

Abstract
Background
Selinexor (KPT-330) is an oral agent that has been shown to inhibit the nuclear exporter XPO1. Given the pressing need for novel therapies for triple-negative breast cancer (TNBC), we sought to determine the antitumor effects of selinexor in vitro and in vivo.
Methods
Twenty-six breast cancer cell lines of different breast cancer subtypes were treated with selinexor in vitro. Cell proliferation assays were used to measure the half-maximal inhibitory concentration (IC50) and to test the effects in combination with chemotherapy. In vivo efficacy was tested both as a single agent and in combination therapy in TNBC patient-derived xenografts (PDXs).
Results
Selinexor demonstrated growth inhibition in all 14 TNBC cell lines tested; TNBC cell lines were more sensitive to selinexor (median IC50 44 nM, range 11 to 550 nM) than were estrogen receptor (ER)-positive breast cancer cell lines (median IC50 > 1000 nM, range 40 to >1000 nM; P = 0.017). In multiple TNBC cell lines, selinexor was synergistic with paclitaxel, carboplatin, eribulin, and doxorubicin in vitro. Selinexor as a single agent reduced tumor growth in vivo in four of five different TNBC PDX models, with a median tumor growth inhibition ratio (T/C: treatment/control) of 42% (range 31 to 73%) and demonstrated greater antitumor efficacy in combination with paclitaxel or eribulin (average T/C ratios of 27% and 12%, respectively).
Conclusions
Collectively, these findings strongly suggest that selinexor is a promising therapeutic agent for TNBC as a single agent and in combination with standard chemotherapy.
Electronic supplementary material
The online version of this article (doi:10.1186/s13058-017-0878-6) contains supplementary material, which is available to authorized users.
Keywords: Breast cancer, Selinexor, XPO1, PDX, TNBC
Background
Normal cell homeostasis depends on the cell’s ability to compartmentalize proteins. During carcinogenesis, nuclear export is disrupted and as consequence multiple tumor-suppressor proteins that usually trigger apoptosis and cell cycle arrest in the nucleus are transported to the cytoplasm where they are subsequently targeted for degradation [1]. The nuclear exporter Exportin1 or CRM1 (XPO1) is one of at least seven exportins that mediate the transport of over 200 proteins, including several key regulators of the cell cycle (e.g., p53, STAT3, survivin, FoxO3a, BRCA1, and others) [2, 3]. XPO1 interacts with cargo proteins through a nuclear export sequence (NES), and it is dependent on the small GTPase protein Ran in its guanine triphosphate (GTP)-bound form, which binds to XPO1 together with export cargos [4].
Increased XPO1 expression has been linked to poor prognosis in multiple solid and hematologic malignancies [5–7], and therefore inhibition of XPO1 is being pursued as a promising target for cancer therapy. Selinexor (KPT-330) is an oral agent that has been shown to inhibit XPO1 [8] and is currently in clinical trials for treatment of hematologic and solid malignancies, with promising results.
Approximately 15% of all breast cancers are categorized as triple-negative breast cancer (TNBC) [9]. Given that there is no specific targeted therapy, systemic chemotherapy is usually the first line of treatment [10]. However, currently available chemotherapy regimens for advanced TNBC often fail to achieve disease control. Given the poor prognosis for patients with TNBC, there is a pressing need for novel therapies. XPO1 mediates nuclear export of several proteins that have been linked to TNBC [11], and there have been efforts to show breast cancer growth inhibition by several selective inhibitors of nuclear export [12]. We therefore sought to determine the antitumor effects of selinexor on breast cancer by testing multiple cell lines in vitro and in multiple patient-derived xenograft (PDX) models in vivo, with a particular focus on TNBC. From there, we sought to determine the best possible combination with cytotoxic chemotherapies.
Methods
Cell lines and culture conditions
Twenty-six breast cancer cell lines were obtained from the American Tissue Culture Collection (ATCC, Manassas, VA, USA): BT-20, BT-474, BT-483, BT-549, CAMA-1, HCC-38, HCC-1143, HCC-1395, HCC-1419, HCC-1569, HCC-1806, HCC-1937, HCC-1954, HCC-70, MCF-7, MDA-MB-134vi, MDA-MB-157, MDA-MB-175, MDA-MB-231, MDA-MB-361, MDA-MB-436, MDA-MB-453, MDA-MB-468, SK-BR-3, SUM-159PT, and T-47D. Cells were cultured in Dulbecco’s modified Eagle’s medium/F-12 supplemented with 10% fetal bovine serum at 37 °C and humidified 5% CO2.
Drugs and reagents
Selinexor was obtained from Karyopharm Therapeutics Inc. as a generous gift. For in vivo experiments, selinexor was prepared in 0.6% w/v Pluronic F-68 surfactant and 0.6% w/v PVP K-29/32 polymer. Paclitaxel, carboplatin, and gemcitabine were purchased from Selleck Chemicals. Doxorubicin and eribulin were acquired from the MD Anderson Cancer Center pharmacy. Dimethyl sulfoxide (DMSO) was from Sigma-Aldrich. For in vitro studies, all drugs were dissolved in DMSO.
Cell proliferation assays
Cells were seeded in 96-well plates at densities of 3000 to 6000 cells per well depending on the growth characteristics of each cell line. After cells had adhered overnight, titrating concentrations of the designated drug were added to the wells in triplicate and incubated at 37 °C for 72 hours. Cell viability was then measured using sulforhodamine B (SRB) staining. The half-maximal inhibitory concentration (IC50) was determined based on a dose-response curve generated using GraphPad Prism software, v6.05. All experiments were performed at least three times.
Colony formation assay
Cells were plated at a density of 2 × 103 cells on 60-mm plates in triplicate for each treatment group. Cells were treated the next day with the indicated concentrations of selinexor, paclitaxel, eribulin, vehicle control (DMSO), or a combination of selinexor and paclitaxel or eribulin for 2 weeks. The culture medium was changed every 7 days. The colonies were then fixed in 10% formalin and stained with 0.05% crystal violet in 25% methanol. Percent surface area was measured using NIH ImageJ software, v.1.48.
Western blot analysis
Cells were washed with cold PBS and lysed in Laemmli buffer. The protein was quantified using the Pierce BCA protein assay kit (ThermoFisher) before loading onto the gel. After SDS-PAGE, the protein was transferred to a 0.2-μm nitrocellulose membrane (Bio-Rad Laboratories). Membranes were blocked with 0.1% casein in Tris-buffered saline. Immunoblotting was performed with anti-β-catenin, anti-cyclin D1, anti-XIAP, and anti-survivin (Cell Signaling Technology), anti-β-actin, XPO1 (Santa Cruz Biotechnology), followed by secondary antibody anti-rabbit IgG Alexa fluor 680 (Sigma). The immunoblots were visualized using the Odyssey IR imaging system (Li-Cor Biosciences). Image Studio software, v4.0 was used to analyze the bands. Each band was normalized to its respective β-actin band. Representative blots of at least two independent experiments are shown. Results were analyzed using Student’s unpaired t test, with two-tailed P values. Values are presented as mean ± SEM.
Apoptosis and cell cycle assays
Cells were plated and treated the following day with DMSO or with selinexor and/or paclitaxel in triplicate. After 72 hours, both floating cells and attached cells were collected. Apoptosis was identified by using the annexin V apoptosis kit (Roche cat. no. 11858777001) according to the manufacturer’s protocol. The samples were analyzed by flow cytometry at the Flow Cytometry and Cellular Imaging Core Facility at MD Anderson. For cell cycle analysis, cells were collected after 72 hours of treatment, and DNA content was determined with flow cytometry using propidium iodide (Roche cat. no. 11348639001) following the manufacturer’s protocol. Each experiment was performed at least three times.
In vivo studies
All animal experiments were approved by the Institutional Animal Care and Use Committee of MD Anderson. TNBC PDX models have been previously described [13]. Tumors were implanted into female BALB/c nu/nu mice, 6 to 8 weeks old, under isoflurane anesthesia. A skin incision (approximately 0.3 cm) was made with a subcutaneous pocket on the mid back. One tumor piece (approximately 27 mm3) was inserted into a pocket and the skin was then closed. The mice were treated when the tumor diameter reached at least 200 mm3. The mice were killed when the diameter reached 1.5 cm, and the individual relative tumor volume (RTV) was measured. RTV was defined as Vx/V1, where Vx is the volume in mm3 at a given time and V1 is the volume at the start of treatment [14, 15]. The statistical analyses were performed by comparing RTV in the treatment arms with RTV in the vehicle arm. Tumor growth inhibition ratios (T/C: treatment/control) were calculated using the formula:
[(Median tumor volume of treated group)/(Median tumor volume of control group)] × 100.
Activity was defined as a T/C ratio <40% [16, 17].
The BCX 10 mice were treated with vehicle or selinexor via oral gavage 12.5 mg/kg twice a week (2 weeks on, 1 week off); the BCX 6 and BCX 11 mice were treated with selinexor via oral gavage 12.5 mg/kg once a week. The dose for these experiments was decreased to model a clinical trial that was being run in parallel. Mice in the three models were also treated with paclitaxel 10 mg/kg intravenously weekly (2 weeks on, 1 week off), eribulin 1 mg/kg intravenously weekly (2 weeks on, 1 week off), carboplatin 75 mg/kg intraperitoneally weekly (1 week on, 2 weeks off), or a combination of selinexor with each individual drug.
Results
Selinexor has potent antitumor efficacy in breast cancer cells in vitro
To test the effects of selinexor in breast cancer, we selected 26 breast cancer cell lines representing different subtypes that included TNBC and estrogen receptor (ER)-positive (ER+) and human epidermal growth factor receptor (HER2) + cell lines, with various genomic alterations including PIK3CA, TP53, and PTEN mutations as described in Additional file 1. Cell growth was measured after 72 hours of treatment using the SRB colorimetric assay. Sensitivity was evaluated by calculating the IC50 using isobologram curves. The sensitivities among the cell lines varied, with an overall median IC50 of 66 nM (range 11 to >1000 nM) (Fig. 1). Sensitivity was defined as <1000 nM, which is a physiologically achievable dose based on previous phase I studies [18]. All 14 of the TNBC cell lines tested were significantly more sensitive to selinexor (median IC50 of 44 nM, range 11 to 550 nM) than were ER+ cell lines (median IC50 of >1000 nM, range 40 to >1000 nM) (P = 0.017).
Fig. 1.
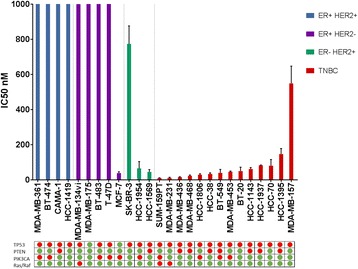
Effects of selinexor on cell proliferation in vitro. Twenty-six breast cancer cell lines representing varying hormone receptor status were treated with selinexor at 10 concentrations based on a fivefold dilution series (range 0 to 100 μM). Cell growth was measured after 72 hours of treatment using the sulforhodamine B assay, and half-maximal inhibitory concentration (IC 50) was then calculated using isobologram curves, with an overall median IC50 of 66 nM (range 11 to >1000 nM). Sensitivity was defined as IC50 < 1000 nM, a physiologically achievable dose based on previous phase I studies. TP53, PTEN, PIK3CA, and Ras/Raf status is shown. Green circle wild type, red circle mutation/deletion or frameshift. ER estrogen receptor, HER2 human epidermal growth factor receptor, TNBC triple-negative breast cancer
To assess the effects of selinexor on XPO1 expression, we chose an array of breast cancer cell lines representing varying hormone receptor status and representing different levels of sensitivity to selinexor. Seventy-two hours after treatment with or without 800 nM selinexor (a physiologically achievable concentration [18]), cells were lysed and immunoblotted. The intensities of XPO1 bands were quantified and normalized to their respective β-actin bands. Selinexor reduced the levels of XPO1 and of survivin in all cell lines. XIAP (an inhibitor of apoptosis protein) and β-catenin (a known regulator of survivin [19, 20]) were lower in MDA-MB-436 and MDA-MB-468, both TNBC cell lines that are sensitive to selinexor (Fig. 2). These effects were more robust when the dose of selinexor was increased to 5 μM (Additional file 2).
Fig. 2.
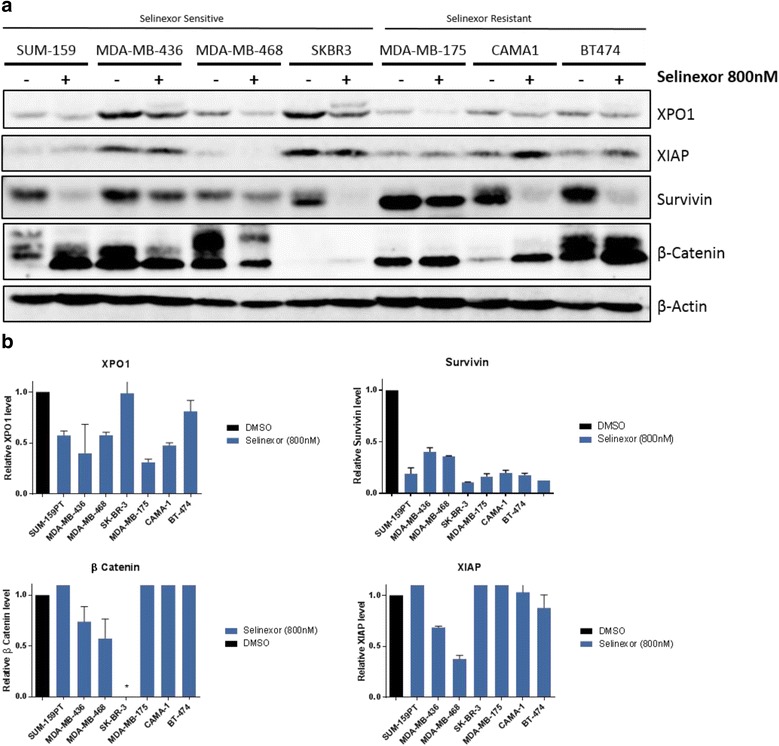
Effects of selinexor in breast cancer models. a Seven breast cancer cell lines were treated with vehicle or 800 nM selinexor, a physiologically achievable dose. Cells were lysed and blotted with the indicated antibodies. b The bands were quantified and normalized to their respective β-actin controls. Values are shown as relative protein levels vs vehicle control of cell lines treated with selinexor
Selinexor in combination with standard chemotherapy enhanced antitumor efficacy in vitro
To determine the effects of selinexor in combination with standard chemotherapy, we selected several commonly used agents: paclitaxel (a microtubule stabilizer), carboplatin (DNA-binding alkylating agent), eribulin (microtubule inhibitor), doxorubicin (topoisomerase inhibitor), and gemcitabine (pyrimidine analogue), all with various mechanisms of action. Four TNBC cell lines with different genomic backgrounds were studied: SUM-159PT (HRAS, PIK3CA, and TP53 mutant), MDA-MB-436 (PTEN and BRCA mutant), MDA-MB-231 (KRAS and TP53 mutant), and MDA-MD-157 (TP53 mutant); all cell lines were XPO1 wild-type. Cells were treated with serial concentration dilutions of selinexor in combination with serial concentration dilutions of the various chemotherapy agents. After 72 hours of treatment, growth inhibition was assessed using the SRB assay and IC50 was calculated for single-agent treatment alone and the combinations. Combination index (CI) values were then calculated using the method of Chou and Talalay, where a CI value <0.8 indicates synergism, a CI of 0.8 to 1.2 indicates addition, and a CI >1.2 indicates antagonism [21, 22]. Synergistic effects were found for all four cell lines when selinexor was combined with doxorubicin and paclitaxel and for SUM-159PT and MDA-MB-231 when combined with carboplatin or eribulin (Fig. 3a).
Fig. 3.
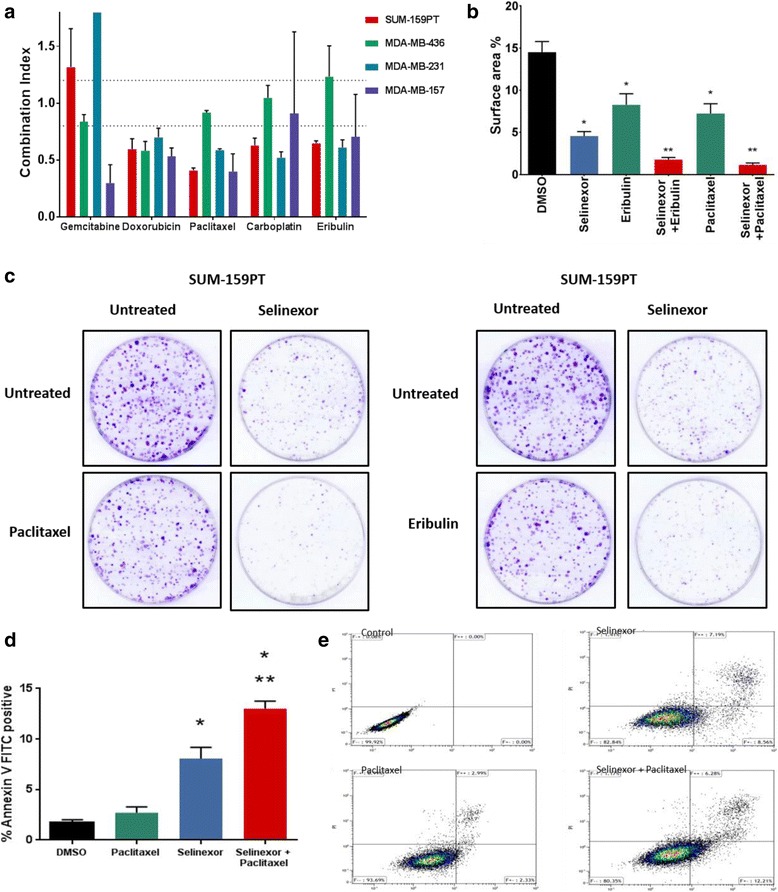
Effects of selinexor in combination with standard chemotherapy in vitro. a Four different triple-negative breast cancer cell lines were treated with selinexor in combination with paclitaxel, doxorubicin, gemcitabine, carboplatin, and eribulin. Cell growth was measured after 72 hours of treatment using the Sulforhodamine B (SRB) assay, and the combination index (CI) was then calculated using the method of Chou and Talalay [20]. A CI value <0.8 indicates synergism, a value equal to 1 indicates addition, and a CI significantly >1.2 indicates antagonism. b, c SUM-159PT cells were trypsinized, counted, and plated at a density of 2 × 103 cells in 60-mm plates in triplicate for each treatment group. Cells were treated for 2 weeks with vehicle, selinexor (50 nM), paclitaxel (0.5 nM), or eribulin (1 nM) or in combinations of selinexor with paclitaxel and selinexor with eribulin. Colonies were then fixed and stained with crystal violet. Percent surface area was calculated using NIH Image J v.1.48 software. Values are presented as mean ± SEM (*P < 0.005 vs control, **P < 0.001 combination paclitaxel vs selinexor alone, P = 0.0013 combination eribulin vs selinexor alone). d, e SUM-159PT cells were treated with 0.5 nM paclitaxel alone, 400 nM selinexor alone, and the combination of both. After 72 hours, annexin-V-positive cells were determined by fluorescence-activated cell sorting FACS analysis. (*P < 0.001 selinexor alone vs vehicle control, **P = 0.004 combination vs selinexor alone). Each individual experiment was performed in triplicate. DMSO dimethyl sulfoxide, FITC, fluorescein isothiocyanate
Using the colony formation assay, we then assessed the effects of selinexor in combination with paclitaxel and with eribulin. The TNBC cell line SUM-159PT was treated with either vehicle control (DMSO), 50 nM selinexor, 0.5 nM paclitaxel, 1 nM eribulin, or with a combination of selinexor with paclitaxel or selinexor with eribulin. After 2 weeks of treatment, cell colonies were stained with crystal violet. Plates were then scanned and the percentage of stained surface area was quantified (Fig. 3b and c). Treatment with selinexor in combination with paclitaxel and in combination with eribulin resulted in a dramatic decline in cell growth compared with treatment with either drug alone (P < 0.001 for combination paclitaxel vs selinexor alone and P = 0.0013 for combination eribulin vs selinexor alone).
Once we established that there was significant synergy between selinexor and paclitaxel, we wanted to determine whether apoptosis was induced. We treated SUM-159PT cells with selinexor, paclitaxel, or selinexor in combination with paclitaxel for 72 hours with a physiologically achievable dose of the compounds. To assess for apoptosis, the harvested cells were analyzed by fluorescence-activated cell sorting (FACS) using the annexin V apoptosis kit. Notably, our results showed an increased number of annexin-V-positive cells when treated with selinexor alone compared with vehicle control (P < 0.001). The percentage of cells that were positive for annexin V was significantly higher when selinexor was used in combination with paclitaxel compared with vehicle control (P < 0.001) and compared with selinexor alone (P = 0.004) (Fig. 3d and e). In a similar fashion, cell cycle analysis was performed on SUM-159PT and MDA-MB-468 cells. The results showed a significantly greater number of cells in sub-G1 phase (P = 0.003) when selinexor was used in combination with paclitaxel compared with either treatment alone, as shown in Additional file 3.
Selinexor has in vivo efficacy in TNBC PDX models
To test the effects of selinexor in vivo, we selected five different TNBC PDX models: BCX 6, BCX 10, BCX 11, BCX 22, and BCX 51, with varying status for TP53 and PIK3CA [13] and gene expression subtypes. Molecular characteristics of these PDX models are listed in Additional file 4 (detailed manuscript on PDXs under review). Mice implanted with these PDX models were treated either with vehicle or selinexor via oral gavage 12.5 mg/kg twice a week (2 weeks on and 1 week off). We observed that, in four of five PDX models, mice treated with selinexor had significantly lower tumor growth (P < 0.001), with a median T/C score of 42% (range 31 to 73%) (Fig. 4). The mice tolerated the drug well, with no weight loss during treatment (Additional file 5).
Fig. 4.
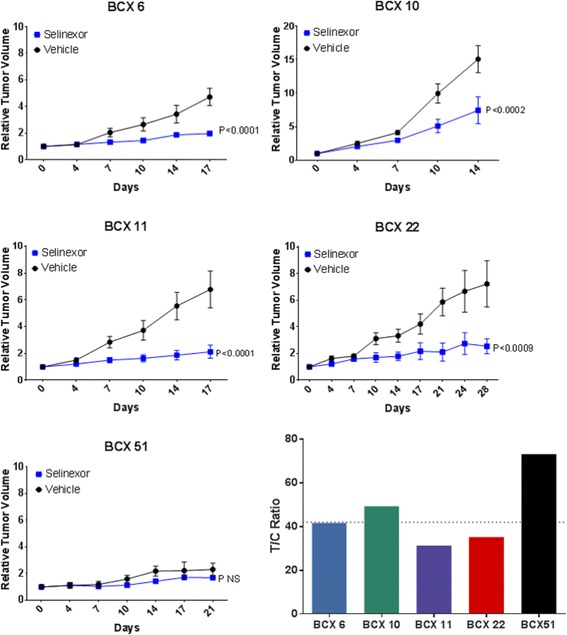
Effects of selinexor on tumor growth of triple-negative breast cancer (TNBC) patient-derived xenograft (PDX) models. Mice bearing BCX 6, BCX 10, BCX 11, BCX 22, and BCX 51 TNBC patient-derived xenografts were treated with vehicle or selinexor 12.5 mg/kg twice a week. Values are presented as mean ± SEM of relative tumor volume. The tumor volumes at the conclusion of experiment were compared with vehicle, and data were analyzed by two-way analysis of variance to determine statistical significance. Tumor growth inhibition (T/C) ratios were calculated using the formula: [(Median tumor volume of treated group)/(Median tumor volume of control group)] × 100. Activity was defined as a T/C % ratio <40%
Next, we compared the effects of selinexor in vivo in combination with standard chemotherapy agents, including paclitaxel, eribulin, and carboplatin, on BCX 6, BCX 10, and BCX 11 models, all with various degrees of sensitivity to these agents (Fig. 5). In vivo, BCX 10 and BCX 6 were relatively resistant to carboplatin, and although the combination with selinexor was significantly more effective than carboplatin alone (P < 0.02), it was not significantly more effective than selinexor alone. However, selinexor in combination with paclitaxel or eribulin had significant antitumor efficacy, with T/C ratios <40% for all three PDX models. For BCX 11, selinexor in combination with paclitaxel compared with either agent alone was significantly more effective (P < 0.003), and selinexor in combination with eribulin was significantly more effective than either agent alone in BCX 10 (P < 0.001), and BCX 11 (P = 0.005).
Fig. 5.
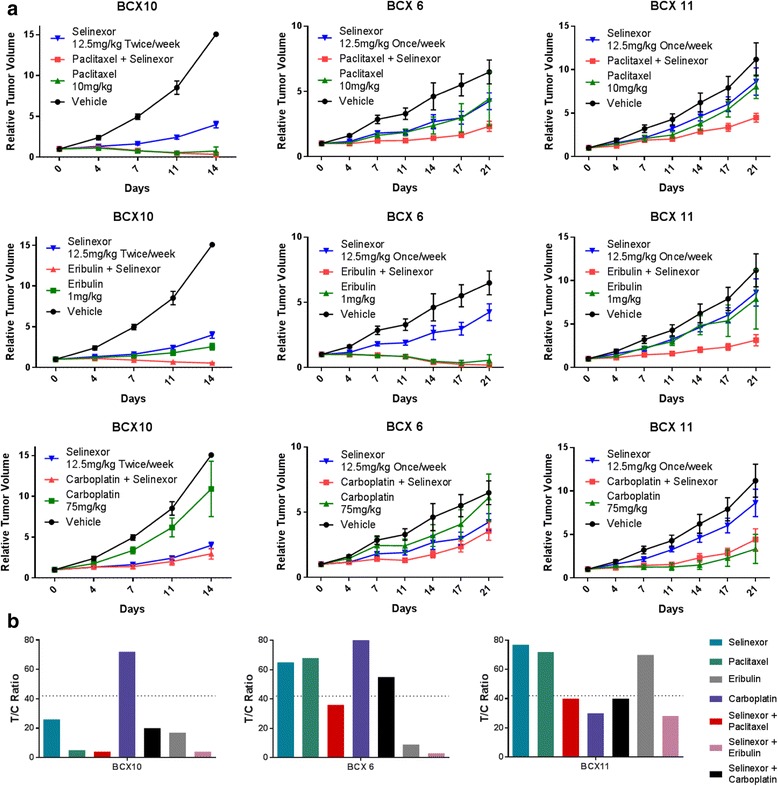
Effects of selinexor on tumor growth in combination with standard chemotherapy. Mice bearing three different triple-negative breast cancer patient-derived xenografts were treated with vehicle, selinexor 12.5 mg/kg twice a week for BCX 10 and once weekly for BCX 6 and BCX 11, paclitaxel 10 mg/kg weekly, eribulin 1 mg/kg, carboplatin 75 mg/kg weekly, and combinations of selinexor with each chemotherapy agent. a Values are presented as mean ± SEM. The tumor volumes at the conclusion of the experiment were compared with vehicle, and the data were analyzed by two-way analysis of variance to determine statistical significance. b Tumor growth inhibition (T/C) ratios were calculated using the formula: [(Median tumor volume of treated group)/(Median tumor volume of control group)] × 100. Activity was defined as a T/C % ratio <40%
Discussion
TNBC occurs in approximately 15 to 20% of patients with breast cancer and is associated with an unfavorable prognosis [10]. Although patients with TNBC who achieve complete pathological response have better survival [23], patients who have significant residual disease are at high risk of relapse and have limited options upon recurrence. Therefore there is a great need for superior therapy options for TNBC. Here we showed the antitumor efficacy of selinexor, as a novel approach for TNBC therapy.
XPO1 compartmentalizes tumor suppressors and cell cycle regulators, which are dependent on location to exert their apoptotic/proliferative functions [24]. Many of these have been linked to the elusive molecular network in TNBC [11], providing a rationale for exploring XPO1 inhibition as a potential target for cancer therapy. Our findings have established the antitumor efficacy of selinexor, a selective inhibitor of nuclear export, on breast cancer. We have shown that selinexor effectively inhibits cell proliferation and cell growth both in vitro and in vivo at biologically relevant drug concentrations. Our experiments suggest that there is enhanced sensitivity of TNBC cell lines to selinexor compared with ER+ cells. Treatment with selinexor as a single agent resulted in enhanced tumor growth inhibition in four of five TNBC PDX models in vivo. We also demonstrated that treatment with selinexor induced apoptosis. Concordantly, treatment with selinexor decreased levels of survivin, XIAP, and β-catenin. Furthermore, while survivin has been previously reported as a possible explanation for the mechanism of action of selinexor [3, 25], by testing a wide array of breast cancer cell lines in this study, we have shown that selinexor decreases XPO1 expression and survivin expression not only in selinexor-sensitive cell lines but also in selinexor-resistant lines. We have also shown that selinexor is effective independent of PTEN, PIK3CA, TP53, or Ras/Ras status. Although we did see greater sensitivity in TNBC cell lines compared to other subtypes, we have not tested the sensitivity of ER+ and HER+ cancers in vivo, and we have not determined the mechanism behind the differential sensitivity between subtypes in vitro. Further studies are needed to identify pharmacodynamic markers of response and mechanisms of intrinsic resistance. The inherent complexity of the mechanism behind XPO1 inhibition involves the ability of XPO1 to interact with several different tumor suppressors and cell cycle regulators, therefore potentially targeting multiple pathways. One of the most common examples is the role of XPO1 inhibition leading to the accumulation of TP53 in the nucleus, an exciting finding of a possible mechanism explaining its anti-proliferative effects in leukemia, lymphoma, prostate cancer, melanoma, and hepatocellular carcinoma [26–28]. In our study, however, most cell lines tested were TP53 mutant yet differed in their selinexor sensitivity. Thus, XPO1 inhibition is effective independent of the TP53 status as we have shown here, consistent with what has been previously shown for sarcoma, non-small cell lung cancer, multiple myeloma, and mesothelioma [29–32].
Selinexor also had enhanced efficacy when combined with standard chemotherapy agents. Four different TNBC cell lines showed notable synergy in vitro, in particular with doxorubicin and paclitaxel (median CI values of 0.6 and 0.5, respectively), agents used commonly in the neoadjuvant or adjuvant treatment of TNBC, and to some extent with carboplatin and eribulin (median CI values of 0.8 and 0.7, respectively). At the same time, the combination of selinexor with paclitaxel enhanced apoptosis. Further, selinexor showed enhanced antitumor activity in vivo when combined with paclitaxel and eribulin, with T/C ratios <40% in three different TNBC PDX models.
Conclusions
Altogether, these results provide preclinical evidence that selinexor is a promising agent in the treatment of TNBC, with enhanced antitumor activity in combination with chemotherapy. The safety and tolerability of selinexor combined with chemotherapy are being explored in ongoing clinical trials (NCT02419495), with planned expansions in breast cancer.
Additional files
PIK3CA, PTEN, TP53 and Ras/Raf status of cell lines in panel. A panel of breast cancer cell lines was tested with selinexor, and cell growth was measured after 72 hours of treatment using the SRB assay. IC50 was then calculated using isobologram curves. The mutation statuses for PIK3CA, PTEN, TP53, and Ras/Raf status are reported. (DOCX 23 kb)
Effects of selinexor in breast cancer models. A Eight breast cancer cell lines were treated with vehicle or 5 μM selinexor. Cells were lysed and blotted with the indicated antibodies. B The bands were quantified and normalized to their respective β-actin controls. Data are shown as relative protein levels vs vehicle control of cell lines treated with selinexor. (DOCX 397 kb)
Effects of selinexor on the cell cycle. SUM-159PT and MDA-MB-468 cells were treated with 1 nM paclitaxel alone (2 nM for MDA-MB-468), 400 nM selinexor alone, and the combination of both. After 72 hours, cells were stained with propidium iodide and analyzed via flow cytometry. (DOCX 514 kb)
Actionable DNA alterations in PDXs. BCX 6, BCX 10, BCX 11, and BCX 22 were analyzed by targeted exome sequencing of 202 cancer-relevant targets and whole-exome sequencing. BCX 51 was analyzed using targeted exome sequencing of 265 genes that included XPO1. HDEL: copy number <1, HAMP: copy number >4. (DOCX 15 kb)
Tolerance of selinexor in vivo. Body weight–time curve is shown. Data are presented as mean ± SEM. (DOCX 126 kb)
Acknowledgements
Not applicable.
Funding
This work was supported by National Institutes of Health T32 CA009599 (NPA, FMB), Susan G. Komen Foundation for the Cure grant SAC10006 (FMB), MD Anderson Women’s Cancers Moonshot Program (KWE, FMB), the Nellie B. Connally Breast Cancer Research Endowment (FMB), the Barr funds, and the MD Anderson Cancer Center support grant (P30 CA016672).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Accession number to microarray
Not applicable.
Clinical trial registration number and date
Not applicable.
Abbreviations
- CI
Combination index
- DMSO
Dimethyl sulfoxide
- ER
Estrogen receptor
- FACS
Fluorescence-activated cell sorting
- IC50
Half-maximal inhibitory concentration
- NES
Nuclear export sequence
- PBS
Phosphate-buffered saline
- PDX
Patient-derived xenografts
- SRB
Sulforhodamine B
- T/C ratio
Tumor growth inhibition ratio
- TNBC
Triple-negative breast cancer
- XPO
Exportin1 or CRM1
Authors’ contributions
FMB is the senior author and conceived and designed the study and edited the manuscript. NPA performed and analyzed the in vitro and in vivo experiments, interpreted and analyzed the data, constructed the figures, and wrote the manuscript. EY, KWE, and SS performed in vivo experiments. CK and MZ conducted in vitro viability assays. AMG, FJ, NTU, DT, AN, and AA conceived the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Ethics approval
All animal experiments were approved by the Institutional Animal Care and Use Committee at MD Anderson (ACUF protocol # 00001113 and 00001405). This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Consent for publication
All authors consent for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13058-017-0878-6) contains supplementary material, which is available to authorized users.
References
- 1.Gravina GL, Senapedis W, McCauley D, Baloglu E, Shacham S, Festuccia C. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol. 2014;7:85. doi: 10.1186/s13045-014-0085-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abraham SA, Holyoake TL. Redirecting traffic using the XPO1 police. Blood. 2013;122(17):2926–8. doi: 10.1182/blood-2013-09-523670. [DOI] [PubMed] [Google Scholar]
- 3.Cheng Y, Holloway MP, Nguyen K, McCauley D, Landesman Y, Kauffman MG, Shacham S, Altura RA. XPO1 (CRM1) inhibition represses STAT3 activation to drive a survivin-dependent oncogenic switch in triple-negative breast cancer. Mol Cancer Ther. 2014;13(3):675–86. doi: 10.1158/1535-7163.MCT-13-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishizawa J, Kojima K, Hail N, Jr, Tabe Y, Andreeff M. Expression, function, and targeting of the nuclear exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol Ther. 2015;153:25–35. doi: 10.1016/j.pharmthera.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyake T, Pradeep S, Wu SY, Rupaimoole R, Zand B, Wen Y, Gharpure KM, Nagaraja AS, Hu W, Cho MS, et al. XPO1/CRM1 inhibition causes antitumor effects by mitochondrial accumulation of eIF5A. Clin Cancer Res. 2015;21(14):3286–97. doi: 10.1158/1078-0432.CCR-14-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranganathan P, Yu X, Santhanam R, Hofstetter J, Walker A, Walsh K, Bhatnagar B, Klisovic R, Vasu S, Phelps MA, et al. Decitabine priming enhances the antileukemic effects of exportin 1 (XPO1) selective inhibitor selinexor in acute myeloid leukemia. Blood. 2015;125(17):2689–92. doi: 10.1182/blood-2014-10-607648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83(8):1021–32. doi: 10.1016/j.bcp.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, Ellis JJ, Landesman Y, Eisfeld AK, Gabrail NY, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph + leukemias. Blood. 2013;122(17):3034–44. doi: 10.1182/blood-2013-04-495374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mo W, Liu Q, Lin CC, Dai H, Peng Y, Liang Y, Peng G, Meric-Bernstam F, Mills GB, Li K, et al. mTOR inhibitors suppress homologous recombination repair and synergize with PARP inhibitors via regulating SUV39H1 in BRCA-proficient triple-negative breast cancer. Clin Cancer Res. 2016;22(7):1699–712. doi: 10.1158/1078-0432.CCR-15-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 11.Ossovskaya V, Wang Y, Budoff A, Xu Q, Lituev A, Potapova O, Vansant G, Monforte J, Daraselia N. Exploring molecular pathways of triple-negative breast cancer. Genes Cancer. 2011;2(9):870–9. doi: 10.1177/1947601911432496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen SH, Murphy DA, Lassoued W, Thurston G, Feldman MD, Lee WM. Activated STAT3 is a mediator and biomarker of VEGF endothelial activation. Cancer Biol Ther. 2008;7(12):1994–2003. doi: 10.4161/cbt.7.12.6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, Zhao H, Eterovic AK, Sangai T, Holder AM, Sharma C, et al. Ability to generate patient-derived breast cancer xenografts is enhanced in chemoresistant disease and predicts poor patient outcomes. PLoS One. 2015;10(9) doi: 10.1371/journal.pone.0136851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanceau J, Poupon MF, Delattre O, Sastre-Garau X, Wietzerbin J. Strong inhibition of Ewing tumor xenograft growth by combination of human interferon-alpha or interferon-beta with ifosfamide. Oncogene. 2002;21(50):7700–9. doi: 10.1038/sj.onc.1205881. [DOI] [PubMed] [Google Scholar]
- 15.Xu S, Li S, Guo Z, Luo J, Ellis MJ, Ma CX. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol Cancer Ther. 2013;12(8):1665–75. doi: 10.1158/1535-7163.MCT-13-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alley MC, Hollingshead MG, Dykes D, Waud W. Human tumor xenograft models in NCI drug development. In: Teicher BA, editor. Cancer drug discovery and development: anticancer drug development guide: preclinical screening, clinical trials, and approvals. 1. Andrews: Humana Press; 2004. pp. 125–52. [Google Scholar]
- 17.Rad FH, Le Buanec H, Paturance S, Larcier P, Genne P, Ryffel B, Bensussan A, Bizzini B, Gallo RC, Zagury D, et al. VEGF kinoid vaccine, a therapeutic approach against tumor angiogenesis and metastases. Proc Natl Acad Sci USA. 2007;104(8):2837–42. doi: 10.1073/pnas.0611022104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdul Razak AR, Mau-Soerensen M, Gabrail NY, Gerecitano JF, Shields AF, Unger TJ, Saint-Martin JR, Carlson R, Landesman Y, McCauley D, et al. First-in-class, first-in-human phase I study of Selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J Clin Oncol. 2016;34(34):4142–50. doi: 10.1200/JCO.2015.65.3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang T, Otevrel T, Gao Z, Gao Z, Ehrlich SM, Fields JZ, Boman BM. Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res. 2001;61(24):8664–7. [PubMed] [Google Scholar]
- 20.Fernandez JG, Rodriguez DA, Valenzuela M, Calderon C, Urzua U, Munroe D, Rosas C, Lemus D, Diaz N, Wright MC, et al. Survivin expression promotes VEGF-induced tumor angiogenesis via PI3K/Akt enhanced beta-catenin/Tcf-Lef dependent transcription. Mol Cancer. 2014;13:209. doi: 10.1186/1476-4598-13-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 22.Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB, Meric-Bernstam F. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004;10(20):7031–42. doi: 10.1158/1078-0432.CCR-04-0361. [DOI] [PubMed] [Google Scholar]
- 23.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 24.Gerecitano J. SINE (selective inhibitor of nuclear export)–translational science in a new class of anti-cancer agents. J Hematol Oncol. 2014;7:67. doi: 10.1186/s13045-014-0067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nair JS, Musi E, Schwartz GK. Selinexor (KPT-330) induces tumor suppression through nuclear sequestration of IkappaB and down-regulation of survivin. Clin Cancer Res. 2017;1–11. [DOI] [PubMed]
- 26.Yang J, Bill MA, Young GS, La Perle K, Landesman Y, Shacham S, Kauffman M, Senapedis W, Kashyap T, Saint-Martin JR, et al. Novel small molecule XPO1/CRM1 inhibitors induce nuclear accumulation of TP53, phosphorylated MAPK and apoptosis in human melanoma cells. PLoS One. 2014;9(7), e102983. [DOI] [PMC free article] [PubMed]
- 27.Yoshimura M, Ishizawa J, Ruvolo V, Dilip A, Quintas-Cardama A, McDonnell TJ, Neelapu SS, Kwak LW, Shacham S, Kauffman M, et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma. Cancer Sci. 2014;105(7):795–801. doi: 10.1111/cas.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol. 2014;74(3):487–95. doi: 10.1007/s00280-014-2495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayama R, Zhang YX, Czaplinski JT, Anatone AJ, Sicinska ET, Fletcher JA, Demetri GD, Wagner AJ. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget. 2016;7(13):16581–92. [DOI] [PMC free article] [PubMed]
- 30.De Cesare M, Cominetti D, Doldi V, Lopergolo A, Deraco M, Gandellini P, Friedlander S, Landesman Y, Kauffman MG, Shacham S, et al. Anti-tumor activity of selective inhibitors of XPO1/CRM1-mediated nuclear export in diffuse malignant peritoneal mesothelioma: the role of survivin. Oncotarget. 2015;6(15):13119–32. [DOI] [PMC free article] [PubMed]
- 31.Sun H, Hattori N, Chien W, Sun Q, Sudo M, E-Ling GL, Ding L, Lim SL, Shacham S, Kauffman M, et al. KPT-330 has antitumour activity against non-small cell lung cancer. Br J Cancer. 2014;111(2):281–91. doi: 10.1038/bjc.2014.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, Tannenbaum D, Cagnetta A, Reagan M, Munshi AA, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28(1):155–65. doi: 10.1038/leu.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PIK3CA, PTEN, TP53 and Ras/Raf status of cell lines in panel. A panel of breast cancer cell lines was tested with selinexor, and cell growth was measured after 72 hours of treatment using the SRB assay. IC50 was then calculated using isobologram curves. The mutation statuses for PIK3CA, PTEN, TP53, and Ras/Raf status are reported. (DOCX 23 kb)
Effects of selinexor in breast cancer models. A Eight breast cancer cell lines were treated with vehicle or 5 μM selinexor. Cells were lysed and blotted with the indicated antibodies. B The bands were quantified and normalized to their respective β-actin controls. Data are shown as relative protein levels vs vehicle control of cell lines treated with selinexor. (DOCX 397 kb)
Effects of selinexor on the cell cycle. SUM-159PT and MDA-MB-468 cells were treated with 1 nM paclitaxel alone (2 nM for MDA-MB-468), 400 nM selinexor alone, and the combination of both. After 72 hours, cells were stained with propidium iodide and analyzed via flow cytometry. (DOCX 514 kb)
Actionable DNA alterations in PDXs. BCX 6, BCX 10, BCX 11, and BCX 22 were analyzed by targeted exome sequencing of 202 cancer-relevant targets and whole-exome sequencing. BCX 51 was analyzed using targeted exome sequencing of 265 genes that included XPO1. HDEL: copy number <1, HAMP: copy number >4. (DOCX 15 kb)
Tolerance of selinexor in vivo. Body weight–time curve is shown. Data are presented as mean ± SEM. (DOCX 126 kb)
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.