

Abstract
Background
DNA methylation is an important epigenetic modification. It can regulate the expression of many key genes without changing the primary structure of the genomic DNA, and plays a vital role in the growth and development of the organism. The genome-wide DNA methylation profile of the cytoplasmic male sterile (CMS) line in soybean has not been reported so far.
Results
In this study, genome-wide comparative analysis of DNA methylation between soybean CMS line NJCMS5A and its maintainer NJCMS5B was conducted by whole-genome bisulfite sequencing. The results showed 3527 differentially methylated regions (DMRs) and 485 differentially methylated genes (DMGs), including 353 high-credible methylated genes, 56 methylated genes coding unknown protein and 76 novel methylated genes with no known function were identified. Among them, 25 DMRs were further validated that the genome-wide DNA methylation data were reliable through bisulfite treatment, and 9 DMRs were confirmed the relationship between DNA methylation and gene expression by qRT-PCR. Finally, 8 key DMGs possibly associated with soybean CMS were identified.
Conclusions
Genome-wide DNA methylation profile of the soybean CMS line NJCMS5A and its maintainer NJCMS5B was obtained for the first time. Several specific DMGs which participated in pollen and flower development were further identified to be probably associated with soybean CMS. This study will contribute to further understanding of the molecular mechanism behind soybean CMS.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3962-5) contains supplementary material, which is available to authorized users.
Keywords: Soybean (Glycine max (L.) Merr.), Cytoplasmic male sterility, DNA methylation, Differentially methylated gene, Gene expression
Background
Cytoplasmic male sterility (CMS) is a common maternally inherited phenomenon that prevents the production of functional pollen [1]. In most cases of CMS, male-fertility can be recovered by specific gene named as restorer-of-fertility (RF) in the nuclear genome [2, 3]. At present, CMS has been present in more than 200 species of plants [4] and was widely used in crops hybrid breeding. Although the molecular mechanism of soybean CMS has been reported in transcriptomics [5], proteomics [6], microRNA [7] and mitochondrial genome [8] studies, the epigenetic regulation of the CMS remains poorly understood.
DNA methylation, a conserved epigenetic silencing mechanism in most eukaryote, could regulate the expression of many key genes, for example histone methylation along with gene silencing [9], RNA-directed DNA methylation repressing the LDMAR gene expression [10], and FLC chromatin with methylated modification delaying flowering [11]. In plant and animal, DNA methylation occurs predominately in CG enriched region of the genome, especially at CG island. However, cytosine methylation has also been observed in the CHG and CHH contexts with a low level in plant genome [12]. According to statistics, DNA methylation level varies from 4.6% to 30% in plant, which is relatively higher than that in animal [13]. No matter animal, plant or fungi, active genes are generally unmethylated, while transposable elements (TEs) are heavily methylated. So it was proposed that DNA methylation is roughly positively correlated with TE abundance, but negatively related to gene expression [14]. Within promoter region, DNA methylation is supposed to impact the transcriptional level of genes by silencing TE; whereas, within the gene body, DNA methylation may be associated with highly-expressed genes [15]. In addition, the establishment of DNA methylation mainly depends on DNA methyltransferases (DNMT), and DNA methylation regulation also relies on DNMT. To date, genes that encode DNMT have been isolated from a variety of plants, including rice, tobacco, corn and soybean [16–20].
Whole-genome sequencing and methylation profile has been widely reported in many plants, such as Arabidopsis [15], rice [21], soybean [14] and cotton [22]. Chen et al. [23] studied differentially methylation levels between the rice male sterile line PA64s and its fertile plant, and found the methylation level was higher in the sterile line than that of the fertile line. In addition, they also identified a differentially methylated gene (DMG) that was probably related to male sterility [24]. Liu et al. [25] proposed that the DNA methylation levels were higher in the corn CMS line than that in the maintainer line, and inferred that the changes of DNA methylation level in maize tassel may be associated with CMS. Although DNA methylome has also been used to analyze the distribution and average level of methylation in soybean root, stem, leaf and cotyledon [14], how DNA methylation may regulate the soybean CMS has no related report so far.
The soybean CMS line NJCMS5A was developed through consecutive backcross procedures with NJCMS1A [26–28] as the donor parent and Wandou 28 (designated as NJCMS5B afterwards) as recurrent parent in our laboratory. In this study, genome-wide comparative analysis of DNA methylation between soybean CMS line NJCMS5A and its maintainer NJCMS5B was conducted by whole-genome bisulfite sequencing. This is the first time to exploit epigenetic variation, and how gene expression is regulated in the whole genome of the soybean CMS.
Results
Analysis of genome-wide DNA methylation data of NJCMS5A and NJCMS5B
To study the genome-wide DNA methylation pattern of soybean, we collected flower buds from the soybean CMS line NJCMS5A and its maintainer NJCMS5B for constructing genomic DNA libraries. In total, 383,901,574 (NJCMS5A) and 398,207,546 (NJCMS5B) raw reads were generated from the two DNA library samples by whole-genome bisulfite sequencing (Table 1). After removal of related adapters, low-quality reads and containing Ns, 147,400,718 in NJCMS5A and 150,714,851 in NJCMS5B clean reads were collected (Table 1), of which 65.08% (NJCMS5A) and 62.10% (NJCMS5B) were uniquely mapped to the reference soybean genome of Williams82 (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Gmax) (Table 1). Over 88% cytosines in soybean genome were covered by at least 5-fold coverage (Additional file 1). Meanwhile, more than 99% cytosines were converted, which indicates a high conversion rate in this study (Table 1).
Table 1.
Summary of genome-wide methylation sequencing data
| Sample | Raw reads | Clean reads | Total unique mapped reads | Percentage of mapped reads in total reads | The coverage of 5 × reads in total reads | Bisulfite sequencing conversion rate |
|---|---|---|---|---|---|---|
| NJCMS5A | 383,901,574 | 147,400,718 | 95,930,507 | 65.08% | 88.75% | 99.75% |
| NJCMS5B | 398,207,546 | 150,714,851 | 93,590,676 | 62.10% | 88.48% | 99.39% |
The difference of DNA methylation level in the genome-wide between NJCMS5A and NJCMS5B was not significant. We detected 14.60% and 14.26% of methylated cytosines in NJCMS5A and NJCMS5B (Fig. 1a). Meanwhile, the average level of methylated cytosines in each context was also calculated for them. There were 48.58% of mCG (mCG/CG), 39.10% of mCHG (mCHG/CHG) and 6.74% of mCHH (mCHH/CHH) in NJCMS5A. Correspondingly, 47.37%, 37.27% and 6.73% of cytosines were methylated in CG, CHG and CHH contexts of NJCMS5B, respectively (Fig. 1a). Clearly, the average levels of mCG and mCHG were much higher than that of mCHH (Fig. 1a). And this distribution trend in three contexts in flower bud is generally similar to the trend reported previously in soybean root, steam, leaf and cotyledon [14]. Surprisingly, though the percent of mCHG was much lower than that of mCG and mCHG, the number of methylated cytosine sites in the three contexts was very similar. In NJCMS5A, we detected 48,495,434 mC sites, 15,497,738 mCG cites (32.0% of all mC), 15,425,507 mCHG sites (31.8% of all mC), 17,572,189 mCHH sites (36.2% of all mC), respectively; similarly, there were 31.9% mCG, 31.1% mCHG, and 37.0% mCHH in NJCMS5B (Fig. 1b). These results are consistent with the previous report about the DNA methylome of soybean LD00-2817P (“LD”) germplasm (Fig. 1b) [29]. So, we inferred there was a sizable proportion in of non-CG (CHG and CHH) in the plant cell.
Fig. 1.
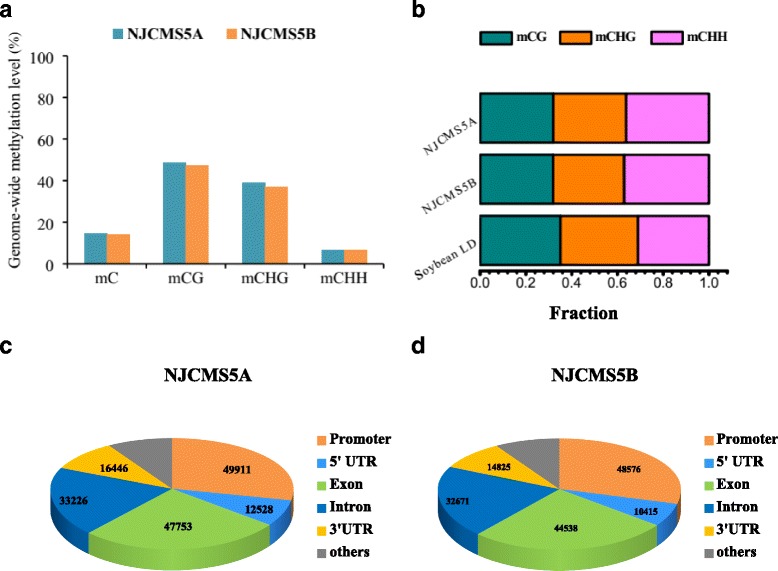
Feature of soybean genome-wide methylation between NJCMS5A and NJCMS5B. a Fraction of genome-wide methylated cytosines (mC) level and fraction of mC level in CG, CHG and CHH contexts. The blue column represented NJCMS5A, and red column represented NJCMS5B; b Distribution of methylated cytosines in the three contexts of NJCMS5A, NJCMS5B and soybean LD (soybean germplasm line LD00-2817P); c Number of methylated genes in the promoter, 5′UTR, exon, intron and 3′UTR region of NJCMS5A; d Number of methylated genes in the promoter, 5′UTR, exon, intron and 3′UTR region of NJCMS5B
Profile of DNA methylation in gene and transposable element (TE) region
The density distribution of methylated cytosines was tested to detect DNA methylation region. Firstly, from the chromosome level, DNA methylation was enriched mostly in the centromeric region, but with a little methylation was present at both ends of the chromosome (Additional file 2). Secondly, to better understand the relationship of DNA methylation and gene expression, we divided the genome into some functional regions, namely promoter region defined as the 2 kb region upstream of a transcription start site (TSS) and gene body consisting of 5′UTR, exon, intron and 3′UTR. For the promoter region, the DNA methylation level rapidly increased as departing from the TSSs sites in all contexts (Fig. 2). And it showed a low level in the gene body (Fig. 2). Besides this feature, when compared with CHH methylation level, CG and CHG methylation levels were significantly higher in promoter or gene body region (Fig. 2).
Fig. 2.
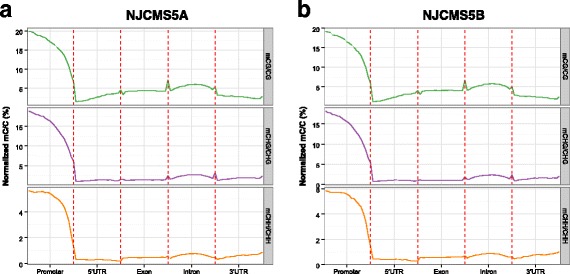
DNA methylation pattern in gene functional region under mC, mCG, mCHG and mCHH contexts. a Average density of DNA methylation in the promoter, 5′UTR, exon, intron and 3′UTR region under three contexts of NJCMS5A; b Average density of DNA methylation in the promoter, 5′UTR, exon, intron and 3′UTR region under three contexts of NJCMS5B. Each functional region was equally divided into 20 bins, and the mean of methylated cytosine density in each bin was defined as methylation density
To further assess the correlations among DNA methylation, TE and gene expression, we collected genome sequences (from our laboratory) and TE sequences from SoyTEdb (http://www.soybase.org/soytedb/) [30, 31] (Fig. 3). On the whole, the pattern of DNA methylation in NJCMS5A was very similar to that in NJCMS5B (Figs. 2 and 3). The level of DNA methylation of the whole soybean genome was just about 14%. Except for C-G nucleotide pairs, most methylation occurred in CHG site (Figs. 1a and 3). In the CG, CHG and CHH contexts, DNA methylation was most abundant in TE-rich and gene-poor expression region in this study (Fig. 3). So DNA methylation may be positively related with TE density and negatively related to the expression level of genes in the soybean genome, which agrees with an idea proposed previously [29]. However, more empirical evidence is required to confirm that this hypothesis could be applied to any mC site detected in this study.
Fig. 3.
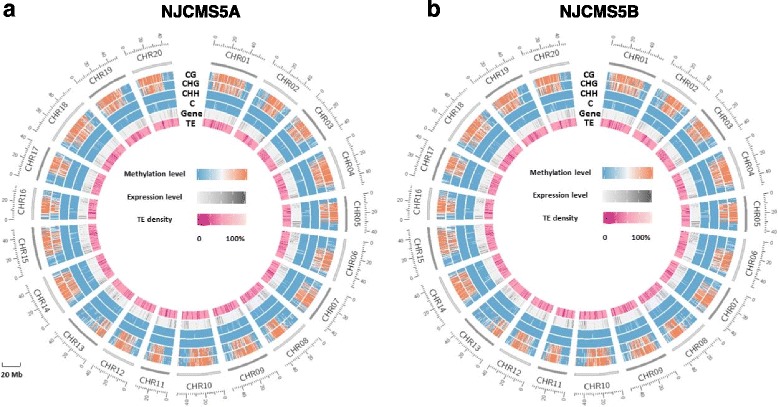
Circle plots of gene expression level, transposable element (TE) density and methylation level in the mC, mCG, mCHG and mCHH contexts of soybean (a) NJCMS5A and (b) NJCMS5B. TE indicated “TE density”; Gene indicated “gene expression level”; C, CG, CHH and CHG indicated “methylation level in mC, mCG, mCHG and mCHH, respectively”. Gene expression level was calculated by fragments per kilobase of exon model per million mapped fragments (RPKM). Data is plotted for 20 Mb windows in all soybean chromosomes (CHR01-CHR20). Gene expression level (from light colour to dark colour) indicates the level from 0 to 100%, and TE density (from dark colour to light colour) indicates the density from 0 to 100%
Effect of DNA methylation on gene expression
Considered that the higher methylation level may reduce gene expression in the promoter region, we performed a further analysis of gene expression in different functional region using RNA-Seq data. Genes were divided into five groups, according to their expression level (Additional file 3): 0–1 (silent gene), 1–3 (low expressed gene), 3–15 (moderately expressed gene), 15–60 (highly expressed gene) and > 60 (gene with highest expression). The results clearly showed the negative correlation between DNA methylation and gene expression level in the promoter (Fig. 1c, d and Additional file 4). Due to large amounts of methylated genes (49,911 of NJCMS5A, 48,576 of NJCMS5B) occurred in promoter region, gene expression level in promoter was obviously lower than that in gene body (Fig. 1c, d and Additional file 4). However, in the gene body, the expression levels of high and middle genes were much high, which indicated no negative or positive correlation between gene expression and DNA methylation (Fig. 1c, d and Additional file 4).
Identification of differentially methylated gene (DMG) between NJCMS5A and NJCMS5B
Differentially methylated region (DMR) is a significant sign of epigenetic variation, which may participate in regulating the DMGs to influence biological processes. By a genome-wide comparison of DNA methylation sequences between NJCMS5A and NJCMS5B, 3527 DMRs (Additional file 5) were identified by swDMR software (https://sourceforge.net/projects/swdmr/) with “FDR ≤ 0.05 and coverage changes ≥ 5” in this study. Then, DMGs were defined as genes overlapping with significant DMRs with at least 1 bp in the promoter and/or gene body. From 739 DMRs (Additional file 6) with genomic feature, we obtained 485 non-repeat DMGs including 281 hypo-methylated genes (57.9%) and 88 hyper-methylated genes (18.2%) in the NJCMS5A relative to NJCMS5B. In addition, 116 methylation-unstable genes (23.9%) that overlapped with different gene functional region (promoter, 5′UTR, exon, intron or 3′UTR) showed both hyper-methylation and hypo-methylation (Fig. 4a). In the gene functional region, there were more promoter-related DMGs (59.2%) than the exon-related (49.3%) or intron-related DMGs (42.9%) (Table 2 and Fig. 4b). In addition, we found the number of hypo-methylated DMGs was almost two to five times more than the number of hyper-methylated DMGs (Fig. 4c), which implied that DNA hypo-methylation often occurred as the epigenetic initial abnormality.
Fig. 4.
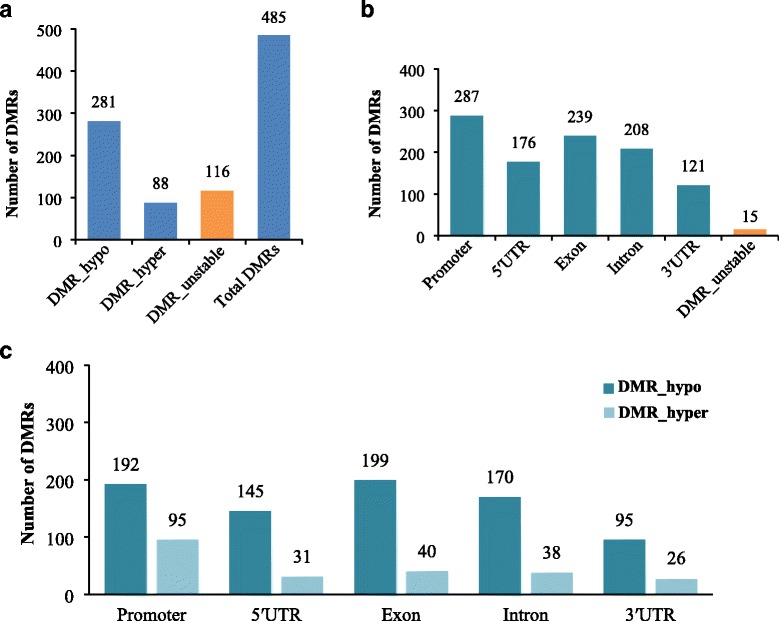
Distribution and proportion of differentially methylated genes (DMGs). a Total DMGs divided into hypo-DMGs and hyper-DMGs and unstable-DMGs (genes overlapping with both hyper- and hypo- DMGs); b Number of DMGs in gene functional region (promoter, 5′UTR, exon, intron and 3′UTR); c Number of hypo-DMGs and hyper-DMGs in gene functional region (promoter, 5′UTR, exon, intron and 3′UTR)
Table 2.
Distribution of differentially methylated genes (DMGs) in gene functional region
| Gene functional region | Promoter | 5′ UTR | Exon | Intron | 3′ UTR |
|---|---|---|---|---|---|
| DMG_hypo | 192 | 145 | 199 | 170 | 95 |
| DMG_hyper | 95 | 31 | 40 | 38 | 26 |
| Total DMG | 287 | 176 | 239 | 208 | 121 |
| DMG rate in genome (%) | 59.2% | 36.3% | 49.3% | 42.9% | 24.9% |
Validation of the whole-genome bisulfite sequencing (WGBS) data by bisulfite treatment
We randomly selected 25 DMRs (16 hypo-DMRs and 9 hyper-DMRs) in NJCMS5A to confirm the WGBS data by bisulfite treatment, including Glyma.04G206000 (OSBP), Glyma.19G144200 (bHLH DNA-binding), Glyma.16G081800 (lipase), Glyma.06G266900 (galacturonosyl transferase) and so on (Figs. 5 and 6). Many of DMGs showed the cytosines in the CG and CHG contexts were more frequently methylated than that in the CHH context (Fig. 5 and Additional file 7). The bisulfite sequencing results showed that 21 DMRs containing of 13 hypo-DMRs and 8 hyper-DMRs were consistent with the WGBS data, which indicated WGBS data were credible in the study (Additional files 7 and 8). In term of CG context, many DMGs were methylated highly, so we also made models of the methylated percentage at each site (Fig. 6), including Glyma.08G317600 (MYB domain protein), Glyma.10G248800 (methyltransferases), Glyma.U028100 (MYB domain protein) and Glyma.U013000 (AGAMOUS). Among them, the gene of Glyma.U013000 exhibited a higher methylation level in NJCMS5A compared NJCMS5B. But Glyma.08G317600, Glyma.10G248800 and Glyma.U028100 were identified to be hypo-methylation in NJCMS5A.
Fig. 5.
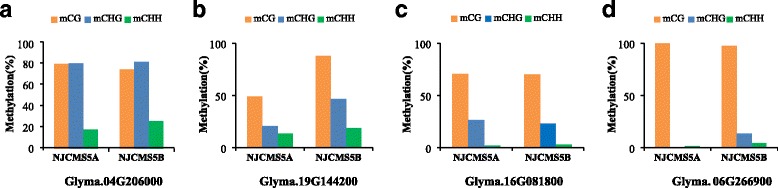
DNA methylation patterns of four differentially methylated genes (DMGs) validated by bisulfite treatment. a Glyma.04G206000 (oxysterol binding protein); b Glyma.19G144200 (basic helix-loop-helix DNA-binding protein); c Glyma.16G081800 (lipase); d Glyma.06G266900 (galacturonosyl transferase). The red, blue and green columns in the histograms refer to the collective methylation levels (in percentage) in CG, CHG and CHH contexts, respectively
Fig. 6.
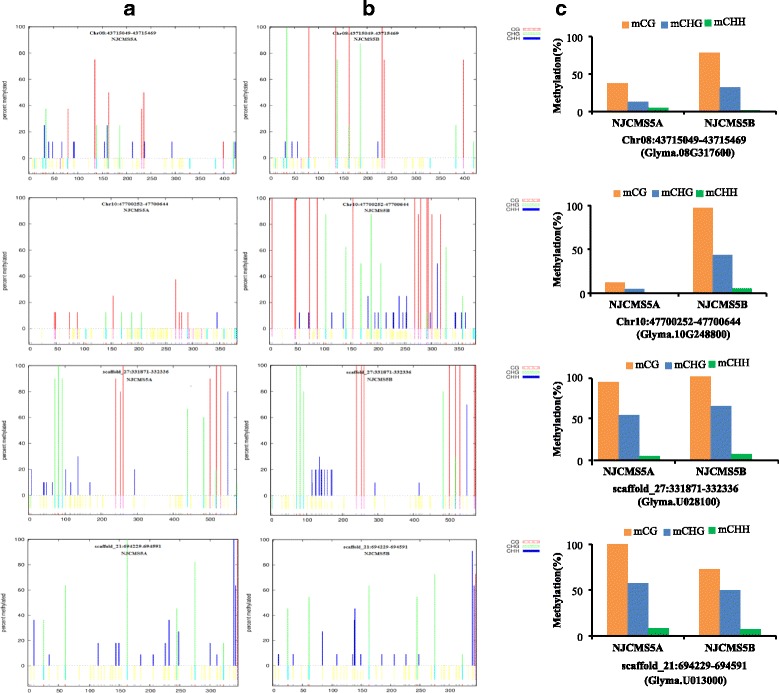
DNA methylation patterns of four typical differentially methylated regions (DMRs) with high methylation site. a Analysis of bisulfite treatment results of NJCMS5A; b Analysis of bisulfite treatment results of NJCMS5B. The colored bars above the x-axis show the methylated percent at each site. The short bars at the bottom of the graph show the position of the cytosines. The average level of methylation was calculated with 8–12 clones by bisulfite treatment and nested PCR. c Methylation patterns of DMRs, including hyper-methylated regions (Chr08:43,715,049–43,715,469, Chr10:47,700,252–47,700,644 and scaffold_27:331,871–332,336), and hypo-methylated region (scaffold_21:694,229–694,591)
Gene ontology (GO) annotation and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis
To describe property of genes and their products, WEGO (http://wego.genomics.org.cn) was used to functionally categorize the DMGs (Fig. 7). In total of 334 DMGs, comprising of 270 hypo-methylated and 139 hyper-methylated DMGs were annotated to 31 functional categories, including, 14 biological processes (BP), 9 cellular components (CC) and 8 molecular functions (MF) (Fig. 7). In the term of BP, we found that the hyper-methylated DMGs were mainly involved in catabolic process (GO: 0044238, GO: 0044237, GO: 0043170), cellular localization (GO: 0051641), macromolecule of localization (GO: 0033036) and transport (GO: 0006810) (Fig. 7). In the term of CC, the hyper-methylated DMGs were mostly participated in nucleoplasm part (GO: 0005654, GO: 0044451), sperm part (GO: 0036126, GO: 0097223, GO: 0097228) and organism membrane (GO: 0033644, GO: 0044218, GO: 0044279). And in the term of MF, the hyper-methylated DMGs were mostly involved in the process of transcription regulation (GO:0060089) and transporter (GO:0005215, GO:0022857) (Fig. 7). In addition, those hyper-methylation genes of NJCMS5A were made for GO enrichment analysis by using the GOseq R package. The result showed many of them appeared to be enriched in macromolecule modification, cellular metabolism, transcription regulation, protein phosphorylation and so on. These corresponding genes were taken into account for significantly down-regulated genes and further were listed (Table 3), including 86 genes for “BP” (36), 15 genes for “CC” (17) and 95 genes for “MF” (41).
Fig. 7.
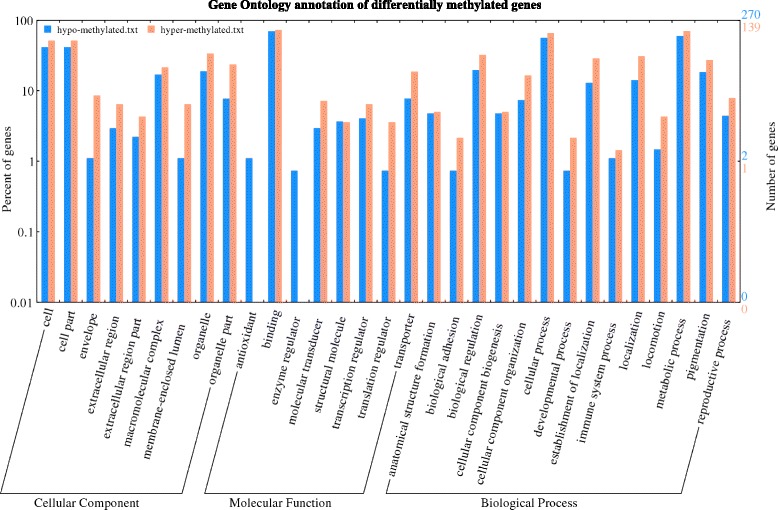
Gene Ontology (GO) annotation of differentially methylated genes (DMGs) between NJCMS5A and NJCMS5B. The blue and red column in the histogram referred to the hypo-methylation and hyper-methylation level (percentage and number), respectively
Table 3.
Significant Gene Ontology (GO) terms of hyper-methylated genes in NJCMS5A
| GO accession | Term type | Description | DMR _ item | BG _ item | P _ value |
|---|---|---|---|---|---|
| GO:0019058 | BP | viral infectious cycle | 6 | 364 | 0.0022802 |
| GO:0006011 | BP | UDP-glucose metabolic process | 1 | 1 | 0.0036546 |
| GO:0015977 | BP | carbon fixation | 2 | 25 | 0.0037648 |
| GO:0016042 | BP | lipid catabolic process | 3 | 109 | 0.0075620 |
| GO:0044403 | BP | symbiosis, encompassing mutualism through parasitism | 8 | 789 | 0.0086707 |
| GO:0071704 | BP | organic substance metabolic process | 76 | 16,860 | 0.0089794 |
| GO:0044419 | BP | interspecies interaction between organisms | 8 | 796 | 0.0091147 |
| GO:0006099 | BP | tricarboxylic acid cycle | 2 | 40 | 0.0094446 |
| GO:0000723 | BP | telomere maintenance | 5 | 350 | 0.0094633 |
| GO:0032200 | BP | telomere organization | 5 | 350 | 0.0094633 |
| GO:0060249 | BP | anatomical structure homeostasis | 5 | 350 | 0.0094633 |
| GO:0019067 | BP | viral assembly, maturation, egress, and release | 3 | 130 | 0.0121930 |
| GO:0009060 | BP | aerobic respiration | 2 | 46 | 0.0123550 |
| GO:0016032 | BP | viral process | 7 | 725 | 0.0175200 |
| GO:0044238 | BP | primary metabolic process | 72 | 16,245 | 0.0190260 |
| GO:0009225 | BP | nucleotide-sugar metabolic process | 1 | 6 | 0.0217300 |
| GO:0034219 | BP | carbohydrate transmembrane transport | 1 | 6 | 0.0217300 |
| GO:0071806 | BP | protein transmembrane transport | 2 | 66 | 0.0244210 |
| GO:0006367 | BP | transcription initiation from RNA polymerase II promoter | 2 | 67 | 0.0251130 |
| GO:0030150 | BP | protein import into mitochondrial matrix | 1 | 7 | 0.0253060 |
| GO:0044764 | BP | multi-organism cellular process | 7 | 785 | 0.0256590 |
| GO:0015074 | BP | DNA integration | 2 | 69 | 0.0265210 |
| GO:0044237 | BP | cellular metabolic process | 67 | 15,179 | 0.0286360 |
| GO:0072519 | BP | parasitism | 1 | 9 | 0.0324180 |
| GO:0018149 | BP | peptide cross-linking | 1 | 10 | 0.0359550 |
| GO:0006464 | BP | cellular protein modification process | 19 | 3357 | 0.0370220 |
| GO:0036211 | BP | protein modification process | 19 | 3357 | 0.0370220 |
| GO:0006468 | BP | protein phosphorylation | 15 | 2522 | 0.0427720 |
| GO:0019068 | BP | virion assembly | 2 | 90 | 0.0430940 |
| GO:0043170 | BP | macromolecule metabolic process | 57 | 12,853 | 0.0449750 |
| GO:0043412 | BP | macromolecule modification | 20 | 3665 | 0.0451580 |
| GO:0032940 | BP | secretion by cell | 3 | 221 | 0.0477090 |
| GO:0046903 | BP | secretion | 3 | 221 | 0.0477090 |
| GO:0006807 | BP | nitrogen compound metabolic process | 41 | 8794 | 0.0487850 |
| GO:0000079 | BP | regulation of cyclin-dependent protein serine/threonine kinase activity | 1 | 14 | 0.0499750 |
| GO:0071900 | BP | regulation of protein serine/threonine kinase activity | 1 | 14 | 0.0499750 |
| GO:0016592 | CC | mediator complex | 4 | 183 | 0.0046636 |
| GO:0036126 | CC | sperm flagellum | 1 | 2 | 0.0072960 |
| GO:0097223 | CC | sperm part | 1 | 2 | 0.0072960 |
| GO:0097228 | CC | sperm principal piece | 1 | 2 | 0.0072960 |
| GO:0005654 | CC | nucleoplasm | 5 | 384 | 0.0136830 |
| GO:0044451 | CC | nucleoplasm part | 5 | 384 | 0.0136830 |
| GO:0071203 | CC | WASH complex | 2 | 61 | 0.0210820 |
| GO:0033644 | CC | host cell membrane | 2 | 67 | 0.0251130 |
| GO:0044218 | CC | other organism cell membrane | 2 | 67 | 0.0251130 |
| GO:0044279 | CC | other organism membrane | 2 | 67 | 0.0251130 |
| GO:0044441 | CC | cilium part | 1 | 7 | 0.0253060 |
| GO:0036128 | CC | catSper complex | 1 | 9 | 0.0324180 |
| GO:0030658 | CC | transport vesicle membrane | 2 | 87 | 0.0405380 |
| GO:0019031 | CC | viral envelope | 4 | 354 | 0.0413760 |
| GO:0036338 | CC | viral membrane | 4 | 354 | 0.0413760 |
| GO:0005891 | CC | voltage-gated calcium channel complex | 1 | 13 | 0.0464890 |
| GO:0034704 | CC | calcium channel complex | 1 | 13 | 0.0464890 |
| GO:0004675 | MF | transmembrane receptor protein serine/threonine kinase activity | 2 | 18 | 0.0019525 |
| GO:0008964 | MF | phosphoenolpyruvate carboxylase activity | 2 | 20 | 0.0024130 |
| GO:0008234 | MF | cysteine-type peptidase activity | 5 | 273 | 0.0033820 |
| GO:0019199 | MF | transmembrane receptor protein kinase activity | 2 | 25 | 0.0037648 |
| GO:0001076 | MF | RNA polymerase II transcription factor binding transcription factor activity | 4 | 180 | 0.0043995 |
| GO:0001104 | MF | RNA polymerase II transcription cofactor activity | 4 | 180 | 0.0043995 |
| GO:0017111 | MF | nucleoside-triphosphatase activity | 17 | 2371 | 0.0059866 |
| GO:0000988 | MF | protein binding transcription factor activity | 5 | 320 | 0.0065718 |
| GO:0004611 | MF | phosphoenolpyruvate carboxykinase activity | 2 | 34 | 0.0068906 |
| GO:0016462 | MF | pyrophosphatase activity | 17 | 2410 | 0.0070057 |
| GO:0004197 | MF | cysteine-type endopeptidase activity | 4 | 221 | 0.0089745 |
| GO:0016818 | MF | hydrolase activity, acting on acid anhydrides, in phosphorus-containing anhydrides | 17 | 2498 | 0.0098272 |
| GO:0000989 | MF | transcription factor binding transcription factor activity | 4 | 228 | 0.0099793 |
| GO:0003712 | MF | transcription cofactor activity | 4 | 228 | 0.0099793 |
| GO:0016787 | MF | hydrolase activity | 38 | 7372 | 0.0141010 |
| GO:0016817 | MF | hydrolase activity, acting on acid anhydrides | 17 | 2626 | 0.0154860 |
| GO:0005516 | MF | calmodulin binding | 2 | 56 | 0.0179520 |
| GO:0000149 | MF | SNARE binding | 1 | 5 | 0.0181410 |
| GO:0019905 | MF | syntaxin binding | 1 | 5 | 0.0181410 |
| GO:1,901,363 | MF | heterocyclic compound binding | 59 | 12,823 | 0.0194820 |
| GO:0097159 | MF | organic cyclic compound binding | 59 | 12,831 | 0.0197370 |
| GO:0004386 | MF | helicase activity | 9 | 1116 | 0.0217290 |
| GO:0004308 | MF | exo-alpha-sialidase activity | 1 | 6 | 0.0217300 |
| GO:0004683 | MF | calmodulin-dependent protein kinase activity | 1 | 6 | 0.0217300 |
| GO:0016997 | MF | alpha-sialidase activity | 1 | 6 | 0.0217300 |
| GO:0034062 | MF | RNA polymerase activity | 5 | 448 | 0.0247360 |
| GO:0070403 | MF | NAD+ binding | 1 | 7 | 0.0253060 |
| GO:0016779 | MF | nucleotidyltransferase activity | 7 | 785 | 0.0256590 |
| GO:0003676 | MF | nucleic acid binding | 33 | 6489 | 0.0271790 |
| GO:0003852 | MF | 2-isopropylmalate synthase activity | 1 | 8 | 0.0288680 |
| GO:0008083 | MF | growth factor activity | 2 | 73 | 0.0294300 |
| GO:0003899 | MF | DNA-directed RNA polymerase activity | 4 | 319 | 0.0299720 |
| GO:0003677 | MF | DNA binding | 23 | 4224 | 0.0332160 |
| GO:0016298 | MF | lipase activity | 3 | 197 | 0.0359010 |
| GO:0004325 | MF | ferrochelatase activity | 1 | 10 | 0.0359550 |
| GO:0038023 | MF | signaling receptor activity | 4 | 347 | 0.0389240 |
| GO:0047750 | MF | cholestenol delta-isomerase activity | 1 | 11 | 0.0394790 |
| GO:0003678 | MF | DNA helicase activity | 6 | 693 | 0.0423700 |
| GO:0016863 | MF | intramolecular oxidoreductase activity, transposing C = C bonds | 1 | 12 | 0.0429910 |
| GO:0046915 | MF | transition metal ion transmembrane transporter activity | 3 | 221 | 0.0477090 |
| GO:0046789 | MF | host cell surface receptor binding | 1 | 14 | 0.0499750 |
BP biological process; CC cellular component; MF molecular functional; DMR differentially methylated region; BG: background
Among the DMGs, 51 DMGs (Additional file 9) were predicted to be enriched in 35 biochemical metabolic pathways in the KEGG database (http://www.genome.jp/kegg/), including plant hormone signal transduction (ko04075), glycolysis / gluconeogenesis (ko00010), regulation of actin cytoskeleton (ko04810), RNA degradation (ko03018), cysteine and methionine metabolism (ko00270) and oxidative phosphorylation (ko00190). Based on the GO and KEGG pathway analysis, 8 genes were determined to be potentially related to soybean CMS (Table 4). For example, anther wall tapetum development gene Glyma.U015500, ATPase activity genes Glyma.16G195100 and Glyma.06G248800, proteolysis gene Glyma.U045200, regulation of transcription genes Glyma.08G317600, Glyma.U028100 and Glyma.U040000 and mitochondrion structural gene Glyma.14G212600.
Table 4.
Identified eight genes potentially related to cytoplasmic male sterility (CMS)
| NO. | Gene ID | DMR region | Methylation status (NJCMS5A vs. NJCMS5B) |
Description |
|---|---|---|---|---|
| 1 | Glyma.U015500 | promoter | Hypo | bHLH DNA-binding super family protein |
| 2 | Glyma.16G195100 | gene body | Hypo | mitochondrial mRNA modification |
| 3 | Glyma.06G248800 | gene body | Hypo | ABC-2 type transporter family protein |
| 4 | Glyma.U045200 | gene body promoter |
Hypo | cysteine proteinases super family protein |
| 5 | Glyma.08G317600 | promoter | Hypo | MYB domain protein 97 |
| 6 | Glyma.U028100 | promoter | Hypo | MYB domain protein 98 |
| 7 | Glyma.U040000 | promoter | Hyper | AP2/B3-like transcriptional factor family protein |
| 8 | Glyma.14G212600 | gene body promoter |
Hyper | PPR super family protein |
Hyper DNA methylation level in NJCMS5A was higher than that in NJCMS5B; Hypo DNA methylation level in NJCMS5A was lower than that in NJCMS5B
Validation of target DMGs by quantitative real-time PCR (qRT-PCR)
We also tried to conduct qRT-PCR to validate the relationship between DNA methylation and gene expression with 9 DMGs. As a result, 8 of 9 DMGs showed a negative correlation between DNA methylation and gene expression (Fig. 8). When the NJCMS5A methylation level of a DMG was significantly higher than that in NJCMS5B (P < 0.001) (Fig. 8b), the expression of the gene showed down-regulated in NJCMS5A, including Glyma.14G212600 (PPR protein), Glyma.U029400 (adenosine kinase) and Glyma.U040000 (AP2/B3 transcriptional factor protein). In addition, when the methylation level of a DMG in NJCMS5A was lower than that in NJCMS5B, the expression of the gene showed up-regulated in NJCMS5A, such as Glyma.06G248800 (ABC transporter protein), Glyma.U045200 (cysteine proteinases protein), Glyma.06G266900 (galacturonosyl transferase), Glyma.08G305500 (fimbrin-like protein) and Glyma.16G195100 (mitochondrial mRNA modification). Only gene Glyma.U013000 (AGAMOUS) had a high DNA methylation level and showed a high expression in NJCMS5A, which may be related to its function of inhibiting the down-stream gene expression, and it would be verified through the further research.
Fig. 8.
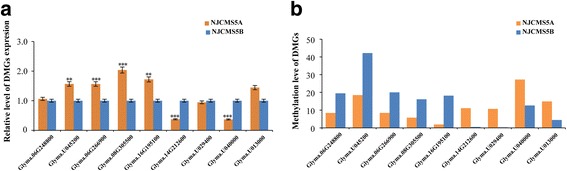
Expression level of nine DMGs validated by qRT-PCR. a qRT-PCR analysis of nine DMGs in NJCMS5A and NJCMS5B. X-axis represented gene name, the red column represented NJCMS5A, and blue column represented CK (NJCMS5B); Y-axis represented the relative level of gene expression. All qRT-PCR reactions were performed with three biological replicates; b DNA methylation level of nine DMGs in NJCMS5A and NJCMS5B
Discussion
Although genome-wide DNA methylation map has been reported in many organism [15, 21, 22], DNA methylome of soybean CMS has few related studies. In this study, we apply DNA methylation sequencing on the soybean CMS line NJCMS5A and its maintainer NJCMS5B for the first time. And the methylation profile of flower bud was in line with the previously reported methylation profile of soybean root, steam, cotyledon and leaf [14]. The genomic DNA methylation data were obtained by whole-genome bisulfite sequencing from NJCMS5A and NJCMS5B (Table 1 and Fig. 1a), and it revealed that DNA methylation often occurred not only in CG context but also in non-CG (CHG and CHH) context throughout all chromosomes or genome functional region of soybean (Fig. 2), which is consistent with the DNA methylation level reported in other plants [14, 15, 23, 29].
DNA methylation is treated as an important epigenetic process that influences gene expression [9, 11]. In this study, the global methylation patterns of the two soybean lines are in agreement with previous observations [14, 29]. In addition, to further verify DNA methylation could silence gene expression in NJCMS5A, we select 9 DMRs to verify the negative correlation between DNA methylation and gene expression by qRT-PCR (Fig. 8). In addition, we identified 739 DMRs (Additional file 6) with genomic feature and 485 DMGs with 281 hypo-methylated genes (57.9%) and 88 hyper-methylated genes (18.2%) in the NJCMS5A relative to NJCMS5B (Fig. 4a). According to the gene region annotation, gene function, GO and KEGG pathway analysis, as well as previously reported studies of male sterility in plant, 8 key DMGs (Table 4) that may be related to soybean CMS were identified. And they were further discussed as follows.
DMGs involved in regulation of pollen development potentially related to CMS
Numerous studies have shown that the pollen wall plays an important part in protecting pollen development [32]. The innermost layer of the anther wall (called tapetum) directly wrapped the microspore mother cell and microspore, and provided nutrition for microspore development by secreting large amounts of carbohydrate and lipid [33]. Therefore, tapetum played an important role in the process of pollen development, and the tapetum mutation may lead to pollen abortion [34–37]. The dysfunctional Tapertum1 gene (DYT1), encoding a basic helix-loop-helix (bHLH) TF, participated in tapetum development and protection [38]. In Arabidopsis Dyt1 mutant, because of the thicker callose wall, the pollen mother cell was unable to produce spores, leading to pollen abortion. In addition, the transcriptome analysis of Arabidopsis wild-type and dyt1 mutant showed dyt1 gene was in upstream of at least 22 coding bHLH TF genes, participating in regulating many specific metabolic pathways [39]. In this study, a DMG Glyma.U015500 was found to encode a bHLH TF, which was a homolog of DYT1 in Arabidopsis. We supposed that Glyma.U015500 may participate in the formation of pollen wall and may have an impact on pollen development in soybean.
The pollen coat is the outermost layer of the anther and it is mainly composed of sterol esters to protect the pollen [40]. The ATP binding cassette (ABC) transporter is in charge of transporting the coat materials for male gametophyte development [41]. In this study, a DMG Glyma.06G248800 was supposed to encode an ABC-2 type transporter family protein, which was a homolog of ABCG9 of Arabidopsis. ABCG9 mainly participates in formation of the pollen coat with a high expression level in tapetum. However, mutations of abcg9–1 and abcg31–1 produced a lot of distorted and shrunken pollen grains, without mature pollen released [42]. So we considered Glyma.06G248800 may contribute to the accumulation of sterol esters on the surface of the soybean pollen.
Together, abnormal methylation levels of Glyma.U015500 and Glyma.06G248800 in NJCMS5A may influence the development of the pollen wall, which may be a factor in the soybean CMS.
DMGs involved in carbohydrate and energy metabolism potentially related to CMS
In the early stage of development, pollen is surrounded by tapetum. And in the late stage, tapetum begins to degrade [43]. Tapetum programmed cell death (PCD) plays a vital role in developing pollen. Cysteine Protease participates in plant PCD as the most common hydrolytic enzyme [44]. In this study, a DMG Glyma.U045200 was predicted to encode a homolog of Cysteine Protease CEP1 of Arabidopsis. CEP1 participates in tapetum PCD and regulates the expression of FLOWERING LOCUS T (FT) in Arabidopsis. Excessive expression of CEP1 led to tapetum PCD degeneration in advance and pollen abortion [45], so proper CEP1 expression contributed to tapetum PCD for releasing the fertile pollen. Glyma.U045200 showed a lower methylation level in NJCMS5A compared with NJCMS5B, which may promote the expression of gene Glyma.U045200 in NJCMS5A, and eventually affected normal tapetum PCD, leading to pollen abortion.
Transcription factor (TF) potentially related to CMS
In plant, TF plays a critical role in the regulation of plant metabolism and development by modifying the expression of their target genes [46]. For example, R2R3-type MYB gene could control many aspects of plant in secondary metabolism, as well as regulation of plant cell fate and identity [47]. MYB98, a member of the R2R3-MYB family TF, mainly regulates of transcriptional events in the synergid cell [48]. So it is necessary for pollen tube guidance and successful fertilization in flowering plant [49]. In the mature pollen of Arabidopsis, the expression of MYB97 was much higher than normal, and a triple mutant of myb97–1, myb101–2 and myb120–3 caused overgrowth of the pollen tube into the embryo sac and disrupted sperm cell discharge, leading the failure of fertilization [50]. In this study, the DMG Glyma.U028100 was predicted to encode a homolog of MYB98, and Glyma.08G317600 was also predicted to encode a MYB TF protein. The promoter region of both DMGs showed a lower methylation levels in NJCMA5A compared with NJCMS5B, which implied that the abnormal expression of these two genes may disorder the development of floral organ, resulting in male sterility in NJCMS5A.
The MADS-box TF family is widespread in plant and animal, which involves in diverse and important biological functions [51], especially in floral organ process. Recently, soybean GmMADS28 which encodes a MADS-box protein has been reported [52]. The genetically modified anther of 35S:GmMADS28 was not dehiscent and failed to release pollen, which largely led to plant male sterility [52]. In this study, the DMG Glyma.U040000 was annotated as an AP2/B3-like TF family protein contained a B3 domain. In Arabidopsis, VRN1, as a member of the REM subfamily, contained two B3 DNA-binding domains. It could interact with AGAMOUS-like 20 (AGL20), flowering locus T (FT) and MADS-box protein flowering locus C (FLC), as well as with other regulators to restrain flowering [53]. The promoter region of Glyma.U040000 with abnormal methylation level in NJCMS5A was supposed to negatively regulate gene expression, which may indirectly influence flower structure and flowering mechanism, leading to male sterility in NJCMS5A.
DMGs encoding mitochondrial protein potentially related to CMS
Recently, it has also been proposed that the fertility of CMS plant could be restored by corresponding nuclear fertility restoration (RF) gene. And RF genes that were encoded by pentatricopeptide repeat (PPR) protein have been isolated from rice, carrot, and pepper [54–56]. RNA processing factor 1 gene (RPF1) belongs to a subgroup of PPR protein, which could guide RF gene product to restore CMS. In this study, the DMGs Glyma.14G212600 and Glyma.16G195100 were placed in the mitochondria. Glyma.16G195100 was homologous with RPF1, which was supposed to play a role in fertility recovery in CMS plant [57]. In addition, the promoter of Glyma.14G212600 was hyper-methylated in NJCMS5A compared with NJCMS5B, implying the expression of Glyma.14G212600 may be suppressed. So, Glyma.14G212600 and Glyma.16G195100 were predicted to encode two PPR proteins, which may be associated with editing mitochondrial gene to impact the expression of the CMS-related genes in the mitochondria, resulting in CMS in NJCMS5A.
Conclusion
In the study, the genome-wide methylation profiles of the soybean CMS line NJCMS5A and its maintainer NJCMS5B were obtained from whole-genome bisulfite sequencing. As a result, 739 DMRs and 485 DMGs, including 353 high-credible methylated genes, 56 methylated genes coding unknown protein and 76 novel methylated genes with no known function were identified. In addition, those valid methylated genes were identified as 281 hypo-methylated genes and 88 hyper-methylated genes in NJCMS5A relative to NJCMS5B. According to the gene region annotations, gene function, GO and KEGG pathway analysis, as well as the previous articles reported studies of male sterility in the plant, 8 key DMGs that may be related to soybean CMS were discussed, which mainly involved in participating in pollen development, encoding TF and the mitochondrial genome. This study provides DNA methylome of soybean CMS line and its maintainer line for the first time, which will contribute to further understanding the methylation mechanism in soybean CMS.
Methods
Plant materials
The soybean [Glycine max (L.) Merr.] CMS line NJCMS5A was developed through consecutive backcross procedures with NJCMS1A [26–28] as donor parent and Wandou28 (designated as NJCMS5B afterwards) as the recurrent parent. Both NJCMS5A and NJCMS5B were planted in the summer of 2014 at the Jiangpu Experimental Station, National Center for Soybean Improvement, Nanjing Agricultural University, Nanjing, Jiangsu, China. The male-sterile plant was identified through three kinds of methods including the dehiscence of anthers, germination rate of pollen, and performance of plant at maturity. Because it is very difficult to judge the precise development stage of pollen from the appearance of the flower buds in soybean, so during flowering period, the flower buds in different sizes were collected and pooled from NJCMS5A and NJCMS5B plant respectively, then immediately frozen in liquid nitrogen and stored at −80 °C for further use.
Total DNA extraction and DNA library construction
Genomic DNA was extracted from the flower bud of NJCMS5A and NJCMS5B using DNeasy Plant Mini Kit (Qiagen Valencia, CA, USA), respectively, according to the manufacturer’s instruction. First, the purity of DNA sample was detected using the NanoPhotometer Spectrophotometer (Implen, CA, USA), and the concentration of DNA sample was measured using UV-Vis Spectrophotometer (Thermo Scientific, MA, USA). Subsequently, DNA sample was randomly sonicated to 200–300 bp using Covaris S220 (Thermo Scientific, MA, USA). After end repair and adenylation, the sonicated DNA fragments were ligated to cytosine-methylated barcodes according to manufacturer’s instruction. DNA fragment was treated twice with bisulfite using EZ DNA Methylation-Gold Kit (Zymo Research, CA, USA). After that, DNA library was constructed by PCR amplification. DNA library concentration was quantified by Qubit2.0 Fluorometer (Life Technologies, CA, USA), and the size of the insert fragment was tested by Biological Analyzer Agilent 2100 (Agilent, CA, USA). Finally, the qualified DNA library was sequenced on an Illumina Hiseq 2500 platform with paired-end reads by Novogene Bioinformatics Technology Co.Ltd. (Beijing, China).
Bioinformatics analysis of DNA methylation sequencing data
After sequencing, the raw reads were filtered to remove adapters, Ns and low quality reads. The remaining reads called clean reads were stored in FASTQ format. Clean reads were aligned to the reference genome of soybean (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Gmax) using Bismark software (version 0.12.5) with the default parameters [58]. The genetic structure annotation file was obtained from the public fit site of Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Gmax). Annotations of transposable element (TE) were downloaded from the SoyTEdb database (http://www.soybase.org/soytedb/) and TEs were plotted in 20 kb windows along the chromosome. Finally, only reads that were uniquely mapped to soybean reference genome were used for further analysis. mC percent (%) was calculated as the percentage of methylated C sites in the whole genome. mCG percent (%), mCHG percent (%) and mCHH percent (%) were calculated as the percentage of methylated C site in the C, CG and CHH contexts, respectively.
To identify the methylation site, we modeled the sum s + i,j of methylated counts as a binomial (Bin) random variable with methylation rate r i,j, s + i,j ~ Bin ( s + i,j + s − i,j, ,r i,j ). Then we employed a sliding-window approach, which is conceptually similar to the approaches used for bulk BS-Seq (http://www.bioconductor.org/packages/2.13/bioc/html/bsseq.html). With window size w = 3000 bp, step size = 600 bp [59] and sequencing depth ≥ 5, q-value < 0.05, the sum of methylated and unmethylated counts in every window were calculated. Else, the methylation level (ML) for each C site was defined as ML (C) = reads (mC)/ reads (mC + umC). The calculated ML was further corrected using with non-conversion rate r, ML (corrected) = ( ML – r )/(1 - r ) [60]. Density was calculated by sites (mC)/ sites (mC + umC). ML and density were both used for the analysis of the reads distribution in soybean chromosome and in the different genome functional region (promoter, 5′UTR, exon, intron, and 3′UTR) under three different contexts (CG, CHG and CHH).
Differentially methylated regions (DMRs) between NJCMS5A and NJCMS5B were identified using the swDMR software (https://sourceforge.net/projects/swdmr/) with a sliding-window approach [61]. The window is set to 1000 bp and step length is 100 bp. Fisher test is used to detect the DMRs with “FDR ≤ 0.05 and coverage changes ≥ 5”. Then the differentially methylated genes (DMGs) were collected from DMRs that overlapped gene functional region (promoter, 5′UTR, exon, intron and 3′UTR) with at least 1 bp. Next, Gene Ontology (GO) enrichment analysis (http://www.geneontology.org/) was performed for all the identified DMGs using the GOseq R package (P-value < 0.05) [62]. Finally, the hypergeometric test statistical method was used for the metabolic pathway analysis of all identified DMGs in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (http://www.genome.jp/kegg/) using KOBAS software [63].
Validation of the sequencing data by bisulfite treatment
Bisulfite genomic sequencing is regarded as a gold-standard for detection of DNA methylation, because it provides a qualitative and quantitative method to apply to a limit number of loci [64]. Twenty-five DMRs were selected randomly to validate the reliability of the sequencing data using a nest PCR (nPCR). All the primers (Additional file 10) were designed using the website of Kismeth (http://katahdin.mssm.edu/kismeth/revpage.pl), and synthesized commercially (Invitrogen, Shanghai, China). Briefly, 1 μg of genomic DNA was treated by bisulfite according to the protocol of the EZ DNA Methylation-Gold Kit (Zymo Research, CA, USA), and used as the template for the following nPCR amplification. Then, the bisulfite treatment PCR (BS-PCR) was performed in a 50 μl reaction mixture, containing 25 μl premix EX Taq DNA polymerase (TaKaRa, Osaka, Japan), 25 μg bisulfite-treated DNA and 0.2 μM of each pair of primers with a nest PCR. Next, products were purified using Gel Extraction Kit (Axygen, CA, USA), and cloned into the pMD19-T Simple Vector (TaKaRa, Osaka, Japan). Each DMR amplified with 10–14 clones was sequenced by Invitrogen Trading Shanghai Co.Ltd. (Shanghai, China), and the sequencing results were spliced and edited by BioEdit and vector NTI8 software, then analyzed in the Kismeth website.
Validation of candidate DMGs by quantitative real-time PCR (qRT-PCR)
qRT-PCR was used to verify the methylated gene expression. All the primers (Additional file 11) were designed by Primer-BLAST, and synthesized commercially (Invitrogen, Shanghai, China). Firstly, 1 μg of total RNA was reverse-transcribed by reverse transcriptase according to the protocol of iScript cDNA Synthesis Kit (Bio-Rad, CA, USA), and used as the template for the following qRT-PCR amplification. Then, qRT-PCR was performed on the Bio-Rad CFX96 Touch q-PCR System (Bio-Rad, CA, USA) with iTaq Universal SYBR Green Super mix (Bio-Rad, CA, USA). Each reaction was replicated three times, and β-actin gene was used as the internal control gene. The relative level of gene expression was evaluated by the 2–ΔΔCt method, NJCMS5B served as the control. The relative level of gene expression greater than 1 was regarded as up-regulated and less than 1 was regarded as down-regulated. Student’s t-test was adopted to test the difference between the relative level of gene expression of NJCMS5A and the control NJCMS5B. The means of 2–ΔΔCt were considered significantly different at P < 0.05.
Differential expression analysis of mRNA sequencing data
All the mRNA-seq analysis was based on the clean data with high quality. The expression quantity of each gene (fragments per kilobase of exon model per million mapped fragments, FPKM) was estimated by Cuffdiff software [65]. “FDR (False Discovery Rate) ≤ 0.05 [66, 67] and |Log2FC (Fold Change)| ≥ 1” were used as the threshold for judging the significant of gene expression difference.
Additional files
Data analysis of soybean genome-wide methylation sequencing. (PDF 207 kb)
Density distribution of genome-wide methylation in soybean chromosome. (PDF 1510 kb)
Gene expression level of transcriptome data. (XLS 8687 kb)
Graph of gene expression level in gene functional region. (PDF 146 kb)
Number of differentially methylated regions (DMRs) between NJCMS5A and NJCMS5B. (XLS 695 kb)
Number of differentially methylated genes (DMGs) between NJCMS5A and NJCMS5B. (XLS 165 kb)
Result of target DMRs in the CG, CHG and CHH context. (DOCX 22 kb)
Validation of sequencing data by bisulfite treatment. (DOCX 16 kb)
DMGs predicted to be enriched in KEGG pathways. (DOCX 18 kb)
Primer pairs used for bisulfite treatment. (DOCX 21 kb)
Primer pairs used for quantitative real-time PCR (qRT-PCR). (DOCX 17 kb)
Acknowledgements
We would thank Novogene Bioinformatics Technology Co.Ltd. (Beijing, China) for conducting the whole-genome methylation sequencing.
Funding
This work was supported by the National Key R&D Program of China (2016YFD0101500, 2016YFD0101504), the National Hightech R&D Program of China (2011AA10A105), and the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT13073).
Availability of data and materials
All whole-genome bisulfite sequencing raw data were deposited in the National Center for Biotechnology Information (NCBI) under the accession number GSE101652 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101652). The transcriptome sequencing data were unpublished.
Abbreviations
- bHLH
basic helix-loop-helix
- BP
biological process
- CC
cellular component
- CMS
cytoplasmic male sterility
- DMG
differentially methylated gene
- DMR
differentially methylated region
- DNMT
DNA methyltransferase
- DYT1
dysfunctional tapetum 1
- GO
Gene Ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- mC
methylated cytosine
- MF
molecular function
- PCD
programmed cell death
- PPR
pentatricopeptide repeat-containing
- qRT-PCR
quantitative real-time PCR
- RF
fertility restoration
- RPF1
RNA processing factor 1
- TE
transposable element
- TF
transcription factor
- WGBS
whole-genome bisulfite sequencing
Authors’ contributions
SPY and YWL conceived and designed the experiments. YWL performed the experiments and analyzed the data. YWL, XLD, XW, TTH, HZ, LSY, TLW and LFC contributed reagents/materials/analysis tools. YWL and SPY wrote the paper. SPY, JYG and YWL revised the paper. All authors read and approved the final manuscript.
Authors’ information
All authors came from Soybean Research Institute, National Center for Soybean Improvement, Key Laboratory of Biology and Genetic Improvement of Soybean (General, Ministry of Agriculture), State Key Laboratory of Crop Genetics and Germplasm Enhancement, Jiangsu Collaborative Innovation Center for Modern Crop Production, Nanjing Agricultural University, Nanjing 210095, China.
Ethics approval and consent to participate
The plant materials came from Soybean Research Institute, National Center for Soybean Improvement, Key Laboratory of Biology and Genetic Improvement of Soybean (General, Ministry of Agriculture), Nanjing Agricultural University, Nanjing 210095, China. The use of plant materials complied by the ethics of the People’s Republic of China.
Consent for publication
Not applicable.
Competing interests
The authors declared that they had no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-3962-5) contains supplementary material, which is available to authorized users.
Contributor Information
Yanwei Li, Email: wwyixiao2013@163.com.
Xianlong Ding, Email: xlding2012@163.com.
Xuan Wang, Email: sink0828@163.com.
Tingting He, Email: ht142857@163.com.
Hao Zhang, Email: haozhang1223@163.com.
Longshu Yang, Email: dashu6090@163.com.
Tanliu Wang, Email: tanliuWTL@126.com.
Linfeng Chen, Email: linfeng_chen2015@163.com.
Junyi Gai, Email: sri@njau.edu.cn.
Shouping Yang, Email: spyung@126.com.
References
- 1.Hanson MR. Plant mitochondrial mutations and male sterility. Annu Rev Genet. 1991;25:461–486. [DOI] [PubMed]
- 2.Chen L, Liu YG. Male sterility and fertility restoration in crops. Annu Rev Plant Biol. 2014;65:579–606. [DOI] [PubMed]
- 3.Carlsson J, Glimelius K. Cytoplasmic male-sterility and nuclear encoded fertility restoration. Plant Biol. 2010;1:469–91.
- 4.Hu J, Wang K, Huang WC, Liu G, Gao Y, Wang JM, et al. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell. 2012;24(1):109–122. [DOI] [PMC free article] [PubMed]
- 5.Li JJ, Han SH, Ding XL, He TT, Dai JY, Yang SP, et al. Comparative transcriptome analysis between the cytoplasmic male sterile line NJCMS1A and its maintainer NJCMS1B in soybean (Glycine max (L.) Merr.). PloS One. 2015;10(5):e0126771. [DOI] [PMC free article] [PubMed]
- 6.Li JJ, Ding XL, Han SH, He TT, Zhang H, Yang LS, et al. Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J Proteome. 2016;138:72–82. doi: 10.1016/j.jprot.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 7.Ding XL, Li JJ, Zhang H, He TT, Han SH, Li YW, et al. Identification of miRNAs and their targets by high-throughput sequencing and degradome analysis in cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B of soybean. BMC Genomics. 2016;17:24. [DOI] [PMC free article] [PubMed]
- 8.Chang SX, Wang YK, Lu JJ, Gai JY, Li JJ, Chu P, et al. The mitochondrial genome of soybean reveals complex genome structures and gene evolution at intercellular and phylogenetic levels. PLoS One. 2013;8(2):e56502. [DOI] [PMC free article] [PubMed]
- 9.Cao R, Wang LJ, Wang HB, Xia L, Zhang Y, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298(5595):1039–43. [DOI] [PubMed]
- 10.Ding JH, Shen JQ, Mao HL, Xie WB, Li XH, Zhang QF. RNA-directed DNA methylation is involved in regulating photoperiod-sensitive male sterility in rice. Mol Plant. 2012;5(6):1210–1216. [DOI] [PubMed]
- 11.Schmitz RJ, Sung S, Amasino RM. Histone arginine methylation is required for vernalization-induced epigenetic silencing of FLC in winter-annual Arabidopsis thaliana. Proc Natl Acad Sci USA. 2008;105(2):411–16. [DOI] [PMC free article] [PubMed]
- 12.Takuno S, Gaut BS. Gene body methylation is conserved between plant orthodoxs and is of evolutionary consequence. Proc Natl Acad Sci USA. 2013;110(5):1797–1802. [DOI] [PMC free article] [PubMed]
- 13.Finnegan EJ, Peacock WJ, Dennis ES. DNA methylation, a key regulator ofplant development and other processes. Curr Opin Genet Dev. 2000;10(2):217–223. [DOI] [PubMed]
- 14.Song QX, Lu X, Li QT, Chen H, Hu XY, Ma B, et al. Genome-wide analysis of DNA methylation in soybean. Mol Plant. 2013;6(6):1961–74. [DOI] [PubMed]
- 15.Zhang X, Yazaki J, Sundaresan A, Cokus S, Chan SW, Chen H, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126(6):1189–1201. [DOI] [PubMed]
- 16.Cao X, Aufsatz W, Zilberman D, Mette MF, Huang MS, et al. Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr Biol. 2003;13(24):2212–17. [DOI] [PubMed]
- 17.Henderson IR, Deleris A, Wong W, Zhong X, Chin HG, Horwitz GA, et al. The de novo cytosine methyltransferase DRM2 requires intact UBA domains and a catalytically mutated paralog DRM3 during RNA-directed DNA methylation in Arabidopsis thaliana. PLoS Genet. 2010;6(10):e1001182. [DOI] [PMC free article] [PubMed]
- 18.Dalakouras A, Moser M, Zwiebel M, Krczal G, Hell R, Wassenegger M. Ahairpin RNA construct residing in an intron efficiently triggered RNA-directed DNA methylation in tobacco. Plant J. 2009;60(5):840–51. [DOI] [PubMed]
- 19.Arteagavazquez M, Sidorenko L, Rabanal FA, Shrivistava R, Nobuta K, Green PJ, et al. RNA-mediated trans-communication can establish paramutation at the b1 locus in maize. Proc Natl Acad Sci USA. 2010;107(29):12986–991. [DOI] [PMC free article] [PubMed]
- 20.Wu T, Yuan T, Tsai SN, Wang C, Sun SM, Lam HM, et al. Mass spectrometry analysis of the variants of histone H3 and H4 of soybean and their post-translational modifications. BMC Plant Biol. 2009;9:98. [DOI] [PMC free article] [PubMed]
- 21.Yan HH, Kikuchi SJ, Neumann P, Zhang WL, Wu YF, Chen F, et al. Genome-wide mapping of cytosine methylation revealed dynamic DNA methylation patterns associated with genes and centromeres in rice. Plant J. 2010;63(3):353–65. [DOI] [PubMed]
- 22.Zhao YL, Yu SX, Ye WW, Wang HM, Wang JJ, Fang BX. Study on DNA cytosine methylation of cotton (Gossypium hirsutum L.) genome and its implication for salt tolerance. Agric Sci China. 2010;09(6):783–791.
- 23.Chen XJ, Hu JH, Zhang HY, Ding Y. DNA methylation changes in photoperiod-thermo-sensitive male sterile rice PA64S under two different conditions. Gene. 2014;537(1):143–8. [DOI] [PubMed]
- 24.Hu JH, Chen XJ, Zhang HY, Ding Y. Genome-wide analysis of DNA methylation in photoperiod- and thermo-sensitive male sterile rice Peiai 64S. BMC Genomics. 2015;16:102. [DOI] [PMC free article] [PubMed]
- 25.Lu YL, Liu Y, Wang J, Cao M, Rong TZ. Variation and patterns of DNA methylation in maize C-type CMS lines and their maintainers. J Plant Biochem Biotechnol. 2009;19(1):43–50.
- 26.Gai JY, Cui ZL, Ji DF, Ren ZJ, Ding DR. A report on the nuclear cytoplasmic male sterility from a cross between two soybean cultivars. Soy Genet Newsl. 1995;22:55–58.
- 27.Ding DR, Gai JY, Cui ZL, Yang SP, Qiu JX. Development and verification of the cytoplasmic-nuclear male sterile soybean line NJCMS1A and its maintainer NJCMS1B. Chin Sci Bull. 1999;44(2):191–192.
- 28.Gai JY, Ding DR, Cui ZL, Qiu JX. Development and performance of the cytoplasmic-nuclear male sterile line NJCMS1A of soybean. Sci Agric Sin. 2000;(1):41–47.
- 29.Schmitz RJ, He YP, Khan SM, Joshi T, Urich MA, et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013;23(10):1663–74. [DOI] [PMC free article] [PubMed]
- 30.Du J, Grant D, Tian Z, Nelson RT, Zhu L, Shoemaker RC, et al. SoyTEdb: a comprehensive database of transposable elements in the soybean genome. BMC Genomics. 2010;11:113. [DOI] [PMC free article] [PubMed]
- 31.Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463(7278):178–83. [DOI] [PubMed]
- 32.Xu J, Yang CY, Yuan Z, Zhang DS, Gondwe MY, Ding Z, et al. Aborted microspores regulatory network is required for postmeiotic male reproductive development in Arabidopsis thaliana. Plant Cell. 2010;22(1):91–107. [DOI] [PMC free article] [PubMed]
- 33.Scott RJ, Spielman M, Dickinson HG. Stamen structure and function. Plant Cell. 2004;16(Suppl):S46–60. [DOI] [PMC free article] [PubMed]
- 34.Wilson ZA, Morroll SM, Dawson J, Swarup R, Tighe PJ. The Arabidopsis male sterility1 ( MS1 ) gene is a transcriptional regulator of male gametogenesis, with homology to the PHD-finger family of transcription factors. Plant J. 2001;28(1):27–39. [DOI] [PubMed]
- 35.Kapoor S, Kobayashi A, Takatsuji H. Silencing of the tapetum-specific zinc finger gene TAZ1 causes premature degeneration of tapetum and pollen abortion in petunia. Plant Cell. 2002;14(10):2353–67. [DOI] [PMC free article] [PubMed]
- 36.Sorensen AM, Kröber S, Unte US, Huijser P, Dekker K, Saedler H. The Arabidopsis aborted microspores ( AMS ) gene encodes a MYC class transcription factor. Plant J. 2003;33(2):413–23. [DOI] [PubMed]
- 37.Higginson T, Li SF, Parish RW. AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana. Plant J. 2003;35(2):177–92. [DOI] [PubMed]
- 38.Zhang W, Sun Y, Timofejeva L, Chen C, Grossniklaus U, Ma H. Regulation of Arabidopsis Tapetum development and function by dysfunctional tapetum1 (DYT1) encoding a putative bHLH transcription factor. Development. 2006;133(16):3085–95. [DOI] [PubMed]
- 39.Feng BM, Lu DH, Ma X, Peng YB, Sun YJ, Ning G, et al. Regulation of the Arabidopsis anther transcriptome by DYT1 for pollen development. Plant J. 2012;72(4):612–24. [DOI] [PubMed]
- 40.Hsieh K, Huang AH. Tapetosomes in brassica tapetum accumulate endoplasmic reticulum-derived flavonoids and alkanes for delivery to the pollen surface. Plant Cell. 2007;19(2):582–96. [DOI] [PMC free article] [PubMed]
- 41.Choi H, Jin JY, Choi S, Hwang JU, Kim YY, Mi CS, et al. An ABCG/WBC-type ABC transporter is essential for transport of sporopollenin precursors for exine formation in developing pollen. Plant J. 2011;65(2):181–93. [DOI] [PubMed]
- 42.Choi H, Ohyama K, Kim YY, Jin JY, Lee SB, Yamaoka Y, et al. The role of ArabidopsisABCG9 and ABCG31 ATP binding cassette transporters in pollen fitness and the deposition of steryl glycosides on the pollen coat. Plant Cell. 2014;26(1):310–24. [DOI] [PMC free article] [PubMed]
- 43.Parish RW, Li SF. Death of a tapetum. A programme of developmental altruism. Plant Sci. 2010;178(2):73–89.
- 44.Solomon M, Belenghi B, Delledonne M, Menachem E, Levine A. The involvement of cysteine proteases and protease inhibitor genes in the regulation of programmed cell death in plants. Plant Cell. 1999;11(3):431–44. [DOI] [PMC free article] [PubMed]
- 45.Zhang DD, Liu D, Lv XM, Wang Y, Xun ZL, Liu ZX, et al. The cysteine protease CEP1, a key executor involved in tapetal programmed cell death, regulates pollen development in Arabidopsis. Plant Cell. 2014;26(7):2939–61. [DOI] [PMC free article] [PubMed]
- 46.Martin C. Transcription factors and the manipulation of plant traits. Curr Opin Biotechnol. 1996;7(7):130–38.
- 47.Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010;15(10):573–81. [DOI] [PubMed]
- 48.Eckardt NA. Elucidating the function of synergid cells: a regulatory role for MYB98. Plant Cell. 2007;19(8):2320–21.
- 49.Kasahara RD, Portereiko MF, Sandaklie-Nikolova L, Rabiger DS, Drews GN. MYB98 is required for pollen tube guidance and synergid cell differentiation in Arabidopsis. Plant Cell. 2005;17(11):2981–92. [DOI] [PMC free article] [PubMed]
- 50.Liang Y, Tan ZM, Zhu L, Niu QK, Zhou JJ, Li M, et al. MYB97, MYB101 and MYB120 function as male factors that control pollen tube-synergid interaction in Arabidopsis thaliana fertilization. PLoS Genet. 2013;9(11):e1003933. [DOI] [PMC free article] [PubMed]
- 51.Messenguy F, Dubois E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene. 2003;316:1–21. [DOI] [PubMed]
- 52.Huang F, Xu GL, Chi YJ, Liu HC, Xue Q, Zhao TJ, et al. A soybean MADS-box protein modulates floral organ numbers, petal identity and sterility. BMC Plant Biol. 2014;14:89. [DOI] [PMC free article] [PubMed]
- 53.Levy YY, Mesnage S, Mylne JS, Gendall AR, Dean C. Multiple roles of Arabidopsis VRN1 in vernalization and flowering time control. Science. 2002;297(5579):243–46. [DOI] [PubMed]
- 54.Huang WC, Hu J, Yu CC, Huang Q, Wan L, Wang LL, et al. Two non-allelic nuclear genes restore fertility in a gametophytic pattern and enhance abiotic stress tolerance in the hybrid rice plant. Theor Appl Genet. 2012;124(5):799–807. [DOI] [PubMed]
- 55.Yasumoto K, Terachi T, Yamagishi H. A novel Rf gene controlling fertility restoration of Ogura male sterility by RNA processing of orf138 found in Japanese wild radish and its STS markers. Genome. 2009;52(6):495–504. [DOI] [PubMed]
- 56.Jo YD, Kim YM, Park MN, Yoo JH, Park MK, Kim BD, et al. Development and evaluation of broadly applicable markers for restorer-of-fertility in pepper. Mol Breed. 2010;25(2):187–201.
- 57.Holzle A, Jonietz C, Torjek O, Altmann T, Binder S, Forner J. A restorer of fertility-like PPR gene is required for 5′-end processing of the nad4 mRNA in mitochondria of Arabidopsis thaliana. Plant J. 2011;65(5):737–44. [DOI] [PubMed]
- 58.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–72. [DOI] [PMC free article] [PubMed]
- 59.Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peet J, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11(8):817–20. [DOI] [PMC free article] [PubMed]
- 60.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. [DOI] [PMC free article] [PubMed]
- 61.Wang Z, Li XF, Jiang Y, Shao QZ, Liu Q, Chen BY, Huang DS. swDMR: A sliding window approach to identify differentially methylated regions based on whole genome bisulfite sequencing. PloS One. 2015;10(7):e0132866. [DOI] [PMC free article] [PubMed]
- 62.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. [DOI] [PMC free article] [PubMed]
- 63.Mao XZ, Cai T, Olyarchuk JG, Wei LP. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787–93. [DOI] [PubMed]
- 64.Li YY, Tollefsbol TO. DNA methylation detection: bisulfite genomic sequencing analysis. Methods Mol Biol. 2011;791:11–21. [DOI] [PMC free article] [PubMed]
- 65.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31(1):46–53. [DOI] [PMC free article] [PubMed]
- 66.Benjamini BY, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29:1165–88.
- 67.Benjamini BY, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data analysis of soybean genome-wide methylation sequencing. (PDF 207 kb)
Density distribution of genome-wide methylation in soybean chromosome. (PDF 1510 kb)
Gene expression level of transcriptome data. (XLS 8687 kb)
Graph of gene expression level in gene functional region. (PDF 146 kb)
Number of differentially methylated regions (DMRs) between NJCMS5A and NJCMS5B. (XLS 695 kb)
Number of differentially methylated genes (DMGs) between NJCMS5A and NJCMS5B. (XLS 165 kb)
Result of target DMRs in the CG, CHG and CHH context. (DOCX 22 kb)
Validation of sequencing data by bisulfite treatment. (DOCX 16 kb)
DMGs predicted to be enriched in KEGG pathways. (DOCX 18 kb)
Primer pairs used for bisulfite treatment. (DOCX 21 kb)
Primer pairs used for quantitative real-time PCR (qRT-PCR). (DOCX 17 kb)
Data Availability Statement
All whole-genome bisulfite sequencing raw data were deposited in the National Center for Biotechnology Information (NCBI) under the accession number GSE101652 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101652). The transcriptome sequencing data were unpublished.