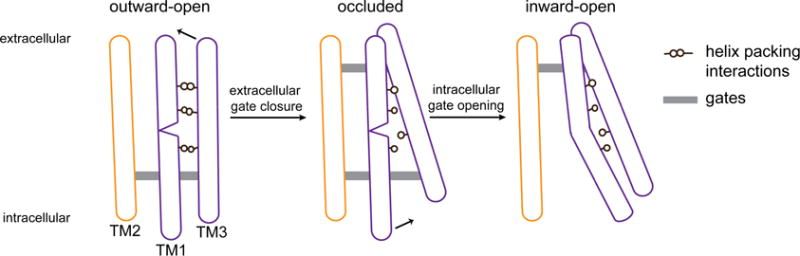
TM2 of one protomer (yellow) and TM1 and TM3 of the opposing protomer (purple) are shown. The other, identical set of three helices is omitted for clarity. Gray bars represent closed extracellular and intracellular gates, and black circles represent helical packing interactions. In the outward open state (left), TM1—which, if isolated, tends to kink about a centrally located proline residue—instead assumes a straightened conformation stabilized by packing interactions with TM3. Closure of the extracellular gate, which involves a substantial motion of TM3, leads to a loss of packing interactions between TM1 and TM3. In this more ‘isolated’ environment, TM1 kinks, opening the intracellular gate. This cartoon is intended as a two-dimensional schematic rather than an accurate representation of SemiSWEET’s three-dimensional geometry. The inward motion of the extracellular end of TM3 upon closure of the extracellular gate also causes TM1 to shift slightly away from the center of the helical bundle. This smaller motion is not illustrated in the cartoon but appears to contribute to the loss of TM1–TM3 packing interactions upon extracellular gate closure.
In the occluded state—when both gates are closed—TM1 is in an unstable configuration, as it is neither kinked nor packed well against TM3. The intracellular gate is driven to open such that TM1 can kink, and the extracellular gate is driven to open such that TM1 can pack more favorably against TM3. Once one gate is open, however, there is no driving force for the other gate to open, as a kinked TM1 cannot pack favorably against TM3. As a result, the two gates do not open simultaneously.
We validated these results through several mutagenesis experiments. Our simulations indicated that Ile60, Tyr51, and Phe17 play a critical role in the gating process (Figure 4A). In particular, transition of the protein backbone from the outward-open to the occluded state was tightly coupled with the formation of cross-protomer packing contacts between Tyr51 and Ile60, and Phe17 is the primary residue whose motion is responsible for opening a pathway to the substrate-binding pocket on the intracellular side. Indeed, we found that alanine substitution of each of these three residues ablated glucose uptake (Figure 4C). An additional mutant, Y38A, also significantly reduced glucose transport, perhaps because Tyr38 might stabilize the closed conformation of Phe17 by forming a hydrophobic packing interface in the core of the transporter. By contrast, alanine mutation of several other residues that form part of the crystallographically observed extracellular and intracellular gates, such as Asp57, Arg55, Met37, and Phe41, had no significant effect on transporter activity. Indeed, in simulations of D57A and I60A mutants initiated in the outward-open conformation (five simulations of each construct, each 3 μs in duration; see Table S2), we observed two transitions to the occluded state for D57A but none for I60A. This is consistent with the observation that a bulky, hydrophobic residue at position 60 facilitates closure of the extracellular gate.
Structural basis for substrate translocation
Perhaps our most striking observation about the substrate translocation process is what the substrate does not do. Comparing simulations with and without substrate bound indicated that the transporter visits the same conformational states and undergoes the same conformational transitions with and without substrate bound (Figure 6 and Figure S4). In other words, the substrate appears to take a “free ride” across the membrane as the transporter undergoes the same conformational changes it would have done even in the absence of substrate, although the substrate might alter the relative energies of the various conformations or the transition rates between them.
How does glucose interact with SemiSWEET? In the outward-open crystal structure, the glucose ring lies in a vertical plane, off-center from the two-fold axis of the protein and partially sandwiched between the pyrrole rings of Trp48 on each protomer. Asn64 and Thr13 on only one protomer directly coordinate substrate hydroxyl groups (Figure 7A). Compared with other sugar transporter structures, in which multiple residues appear to directly coordinate with substrate (Cao et al., 2011; Deng et al., 2014; Faham et al., 2008; Kumar et al., 2014; Oldham et al., 2007), SemiSWEET appears to form fewer direct hydrogen bonds with glucose in this outward-open state.
Figure 7. Substrate recognition and translocation.
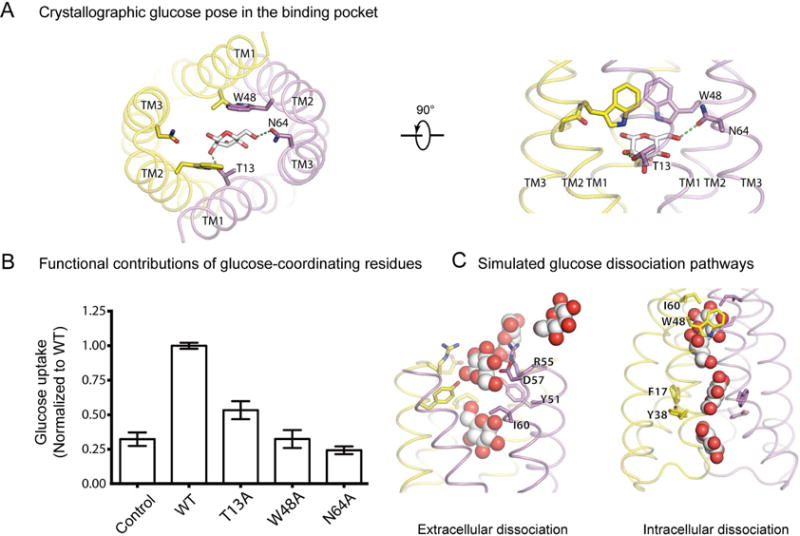
(A) Glucose coordination by substrate recognition residues in the outward-open crystal structure. Glucose is shown in silver sticks. Hydrogen bonds are indicated by green dotted lines. (B) Glucose (14C) uptake activities for LbSemiSWEET with alanine point mutations of substrate-binding residues compared to wild-type SemiSWEET and control (empty vector) (±s.d., n=3). (C) Extracellular (left) and intracellular (right) glucose dissociation paths shown using overlaid, down-sampled frames taken from Simulation 3 and Simulation 9, respectively. See also Figure S7 and Table S2.
In simulations of the glucose-bound outward-open crystal structure, glucose was quite mobile. It maintained its vertical orientation and its interactions with both Trp48 residues, but shifted within its vertical plane. It alternated frequently between hydrogen bonding directly with one Asn64 residue or the other, often forming a water-mediated interaction with the second Asn64 residue. The glucose molecule formed only weak and intermittent interactions with Thr13.
Simulations indicate that, in the occluded and inward-open states, the substrate-binding pocket narrows, such that glucose forms hydrogen bonds with both Asn64 residues simultaneously (Figure 6B). As a result, glucose becomes less mobile, particularly in the occluded state. Glucose maintains its interactions with both Trp48 residues and continues to interact intermittently with Thr13. In the absence of substrate, water molecules form extensive hydrogen bonds with the binding pocket, substituting for the hydrogen bonds formed by glucose when it is present (Figure S5).
When glucose escaped from the outward-open state in simulation, it moved directly into the extracellular bulk solvent. By contrast, to escape from the inward-open state, it first rotated roughly 90° around the membrane axis to move between the Phe17 gate residues, before continuing further down to contact the intracellular loops (Figure 7C). Both the interactions the substrate forms in the substrate-binding pocket (particularly in the occluded and inward-open states, when the binding pocket narrows) and the pathway the substrate follows to escape the transporter may place geometric constraints on the types of molecules that can diffuse through SemiSWEET.
We validated the significance of key binding pocket residues through additional site-directed mutagenesis experiments. Our simulations suggest that Trp48 and Asn64 play a particularly critical role in coordinating the substrate. Indeed we found experimentally that alanine substitution at Trp48 and Asn64 ablated glucose uptake (Figure 7B), in agreement with previous studies (Xu et al., 2014). Notably, Trp48 and Asn64 are conserved across SemiSWEETs, consistent with a critical role in transport (Xu et al., 2014). Our simulations indicate that Thr13 interacts with the substrate less frequently than Asn64 interacts with substrate, and indeed, mutating it to alanine only partially ablated glucose uptake.
DISCUSSION
In an attempt to characterize the dynamic mechanism of an alternating access transporter in atomic detail, we studied a “minimal” transporter, LbSemiSWEET. To our surprise, we were able to capture the process of substrate translocation—including the transporter’s spontaneous transition from an outward-open to an inward-open conformation—using unbiased atomic-level simulations. This was most likely possible not only because we performed extensive, computationally demanding simulations—more than 175 μs in total—but also because LbSemiSWEET apparently undergoes this central conformational transition on shorter timescales than many other transporters. (We observed approximately one transition to the inward-open state for every ∼14 μs of simulation of the outward-open and occluded states.)
To enable these simulations, we solved a glucose-bound, outward-open crystal structure of LbSemiSWEET. Importantly, simulations initiated from the outward-open structure arrived spontaneously at conformational states closely matching both our newly determined inward-open crystal structure and a previously reported occluded-state structure, even though the simulations were completely unguided and did not draw on any structural information other than their initial conformation.
Our simulations, together with the crystal structures and mutagenesis experiments, suggest that several principles underlie alternating access transport in LbSemiSWEET—and, possibly, in various other alternating access transporters. First, the favorable interactions between gate residues when either gate closes represent one of the main driving forces for conformational change in LbSemiSWEET. That is, our data suggests that the gate residues play a dual role; they appear not only to block passage of the substrate but also to stabilize the closed conformation of the gate. In SemiSWEET, which consists of two identical and symmetric protomers, these gate interactions can also lead to cooperativity between the two protomers: the gate residues of one protomer are more likely to adopt a closed conformation when the corresponding gate residues of the other protomer do so, because many of the favorable interactions between these residues take place only when both halves of a gate are closed. Favorable interactions between gate residues may drive conformational change even in transporters that consist of dissimilar or asymmetric units (Fowler et al., 2015; Quistgaard et al., 2016; Stelzl et al., 2014).
Second, the crystallographically observed occluded state of LbSemiSWEET appears to be an on-pathway intermediate between the outward-open and inward-open states. Crystal structures and biophysical measurements have revealed occluded states in multiple transporter families (Fukuda et al., 2015; Kaback, 2015; Kazmier et al., 2014; Madej et al., 2012; Pedersen et al., 2013; Sun et al., 2012; Yamashita et al., 2005; Yin et al., 2006), and these are generally hypothesized to represent on-pathway intermediates (Fukuda et al., 2015; Kaback, 2015; Kazmier et al., 2014; Madej et al., 2012). Demonstrating that this is in fact the case has been difficult, however, because it requires time-resolved measurements (Akyuz et al., 2015). Several previous simulation studies pointed in this direction (Adelman et al., 2011; Holyoake and Sansom, 2007; Koldso et al., 2011; Stelzl et al., 2014; Zhao et al., 2012). Our unbiased atomic-level simulations not only provide direct observation of an on-pathway occluded state but also reveal the mechanism of transitions into and out of this state. Furthermore, our simulations suggest that this metastable intermediate state lowers the activation energy for transitions between the outward-open and inward-open states by allowing some favorable interactions to form early in the transition process, potentially accelerating transport.
Third, the occluded state plays a critical role in coordinating motions of the inner and outer gates: the gates are only driven to open in the occluded state, because TM1 assumes an unstable configuration that can be stabilized by the opening of either gate. The occluded state thus acts as an “air lock” that ensures that one gate will generally close before the other opens, and thus that the two will generally not be open simultaneously (Forrest, 2013; Stelzl et al., 2014). Here we provide a possible answer to the long-standing question of the mechanism underlying negative cooperativity between gates in a transporter.
Finally, our results support a model in which glucose takes a “free ride” through LbSemiSWEET, with the transporter adopting the same conformations and undergoing the same transitions in the presence and absence of the substrate. This is in contrast to the conformational changes previously described for a number of coupled transporters (e.g., symporters and antiporters), which require coordinated movement of two or more substrates (Ewers et al., 2013; Kaback, 2015; Masureel et al., 2014; Moradi et al., 2015; Verdon et al., 2014). Unlike many coupled transporters, a uniporter does not require cooperativity between two binding sites, so there may not be a need for the substrate to influence the transporter conformation. Whether and how substrate affects the relative energies of the conformational states of SemiSWEET or the transition rates between them represents an interesting direction for future research.
Our simulations tend to progress from the outward-open and occluded states towards the inward-open state, suggesting a relatively more stable inward-open state in silico, which warrants future experimental examination. A slower transition from the inward- to outward-open state would slow the overall transport process and might help to set a physiologically optimal transport rate. We note, however, that such relative stabilities can be difficult to estimate accurately in simulation, and that we performed a limited amount of simulation of the inward-open state.
Several caveats are in order. First, we did not simulate the inward-open to outward-open transition. We did, however, simulate the outward-open to inward-open transition both in the presence and absence of glucose; at equilibrium, the forward and reverse pathways for each transition are the same due to microscopic reversibility. Second, we also did not capture the final release of substrate from the intracellular loops into cytosol. Third, although we observed a consistent sequence of events in the eight simulations that transitioned from the outward-open state to the inward-open state, with extracellular gate closure preceding intracellular gate opening, other sequences of events may occur occasionally. Finally, we have examined only a single alternating access transporter. Our findings for this transporter may provide a foundation for understanding alternating access transport more generally, but substantial structural, biophysical, and computational investigation will be required to determine the extent to which the principles underlying transport in SemiSWEET carry over to other transporter systems.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Liang Feng (liangf@stanford.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
LbSemiSWEET for crystallography and liposome reconstitution was overexpressed in E.coli BL21 (DE3) grown in LB. Protein expression was induced by 0.2 mM IPTG and cells were incubated overnight at 22°C. Cell-based radioactive glucose uptake assays were performed using E. coli strain JM1100, a glucose-transporter deficient strain. Cells were incubated at 37°C and induced by IPTG for 30 minutes.
METHOD DETAILS
Expression and purification of SemiSWEET
LbSemiSWEET with a C-terminal 3C protease recognition sequence and 10xHis-tag was cloned into pJexpress411 (DNA2.0 Inc.). Transformed E. coli BL21 (DE3) cells were grown at 37°C until the OD600 reached 0.6 and the temperature was lowered to 22°C. Overexpression was induced by 0.2 mM isopropyl β-D-thiogalactoside (IPTG) and incubated overnight. The pelleted cells were resuspended in a TBS containing 50 mM Tris-HCl, pH 8.0, and 150 mM NaCl and were lysed by sonication. The lysate was solubilized in 2% (w/v) dodecyl-β-D-maltopyranoside (DDM; Anatrace) for two hours at 4°C and then centrifuged at 16,000 rpm to pellet insoluble debris. SemiSWEET was isolated from the supernatant by affinity chromatography using HisPur cobalt resin (ThermoScientific) pre-equilibrated with TBS with 0.1% DDM. SemiSWEET was eluted from the column by cleaving the His-tag off using 3C protease. Protein was further purified by gel filtration (Superdex-200, GE Healthcare) in a buffer containing 10 mM Tris-HCl, pH8.0, 150 mM NaCl, and 0.03% DDM. Peak fractions were pooled, concentrated, flash frozen in liquid N2, and stored at −80°C.
Crystallization
The LbSemiSWEET mutants D57A and Q20A were reconstituted in a 1:1.5 (wt/wt) ratio with the lipid 1-(11Z-octadecenoyl)-rac-glycerol (monovaccenin, 11.7 MAG) and 1-(9Z-hexadecenoyl)-rac-glycerol (monopalmitolein, 9.7 MAG), respectively (NU-CHEK PREP Inc). Approximately 25 nL of reconstituted protein solution was deposited onto glass plates and coated with 650 nL of precipitant. D57A crystals grew within one week in a precipitant solution consisting of 50 mM tris pH 8, 100 mM LiSO4, 35% PEG 400, and 40 mM glucose. Q20A crystals appeared within two days in a precipitant solution containing 50 mM sodium acetate pH 4.5, 100 mM ZnCl2, and 16% PEG 400. Crystals were harvested after one to two weeks using mesh grid loops (MiTeGen) and flash frozen in liquid N2.
Structure determination
Diffraction data collection was carried out at the Advanced Photon Source beamline 23ID-D. LbSemiSWEET D57A data processing was done using the HKL2000 software package (Otwinowski and Minor, 1997). LbSemiSWEET Q20A data from multiple crystals were integrated by XDS (Kabsch, 2010) implemented in RAPD (https://github.com/RAPD/RAPD). Initial phases were determined by molecular replacement using the occluded-state LbSemiSWEET structure [PDB: 4QNC] as the search model in PHASER (McCoy, 2007). For the Q20A structure, intracellular loops were removed from the search model prior to molecular replacement. In each case, an initial round of deformable elastic network (DEN) refinement was performed using 4QNC as a reference (Schroder et al., 2010), which led to significant improvement of the model with substantial conformational changes. For both crystal structures, iterative manual model building in Coot and refinement in Phenix were performed to generate the final models (Adams et al., 2010; Emsley and Cowtan, 2004). All images of crystal structures were rendered in PyMOL (DeLano, 2008).
One asymmetric unit cell of the Q20A crystal contains four copies of protomers; two form near-identical crystallographic homodimers (conformation I) while the other two associate into a dimer in a slightly different conformation (conformation II). At the final stage of model building and refinement, it became evident that that TM1 was swapped between two layers of oppositely stacking SemiSWEET dimers (Figure S2B). While all three transporters are in an apparent inward-facing conformation, the transporters in conformation I closely resemble a previously published inward-open E.coli SemiSWEET structure (Figure S2C). In conformation II, a non-native c-terminal 3C “scar” extends into the central pore, forming crystal lattice contacts and potentially distorting the local structure (Figure S2D). For this reason, our structural analysis focuses on conformation I. While the protein was crystalized in a domain-swapped configuration, it overlaid well with an inward-open SemiSWEET from E. coli (Cα RMSD = 0.86 Å). Within a protomer, TM helices pack in the same manner as observed in other conformations. Furthermore, this inward open crystal structure matches well with simulated inward-open conformations (Cα RMSD = 1.4 Å).
Although the inward-open and outward-open crystal structures were determined for mutant transporters, they appear to represent conformations of the wild-type transporter. First, these structures closely match the conformations of wild-type crystal structures of other SemiSWEET orthologs [in particular, the outward-open and inward-open conformations of E. coli SemiSWEET (PDB entries 4X5N and 4X5M) (Lee et al., 2015); backbone Cα RMSDs are 0.86 Å and 0.57 Å, respectively]. Second, as discussed previously, MD simulations of the LbSemiSWEET crystal structures reverted to the wild-type sequence reveal that these conformational states are stable on timescales of at least hundreds of nanoseconds.
Cell-based uptake assay
The LbSemiSWEET mutants were cloned into pQE-80L (Qiagen) and transformed into the glucose transporter-deficient E. coli strain JM1100 (Yale Coli Genetic Stock Center). An empty pQE-80L plasmid was used as a control. Fresh cultures were induced at an OD600 of 1.5 with 50 μM IPTG for 30 minutes at 37°C. Cells were washed twice with a buffer containing 5 mM MES pH 6.5 and 150 mM KCl (MK buffer). Cells were resuspended in MK buffer to an OD600 of 2.0. Glycerol was added to a final concentration of 20 mM to energize cells for five minutes at room temperature. A mixture of 1 mM D-glucose and 2 μM D-[14C]glucose was then added to start the uptake. After a one-minute incubation, cells were collected on a 0.22 μm filter, washed three times quickly with ice-cold MK buffer, and subjected to scintillation quantification. All inactive mutants were tested for expression by Western blot unless previously confirmed to express at the wild-type level. Mutants that showed low levels of expression compared to wild type were additionally tested by the liposome-based uptake assay (Figure S7, A and B).
Liposome-based uptake assay
Purified protein of LbSemiSWEET and its mutant was reconstituted into E. coli polar lipid (Avanti) at an 80:1 lipid-to-protein ratio according to a previously published protocol (Veenhoff and Poolman, 1999). Proteoliposomes were extruded through a 0.4 μm membrane, collected by ultracentrifugation at 100,000 g for one hour, and washed twice with 50 mM potassium phosphate, 2 mM MgSO4, pH 4.5 (KPM buffer). Liposomes were resuspended in KPM buffer at 100 mg/mL. Uptake was initiated by the addition of 2 μL liposome to 100 μL of KPM buffer containing 2 mM glucose and 2 μM of D-[14C]glucose. After a five-minute incubation, the liposomes were collected on 0.22 μm filters (Millipore) and washed with 1 mL cold KPM buffer twice. Filters were then incubated with 4 mL scintillation fluid for quantification.
Western blot
E. coli cells expressing wild-type LbSemiSWEET and mutants were prepared the same way as for the cell-based uptake assay. Cell pellets were resuspended in TBS and lysed by milling with 0.5 mm glass beads (BioSpec). Lysates were transferred to a new tube and DDM was added to a final concentration of 2%. Following two hours of solubilization at 4°C, the lysates were spun at 16,000 g and the supernatants were collected and quantified. Lysates were brought to an equal total protein concentration based on OD280 before being loaded onto an SDS-PAGE gel. The protein was transferred to a nitrocellulose membrane and blocked for one hour in TBS with 5% BSA (Fisher Scientific) and 0.2% Tween-20 (AMRESCO). The membrane was incubated with anti-His mouse primary antibody (QIAGEN) overnight. Following washing with TBS-BSA-Tween buffer, the membrane was incubated with peroxidase-conjugated anti-mouse secondary antibody (Sigma) for one hour before washing and addition of peroxidase substrate (Thermo Scientific). The membrane was visualized using a CCD imager.
System setup for molecular dynamics simulations
Simulations of LbSemiSWEET were initiated from the glucose-bound, outward-open crystal structure described in this manuscript and from the previously published occluded state crystal structure (PDB ID: 4QNC) (Xu et al., 2014). Simulations of E. coli SemiSWEET (EcSemiSWEET) were initiated from a previously published inward-open crystal structure (PDB ID: 4X5M) (Lee et al., 2015). Wild-type SemiSWEET was simulated under five conditions: (1) the glucose-bound, outward-open crystal structure, reported in this manuscript; (2) the same outward-open crystal structure reported here but with glucose removed; (3) the occluded state crystal structure, without glucose; (4) the occluded-state crystal structure with glucose bound, prepared by aligning the occluded and outward-open crystal structures to each other and then inserting glucose in its outward-open orientation into the occluded-state crystal structure; and (5) the inward-open crystal structure from EcSemiSWEET. We also performed simulations of two SemiSWEET mutants, D57A and I60A, starting from the glucose-bound, outward-open structure described here. Finally, we performed simulations of an isolated transmembrane helix 1 (residues 1–28 of chain A) starting from its conformation in the outward-open structure. All proteins simulated, including the isolated transmembrane helix 1, were embedded in a hydrated lipid bilayer, as described below. We did not perform simulations of the LbSemiSWEET inward-open crystal structure, as the domain swap left uncertainty in the structure of the intracellular loop regions. We performed multiple replicates for each simulation condition (Table S2). For each replicate, initial atom velocities were assigned randomly and independently.
Coordinates were prepared by removing all non-substrate and non-protein molecules for both structures. Prime (Schrödinger, Inc.) was used to model missing side chains, and protein chain termini were capped with neutral acetyl and methylamide groups. Titratable residues remained in their dominant protonation state at pH 7. The D57A mutation present in the outward-open crystal structure was reverted to its wild-type identity, Asp57, for simulations of wild-type SemiSWEET. For simulations of the occluded crystal structure, Dowser software was used to add waters to cavities within the protein structure (Hermans et al., 2003; Zhang and Hermans, 1996), including the glucose-binding pocket. In the second round of simulations, as denoted in Table S2, Dowser software was used to add waters to wild-type and mutant simulations started from the outward-open structure.
The prepared protein structures were then aligned to the Orientations of Proteins in Membranes (Lomize et al., 2006) entry for 4QNC (for LbSemiSWEET) and 4X5M (for EcSemiSWEET) using MacPyMOL. Each prepared structure was inserted into a hydrated, pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine bilayer. Sodium and chloride ions were added to neutralize the system at a concentration of 150 mM. Membrane dimensions were chosen to maintain approximately a 35 Å buffer between protein images in the x–y plane and a 20 Å buffer between protein images in the z dimension. Final system dimensions are listed in Table S2 for each simulation condition.
MD simulation protocols
For all simulations, we used the CHARMM36 force field for proteins, lipids, ions, and glucose and the TIP3P model for waters (Best et al., 2012a; Best et al., 2012b; Guvench et al., 2009; Huang and MacKerell, 2013; Klauda et al., 2010).
We performed each simulation with 2.5 fs time steps using the Compute Unified Device Architecture (CUDA) version of Particle-Mesh Ewald Molecular Dynamics (PMEMD) in AMBER on two graphics processing units (GPUs) (Case et al.; Le Grand et al., 2013; Salomon-Ferrer et al., 2013). Initial simulations were performed using AMBER14. The second round of simulations, as denoted in table S2, was performed using AMBER15 software. Systems were heated from 0K to 100K in the NVT ensemble over 12.5 ps and then from 100K to 310K in the NPT ensemble over 125 ps, using 10.0 kcal mol−1 Å−2 harmonic restraints applied to lipids, protein, and substrate. Systems were then equilibrated at 310 K in the NPT ensemble at 1 bar, with harmonic restraints on all protein heavy atoms tapered off by 1.0 kcal mol−1 Å−2 starting at 5.0 kcal mol−1 Å−2 for six consecutive steps, each for 2.0 ns. Production simulations were performed in the NPT ensemble at 310K and 1 bar, using a Langevin thermostat for temperature coupling and either a Monte Carlo or Berendsen barostat for pressure coupling.
Simulations used periodic boundary conditions. Bond lengths to hydrogen atoms were constrained with SHAKE. The non-bonded interaction cut-off for electrostatics calculations was set to 9.0 Å, and Particle-Mesh Ewald was used to compute long-range electrostatics interactions, with an Ewald coefficient of approximately 0.30 Å−1 and a B-spline interpolation of order 4. The Fourier transform grid size was chosen such that the width of a grid cell was approximately 1 Å.
Analysis protocols for MD simulation
Trajectory snapshots were saved every 100 ps during production simulations. The AmberTools14 CPPTRAJ package was used to reimage and center trajectories (Roe and Cheatham, 2013). Simulations were visualized and analyzed using Visual Molecular Dynamics (VMD) (Humphrey et al., 1996). Plots were generated using VMD and smoothed using a moving average with a window size of 10 ns unless otherwise indicated and visualized using the PyPlot package from Matplotlib (Hunter, 2007).
For measuring the inward displacement of TM3 during occlusion of the extracellular gate, we determined the movement of the same atom (Ile60-Cα) in each protomer of the protein by computing the distance of those coordinates in time to a point along the axis orthogonal to the membrane plane. We defined the reference point as the original midpoint between the two atoms in the outward-open crystal structure, and we projected the current atom position onto the line defined by the original separation of the two atoms in the crystal structure.
We computed the average Cα RMSD of the simulated transporter to a given crystal structure at each point in time over the course of each simulation, using the conformations of the extracellular and intracellular gate to mark the transition between distinct conformational states (i.e., outward-open, partially occluded, occluded, and inward-open). We classified an extracellular gate as closed if each Ile60 residue had moved at least 1.5 Å inward towards the center of the helical bundle, and as open otherwise. We classified the intracellular gate as open if the X1 dihedral angle (C-Cα-Cβ-Cγ) of each Phe17 residue had rotated at least 90° from its position in the outward-open crystal structure. We then used the classified frames to compute ensemble average structures representing each of the three conformational states with and without glucose. The RMSD of the ensemble average structure to each corresponding crystal structure is reported in Figure S4. The average structures were generated using only simulations that transitioned from outward-open to inward-open in the first round of simulations (simulations 4, 9, 22 and 30).
To estimate the frequency of outward-open to inward-open transitions, we summed the time spent in the outward-open state across all simulations initiated from the outward-open state. The estimated transition timescale (13.8 μs) corresponds to the average amount of simulation time in the outward-open or occluded states to see a transition to the inward-open state.
To quantify interhelical packing between TM1 and TM3, we used the built-in VMD contacts function to count the number of contacts between all non-hydrogen atoms in the region of TM1 near the proline-mediated kink (residues 10–22) and all non-hydrogen atoms in TM3 (residues 57–80). We also used the built-in VMD hydrogen bond finding function with an angle cutoff of 60° and a heavy atom donor-acceptor distance of 3.5 Å to detect hydrogen bonds along the backbone of TM1. Finally, to calculate the global bend angle in TM1, we identified three vertices using the center of mass of Cα atoms at the top of TM1, at the bending point in TM1 and at the base of TM1 and used the law of cosines to compute the angle connecting those points.
To identify binding pocket waters, we first calculated the average position of glucose during a stable portion of occluded simulation (Simulation 9, 0.3–0.8 μs), chosen because glucose stably resides in the binding pocket in that state. Across all simulations, we counted a water molecule as present in the binding pocket if it fell within 5 Å of glucose in the previously determined average position, after aligning each simulation on an averaged structure of the occluded state, which required use of the TopoTools VMD plugin 1.7.
QUANTIFICATION AND STATISTICAL ANALYSIS
The bar graphs in Figure 4, 7 and S7 represent mean ± SD. Each measurement was repeated at least three times independently. The statistical analyses of the structural models were given in Table S1.
DATA AND SOFTWARE AVAILABILITY
The crystal structures of LbSemiSWEET have been deposited in protein data bank, with accession number 5UHS (outward-open) and 5UHQ (inward-open). The standard crystallography and MD simulation software used in this study is available from web servers listed in the key resource table.
Supplementary Material
Figure S1. Comparison of outward-open structures of EcSemiSWEET and VsSemiSWEET, Related to Figure 1. The structure of Vibrio sp. SemiSWEET (VsSemiSWEET) in an outward-open conformation (blue, PDB: 4QND) was superimposed onto that of EcSemiSWEET (red, PDB: 4X5M). The non-conserved additional N-terminal 11 residues in VsSemiSWEET are removed for clarity.
Figure S2. Strategies to trap LbSemiSWEET in distinct conformational states and the crystal structure of the Q20A mutant, Related to Figure 1 and Figure 3. (A) Based on an occluded-state structure of LbSemiSWEET, cross protomer interactions are predicted to stabilize the closed states of the extracellular and intracellular gates (upper right and lower right, respectively). (B) Crystal lattice structure of the Q20A mutant is shown in ribbon representation. Two SemiSWEET dimer molecules are colored yellow, violet, red, and green by chain, with each chain contributing to both transporter molecules. (C) Comparison of the two inward-open conformations of Q20A (colored yellow and violet by chain) to that of EcSemiSWEET (silver; PDB: 4X5M). (D) Crystal packing of conformation II showing the C-terminus of one transporter extend into the pore of its crystallographic symmetry mate (left). A zoomed-in view of the boxed area shows hydrophobic residues of the C-terminus pack against key residues in the pore.
Figure S3. Quantitative evidence for match between conformations spontaneously adopted in simulations and crystal structures, Related to Figure 3. (A) (Left) Comparison between a representative frame of LbSemiSWEET to a representative simulated frame of the inward-open state of EcSemiSWEET (4X5M). (Right) The purple trace shows the RMSD of a simulation of EcSemiSWEET (Simulation 55) to the LbSemiSWEET inward-open crystal structure and the blue trace shows the RMSD of a simulation that transitions between outward-open and inward-open states (Simulation 4) to the LbSemiSWEET inward-open crystal structure. The sliding average for each plot is computed over 10 ns (see Methods). (B) Table showing the Cα RMSD of the ensemble average structure for each conformational state, with and without glucose, to each corresponding crystal structure (see Methods).
Figure S4. Gate residue dynamics during the outward-open to inward-open conformational transition, Related to Figure 4. For each of the four simulations that spontaneously transitioned from the outward-open to the inward-open states in the first round of simulations, the backbone Cα RMSD of each frame to the occluded and inward-open LbSemiSWEET crystal structures is plotted over time. Residues 27 to 35 are omitted in the inward-open calculation due to the domain swap. Changes to gate residue position are plotted, as in main text Figure 4B. The sliding average for each plot is computed over 10 ns (see Methods). The outward-open simulations that transitioned are: (A) Simulation 4 with glucose (B) Simulation 9 with glucose (C) Simulation 22 without glucose, and (D) Simulation 30 without glucose. In Simulation 22, the simulation transitions rapidly from outward-open to inward-open, populating an occluded state for approximately 20 ns. In simulations 9, 22 and 35, Phe17 on one of the two protomers exhibited a small amount of instability preceding extracellular gate closure but only stably populated the ∼60° Χ1 angle after the extracellular gate closed.
Figure S5. Key molecular interactions and conformational changes associated with the outward-open to inward-open transition, Related to Figure 4. (A) Compared to the outward-open state (left), relatively fewer TM1-TM3 interhelical contacts form in the occluded (middle) and inward-open (right) states (top panel) (see Methods). Plots represent traces and their sliding means (window of 10 ns) from fragments of simulation in which the protein remained in a given conformational state for at least 1 μs, taken from protomer A of Simulation 1, Simulation 43, and Simulation 4, respectively. The change in interhelical packing is depicted in the representative outward-open (gray) and occluded (purple) snapshots from Simulation 16 (left). Arrows indicate helical motion during simulation. Backbone and backbone-side chain hydrogen bonds are shown for residues in TM1 (residues 13–19) for the same fragments of simulation as in the top panel (bottom panel). In the inward-open state, two i, i+4 backbone hydrogen bonds form, favoring the kinked state of TM1. (B) In simulations of an isolated TM1 helix, starting from its outward-open conformation, the helix immediately kinks around a proline (Pro19) in the region near the Phe17 gating residue, and the global bend angle in TM1 increases to ∼18° (trace and sliding mean with a window of 100 ns, left). The favored rotameric state of Phe17 decreases from ∼160° to ∼ 60° (trace and sliding mean with a window of 10 ns, right). A representative snapshot (purple) is taken from simulation 68 and shows both the kinked state of TM1 and the change in the preferred Phe17 rotamer compared to that seen in the outward open state (gray).
Figure S6. Water coordination in the substrate recognition pocket, Related to Figure 6. (A) The glucose-binding pocket, including waters, as observed in randomly-selected simulation frames from the three major conformational states in simulations with (top panel) and without (lower panel) glucose. Dotted lines indicated potential hydrogen bonds between nearby atoms, as determined using the polar contact finder in PyMOL. Frames were taken from simulations 1(left, top) and 9 (middle and right, top) and from simulations 22 (left and right, bottom) and 30 (middle, bottom). (B) Counts of binding pocket waters for simulations performed with (left) and without (right) glucose, computed for every 1 ns of simulation (see Methods). In the occluded state, the number of total binding pocket waters decreased in both glucose-bound and glucose-free conditions. The spike at the beginning of the left, blue trace (Simulation 9) corresponds to glucose momentarily moving up toward the extracellular gate, leading to an increase in binding pocket waters. The blue trace on right is from Simulation 30, and the red traces correspond to Simulations 1 (left) and 21 (right).
Figure S7. Expression level of inactive mutants and glucose uptake in a liposome-based assay, Related to Figure 4 and Figure 7. (A) The expression level of wild-type LbSemiSWEET and inactive mutants in a cell-based uptake assay is shown by Western blot. The lane containing the purified LbSemiSWEET is placed next to the marker as the reference. (B) Glucose uptake activities of I60A mutant compared with wild type and control in a proteoliposome-based radioactive glucose (14C) uptake assay (±s.d., n=3).
Table S1. Statistics for crystallography data collection and refinement, Related to Figure 1 and 3.
Table S2. Table of simulations performed, Related to Figure 2, 3, 4, 6 and 7. Simulations are numbered across all conditions. Box size was estimated from the production stages of simulation. In Round 2 of the outward open simulations, the box size was slightly increased. The final conformational state of the simulation refers to the state of the protein at the final frame of the simulation, except for TM1, whose conformation is marked at not applicable (N.A.). For simulations in which glucose diffused away from the binding pocket into the bulk solvent, we have indicated the time at which it dissociated. We also indicate a simulation in which glucose unbinds from the binding pocket but does not dissociate into the bulk.
Highlights.
Crystal structure of SemiSWEET in an outward-open state with glucose bound
Unguided MD simulations capture spontaneous outward-open to inward-open transitions
Gates driven to open only in occluded state, preventing their simultaneous opening
Simulations with and without substrate visit closely matching conformational states
In Brief.
Unguided simulations initiated from an outward-open crystal structure of a transporter spontaneously reach occluded and inward-open conformations that closely match crystal structures, providing an atomic-level description of the process by which an alternating-access transporter carries a substrate across the cell membrane.
Acknowledgments
We thank the staff at Beamline 23ID-B/D and 24ID-C/E (APS, Argonne National Laboratory), at Beamline 12-2 (SSRL, SLAC National Laboratory), and at Beamline 5.0.2 (Advanced Light Source), particularly K. Perry, for assistance at the synchrotron. This work was made possible by support from Stanford University, the Harold and Leila Y. Mathers Charitable Foundation, the Alfred P. Sloan Foundation, NIH (R01 GM117108) to L.F., the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences at the US Department of Energy under grant number DE-FG02-04ER15542 to W.B.F., a Stanford Terman Faculty Fellowship to R.O.D., and the Stanford Bio-X Interdisciplinary Initiative Program (IIP8-70) to L.F. and R.O.D. N.M.F. and N.R.L. were supported by National Science Foundation Graduate Research Fellowships. L.F. was an NIH Director’s New Innovator awardee. We thank M. Maduke for helpful comments, Y. Xu, Z. He, and J. Fang for technical assistance, and R. Betz for assistance with MD simulation analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental information includes seven figures and two tables.
AUTHOR CONTRIBUTIONS
N.R.L., N.M.F., R.O.D. and L.F. designed the research. N.R.L. performed simulations. N.M.F. performed crystallography, mutagenesis, and functional characterization experiments. N.R.L, A.J.V., and R.O.D analyzed simulations, and N.M.F., W.B.F. and L.F. analyzed experimental data. N.R.L., N.M.F., R.O.D. and L.F. wrote the manuscript, with input from A.J.V. and W.B.F. N.R.L. and N.M.F. contribute equally. R.O.D. and L.F. contribute equally.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman JL, Dale AL, Zwier MC, Bhatt D, Chong LT, Zuckerman DM, Grabe M. Simulations of the alternating access mechanism of the sodium symporter Mhp1. Biophys J. 2011;101:2399–2407. doi: 10.1016/j.bpj.2011.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman JL, Ghezzi C, Bisignano P, Loo DD, Choe S, Abramson J, Rosenberg JM, Wright EM, Grabe M. Stochastic steps in secondary active sugar transport. Proc Natl Acad Sci USA. 2016 doi: 10.1073/pnas.1525378113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akyuz N, Georgieva ER, Zhou Z, Stolzenberg S, Cuendet MA, Khelashvili G, Altman RB, Terry DS, Freed JH, Weinstein H, et al. Transport domain unlocking sets the uptake rate of an aspartate transporter. Nature. 2015;518:68–73. doi: 10.1038/nature14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EN, Hubbard RE. Hydrogen bonding in globular proteins. Prog Biophys Mol Biol. 1984;44:97–179. doi: 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- Best RB, Mittal J, Feig M, MacKerell AD., Jr Inclusion of many-body effects in the additive CHARMM protein CMAP potential results in enhanced cooperativity of alpha-helix and beta-hairpin formation. Biophys J. 2012a;103:1045–1051. doi: 10.1016/j.bpj.2012.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, Mackerell AD., Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput. 2012b;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Jin X, Levin EJ, Huang H, Zong Y, Quick M, Weng J, Pan Y, Love J, Punta M, et al. Crystal structure of a phosphorylation-coupled saccharide transporter. Nature. 2011;473:50–54. doi: 10.1038/nature09939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE. AMBER. Vol. 14. University of California; San Francisco: p. 2014. [Google Scholar]
- Chen LQ, Cheung LS, Feng L, Tanner W, Frommer WB. Transport of sugars. Annu Rev Biochem. 2015;84:865–894. doi: 10.1146/annurev-biochem-060614-033904. [DOI] [PubMed] [Google Scholar]
- Chen LQ, Hou BH, Lalonde S, Takanaga H, Hartung ML, Qu XQ, Guo WJ, Kim JG, Underwood W, Chaudhuri B, et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature. 2010;468:527–532. doi: 10.1038/nature09606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng MH, Bahar I. Complete mapping of substrate translocation highlights the role of LeuT N-terminal segment in regulating transport cycle. PLoS Comput Biol. 2014;10:e1003879. doi: 10.1371/journal.pcbi.1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; Palo Alto, CA, USA: 2008. http://www.pymol.org. [Google Scholar]
- Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- Drew D, Boudker O. Shared Molecular Mechanisms of Membrane Transporters. Annu Rev Biochem. 2016 doi: 10.1146/annurev-biochem-060815-014520. [DOI] [PubMed] [Google Scholar]
- Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE. Biomolecular simulation: a computational microscope for molecular biology. Ann Rev Biophys. 2012;41:429–452. doi: 10.1146/annurev-biophys-042910-155245. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ewers D, Becher T, Machtens JP, Weyand I, Fahlke C. Induced fit substrate binding to an archeal glutamate transporter homologue. Proc Natl Acad Sci USA. 2013;110:12486–12491. doi: 10.1073/pnas.1300772110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, Wright EM, Abramson J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science. 2008;321:810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraldo-Gomez JD, Forrest LR. Modeling and simulation of ion-coupled and ATP-driven membrane proteins. Curr Opin Struct Biol. 2011;21:173–179. doi: 10.1016/j.sbi.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Forrest LR. Structural biology. (Pseudo-)symmetrical transport. Science. 2013;339:399–401. doi: 10.1126/science.1228465. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Kramer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta. 2011;1807:167–188. doi: 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Fowler PW, Orwick-Rydmark M, Radestock S, Solcan N, Dijkman PM, Lyons JA, Kwok J, Caffrey M, Watts A, Forrest LR, et al. Gating topology of the proton-coupled oligopeptide symporters. Structure. 2015;23:290–301. doi: 10.1016/j.str.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Takeda H, Kato HE, Doki S, Ito K, Maturana AD, Ishitani R, Nureki O. Structural basis for dynamic mechanism of nitrate/nitrite antiport by NarK. Nat Commun. 2015;6:7097. doi: 10.1038/ncomms8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Shrivastava IH, Amara SG, Bahar I. Molecular simulations elucidate the substrate translocation pathway in a glutamate transporter. Proc Natl Acad Sci USA. 2009;106:2589–2594. doi: 10.1073/pnas.0812299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvench O, Hatcher ER, Venable RM, Pastor RW, Mackerell AD. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J Chem Theory Comput. 2009;5:2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans J, Xia X, Zhang L, Cavanaugh, Dowser D. Dowser program 2003 [Google Scholar]
- Holyoake J, Sansom MS. Conformational change in an MFS protein: MD simulations of LacY. Structure. 2007;15:873–884. doi: 10.1016/j.str.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Huang J, MacKerell AD., Jr CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng. 2007;9:90–95. [Google Scholar]
- Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Kaback HR. A chemiosmotic mechanism of symport. Proc Natl Acad Sci USA. 2015;112:1259–1264. doi: 10.1073/pnas.1419325112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmier K, Sharma S, Quick M, Islam SM, Roux B, Weinstein H, Javitch JA, McHaourab HS. Conformational dynamics of ligand-dependent alternating access in LeuT. Nat Struct Mol Biol. 2014;21:472–479. doi: 10.1038/nsmb.2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SC, Wilson TH. Towards an understanding of the structural basis of ‘forbidden’ transport pathways in the Escherichia coli lactose carrier: mutations probing the energy barriers to uncoupled transport. Mol Microbiol. 1990;4:1433–1438. doi: 10.1111/j.1365-2958.1990.tb02053.x. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldso H, Noer P, Grouleff J, Autzen HE, Sinning S, Schiott B. Unbiased simulations reveal the inward-facing conformation of the human serotonin transporter and Na(+) ion release. PLoS Comput Biol. 2011;7:e1002246. doi: 10.1371/journal.pcbi.1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kasho V, Smirnova I, Finer-Moore JS, Kaback HR, Stroud RM. Structure of sugar-bound LacY. Proc Natl Acad Sci USA. 2014;111:1784–1788. doi: 10.1073/pnas.1324141111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S, Götz AW, Walker RC. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput Phys Commun. 2013;184:374–380. [Google Scholar]
- Lee S, Swanson JM, Voth GA. Multiscale Simulations Reveal Key Aspects of the Proton Transport Mechanism in the ClC-ec1 Antiporter. Biophys J. 2016;110:1334–1345. doi: 10.1016/j.bpj.2016.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Nishizawa T, Yamashita K, Ishitani R, Nureki O. Structural basis for the facilitative diffusion mechanism by SemiSWEET transporter. Nat Commun. 2015;6:6112. doi: 10.1038/ncomms7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine MV, Cuendet MA, Khelashvili G, Weinstein H. Allosteric Mechanisms of Molecular Machines at the Membrane: Transport by Sodium-Coupled Symporters. Chem Rev. 2016;116:6552–6587. doi: 10.1021/acs.chemrev.5b00627. [DOI] [PubMed] [Google Scholar]
- Li J, Wen PC, Moradi M, Tajkhorshid E. Computational characterization of structural dynamics underlying function in active membrane transporters. Curr Opin Struct Biol. 2015;31:96–105. doi: 10.1016/j.sbi.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ke M, Gong H. Protonation of Glu(135) Facilitates the Outward-to-Inward Structural Transition of Fucose Transporter. Biophys J. 2015;109:542–551. doi: 10.1016/j.bpj.2015.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher KP. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol. 2016;23:487–493. doi: 10.1038/nsmb.3216. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Madej MG, Soro SN, Kaback HR. Apo-intermediate in the transport cycle of lactose permease (LacY) Proc Natl Acad Sci USA. 2012;109:E2970–2978. doi: 10.1073/pnas.1211183109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masureel M, Martens C, Stein RA, Mishra S, Ruysschaert JM, McHaourab HS, Govaerts C. Protonation drives the conformational switch in the multidrug transporter LmrP. Nat Chem Biol. 2014;10:149–155. doi: 10.1038/nchembio.1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell P. A general theory of membrane transport from studies of bacteria. Nature. 1957;180:134–136. doi: 10.1038/180134a0. [DOI] [PubMed] [Google Scholar]
- Moradi M, Enkavi G, Tajkhorshid E. Atomic-level characterization of transport cycle thermodynamics in the glycerol-3-phosphate:phosphate antiporter. Nat Commun. 2015;6:8393. doi: 10.1038/ncomms9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi M, Tajkhorshid E. Mechanistic picture for conformational transition of a membrane transporter at atomic resolution. Proc Natl Acad Sci USA. 2013;110:18916–18921. doi: 10.1073/pnas.1313202110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter Charles W., Jr, editor. Methods Enzymol. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Palmgren MG, Nissen P. P-type ATPases. Ann Rev Biophys. 2011;40:243–266. doi: 10.1146/annurev.biophys.093008.131331. [DOI] [PubMed] [Google Scholar]
- Pedersen BP, Kumar H, Waight AB, Risenmay AJ, Roe-Zurz Z, Chau BH, Schlessinger A, Bonomi M, Harries W, Sali A, et al. Crystal structure of a eukaryotic phosphate transporter. Nature. 2013;496:533–536. doi: 10.1038/nature12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quistgaard EM, Low C, Guettou F, Nordlund P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol. 2016;17:123–132. doi: 10.1038/nrm.2015.25. [DOI] [PubMed] [Google Scholar]
- Roe DR, Cheatham TE., 3rd PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Schroder GF, Levitt M, Brunger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–1222. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh SA, Li J, Enkavi G, Wen PC, Huang Z, Tajkhorshid E. Visualizing functional motions of membrane transporters with molecular dynamics simulations. Biochemistry. 2013;52:569–587. doi: 10.1021/bi301086x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T, Weyand S, Beckstein O, Rutherford NG, Hadden JM, Sharples D, Sansom MS, Iwata S, Henderson PJ, Cameron AD. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010;328:470–473. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl LS, Fowler PW, Sansom MS, Beckstein O. Flexible gates generate occluded intermediates in the transport cycle of LacY. J Mol Biol. 2014;426:735–751. doi: 10.1016/j.jmb.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature. 2012;490:361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- Tao Y, Cheung LS, Li S, Eom JS, Chen LQ, Xu Y, Perry K, Frommer WB, Feng L. Structure of a eukaryotic SWEET transporter in a homotrimeric complex. Nature. 2015;527:259–263. doi: 10.1038/nature15391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenhoff LM, Poolman B. Substrate recognition at the cytoplasmic and extracellular binding site of the lactose transport protein of Streptococcus thermophilus. J Biol Chem. 1999;274:33244–33250. doi: 10.1074/jbc.274.47.33244. [DOI] [PubMed] [Google Scholar]
- Verdon G, Oh S, Serio RN, Boudker O. Coupled ion binding and structural transitions along the transport cycle of glutamate transporters. eLife. 2014;3:e02283. doi: 10.7554/eLife.02283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yan C, Li Y, Hirata K, Yamamoto M, Yan N, Hu Q. Crystal structure of a bacterial homologue of SWEET transporters. Cell Res. 2014;24:1486–1489. doi: 10.1038/cr.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A, Choe S, Chaptal V, Rosenberg JM, Wright EM, Grabe M, Abramson J. The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature. 2010;468:988–991. doi: 10.1038/nature09580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright EM. Glucose transport families SLC5 and SLC50. Mol Aspects Med. 2013;34:183–196. doi: 10.1016/j.mam.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Xu Y, Tao Y, Cheung LS, Fan C, Chen LQ, Xu S, Perry K, Frommer WB, Feng L. Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature. 2014;515:448–452. doi: 10.1038/nature13670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan YH, Hu YB, Chen LQ, Sosso D, Ducat DC, Hou BH, Frommer WB. Functional role of oligomerization for bacterial and plant SWEET sugar transporter family. Proc Natl Acad Sci USA. 2013;110:E3685–3694. doi: 10.1073/pnas.1311244110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, Radestock S, Shuster Y, Forrest LR, Schuldiner S. Identification of molecular hinge points mediating alternating access in the vesicular monoamine transporter VMAT2. Proc Natl Acad Sci USA. 2013;110:E1332–1341. doi: 10.1073/pnas.1220497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Yan N. Structural Biology of the Major Facilitator Superfamily Transporters. Ann Rev Biophys. 2015;44:257–283. doi: 10.1146/annurev-biophys-060414-033901. [DOI] [PubMed] [Google Scholar]
- Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312:741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hermans J. Hydrophilicity of cavities in proteins. Proteins. 1996;24:433–438. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Zhao C, Stolzenberg S, Gracia L, Weinstein H, Noskov S, Shi L. Ion-controlled conformational dynamics in the outward-open transition from an occluded state of LeuT. Biophys J. 2012;103:878–888. doi: 10.1016/j.bpj.2012.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparison of outward-open structures of EcSemiSWEET and VsSemiSWEET, Related to Figure 1. The structure of Vibrio sp. SemiSWEET (VsSemiSWEET) in an outward-open conformation (blue, PDB: 4QND) was superimposed onto that of EcSemiSWEET (red, PDB: 4X5M). The non-conserved additional N-terminal 11 residues in VsSemiSWEET are removed for clarity.
Figure S2. Strategies to trap LbSemiSWEET in distinct conformational states and the crystal structure of the Q20A mutant, Related to Figure 1 and Figure 3. (A) Based on an occluded-state structure of LbSemiSWEET, cross protomer interactions are predicted to stabilize the closed states of the extracellular and intracellular gates (upper right and lower right, respectively). (B) Crystal lattice structure of the Q20A mutant is shown in ribbon representation. Two SemiSWEET dimer molecules are colored yellow, violet, red, and green by chain, with each chain contributing to both transporter molecules. (C) Comparison of the two inward-open conformations of Q20A (colored yellow and violet by chain) to that of EcSemiSWEET (silver; PDB: 4X5M). (D) Crystal packing of conformation II showing the C-terminus of one transporter extend into the pore of its crystallographic symmetry mate (left). A zoomed-in view of the boxed area shows hydrophobic residues of the C-terminus pack against key residues in the pore.
Figure S3. Quantitative evidence for match between conformations spontaneously adopted in simulations and crystal structures, Related to Figure 3. (A) (Left) Comparison between a representative frame of LbSemiSWEET to a representative simulated frame of the inward-open state of EcSemiSWEET (4X5M). (Right) The purple trace shows the RMSD of a simulation of EcSemiSWEET (Simulation 55) to the LbSemiSWEET inward-open crystal structure and the blue trace shows the RMSD of a simulation that transitions between outward-open and inward-open states (Simulation 4) to the LbSemiSWEET inward-open crystal structure. The sliding average for each plot is computed over 10 ns (see Methods). (B) Table showing the Cα RMSD of the ensemble average structure for each conformational state, with and without glucose, to each corresponding crystal structure (see Methods).
Figure S4. Gate residue dynamics during the outward-open to inward-open conformational transition, Related to Figure 4. For each of the four simulations that spontaneously transitioned from the outward-open to the inward-open states in the first round of simulations, the backbone Cα RMSD of each frame to the occluded and inward-open LbSemiSWEET crystal structures is plotted over time. Residues 27 to 35 are omitted in the inward-open calculation due to the domain swap. Changes to gate residue position are plotted, as in main text Figure 4B. The sliding average for each plot is computed over 10 ns (see Methods). The outward-open simulations that transitioned are: (A) Simulation 4 with glucose (B) Simulation 9 with glucose (C) Simulation 22 without glucose, and (D) Simulation 30 without glucose. In Simulation 22, the simulation transitions rapidly from outward-open to inward-open, populating an occluded state for approximately 20 ns. In simulations 9, 22 and 35, Phe17 on one of the two protomers exhibited a small amount of instability preceding extracellular gate closure but only stably populated the ∼60° Χ1 angle after the extracellular gate closed.
Figure S5. Key molecular interactions and conformational changes associated with the outward-open to inward-open transition, Related to Figure 4. (A) Compared to the outward-open state (left), relatively fewer TM1-TM3 interhelical contacts form in the occluded (middle) and inward-open (right) states (top panel) (see Methods). Plots represent traces and their sliding means (window of 10 ns) from fragments of simulation in which the protein remained in a given conformational state for at least 1 μs, taken from protomer A of Simulation 1, Simulation 43, and Simulation 4, respectively. The change in interhelical packing is depicted in the representative outward-open (gray) and occluded (purple) snapshots from Simulation 16 (left). Arrows indicate helical motion during simulation. Backbone and backbone-side chain hydrogen bonds are shown for residues in TM1 (residues 13–19) for the same fragments of simulation as in the top panel (bottom panel). In the inward-open state, two i, i+4 backbone hydrogen bonds form, favoring the kinked state of TM1. (B) In simulations of an isolated TM1 helix, starting from its outward-open conformation, the helix immediately kinks around a proline (Pro19) in the region near the Phe17 gating residue, and the global bend angle in TM1 increases to ∼18° (trace and sliding mean with a window of 100 ns, left). The favored rotameric state of Phe17 decreases from ∼160° to ∼ 60° (trace and sliding mean with a window of 10 ns, right). A representative snapshot (purple) is taken from simulation 68 and shows both the kinked state of TM1 and the change in the preferred Phe17 rotamer compared to that seen in the outward open state (gray).
Figure S6. Water coordination in the substrate recognition pocket, Related to Figure 6. (A) The glucose-binding pocket, including waters, as observed in randomly-selected simulation frames from the three major conformational states in simulations with (top panel) and without (lower panel) glucose. Dotted lines indicated potential hydrogen bonds between nearby atoms, as determined using the polar contact finder in PyMOL. Frames were taken from simulations 1(left, top) and 9 (middle and right, top) and from simulations 22 (left and right, bottom) and 30 (middle, bottom). (B) Counts of binding pocket waters for simulations performed with (left) and without (right) glucose, computed for every 1 ns of simulation (see Methods). In the occluded state, the number of total binding pocket waters decreased in both glucose-bound and glucose-free conditions. The spike at the beginning of the left, blue trace (Simulation 9) corresponds to glucose momentarily moving up toward the extracellular gate, leading to an increase in binding pocket waters. The blue trace on right is from Simulation 30, and the red traces correspond to Simulations 1 (left) and 21 (right).
Figure S7. Expression level of inactive mutants and glucose uptake in a liposome-based assay, Related to Figure 4 and Figure 7. (A) The expression level of wild-type LbSemiSWEET and inactive mutants in a cell-based uptake assay is shown by Western blot. The lane containing the purified LbSemiSWEET is placed next to the marker as the reference. (B) Glucose uptake activities of I60A mutant compared with wild type and control in a proteoliposome-based radioactive glucose (14C) uptake assay (±s.d., n=3).
Table S1. Statistics for crystallography data collection and refinement, Related to Figure 1 and 3.
Table S2. Table of simulations performed, Related to Figure 2, 3, 4, 6 and 7. Simulations are numbered across all conditions. Box size was estimated from the production stages of simulation. In Round 2 of the outward open simulations, the box size was slightly increased. The final conformational state of the simulation refers to the state of the protein at the final frame of the simulation, except for TM1, whose conformation is marked at not applicable (N.A.). For simulations in which glucose diffused away from the binding pocket into the bulk solvent, we have indicated the time at which it dissociated. We also indicate a simulation in which glucose unbinds from the binding pocket but does not dissociate into the bulk.