

Abstract
By the time the process of oncogenesis has produced an advanced cancer, tumor cells have undergone extensive evolution. The cellular phenotypes resulting from this evolution have been well studied, and include accelerated growth rates, apoptosis resistance, immortality, invasiveness, and immune evasion. Yet with all of our current knowledge of tumor biology, the details of early oncogenesis have been difficult to observe and understand. Where different oncogenic mutations may work together to enhance the survival of a tumor cell, in isolation they are often pro-apoptotic, pro-differentiative or pro-senescent, and therefore often, somewhat paradoxically, disadvantageous to a cell. It is also becoming clear that somatic mutations, including those in known oncogenic drivers, are common in tissues starting at a young age. These observations raise the question: how do we largely avoid cancer for most of our lives? Here we propose that evolutionary forces can help explain this paradox. As humans and other organisms age or experience external insults such as radiation or smoking, the structure and function of tissues progressively degrade, resulting in altered stem cell niche microenvironments. As tissue integrity declines, it becomes less capable of supporting and maintaining resident stem cells. These stem cells then find themselves in a microenvironment to which they are poorly adapted, providing a competitive advantage to those cells that can restore their functionality and fitness through mutations or epigenetic changes. The resulting oncogenic clonal expansions then increase the odds of further cancer progression. Understanding how the causes of cancer, such as aging or smoking, affect tissue microenvironments to control the impact of mutations on somatic cell fitness can help reconcile the discrepancy between marked mutation accumulation starting early in life and the somatic evolution that leads to cancer at advanced ages or following carcinogenic insults.
1. Introduction
1.1 The Process of Oncogenesis
The evolution of multicellularity created the ability of malignant cellular expansions to disrupt organismal fitness. However, the mechanisms by which cancer arises are yet to be fully understood [1]. Since Hermann Muller's demonstration that X-rays could induce heritable phenotypic changes in Drosophila, cancer research has focused on understanding the origins of mutations and the role they play within tumors [2–4]. Given that cancer biologists frequently focus on understanding the effects that a mutated gene can have on cell behavior, the logical leap is often made that cancer is a disease limited by mutations [5]. Thus, it is widely accepted that the rate-limiting step in the process of oncogenesis is mutation incidence, as oncogenic mutations confer cell traits often referred to as the “hallmarks of cancer” [6]. While it is relatively easy to observe and study fully-developed cancers due to their size, observing early oncogenesis, where cell numbers are much smaller, has proven challenging. This challenge has largely left knowledge of the processes that control early oncogenesis at the theoretical stage.
1.2 Phenotypes of Cancer
Heritable genetic changes, both epigenetic and genetic, can lead to phenotypic changes in cells. Among transformed cells there are a number of characteristic changes that often occur, including increased growth rates, apoptosis resistance, immortality, altered metabolism, increased invasion and immune evasion [6]. The utility of these phenotypic changes is easy to comprehend for a fully formed cancer, as a population of cells that uncontrollably divides while resisting apoptosis and immune removal would certainly be challenging to eliminate. Yet the combined functionality of these hallmark changes does little to explain the early steps in their accumulation, a time in cell transformation that is difficult to observe in vivo [7,8].
If we consider the characteristic genetic changes in cancer independently, their benefit to a cell becomes more challenging to explain [9]. As an example, consider MYC: when overexpressed it can be a powerful promoter of uncontrolled cell division, yet without accompanying anti-apoptotic mutations, induces cell death [10,11]. If cell fitness is proportional to the probability that a genotype is maintained in a population, apoptotic susceptibility will likely lower overall cellular fitness levels. There is abundant evidence that oncogenic mutations in healthy tissues frequently reduce cell fitness, often by increasing apoptosis susceptibility or by reducing self-renewal [12]. Without considering the environmental context in which oncogenic mutations occur, it becomes difficult to rationalize how an otherwise detrimental mutation would not lead to the loss of the cell clone.
1.3 Current Theory to Explain Cancer Development
There is a long history of attempts to explain oncogenesis solely through increased cell division rates or mutation rates [2,3,13–15]. These studies have largely focused on mutation incidence, and contribute to the Somatic Mutation Theory (SMT) of cancer to explain the entire process of oncogenesis [16–19]. SMT works under the assumption that oncogenic mutations generally improve cell fitness and contribute to oncogenesis. If oncogenic mutations are typically beneficial to a cell, then transformation could just be a game of chance, where a cell is awaiting the right oncogenic mutations at the right time [14,20,21]. With a low enough mutation rate, these oncogenic mutations could be rare enough to delay cancer incidence by many decades in humans, and thus fit empiric observations of cancer incidence [22,23]. However, by ignoring the context-dependent nature of mutations, SMT struggles to explain a number of highly relevant observations.
As a mutation focused theory, SMT is insufficient to explain the similar cancer incidence rates observed in organisms of disparate sizes [24–26], or why, while most mutations occur early in life during ontogeny, cancers usually do not arise until late in life [27,28]. In addition, in humans, with large pools of dividing cells, known mutation rates should make oncogenic mutations relatively common [29,30]. While there is no doubt that mutation incidence is necessary for tumorigenesis, it seems increasingly unlikely that it is sufficient for initial tumor formation. This begs the question of what other factors are limiting oncogenic clonal expansions, proportionally increasing the risk of subsequent cancer development.
1.4 Evolutionary Theory and Early Oncogenesis
As we know from evolutionary theory, random heritable phenotypic variation is acted upon by selection, such that allele frequencies within a population change based on their fitness in a particular environmental context. When an organism carries genetic changes that result in a phenotype that is beneficial within a particular environment, that organism will be more competitive, more likely to reproduce, and the genetic alleles conferring fitness benefit will increase in frequency in the population. In small populations, genetic drift (random changes in allele frequencies) can play a substantial role in determining the representation of particular alleles, especially for alleles with minor fitness effects. Finally, migration to a new locale can stimulate adaptation to the new environment, and may bring about population bottlenecks that can allow random fixation of genetic alleles.
Where mutation-centric SMT falls short in explaining oncogenesis, roles for other evolutionary forces may be able to fill in the gaps. Just as at the organismal level, if a genetic mutation confers a phenotypic change on a somatic cell that is beneficial to its persistence within a particular microenvironment, that cell will be more likely to survive and contribute to the somatic gene pool of the tissue, and vice versa. Selection, drift, and migration will thus play important roles in determining whether or not a particular somatic cell variant will continue to contribute to the tissue, or instead be eliminated.
Evolutionary theory expands on classical SMT with its consideration of the context in which an oncogenic mutation occurs. As such, an identical oncogenic event may be favored in one microenvironment, while being disadvantageous in another, as is also the case at the organismal level [31]. As a primary risk factor of cancer, aging as a process brings with it many microenvironmental changes including deregulated nutrient sensing, impaired intracellular signaling, increased cellular senescence, and stem cell exhaustion [32]. The age-altered microenvironment will exert different selective forces on the cells it contains. Where an oncogenic mutation in a youthful microenvironment might signal too robustly and push a cell into apoptosis or cause loss of self-renewal, in an aged environment costs may be outweighed by adaptive benefits to favor survival and expansion. Understanding the context-dependence of mutation effects provides a rationale for how oncogenic mutations could at times be detrimental to a cell while at other times be beneficial.
1.5 The Forces of Evolution Sculpt Tumor Heterogeneity
The idea of somatic evolution in cancer is not new; as a process, it is already well described within the context of mature tumors [3,33–36]. Within tumors, cellular adaptation to different microenvironments gives rise to phenotypically diverse cells across the tumor mass. These cells are evolving around a number of different selective pressures including hypoxia, inadequate perfusion, and even physical space [37]. Just as tumor microenvironmental differences may control the population dynamics of tumor cells, so too could the state of the tissue microenvironment dictate the fate of mutation-bearing cells during early oncogenesis.
Tumor microenvironments are pockets where different evolutionary forces such as selection, drift and migration are at work. The particular clones that migrate into and out of a microenvironment, random genetic drift, and selection for or against particular phenotypes are crucial in determining the eventual clonal composition within each microenvironment. After a particular clone has expanded in a given tumor microenvironment, it becomes easier to observe that clone's population dynamics and subsequently the evolutionary forces at play.
1.6 Evolved Tumor Suppression
Just as maladaptive traits in evolution will be missing from the fossil record, as cancer biologists, we are largely left to study the successes of somatic evolution – clinically detectable tumors. When cell numbers are small within a tissue or within a given tumor microenvironment, making them difficult to observe, it is challenging to understand the role that evolutionary forces play in favoring or eliminating cells. Because initial cell numbers are low following a mutational event, observation of evolutionary forces in early oncogenesis provides a significant technical challenge, often causing these forces to be ignored in early oncogenesis.
With low rates of cancer in youth, it is clear that natural selection has done a good job at limiting cancer incidence in animals during times of likely reproductive success. Given the sheer number of cell divisions required to build large animal bodies, such as those of mammals, evasion of mutations as an evolutionary strategy appears to have been impractical, and we argue that evolution has limited early oncogenesis through controlling the microenvironment. Taking a cue from evolution, strategies to manipulate tissue microenvironments could provide a therapeutically relevant approach to not only eliminate cancer, but also to prevent it. With a better understanding of the impact that evolutionary forces have on changing tissue microenvironments, not only will the process of oncogenesis become clearer, but so too will our entire understanding of cancer biology.
2. Mutations and Cancer
2.1 Somatic Mutation Theory of Cancer
Perhaps the most widely accepted theory as to how functional somatic cells become oncogenic is the SMT of cancer [5,6]. A great deal of work has shaped SMT. With time this theory matured into the idea that the consecutive occurrence of randomly occurring rare somatic mutations yields some low but significant probability that a functional gene would be altered in some way that would provide a fitness advantage to the affected cell, such as by increasing cell cycling or survival, leading to progressive cancer development.
Being elegantly simplistic, the idea that random rare somatic mutations occurring over an organism's lifetime could transform a cell and cause cancer, has been widely accepted for over half a century [6,16,38]. By slowly accumulating somatic mutations at a static rate, the probability that a susceptible cell could accumulate the multiple mutations required to generate a cancer would increase exponentially, seemingly providing a good fit to the observed cancer incidence rates for many human cancers [2].
SMT has become so widely accepted in cancer biology that there have been a number of attempts to attribute tissue-specific cancer risk solely to cell division rates [14,39]. In this and other models, aging simply reflects the time required for mutation accumulation. Accordingly, the impact of aging on tissue structure/function, stem cell activity, immune function, hormone levels, etc., are therefore not viewed as critical for understanding the carcinogenic process. However, the idea that mutation incidence and thereby cell division rates are the sole governors of oncogenesis demands that a number of implausible predictions be true.
2.2 Complications with Somatic Mutation Theory
While SMT appears to provide a straightforward mechanism of oncogenesis, there are a number of discrepancies between predictions made by this model and empirical observations of biology. Given that mutations largely originate from internal sources of DNA damage, such as replication errors, cell division rates play a major role in mutation incidence [40]. Were SMT sufficient to predict oncogenesis purely through the incidence of mutations, it would be expected that the more rapidly cells cycle, the greater the risk and incidence of cancer would be. On the surface, this idea appears plausible. For instance, human colorectal cancers are more frequent than osteosarcomas, in accordance with the more frequent division of stem cells in the colorectal tissue compared to the bone and muscle [14]. While this appears consistent with the theory, ontogeny presents a problem. During the development of an organism, stem cells are responsible for an immense number of cell divisions, as a full organism must be built from a single fertilized egg. This substantial amount of replication will result in a corresponding accumulation of mutations. Yet, most human cancers show low incidence rates up to and through much of adulthood, with exponential increases late in life. The substantial accumulation of mutations early in life creates a quandary: why are most cancers relegated to late in life?
A highly influential study by Welch et al [41] has been heavily employed in support of SMT, as the authors show that mutations accumulate in early hematopoietic progenitor cells (HPC) with late-life kinetics that mirror the pattern of leukemia risk. Thus, the late-life incidence of many leukemias could apparently be rate-limited by mutation accumulation. However, we would argue that this analysis of mutations in HPC is insufficient to derive the pattern of mutation accumulation across human lifetimes. First, the study is underpowered, given that HPC from only seven individuals across 80 years are studied, with three HPC sequenced per person (given the expense, analyses of only a few subjects is understandable). High inter-individual and intra-individual variability in mutation number per HPC is clearly evident from the analyses of AML genomes presented in the same paper – AML mutation burden is proposed by the authors to serve as a proxy for mutation accumulation in the HPC from which it originated. Indeed, the pattern for mutation accumulation in AML genomes is very different from the one presented for normal HPC, with about half of mutations accumulating by human maturity (∼18 years of age) [42,43]. Moreover, data from both mice [44] and humans [45] demonstrate that human HPCs (and also probably HSCs) divide very rapidly during ontogeny and early postnatal growth, and then slow to about one division per year for most of adult life. This pattern of cell division is more consistent with other measurements of mutation and epimutation accumulation in hematopoietic cells in mice and humans [42,46,47], which show substantial early life accumulation of mutations. For mutations to primarily accumulate starting in the 4th decade of life, as suggested by Welch et al, one would need to assume substantially higher (10 to 100-fold) higher mutation rates per cell division late in life. Finally, the authors used a filter to remove mutations in HPC that were present in more than 5% of sequenced total blood, which could have removed mutations that occurred early in life that reached higher allele frequency by drift. While this work is often used in support of the deterministic role that mutation rate plays in cancer incidence, it is incomplete, and warrants further exploration.
A recent study by Blokzijl et al provides critical new insight into the numbers of mutations that accumulate in human stem cells [39]. However, this study is similarly underpowered in its attempts to determine mutation accumulation kinetics with age, due to significant gaps in the age range and number of subjects analyzed (again, understandable given costs). While a regression line was drawn to represent a linear accumulation of mutations with age, such a linear relationship cannot be derived from current data. Together with the study by Welch et al [41], this study emphasizes the great need for more data quantifying mutation accumulation in individual stem and progenitor cells in humans. Interestingly, while Blokzijl et al demonstrated that individual stem cells from human liver, small intestine and large intestine each accumulated about 2500 mutations in a lifetime, cancer incidence is 5-30 times higher for the large intestine than the other two tissues. While other factors need to be considered, this result is not consistent with cancer incidence being limited by the lifetime accumulation of mutations. Though data for mice are limited, murine stem cells in the large and small intestines accumulate roughly 250 and 600 mutations in a lifetime [48]. We can thus estimate that each human intestinal stem cell accumulates 5-10-fold more mutations than in a mouse, likely due to their longer lifespans. Given that humans have about 1000-fold more cells than a mouse, assuming that this ratio holds for intestines, the cumulative lifetime mutation load for human intestines is roughly 5,000 to 10,000-fold greater than for mouse intestines. In all, it is difficult to explain differences in cancer risk between different tissues and between different species if cancer is assumed to be limited by mutation accumulation.
SMT essentially set's up Peto's Paradox, in that larger size and longer lifespans should lead to many more mutations accumulating in a lifetime, and yet cancer risk does not scale with either body size or lifespan [13,24]. Per the logic of SMT, the greater the number of cells generated, the greater the probability that any one cell would acquire an oncogenic mutation. Yet larger, longer-lived organisms like whales do not suffer a correspondingly higher cancer rate than do much smaller, short-lived ones like mice. Clearly, evolution has selected for mechanisms that limit cancer despite large sizes and/or long lives. A common suggestion is that larger organisms may simply have evolved novel tumor suppressor mechanisms that can compensate for the increased numbers of mutations [49]. Often cited in support of this idea is the low cancer incidence rate in naked mole rats. This low cancer incidence is proposed to be, at least in part, mediated by the anti-transformation effects of high-molecular-mass hyaluronan [50,51]. Similarly, elephants have been shown to possess around 20 copies of the tumor suppressor gene p53, providing a potential explanation for Peto's paradox in this family [52]. However, analyses of tumor suppressor gene copy numbers across 36 mammalian species revealed that the elephants were the only species to clearly exhibit p53 copy number expansions, and that increased tumor suppressor gene ploidy does not correlate with body size in general. In all, acquisition of extra tumor suppressor genes does not appear to be a general mechanism of cancer suppression in large vertebrates. Finally, the evolution of large multicellular organisms has not come hand-in-hand with reductions in mutation rates – in fact, mutation rates actually appear higher in larger animals relative to simpler animals or to our unicellular relatives [53].
Analogous to the SMT for aging associated cancer risk, carcinogens are believed to increase cancer incidence through an increased mutation rate. Smoking substantially increases the risk of more than a dozen cancers, and indeed cigarette smoke possesses known mutagens [54]. While increasing mutation frequency should contribute to cancer risk, there are disconnects that suggest a more complicated relationship. First, while lung adenocarcinomas associated with smoking exhibit about 4.5 times more mutations than those from nonsmokers, the risk of developing these cancers is more than 20 times higher in smokers. More interestingly, analyses of mutations in the same cancer types from smokers and nonsmokers reveals that for most sites there is no greater burden of mutations in the cancers from smokers relative to nonsmokers (Figure 1). For example, lung squamous cell carcinomas (SCC) do not show higher mutation burden than lung SCC in nonsmokers. And yet the risk of lung SCC is more than 100-fold greater in smokers. For all cancers combined, smoking is only associated with a 1.15 fold increase in mutations relative to these cancers from nonsmokers. While the authors still conclude that smoking causes cancers by increasing mutation burden, the data presented do not support this conclusion. Other factors are clearly at play – smoking does more to tissues than just inducing mutations.
Figure 1. Increase in Cancer Risk Does not Correlate with Mutation Accumulation Rate.
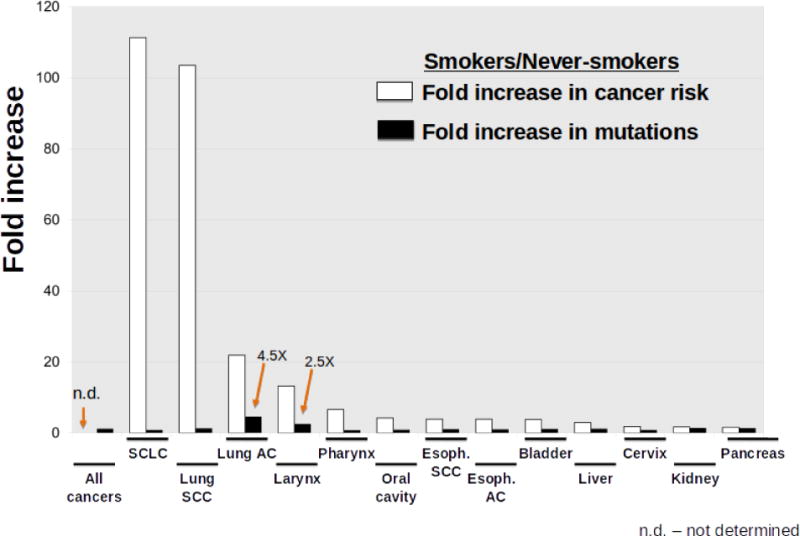
The graph shows the increased risk of certain cancers (white bars) and the increased mutation load within those cancers (black bars), comparing cancers in smokers to those in nonsmokers. Data are graphed from Alexandrov et al [138]. The fold increase in mutations for SCLC is based on only three cancers from nonsmokers, and thus firm conclusions for this cancer will require larger sample size. Abbreviations: Esoph. (esophageal); SCLC (small cell lung cancer); SCC (squamous cell cancer); AC (adenocarcinoma).
2.3 Mutation Incidence and Somatic Genetic Diversity
Mutations, as with other heritable changes, play an important role in tumorigenesis, as they generate phenotypic diversity within somatic cell populations upon which selection can act. Of course, mutations are a primary mechanism of converting proto-oncogenes into functioning oncogenes, and disabling or diminishing the functionality of tumor suppressor genes. Epigenetic modifications can also provide phenotypic diversity for selection, and contribute to cancer phenotypes. It is important to consider that human tissues possess a large amount of epigenetic and genetic diversity, which can inform our understanding of early oncogenesis. Evolutionary forces, such as selection, drift and migration can help reconcile the discrepancy between the kinetics of mutation occurrence, cumulative load of mutations, and cancer incidence.
Past studies have used DNA sequencing to understand the numbers of single nucleotide variants (SNVs) that are present in different human tissues [39,41,55]. However, sequencing or detection technologies are often inadequate to accurately understand total tissue mutation loads across human lifespan. In particular, the failure to detect mutations present in only a fraction of somatic cells cannot be parlayed into conclusions about their absence. One approach to understand mutation loads has been to look at small pieces of tissue in order to increase detection resolution. One group found that in just 1 cm2 of healthy skin biopsies, thousands of mutations can be found, with up to 50 being within known oncogenes [29]. Still, it is important to consider that the detection of mutations in normal tissues is complicated by the limit of detection for allelic variants – only variants that reach some frequency threshold, such as following clonal expansion, will be observed. However, in the absence of technological advances, human mutation loads can be estimated using mathematics in order to reveal how much genetic diversity is present at a given time. Given our good understanding of stem cell numbers and dynamics in this tissue, the hematopoietic system can be used to estimate mutation loads in stem and progenitor cells that have the capability to form a cancer.
While cell cycling, and therefore mutation incidence, will be significantly higher during development [44], for the sake of simplification, adult numbers will be used to estimate mutation loads from maturity to old age, which should represent about half of total mutations accumulated in a lifetime [46]. In human adults, mutation rates in hematopoietic cells during replication are approximately 1.3 × 10-8 mutations per base for stem cell divisions [30], though this number may be as high as 2-4 × 10-7 [56]. Using AML to trace back mutations to a single clone predicts a similar mutation rate of about 5 × 10-9 mutations per base pair [41]. A low estimate for adult hematopoietic stem cell (HSC) division rates is approximately 1.3 times per year [57,58], though this will at times be higher, especially during development. Finally, total numbers of HSCs in the hematopoietic system have been estimated at 11,000 [59] and could be as high as 300,000 [60], a number that appears to increase with age [61]. To account for the increased cell division rates of ontogeny, the total mutation count will be doubled. Using the lowest estimates from the above numbers, a conservative mutation incidence within hematopoietic stem cells can be derived for an average human lifespan.
This result suggests that in an 80 year human lifespan, 6% of the bases in the haploid genome will have been mutated in at least one HSC at some point in time. Using the larger estimated HSC population size (300,000), over 80% of the diploid genome will be mutated at least once over the course of an 80 year human lifespan.
While this number is already quite high, it would be misleading to exclude hematopoietic progenitor cells from this estimate, as they too may be susceptible to malignant transformation. There are about 7.53 × 1011 nucleated cells in the bone marrow [62], of which about 0.02% are common lymphoid progenitors (CLPs) and about 0.02% are common myeloid progenitors (CMPs) [63].
This means that just between the progenitor populations of CMPs and CLPs there are about 300 × 106 cells at any given time. With a hematopoietic progenitor division rate around 2 divisions per day [61,64], the time until every base across the human genome will be mutated somewhere within just the progenitor pool of CLPs and CMPs is a diminutive 1.5 hours.
This number is still likely a substantial underestimation of susceptible cell mutation loads, since cells as differentiated as immature thymocytes are capable of cancer formation, of which there are another 5×1010 cells just in that cell pool [65,66].
If every base in the human genome is mutated at least once every 1.5 hours, over an 80 year lifespan, the entire genome will be mutated some 120,000 times in hematopoietic cells that may be capable of tumor formation. Of course, leukemogenesis may in some cases require initiation in an HSC, in which case the frequency of oncogenic mutations in this compartment becomes lower. Regardless, the question remains as to how a gene pool with this much genetic diversity is capable of largely avoiding oncogenic clonal expansions for the first four decades of human life [67].
3. The Role of Evolution in Oncogenesis
3.1 Evolutionary Forces
Cell number, cell division rate and mutation incidence will certainly play a role in providing the necessary heritable changes for tumorigenesis. However, given that oncogenic mutations are essentially ubiquitous in human tissues, these factors cannot alone explain the age-dependent pattern of oncogenesis. Considering the impact of evolutionary forces such as selection and drift on the genetic composition of a stem and progenitor cell pool may contribute to a more complete understanding of early oncogenesis [68–71].
Natural selection is the process of differential survival and reproductive success among organisms based on their relative fitness, and it typically leads to the overrepresentation of individuals in a population that are better adapted to their environment. Maladaptive traits are eliminated. For a population that is well adapted to its environment, selection will primarily act to maintain the status quo, via stabilizing selection, as most phenotypic changes will reduce adaptation when little room for improvement is left. The history of life on Earth shows periods of long-term stasis for many species (sometimes for millions of years), punctuated by periods of rapid speciation that coincide with major environmental changes [72]. Environmental change is therefore viewed as a major driver of evolutionary change. Similarly, at the cellular level in a young, healthy tissue, cells should be well adapted the tissue microenvironment (on a local peak of a fitness landscape, Figure 2A). With age or upon exposure to environmental toxins or carcinogens, environmental pressures will change. These changes will select for different cellular phenotypes adaptive to the new microenvironments (Figure 2B). While a stem cell will be well adapted to survive in the presence of the evolved levels of survival and stemness signals in a youthful microenvironment, if these signals are altered, selection may now act to select for mutant clones that restore or overcome the emerging limitations.
Figure 2. Changing Landscapes in Oncogenesis.
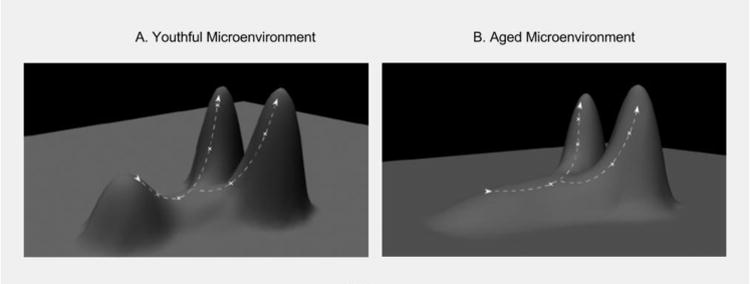
A) In a young and healthy stem cell niche, stem and progenitor cells are well adapted to their environments. On a fitness landscape, this state can be conceptualized as a local fitness peak (short peak). The x-y plane reflects all possible genotypes, and thus different cellular phenotypes change. Phenotypic change results in movement on the fitness landscape. When near the apex of a fitness peak, mutations that result in a phenotypic change will likely be disadvantageous, resulting in downhill movement on the landscape. Because this mutant cell is now less adapted to its environment than its peers, it will likely be eliminated from the gene pool by competition from cellular peers. This elimination will make it improbable for the cell clone to acquire a subsequent oncogenic mutation. Because youthful environments experience strong purifying selection, most phenotypic changes will be detrimental to a cell's survival. B) In an aged individual, stem and progenitor cells are less adapted to their age-altered tissue microenvironment, thus lowering the overall fitness peak of the pool. With this lowered fitness peak purifying selection is relaxed, allowing for the survival of different stem cell phenotypes. This altered fitness landscape increases the probability that a stem cell can achieve a higher position (greater fitness) on the landscape through a single mutational step. Subsequent oncogenic mutations may then allow the original cell clone to ‘climb’ to higher fitness positions, in some cases generating more aggressive cancerous phenotypes.
Drift is another important evolutionary force that reflects the impact of chance on allele frequency distributions in populations. In sufficiently large populations, the allele frequency of a given mutation is not likely to be altered without a change in fitness [69]. Yet when population numbers are reduced, random changes in allele frequencies can play a significant role in fixation within a population. For example, if an environment contains only 5 stem cells, and one of them disappears from the pool, this may change the frequency of particular alleles by 20%. Compared with a stem cell pool containing 10,000 cells, the loss of any single cell would only cause at most 0.01% allele frequency change. The changing impact of drift during human aging is important to consider, as stem cell population sizes can vary considerably over an 80 year lifespan, the most profound difference being between the fetal and early postnatal period and adults.
While both drift and selection affect the organisms or cells already contained within an environment, migration describes the movement of individuals into or out of environments. Some stem cells might randomly migrate from their niches and later end up in a new niche, thus changing the diversity of somatic variants in certain parts of the tissue. This can be an important consideration for some tissues and a negligible factor for others. For example, intestinal crypts contain isolated pockets of stem cells that compete for niche space within each crypt separately, thus representing effectively many small populations where drift has been shown to be a major determinant of stem cell clonal dynamics [73–75]. The hematopoietic and mesenchymal stem cell systems, on the other hand, allow for the migration of stem cells through bloodstream to other bones or parts of the same bone. These stem cells compete for niche space globally as one large population. Thus, unlike intestinal stem cells, migrating stem cells compete with thousands of their peers.
Bacterial evolution of antibiotic resistance provides an illustrative example for how these forces can impact allele frequencies within a population. Mutations causing antibiotic resistance will arise spontaneously within bacterial populations [76]. If an antibiotic is present within the environment, such mutant bacteria will have survival advantage over their peers by escaping the toxic effects of the antibiotic [77,78]. Developing antibiotic resistance is however not without cost. Bacteria often use membrane pumps, or enzymes to deal with antibiotics. Both of these methods expend substantial amounts of ATP, and therefore impose an energetic cost of managing antibiotics [79,80]. In the absence of antibiotic, this added energetic cost might be maladaptive, reducing fitness relative to peers without antibiotic resistance [81]. Thus, it is only when antibiotic is present that resistance mutations become beneficial to a cell, emphasizing the critical role of environment as a determinant of the fitness effects of genetic alleles. Drift and migration can also play an important role in this system as they can introduce new alleles or spontaneously remove alleles from the pool.
Extending these evolutionary principles to early oncogenesis allows an appreciation of the importance of microenvironmental changes mediated by age-related tissue decline, smoking-caused tissue damage, and other insults in carcinogenesis. Tissue changes should be a key link between cancer and its causes.
3.2 Aging-Related Functional Decline of the Microenvironment
One of the primary risk factors for cancer in humans and other organisms is age. If the evolutionary forces of mutation, selection, drift and migration are important in shaping cellular gene pools, then it becomes important to understand how these forces change as an organism ages. We should expect that the many aging-associated changes, such as deregulated nutrient sensing, impaired intracellular signaling, increased cellular senescence, increased inflammation, and stem cell exhaustion [32] should contribute to altered evolutionary pressures in the soma as an organism ages. In a youthful background where stem cells are ideally adapted to their environments, an oncogenic mutation is likely to decrease somatic cell fitness, such as by favoring apoptosis, senescence, or differentiation. In an aged background, particular oncogenic mutations may be beneficial by compensating for some signaling or functional deficiency either in the cell or in its environment (as illustrated in Figure 2). In order to fully understand the process of early oncogenesis, many sources of change must be considered including cell extrinsic microenvironmental functional decline, cell intrinsic functional decline, stem cell population changes, and changes in competition between stem cells.
It is important to understand why physiological aging occurs in humans with age in the first place. For all animals, the strength of natural selection for the maintenance of tissue fitness (the soma) wanes as the odds of reproduction decline, most often due to reduced probability of survival [82,83]. For humans, the chances of reproduction for the vast majority of our evolutionary history rapidly declined in older ages (certainly past age 40, given low rates of survival past this age [84]). Thus, further investment in tissue maintenance past years of likely reproduction would doubtfully pay off in terms of reproductive success (the true measure of fitness) [85]. Thus, evolutionary investment in tissue maintenance has been tuned to maximize reproductive success. With the benefits of modern living, however, human lifespan now extends well past historical reproductive years. Given limited investment in long-term somatic maintenance, aging-related pathologies begin to set in, such as aging-associated inflammation (inflammaging) [86], senescent cell accumulation [87], and dysregulated niche function [88]. As these problems mostly manifest in post-reproductive years, their cost to the fitness value of individual genetic alleles in the population are minimal. These aging-related challenges should be important factors in early oncogenesis, as they must alter the selective pressures to which stem cells are subjected.
The effects of evolved programs for somatic maintenance will clearly impact oncogenesis. First, we argue that natural selection has favored the co-evolution of stem cells and their tissue niches such that the stem cells are well-adapted to the tissue, at least through the youthful periods of probable reproductive success. As such, stabilizing selection in well-adapted stem cell populations should favor the status quo, eliminating cells with phenotype-altering mutations (even when potentially oncogenic). Thus, tumor suppression during youth, which is important for reproductive success, can at least in part result from the lack of selective pressure for somatic cell improvement (Figure 2A). Since we argue that stabilizing selection is an inherent effect of preventing somatic tissue decline, the evolution of any new tumor suppressive innovations was not required for this mechanism. However, the state of our tissues decline as we age (or through exposures such as to tobacco smoke). If stem cells acquire an oncogenic mutation that compensates for any age-related apoptotic or differentiative bias, they may be more likely to outcompete their peers and survive (Figure 2B), just as antibiotic-resistant bacteria would outcompete their peers in the presence of antibiotic.
Evidence for the role of an aged environment in oncogenesis dates back many years. In multiple studies, when tumor cells are introduced into young healthy environments, they fail to engraft, yet when put into an aged background can do so robustly [89–93]. This phenomenon has been correlated with specific age-related microenvironmental changes [94]. Another prominent example of the context-dependence of malignant cell transformation can be observed with Rous Sarcoma Virus (RSV) exposure. While in-vitro RSV transduction of chicken cells results in oncogenic transformation [95], in-vivo injection only results in tumor formation at injury-induced inflammatory sites [92].
Like injury-induced inflammation, aging-associated chronic inflammation may be acting similarly to promote oncogenesis. This aging-associated chronic inflammation might be promoting oncogenesis by providing an environment that is permissive to the expansion of oncogenically initiated cells. In allowing for the expansion of oncogenic cells, chronic inflammation can promote the survival and expansion of the phenotypes that are involved in tumor [75,96,97].
3.3 Stem Cell Functional Decline
The functions of stem and progenitor cells decline in old age, mediated both by microenvironmental and cell-intrinsic changes [98]. In the hematopoietic system, old HSCs are less competitive than young cells, and they have a reduced ability to repopulate a host [99–105]. What is true for HSCs is also true in other tissues such as intestinal stem cells, which are more susceptible to differentiate or apoptose under stress, and possess a reduced regenerative potential [106]. It is often however, unclear whether the stem cell or the niche is the source of the functional decline both in the hematopoietic system and in other tissues [105], and there is clear evidence for cell autonomous and non-autonomous contributions to stem cell aging [98].
Alterations in tissue microenvironments and reductions in stem cell pool fitness can promote oncogenic adaptation. Figure 3 shows a simplistic representation of Fisher's Geometric Model [107], demonstrating how adaptation to a new environment involves selection for new trait values, with adaptive trait changes become smaller and rarer as the population approaches the phenotypic optimum. Within the hematopoietic system, the expression of particular oncogenes (such as activated NRAS) can restore multiple parameters of B-progenitor fitness that become impaired in old age, leading to the expansion of NRAS expressing progenitors in old, but not young, murine bone marrow environments. Importantly, B-progenitor fitness reductions and increased selection for oncogene-bearing clones has been shown to be mediated by the heightened inflammatory environment in aged mice [97]. In order to fully understand early oncogenesis, the mechanisms behind the varied effects of the same oncogenic mutations must be better understood. Age-related tissue decline can help explain some of these varying behaviors of the same oncogenic changes.
Figure 3.
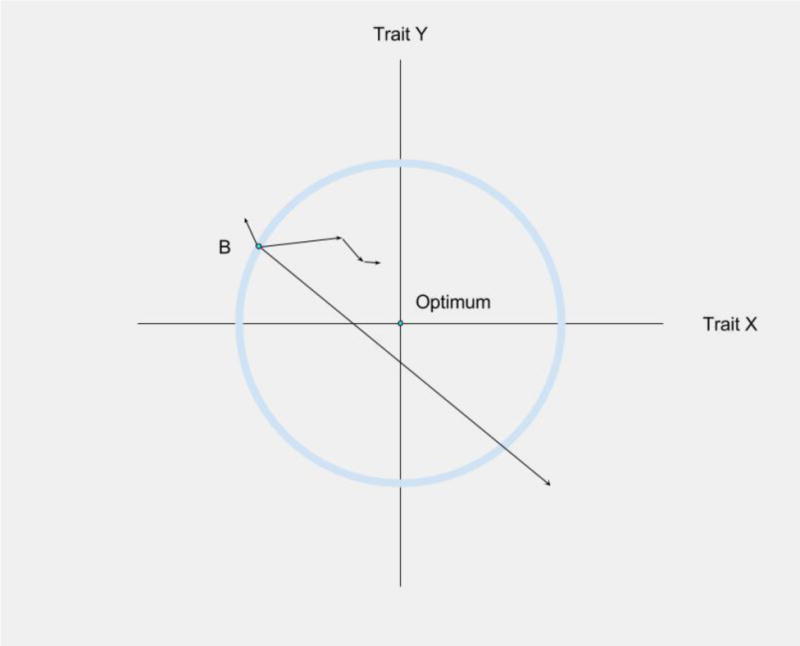
Phenotype Adaptation to a Microenvironment. Fisher's Geometric Model can be used to visualize how a cell's phenotypic changes will impact its fitness In this simplified version of Fisher's Geometric Model [139], involving only two traits, an environmental change has resulted in maladaptation – phenotype B is no longer optimal for either trait (X and Y axes). Mutations will lead to random changes in phenotypes (including for X and Y), and mutations that fall outside the circle will be maladaptive, and mutations leading to phenotypes within the circle will be adaptive. An adaptive walk towards the new phenotypic optimum will involve positive selection for mutations that improve fitness, and theory and experimental studies indicate that earlier mutations will typically exhibit greater effects on phenotype [140,141]. Importantly, the closer a population is to the phenotypic optimum (Optimum), the less likely phenotypic change is to be adaptive, with selection for progressively smaller phenotypic changes selected. Most phenotypic change will now cause movement away from the optimum. Figure adapted from Orr et al [140].
3.4 Aging-Associated Clonality
Given that environmental factors such as functional decline, cytokine signaling, and inflammation can influence the relative selective advantage or disadvantage of a particular oncogenic mutation, it is not surprising that stem cell numbers and clonality change dramatically with age, with important implications for oncogenesis. With age, while hematopoietic cell numbers increase [61,103,108], the genetic diversity drops as the system trends towards clonality [109–111]. This increased clonality with age in itself is associated with an increased risk of cancer [112,113]. Multiple recent studies have demonstrated that clonality within the hematopoietic system increases dramatically in old age, being rarely present before 40 years of age but detectable in up to 15% of people in later decades. The occurrence of clonal expansions is informative about the health of the system, as these clonal expansions typically contain oncogenic mutations and are correlated with the risk of multiple other diseases, including leukemias [114–116]. Of note, these clones typically only contain a single oncogenic mutation indicating that multiple lesions are not necessary for expansion. We can entertain three possible explanations: 1) mutations are largely restricted to ages past 40, 2) expansions start early, but given very minor fitness advantage of the mutations, take decades to reach detectable abundance, and 3) the fitness values of particular mutations are very different in young and old individuals. As over half of mutations accumulate in the hematopoietic system by the time we are 18-20, paralleling the much more rapid HSC division rates during ontogeny (as described in section 2.3), the first explanation is unlikely. While the second explanation cannot be ruled out for all mutations, there is direct evidence that oncogenes can substantially impact the competitive expansion of old hematopoietic progenitors, while not providing any fitness advantage to young progenitors [97], supporting the third explanation. If aging is understood as a process that changes the selective pressures within the hematopoietic system, then with age, particular oncogenic mutations that are maladaptive in early hematopoietic progenitors in youth can become adaptive in progenitors in the aged bone marrow microenvironment, promoting the clonal expansion of HSC and progenitors that experience these mutations.
3.5 Competition
Direct cell-to-cell competition is another important evolutionary mechanism of eliminating alleles from a gene pool based on their competitive fitness. The evolution of multicellularity did not involve the surrendering of individual cell fitness, and instead the cell competition among stem cells can maintain pool fitness just as such competition can maintain the high fitness of yeast or bacteria populations. Competitive fitness can be as simple as a faster cycling rate, or more complicated like induction of apoptosis in neighboring cells [117] or changes in self-renewal odds [118]. One of the earliest examples of direct cell-to-cell competition came from Drosophila experiments showing that elimination of a single copy of the ribosomal gene Minute creates a ‘loser’ phenotype in cells [119,120]. If surrounded by other cells that lack a copy of Minute, cells are able survive. If, however, Minute cells are surrounded by wild-type (WT) cells, they are eliminated from the cell pool. The WT cells are able to outcompete Minute cells for Decapentaplegic (Dpp) binding (Bone Morphogenetic Protein ortholog), resulting in the apoptosis of Minute cells only when WT cells are present to compete for the ligand [121]. Given that Minute cells are less functional (reduced protein synthesis ability), cell competition is an important mechanism for maintaining tissue stem cell pool fitness.
But what about oncogenic mutations? It further appears that rather than just the binary presence or absence of an oncogenic mutation, allele frequency of the mutation within a pool of cells is the important determinant clonal expansion [65]. A good example of this can be seen in the Drosophila tumor suppressor genes Lethal Giant Larvae (lgl) and scribble (scrib). These genes play important roles in cell division and cell polarity [122], and when constitutively overexpressed promote diffuse tumor formation. Yet it WT cells are present, tumor formation is suppressed [123,124], and lgl and scrib mutant cells undergo JNK-driven apoptosis [125]. This outcompetition by WT cells is however dependent on the density of mutant cells. If there are sufficient numbers of mutant cells that they can form pockets or niches without any WT cells, the WT cells are incapable of inducing apoptosis in all of the mutants [126].
The ability of WT cells to eliminate oncogenically initiated clones extends to mammalian systems, such as in the large intestine. When epithelial cells acquire certain oncogenic mutations, they are physically extruded from the epithelial layer into the intestinal lumen, thereby eliminating cells with oncogenic potential [127]. However, this only occurs when WT cells surround the oncogenic cell. The ability for oncogenic cells to initially have such high numbers will be quite rare, given the occurrence of such mutations in individual cells. However, if aging environments are more permissive to oncogenic expansions, this context may allow oncogenic cells to more easily create isolated and contiguous clones, and thereby increase cancer risk. Similarly, drift-driven expansions of oncogenically-initiated clones within small stem cell pools, such as crypts of the large intestine, could facilitate interclonal protection from WT cell mediated elimination.
3.6 Insult Related Changes in Microenvironment
Though aging is a significant risk factor for tumorigenesis, external insults and carcinogens are major contributors to cancer risk. While many carcinogens can directly induce mutation accumulation, the microenvironmental changes induced by carcinogens are often ignored. For instance, while radiation can increase mutation incidence, it can also alter the selective pressures within stem cell pools. As such, a previously irradiated hematopoietic system can promote the survival and expansion of HSC carrying the same mutation that is disadvantageous in a non-irradiated background [128]. Similarly, while loss of p53 is not advantageous under steady-state conditions, acute irradiation of mice leads to potent selection for p53 loss within HSC pools [129,130]. Notably, selection for p53 mutation during chemotherapy has been shown to be relevant for the development of therapy-related acute myeloid leukemia [131].
The effects of changing selection are evident in the lungs of smokers. As described in section 2.2, smoking associated cancers do not show increases in mutation load relative to the same cancers in nonsmokers that sufficiently explain increased cancer risk. These observations suggest, as others have argued [132], that while altered mutation rates are certainly playing a role in lung cancer, they cannot fully explain the large increase in lung cancer risk in smokers (20-100 fold). Instead, the damaging effects of chronic smoking may be altering the selective pressures that would otherwise inhibit clonal expansions of oncogenically-initiated cells. Basically, smoking results in alterations in tissue landscapes that increase selection for mutations that are adaptive to these abnormal conditions. If we can obtain a better understanding of how insults alter tissue microenvironments, we can begin to tease out why some mutations can be both detrimental and beneficial to a cell depending on context.
4. Using Evolution to Inform Cancer Therapies
With a better understanding for how evolutionary forces such as selection, competition, and drift influence tumorigenesis, characterizing tissue microenvironments could be leveraged to improve cancer prognosis and enhance treatments. For example, the degree of clonality in a tissue has been shown to be informative of cancer risk, as well as the risks of other diseases [114,115].
Current treatment strategies are aimed primarily at eliminating all or the bulk of oncogenic clones. However, given that the fitness impact of mutations is dependent on context, perhaps a more effective strategy would be to manipulate the tissue microenvironment in order to alter the selective landscapes in a way that they suppress oncogenic expansion. This strategy could also include improvements to the fitness of more benign tumor cells [133].
Just as with antibiotic resistant bacteria discussed earlier, evolved chemotherapeutic resistance mechanisms are likely to carry an associated cost. As an example, when non-small cell lung cancers are treated with tyrosine kinase inhibitors (TKIs) such as gefitinib or erlotinib to inhibit EGFR signaling, they will inevitably develop secondary EGFR mutations. Chronic TKI administration enhances the relative fitness of cells bearing a secondary EGFR mutation, yet without any TKI these cells are at a relative fitness disadvantage [134]. Can we exploit the cost of resistance, such as by drug holidays or by targeting new dependencies engendered by resistance? For example, if chemotherapeutic doses are intelligently regulated to shape the evolution of cancers, survival rates for tumor-bearing animals can be substantially extended, delaying or preventing relapse with chemotherapy-resistant disease [135–137]. Moreover, if we could understand the microenvironmental contexts that lead to selection for particular oncogenic mutations, we could ask whether restoring particular tissue parameters to more normal (youthful) levels can reverse the adaptiveness of these oncogenic phenotypes. Can we direct the evolutionary trajectory of cancer by modulation of its microenvironment?
5. Conclusion
A great deal of past research has focused on understanding and treating cancers after they have formed sizable tumors. While these studies have been informative and useful, there is still a relative deficiency in our understanding of early oncogenesis. In part this is because it can be tremendously challenging to observe the behavior of small oncogenic clones in vivo. With advances in our ability to observe early oncogenesis, we will be able to observe how the fates of oncogenically-initiated cells change as organisms age or under different contexts (including exposures and diet). While technological improvements will allow for more accurate knowledge of human mutation loads and oncogenic clonal dynamics in healthy tissue, evolutionary theory can already inform and improve our understanding of early tumorigenesis.
The integration of evolutionary theory into our understanding of early oncogenesis will provide a more complete model of the process. Evolutionary theory will facilitate the incorporation of important physiological changes like aging-related declines in tissue functionality and carcinogen induced tissue destruction into our understanding of carcinogenesis. Moreover, evolutionary theory can help explain how differences in tissue hierarchy and structure may be affecting lifetime cancer risk. Not only will an understanding of how evolutionary forces shape the early stages of cancer development be scientifically informative, but it may play an important role in future cancer prevention and treatment strategies.
Acknowledgments
We would like to thank Andrii Rozhok, Mark Gregory and Andrew Weems of the University of Colorado for critical review of the manuscript, and Marco Fragale for Figure 2. These studies were supported by National Cancer Institute grants R01CA180175 and R21CA179501 to J.D. and 1F31CA196231 to L.A.L.
References
- 1.Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 2.Armitage P, Doll R. A two-stage theory of carcinogenesis in relation to the age distribution of human cancer. Br J Cancer. 1957;11:161–169. doi: 10.1038/bjc.1957.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nowell P. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 4.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–173. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg Ra. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Sottoriva A, Kang H, Ma Z, Graham Ta, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, Curtis C. A Big Bang model of human colorectal tumor growth. Nat Genet. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer–a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006 doi: 10.1038/nrc1799. http://www.nature.com/nrc/journal/v6/n2/abs/nrc1799.html. [DOI] [PubMed]
- 9.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Junttila MR, Evan GI. p53—a Jack of all trades but master of none. Nat Rev Cancer. 2009 doi: 10.1038/nrc2728. http://www.nature.com/nrc/journal/v9/n11/abs/nrc2728.html. [DOI] [PubMed]
- 11.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 12.DeGregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene. 2013;32:1869–1875. doi: 10.1038/onc.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peto R, Roe FJ, Lee PN, Levy L, Clack J. Cancer and ageing in mice and men. Br J Cancer. 1975;32:411–426. doi: 10.1038/bjc.1975.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu S, Powers S, Zhu W, Hannun YA. Substantial contribution of extrinsic risk factors to cancer development. Nature. 2016;529:43–47. doi: 10.1038/nature16166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaux DL. In defense of the somatic mutation theory of cancer. Bioessays. 2011;33:341–343. doi: 10.1002/bies.201100022. [DOI] [PubMed] [Google Scholar]
- 17.Soto AM, Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays. 2004;26:1097–1107. doi: 10.1002/bies.20087. [DOI] [PubMed] [Google Scholar]
- 18.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 20.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000;60:6788–6793. [PubMed] [Google Scholar]
- 23.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010 doi: 10.1038/nrm2858. http://www.nature.com/nrm/journal/v11/n3/abs/nrm2858.html. [DOI] [PubMed]
- 24.Caulin AFF, Maley CC. Peto's Paradox: evolution's prescription for cancer prevention. Trends Ecol Evol. 2011;26:175–182. doi: 10.1016/j.tree.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagy JD, Victor EM, Cropper JH. Why don't all whales have cancer? A novel hypothesis resolving Peto's paradox. Integr Comp Biol. 2007;47:317–328. doi: 10.1093/icb/icm062. [DOI] [PubMed] [Google Scholar]
- 26.Roche B, Hochberg ME, Caulin AF, Maley CC, Gatenby RA, Misse D, Thomas F. Natural resistance to cancers: a Darwinian hypothesis to explain Peto's paradox. BMC Cancer. 2012;12:387. doi: 10.1186/1471-2407-12-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank SA. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with …. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.0909343106. http://europepmc.org/abstract/med/19805033. [DOI] [PMC free article] [PubMed]
- 28.Podolskiy DI, Lobanov AV, Kryukov GV, Gladyshev VN. Analysis of cancer genomes reveals basic features of human aging and its role in cancer development. Nat Commun. 2016;7:12157. doi: 10.1038/ncomms12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Al Turki S, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR. UK10K Consortium, M.E. Hurles, Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–133. doi: 10.1038/ng.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rozhok AI, DeGregori J. Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proc Natl Acad Sci U S A. 2015;112:8914–8921. doi: 10.1073/pnas.1501713112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.López-Otín C, Blasco Ma, Partridge L, Serrano M, Kroemer G. The Hallmarks of Aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 34.Lipinski KA, Barber LJ, Davies MN, Ashenden M, Sottoriva A, Gerlinger M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer Res. 2016;2:49–63. doi: 10.1016/j.trecan.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, Mack M, Pipeleers D, In't Veld P, De Baetselier P, Van Ginderachter JA. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 37.Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat Genet. 2016;48:238–244. doi: 10.1038/ng.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curtis HJ. Formal discussion of: somatic mutations and carcinogenesis. Cancer Res. 1965;25:1305–1308. [PubMed] [Google Scholar]
- 39.Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EES, Verstegen MMA, van der Laan LJW, de Jonge J, IJzermans JNM, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, van Boxtel R. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 41.Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, Kandoth C, Fulton RS, McLellan MD, Dooling DJ, Wallis JW, Chen K, Harris CC, Schmidt HK, Kalicki-Veizer JM, Lu C, Zhang Q, Lin L, O'Laughlin MD, McMichael JF, Delehaunty KD, Fulton LA, Magrini VJ, McGrath SD, Demeter RT, Vickery TL, Hundal J, Cook LL, Swift GW, Reed JP, Alldredge PA, Wylie TN, Walker JR, Watson MA, Heath SE, Shannon WD, Varghese N, Nagarajan R, Payton JE, Baty JD, Kulkarni S, Klco JM, Tomasson MH, Westervelt P, Walter MJ, Graubert TA, DiPersio JF, Ding L, Mardis ER, Wilson RK. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rozhok AI, Salstrom JL, DeGregori J. Stochastic modeling indicates that aging and somatic evolution in the hematopoietic system are driven by non-cell-autonomous processes. Aging. 2014;6:1033–1048. doi: 10.18632/aging.100707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J Clin Invest. 2006;116:2808–2816. doi: 10.1172/JCI28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sidorov I, Kimura M, Yashin A, Aviv A. Leukocyte telomere dynamics and human hematopoietic stem cell kinetics during somatic growth. Exp Hematol. 2009;37:514–524. doi: 10.1016/j.exphem.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 46.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vijg J, Busuttil RA, Bahar R, Dollé MET. Aging and genome maintenance. Ann N Y Acad Sci. 2005;1055:35–47. doi: 10.1196/annals.1323.007. [DOI] [PubMed] [Google Scholar]
- 48.Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, Tarpey PS, Roerink S, Blokker J, Maddison M, Mudie L, Robinson B, Nik-Zainal S, Campbell P, Goldman N, van de Wetering M, Cuppen E, Clevers H, Stratton MR. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–425. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seluanov A, Hine C, Bozzella M, Hall A, Sasahara THC, Ribeiro AACM, Catania KC, Presgraves DC, Gorbunova V. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell. 2008;7:813–823. doi: 10.1111/j.1474-9726.2008.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tian X, Azpurua J, Hine C, Vaidya A, Myakishev-Rempel M, Ablaeva J, Mao Z, Nevo E, Gorbunova V, Seluanov A. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature. 2013;499:346–349. doi: 10.1038/nature12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorbunova V, Hine C, Tian X, Ablaeva J, Gudkov AV, Nevo E, Seluanov A. Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc Natl Acad Sci U S A. 2012;109:19392–19396. doi: 10.1073/pnas.1217211109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, Kiso WK, Schmitt DL, Waddell PJ, Bhaskara S, Jensen ST, Maley CC, Schiffman JD. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA. 2015;84112:1. doi: 10.1001/jama.2015.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26:345–352. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 55.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C, Shen D, Harris CC, Dooling DJ, Fulton RS, Fulton LL, Chen K, Schmidt H, Kalicki-Veizer J, Magrini VJ, Cook L, McGrath SD, Vickery TL, Wendl MC, Heath S, Watson MA, Link DC, Tomasson MH, Shannon WD, Payton JE, Kulkarni S, Westervelt P, Walter MJ, Graubert TA, Mardis ER, Wilson RK, DiPersio JF. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vickers MA, Hoy T, Lake H, Kyoizumi S, Boyse J, Hewitt M. Estimation of mutation rate at human glycophorin A locus in hematopoietic stem cell progenitors. Environ Mol Mutagen. 2002;39:333–341. doi: 10.1002/em.10076. [DOI] [PubMed] [Google Scholar]
- 57.Catlin SN, Busque L, Gale RE, Guttorp P, Abkowitz JL. The replication rate of human hematopoietic stem cells in vivo. Blood. 2011;117:4460–4466. doi: 10.1182/blood-2010-08-303537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shepherd BE, Guttorp P, Lansdorp PM, Abkowitz JL. Estimating human hematopoietic stem cell kinetics using granulocyte telomere lengths. Exp Hematol. 2004;32:1040–1050. doi: 10.1016/j.exphem.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 59.Abkowitz JL, Catlin SN, McCallie MT, Guttorp P. Evidence that the number of hematopoietic stem cells per animal is conserved in mammals. Blood. 2002;100:2665–2667. doi: 10.1182/blood-2002-03-0822. [DOI] [PubMed] [Google Scholar]
- 60.Wang J, Doedens M, Dick JE. Primitive human hematopoietic cells are enriched in cord blood compared with adult bone marrow or mobilized peripheral blood as measured by the …. Blood. 1997 http://www.bloodjournal.org/content/89/11/3919.short. [PubMed]
- 61.Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, Vitale L, Pelleri MC, Tassani S, Piva F, Perez-Amodio S, Strippoli P, Canaider S. An estimation of the number of cells in the human body. Ann Hum Biol. 2013;40:463–471. doi: 10.3109/03014460.2013.807878. [DOI] [PubMed] [Google Scholar]
- 63.Mayle A, Luo M, Jeong M, Goodell MA. Flow cytometry analysis of murine hematopoietic stem cells. Cytometry A. 2013;83:27–37. doi: 10.1002/cyto.a.22093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Manesso E, Teles J, Bryder D, Peterson C. Dynamical modelling of haematopoiesis: an integrated view over the system in homeostasis and under perturbation. J R Soc Interface. 2013;10:20120817. doi: 10.1098/rsif.2012.0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martins VC, Busch K, Juraeva D, Blum C, Ludwig C, Rasche V, Lasitschka F, Mastitsky SE, Brors B, Hielscher T, Fehling HJ, Rodewald HR. Cell competition is a tumour suppressor mechanism in the thymus. Nature. 2014;509:465–470. doi: 10.1038/nature13317. [DOI] [PubMed] [Google Scholar]
- 66.Haynes BF, Markert ML, Sempowski GD, Patel DD, Hale LP. The role of the thymus in immune reconstitution in aging, bone marrow transplantation, and HIV-1 infection. Annu Rev Immunol. 2000;18:529–560. doi: 10.1146/annurev.immunol.18.1.529. [DOI] [PubMed] [Google Scholar]
- 67.Rubin H. What keeps cells in tissues behaving normally in the face of myriad mutations? Bioessays. 2006;28:515–524. doi: 10.1002/bies.20403. [DOI] [PubMed] [Google Scholar]
- 68.Charlesworth B. Elements of evolutionary genetics. 2010 [Google Scholar]
- 69.Lande R. Natural Selection and Random Genetic Drift in Phenotypic Evolution. Evolution. 1976;30:314–334. doi: 10.1111/j.1558-5646.1976.tb00911.x. [DOI] [PubMed] [Google Scholar]
- 70.Lacy RC. Loss of genetic diversity from managed populations: interacting effects of drift, mutation, immigration, selection, and population subdivision. Conserv Biol. 1987 http://onlinelibrary.wiley.com/doi/10.1111/j.1523-1739.1987.tb00023.x/full.
- 71.Bagby GC, Fleischman AG. The stem cell fitness landscape and pathways of molecular leukemogenesis. Front Biosci. 2011;3:487–500. doi: 10.2741/s167. [DOI] [PubMed] [Google Scholar]
- 72.Eldredge N. The pattern of evolution. 1999 https://repository.library.georgetown.edu/handle/10822/546337.
- 73.Lopez-Garcia C, Klein AM, Simons BD, Winton DJ. Intestinal stem cell replacement follows a pattern of neutral drift. Science. 2010;330:822–825. doi: 10.1126/science.1196236. [DOI] [PubMed] [Google Scholar]
- 74.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, Clevers H. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 75.Vermeulen L, Morrissey E, van der Heijden M, Nicholson aM, Sottoriva A, Buczacki S, Kemp R, Tavare S, Winton DJ. Defining Stem Cell Dynamics in Models of Intestinal Tumor Initiation. Science. 2013;342:995–998. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- 76.Wang G, Wilson TJ, Jiang Q, Taylor DE. Spontaneous mutations that confer antibiotic resistance in Helicobacter pylori. Antimicrob Agents Chemother. 2001;45:727–733. doi: 10.1128/AAC.45.3.727-733.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baquero F, Blázquez J. Evolution of antibiotic resistance. Trends Ecol Evol. 1997;12:482–487. doi: 10.1016/s0169-5347(97)01223-8. [DOI] [PubMed] [Google Scholar]
- 78.Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andersson DI, Levin BR. The biological cost of antibiotic resistance. Curr Opin Microbiol. 1999 doi: 10.1016/s1369-5274(99)00005-3. http://www.sciencedirect.com/science/article/pii/S1369527499000053. [DOI] [PubMed]
- 80.Dever LA, Dermody TS. Mechanisms of bacterial resistance to antibiotics. Arch Intern Med. 1991;151:886–895. [PubMed] [Google Scholar]
- 81.Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 2010;8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 82.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Medawar PB. UNSOLVED problem of biology. Med J Aust. 1953;1:854–855. doi: 10.5694/j.1326-5377.1953.tb84985.x. [DOI] [PubMed] [Google Scholar]
- 84.Caspari R, Lee SH. Older age becomes common late in human evolution. Proc Natl Acad Sci U S A. 2004;101:10895–10900. doi: 10.1073/pnas.0402857101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kirkwood TBL. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 86.Franceschi C, Bonafè M, Valensin S. Inflamm- aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000 doi: 10.1111/j.1749-6632.2000.tb06651.x. http://onlinelibrary.wiley.com/doi/10.1111/j.1749-6632.2000.tb06651.x/full. [DOI] [PubMed]
- 87.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 88.Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- 89.Boudreau N, Reddy ST, Stoker AW, Fairman C. The embryonic environment and the extracellular matrix suppress oncogenic transformation by rous sarcoma virus in the chick embryo. Mol Cell Differ. 1995 [Google Scholar]
- 90.Illmensee K. Reversion of malignancy and normalized differentiation of teratocarcinoma cells in chimeric mice. Basic Life Sci. 1978;12:3–25. doi: 10.1007/978-1-4684-3390-6_1. [DOI] [PubMed] [Google Scholar]
- 91.Laconi E, Doratiotto S, Vineis P. The microenvironments of multistage carcinogenesis. Semin Cancer Biol. 2008;18:322–329. doi: 10.1016/j.semcancer.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 92.Martins-Green M, Boudreau N, Bissell MJ. Inflammation is responsible for the development of wound-induced tumors in chickens infected with Rous sarcoma virus. Cancer Res. 1994;54:4334–4341. [PubMed] [Google Scholar]
- 93.Park CC, Bissell MJ, Barcellos-Hoff MH. The influence of the microenvironment on the malignant phenotype. Mol Med Today. 2000 doi: 10.1016/s1357-4310(00)01756-1. http://www.sciencedirect.com/science/article/pii/S1357431000017561. [DOI] [PubMed]
- 94.McCullough KD, Coleman WB, Smith GJ, Grisham JW. Age-dependent induction of hepatic tumor regression by the tissue microenvironment after transplantation of neoplastically transformed rat liver epithelial cells into the liver. Cancer Res. 1997;57:1807–1813. [PubMed] [Google Scholar]
- 95.Bishop JM. Viral oncogenes. Cell. 1985;42:23–38. doi: 10.1016/s0092-8674(85)80098-2. [DOI] [PubMed] [Google Scholar]
- 96.Tlsty TD, Hein PW. Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet Dev. 2001;11:54–59. doi: 10.1016/s0959-437x(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 97.Henry CJ, Casás-Selves M, Kim J, Zaberezhnyy V, Aghili L, Daniel AE, Jimenez L, Azam T, McNamee EN, Clambey ET, Klawitter J, Serkova NJ, Tan AC, Dinarello CA, DeGregori J. Aging-associated inflammation promotes selection for adaptive oncogenic events in B cell progenitors. J Clin Invest. 2015;125:4666–4680. doi: 10.1172/JCI83024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Oh J, Lee YD, Wagers AJ. Stem cell aging: mechanisms, regulators and therapeutic opportunities. Nat Med. 2014;20:870–880. doi: 10.1038/nm.3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Geiger H, Zheng Y. Cdc42 and aging of hematopoietic stem cells. Curr Opin Hematol. 2013;20:295–300. doi: 10.1097/MOH.0b013e3283615aba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, Rossi DJ. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci U S A. 2010;107:5465–5470. doi: 10.1073/pnas.1000834107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 102.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192:1273–1280. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Warren LA, Rossi DJ. Stem cells and aging in the hematopoietic system. Mech Ageing Dev. 2009;130:46–53. doi: 10.1016/j.mad.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chambers SM, Goodell MA. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem Cell Rev. 2007;3:201–211. doi: 10.1007/s12015-007-0027-1. [DOI] [PubMed] [Google Scholar]
- 106.Martin K, Kirkwood TB, Potten CS. Age changes in stem cells of murine small intestinal crypts. Exp Cell Res. 1998;241:316–323. doi: 10.1006/excr.1998.4001. [DOI] [PubMed] [Google Scholar]
- 107.Fischer Ma, Moreno-Miralles I, Hunt A, Chyla BJ, Hiebert SW. Myeloid translocation gene 16 is required for maintenance of haematopoietic stem cell quiescence. EMBO J. 2012;31:1494–1505. doi: 10.1038/emboj.2011.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.de Haan G, Van Zant G. Dynamic changes in mouse hematopoietic stem cell numbers during aging. Blood. 1999;93:3294–3301. [PubMed] [Google Scholar]
- 109.Busque L, Mio R, Mattioli J, Brais E, Blais N, Lalonde Y, Maragh M, Gilliland DG. Nonrandom X-inactivation patterns in normal females: lyonization ratios vary with age. Blood. 1996;88:59–65. [PubMed] [Google Scholar]
- 110.Champion KM, Gilbert JG, Asimakopoulos FA, Hinshelwood S, Green AR. Clonal haemopoiesis in normal elderly women: implications for the myeloproliferative disorders and myelodysplastic syndromes. Br J Haematol. 1997;97:920–926. doi: 10.1046/j.1365-2141.1997.1933010.x. [DOI] [PubMed] [Google Scholar]
- 111.Vas V, Senger K, Dörr K, Niebel A, Geiger H. Aging of the microenvironment influences clonality in hematopoiesis. PLoS One. 2012;7:e42080. doi: 10.1371/journal.pone.0042080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, Wei Q, Wang LE, Lee JE, Barnes KC, Hansel NN, Mathias R, Daley D, Beaty TH, Scott AF, Ruczinski I, Scharpf RB, Bierut LJ, Hartz SM, Landi MT, Freedman ND, Goldin LR, Ginsburg D, Li J, Desch KC, Strom SS, Blot WJ, Signorello LB, Ingles SA, Chanock SJ, Berndt SI, Le Marchand L, Henderson BE, Monroe KR, Heit JA, de Andrade M, Armasu SM, Regnier C, Lowe WL, Hayes MG, Marazita ML, Feingold E, Murray JC, Melbye M, Feenstra B, Kang JH, Wiggs JL, Jarvik GP, McDavid AN, Seshan VE, Mirel DB, Crenshaw A, Sharopova N, Wise A, Shen J, Crosslin DR, Levine DM, Zheng X, Udren JI, Bennett S, Nelson SC, Gogarten SM, Conomos MP, Heagerty P, Manolio T, Pasquale LR, Haiman CA, Caporaso N, Weir BS. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner MJ, Cullen M, Epstein CG, Burdett L, Dean MC, Chatterjee N, Sampson J, Chung CC, Kovaks J, Gapstur SM, Stevens VL, Teras LT, Gaudet MM, Albanes D, Weinstein SJ, Virtamo J, Taylor PR, Freedman ND, Abnet CC, Goldstein AM, Hu N, Yu K, Yuan JM, Liao L, Ding T, Qiao YL, Gao YT, Koh WP, Xiang YB, Tang ZZ, Fan JH, Aldrich MC, Amos C, Blot WJ, Bock CH, Gillanders EM, Harris CC, Haiman CA, Henderson BE, Kolonel LN, Le Marchand L, McNeill LH, Rybicki BA, Schwartz AG, Signorello LB, Spitz MR, Wiencke JK, Wrensch M, Wu X, Zanetti KA, Ziegler RG, Figueroa JD, Garcia-Closas M, Malats N, Marenne G, Prokunina-Olsson L, Baris D, Schwenn M, Johnson A, Landi MT, Goldin L, Consonni D, Bertazzi PA, Rotunno M, Rajaraman P, Andersson U, Beane Freeman LE, Berg CD, Buring JE, Butler MA, Carreon T, Feychting M, Ahlbom A, Gaziano JM, Giles CG, Hallmans G, Hankinson SE, Hartge P, Henriksson R, Inskip PD, Johansen C, Landgren A, McKean-Cowdin R, Michaud DS, Melin BS, Peters U, Ruder AM, Sesso HD, Severi G, Shu XO, Visvanathan K, White E, Wolk A, Zeleniuch-Jacquotte A, Zheng W, Silverman DT, Kogevinas M, Gonzalez JR, Villa O, Li D, Duell EJ, Risch HA, Olson SH, Kooperberg C, Wolpin BM, Jiao L, Hassan M, Wheeler W, Arslan AA, Bueno-de-Mesquita HB, Fuchs CS, Gallinger S, Gross MD, Holly EA, Klein AP, LaCroix A, Mandelson MT, Petersen G, Boutron-Ruault MC, Bracci PM, Canzian F, Chang K, Cotterchio M, Giovannucci EL, Goggins M, Hoffman Bolton JA, Jenab M, Khaw KT, Krogh V, Kurtz RC, McWilliams RR, Mendelsohn JB, Rabe KG, Riboli E, Tjønneland A, Tobias GS, Trichopoulos D, Elena JW, Yu H, Amundadottir L, Stolzenberg-Solomon RZ, Kraft P, Schumacher F, Stram D, Savage SA, Mirabello L, Andrulis IL, Wunder JS, Patiño García A, Sierrasesúmaga L, Barkauskas DA, Gorlick RG, Purdue M, Chow WH, Moore LE, Schwartz KL, Davis FG, Hsing AW, Berndt SI, Black A, Wentzensen N, Brinton LA, Lissowska J, Peplonska B, McGlynn KA, Cook MB, Graubard BI, Kratz CP, Greene MH, Erickson RL, Hunter DJ, Thomas G, Hoover RN, Real FX, Fraumeni JF, Jr, Caporaso NE, Tucker M, Rothman N, Pérez-Jurado LA, Chanock SJ. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen Ha, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med. 2014:1–11. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landén M, Höglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, Grönberg H, Hultman CM, McCarroll SA. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, McMichael JF, Schmidt HK, Yellapantula V, Miller CA, Ozenberger BA, Welch JS, Link DC, Walter MJ, Mardis ER, Dipersio JF, Chen F, Wilson RK, Ley TJ, Ding L. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vincent JP, Fletcher AG, Baena-Lopez LA. Mechanisms and mechanics of cell competition in epithelia. Nat Rev Mol Cell Biol. 2013;14:581–591. doi: 10.1038/nrm3639. [DOI] [PubMed] [Google Scholar]
- 118.Fleenor CJ, Rozhok AI, Zaberezhnyy V, Mathew D, Kim J, Tan AC, Bernstein ID, DeGregori J. Contrasting roles for C/EBPα and Notch in irradiation-induced multipotent hematopoietic progenitor cell defects. Stem Cells. 2015;33:1345–1358. doi: 10.1002/stem.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gogna R, Shee K, Moreno E. Cell Competition During Growth and Regeneration. Annu Rev Genet. 2015;49:697–718. doi: 10.1146/annurev-genet-112414-055214. [DOI] [PubMed] [Google Scholar]
- 120.Morata G, Ripoll P. Minutes: mutants of drosophila autonomously affecting cell division rate. Dev Biol. 1975;42:211–221. doi: 10.1016/0012-1606(75)90330-9. [DOI] [PubMed] [Google Scholar]
- 121.Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–759. doi: 10.1038/416755a. [DOI] [PubMed] [Google Scholar]
- 122.Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008;132:583–597. doi: 10.1016/j.cell.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 123.Froldi F, Ziosi M, Garoia F, Pession A, Grzeschik NA, Bellosta P, Strand D, Richardson HE, Pession A, Grifoni D. The lethal giant larvae tumour suppressor mutation requires dMyc oncoprotein to promote clonal malignancy. BMC Biol. 2010;8:33. doi: 10.1186/1741-7007-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T. Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev Cell. 2009;16:458–465. doi: 10.1016/j.devcel.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brumby AM, Richardson HE. scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003;22:5769–5779. doi: 10.1093/emboj/cdg548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Menéndez J, Pérez-Garijo A, Calleja M, Morata G. A tumor-suppressing mechanism in Drosophila involving cell competition and the Hippo pathway. Proc Natl Acad Sci U S A. 2010;107:14651–14656. doi: 10.1073/pnas.1009376107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kajita M, Fujita Y. EDAC: Epithelial defence against cancer—cell competition between normal and transformed epithelial cells in mammals. J Biochem. 2015;158:15–23. doi: 10.1093/jb/mvv050. [DOI] [PubMed] [Google Scholar]
- 128.Marusyk A, Casás-Selves M, Henry CJ, Zaberezhnyy V, Klawitter J, Christians U, DeGregori J. Irradiation alters selection for oncogenic mutations in hematopoietic progenitors. Cancer Res. 2009;69:7262–7269. doi: 10.1158/0008-5472.CAN-09-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bondar T, Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Marusyk A, Porter CC, Zaberezhnyy V, DeGregori J. Irradiation selects for p53-deficient hematopoietic progenitors. PLoS Biol. 2010;8:e1000324. doi: 10.1371/journal.pbio.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL, Shen D, Hundal J, Fulton RS, Heath S, Baty JD, Klco JM, Ding L, Mardis ER, Westervelt P, DiPersio JF, Walter MJ, Graubert TA, Ley TJ, Druley TE, Link DC, Wilson RK. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rodin SN, Rodin AS. Human lung cancer and p53: the interplay between mutagenesis and selection. Proc Natl Acad Sci U S A. 2000;97:12244–12249. doi: 10.1073/pnas.180320897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Maley CC, Reid BJ, Forrest S. Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol Biomarkers Prev. 2004;13:1375–1384. [PubMed] [Google Scholar]
- 134.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, Wang L, Amato KR, Arcila M, Sos ML, Socci ND, Viale A, de Stanchina E, Ginsberg MS, Thomas RK, Kris MG, Inoue A, Ladanyi M, Miller VA, Michor F, Pao W. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Aktipis CA, Boddy AM, Gatenby Ra, Brown JS, Maley CC. Life history trade-offs in cancer evolution. Nat Rev Cancer. 2013;13:883–892. doi: 10.1038/nrc3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Orlando PA, Gatenby RA, Brown JS. Cancer treatment as a game: integrating evolutionary game theory into the optimal control of chemotherapy. Phys Biol. 2012;9:065007. doi: 10.1088/1478-3975/9/6/065007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Enriquez-Navas PM, Kam Y, Das T, Hassan S, Silva A, Foroutan P, Ruiz E, Martinez G, Minton S, Gillies RJ, Gatenby RA. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8:327ra24. doi: 10.1126/scitranslmed.aad7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata T, Campbell PJ, Vineis P, Phillips DH, Stratton MR. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fisher RA, Bennett JH. The Genetical Theory of Natural Selection: A Complete Variorum Edition. OUP Oxford; 1930. [Google Scholar]
- 140.Orr HA. The Population Genetics of Adaptation: The Distribution of Factors Fixed during Adaptive Evolution. Evolution. 1998;52:935–949. doi: 10.1111/j.1558-5646.1998.tb01823.x. [DOI] [PubMed] [Google Scholar]
- 141.Zhao ZM, Zhao B, Bai Y, Iamarino A, Gaffney SG, Schlessinger J, Lifton RP, Rimm DL, Townsend JP. Early and multiple origins of metastatic lineages within primary tumors. Proc Natl Acad Sci U S A. 2016;113:2140–2145. doi: 10.1073/pnas.1525677113. [DOI] [PMC free article] [PubMed] [Google Scholar]