

Abstract
Congenital heart disease is the most common birth defect, and due to major advances in medical and surgical management, there are now more adults living with CHD than children. Until recently, the cause of the majority of CHD was unknown. Advances in genomic technologies have discovered the genetic etiology of a significant fraction of CHD, while at the same time pointing to remarkable complexity in CHD genetics. This review will focus on the evidence for genetic causes underlying CHD and discuss data supporting both monogenic and complex genetic mechanisms underlying CHD. The discoveries from CHD genetic studies draw attention to biological pathways that simultaneously open the door to a better understanding of cardiac development, and impact clinical care of CHD patients. Finally, we address clinical genetic evaluation of patients and families affected by CHD.
Keywords: Congenital Heart Disease; Genetics, diagnostics; Genetics; genome
CHD epidemiology: evidence for genetics underlying CHD
Congenital heart disease (CHD) is a structural abnormality of the heart and/or great vessels that is present at birth1. It is the most common birth defect, affecting approximately 1% of all liveborn infants2. CHD results from perturbation of the normal program of cardiac development (Fig. 1A). Historically, CHD has been categorized based on a combination of final anatomic and physiologic phenotypes (Fig. 1B), such as conotruncal defects that affect the ventricular septum and outflow tract (CTD), defects that lead to obstruction to left ventricular outflow (LVO), defects resulting from abnormal left-right relationships within the heart (heterotaxy, HTX), defects affecting the inflow such as the mitral and tricuspid valve abnormalities seen in atrioventricular canal defect, and a broad range of other defects including isolated atrial or ventricular septal defects3. Approximately one third of patients with CHD have disease that is categorized as severe (comprising univentricular hearts, HTX, CTD, atrioventricular canal defects, total anomalous pulmonary venous return, left ventricular outflow obstruction, and right ventricular outflow obstruction except isolated valvar pulmonary stenosis) and require intervention in the first year of life4. Despite progress in medical and surgical treatments, CHD remains the leading cause of mortality from birth defects in the developed world. Further, among the world’s poorest populations, CHD has a greater contribution to cardiovascular disease associated disability-adjusted life-years than ischemic heart disease or stroke5.
Figure 1.
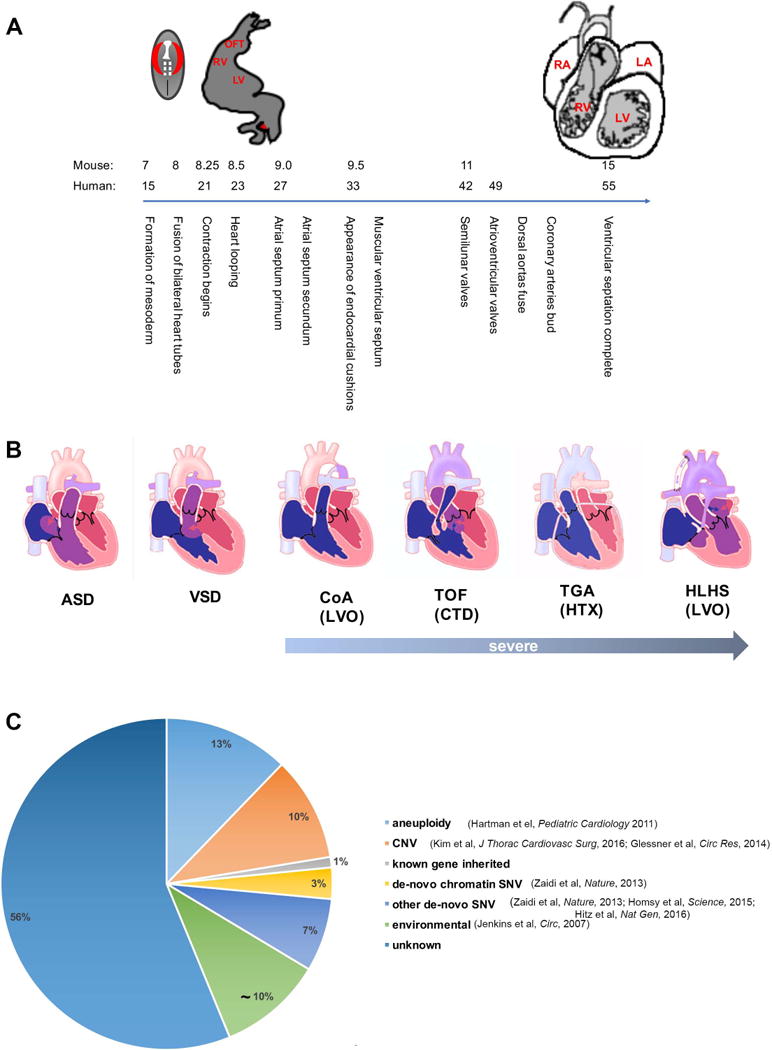
A Outline of human heart development. The X axis displays days of human and mouse gestation. B The spectrum of congenital heart disease from mild to severe. The lesions indicated as “severe” are expected to require intervention in the first year of life. ASD-Atrial Septal Defect, VSD-Ventricular Septal Defect, CoA- Coarctation of the Aorta, TOF-Tetralogy of Fallot, TGA- Transposition of the Great Arteries, HLHS- Hypoplastic Left Heart Syndrome. Classes of CHD based on proposed developmental-genetic mechanisms are indicated in parentheses; LVO-Left Ventricular Outflow Obstruction, CTD- Cono-Truncal Defect, HTX- Heterotaxy. C Genetic causes of CHD identified to date.
The natural history of severe congenital heart disease was altered dramatically by the performance of the first systemic to pulmonary artery shunt procedure by Helen Taussig, Vivien Thomas and Alfred Blalock6. Since then, an almost universally lethal condition has become progressively more approachable through a combination of surgical, catheter-based and medical interventions. In the modern era, patients in developed countries undergoing CHD surgery, including those with complex CHD, have 10-year survival exceeding 80%1. This has resulted in an ever-increasing population of adults who are living with CHD, and there are now more people over the age of 18 with CHD than children with CHD7.
While hemodynamic management of CHD has improved dramatically, many patients with CHD have significant cardiac and extracardiac co-morbidities that impact their quality of life. Patients with repaired or palliated CHD are at risk for developing arrhythmias and myocardial dysfunction; in addition, 13.6% have associated extracardiac structural malformations, compared to 7% in the control population. Potentially the largest impact on quality of life in patients with CHD is from associated neurodevelopmental disabilities (NDD); the prevalence of NDD in the CHD population ranges from 10% in patients with mild CHD to over 50% in patients with severe CHD who require surgery during infancy8.
The underlying causes of CHD remains relatively poorly understood, and although it has long been thought to have both genetic and environmental contributions, the epidemiology of CHD points to genetics contributing to the majority of CHD. The overall incidence of CHD has been very stable at 0.8–1.1% of live births, with small changes in CHD incidence attributable to improved diagnostic methods such as increased detection of small septal defects via echocardiography9, 10. Although there is little evidence of temporal or geographic variation in overall incidence to suggest an environmental trigger, there are small differences in the types of CHD observed in different populations, such as the increased number of LVO lesions amongst white children compared to an increase in RV obstruction amongst Chinese children, which suggests population-specific genetic contributions2, 11.
Evidence supporting the genetic contribution to CHD can be gleaned from a number of sources. There is greater concordance of CHD in monozygotic than dizygotic twins12, 13, although there is evidence that twinning itself increases risk of CHD14,15. The risk of recurrence of related forms of CHD among siblings is elevated, ranging from 3.4 for ASDs to 79.1 for heterotaxy in the Danish national cohort study16; there is a smaller, but still significantly increased risk recurrence for discordant CHD17. Additionally, rare Mendelian forms of CHD, comprising a small fraction of all cases, been described. These include forms of ASD18, HTX19, severe mitral valve prolapse20, 21, and Bicuspid Aortic Valve22. The increased incidence of CHD in populations with high levels of consanguinity suggests a role for recessive genetic contributions23. However, it is striking that a large fraction of CHD, particularly of severely affected subjects, occurs in families with no other history of CHD. This suggests the possibility that a significant fraction of these cases is attributable to de novo genetic events including chromosomal abnormalities, smaller copy number variants and point mutations. The severity of CHD in these instances is likely to impair reproductive fitness, limiting transmission of these large-effect mutations and accounting for the absence of extended pedigrees supporting dominant modes of transmission.
Collectively, these findings point to a major genetic contribution to CHD (Fig. 1C). This observational data does not allow insight into whether CHD in individual subjects is attributable to single loci with large effect, a few loci with epistatic or additive interactions, polygenic effects of many loci, or various combinations of these models together. Additionally, the possibility of gene-environment interaction is an important consideration. The aggregate of genetic contributions to CHD are likely to not only underlie the structural CHD, but also be major contributors to CHD co-morbidities including heart failure, arrhythmia, neurocognitive outcomes, and even to the observation that cancer rates are higher in adults with CHD24. As CHD contributes to an ever-increasing amount of the overall burden of cardiovascular disease25, a thorough understanding of the underlying genetics will become ever more important to improved care of patients with CHD.
Established genetic contributions to CHD
Aneuploidy
Aneuploidies were the earliest identified genetic causes of CHD. Estimates of the proportion of CHD associated with cytogenetic abnormalities range from 9–18%26. The large number of genes that are dysregulated in the setting of aneuploidy results in effects on development that are often pleiotropic and severe, and 98% of fetuses with CHD and cytogenetic abnormalities have at least one extracardiac abnormality27. CHD is observed in 35–50% of liveborns with trisomy 21, 60–80% of liveborns with trisomy 13 and trisomy 18, and 33% with monosomy X. Further, there is a large effect on overall viability, as evidenced by the 33–42% incidence of aneuploidy amongst fetuses with prenatally diagnosed CHD, compared to 9–18% amongst neonates with CHD27. The types of CHD associated with specific aneuploidies covers a broad range of CHD phenotypes, although there are lesions that are more prominently associated with specific chromosomal abnormalities, such as atrioventricular septal defects in trisomy 21. The large numbers of genes with dosage disturbance in aneuploidy make it more challenging to pinpoint the underlying genetic and developmental mechanisms. However, insights have been gleaned from studies of patients with rare segmental trisomies affecting chromosome 21 suggesting that DSCAM and COL6A contribute to Down Syndrome-associated CHD28. Interestingly, overexpression of both DSCAM and COL6A in mice leads to heart abnormalities, while overexpression of either gene alone does not affect heart development29.
Copy number variation
Copy number variation (CNV) refers to structural aberrations consisting of deletions or duplications ranging in size from 1 kb to several megabases and leading to altered dosage of genes encompassed by the CNV. Low-copy repeats and retrotransposons found throughout the genome form the substrate for CNV formation30. CNVs can occur de-novo, or be inherited. Millions of SNPs (Single Nucleotide Polymorphisms), each typically with population allele frequency >1%, can be simultaneously genotyped at very low cost through dense array-based platforms. This technology permits identification of regions of genome duplication and deletion in both coding and non-coding regions of the genome. More recently, CNVs have also been detected through whole exome and genome sequencing data. Comparison of dense array-based platforms with whole-exome sequencing showed that each strategy only identified ~70% of the CNVs that should have been detected,31 and therefore together may provide substantial complementary information.
Several well-characterized large CNVs underlie recognized clinical syndromes that include CHD. Del22q11, a deletion of ~3mB resulting from flanking low-copy repeats, is the most common human microdeletion. It presents with a variable phenotype encompassing congenital heart disease, palate abnormalities, hypocalcemia, immunodeficiency, characteristic facial features, and neurodevelopmental abnormalities including learning disabilities and psychiatric disorders, also known as DiGeorge Syndrome and Velocardio-facial syndrome. Del22q11 includes the T-Box transcription factor TBX1, and haploinsufficiency for TBX1 underlies the cardio-pharyngeal phenotype32–34. Recent work in mice haploinsufficient for Tbx1 delineates regulation of H3K4me1 enrichment, providing an intriguing link between TBX1 and chromatin remodeling in CHD35. Other CHD-associated CNVs that are well characterized include del8p23, which includes the cardiac transcription factor GATA4 and manifests with a range of CHD along with developmental delay36; del7q11, the cause of William Syndrome, wherein the cardiac disease consists of supravalvar aortic and pulmonary stenosis and results from haploinsufficiency for Elastin37, 38; del 11q24-25 resulting in Jacobsen Syndrome39, 40. Recent analyses of larger cohorts of patients with CHD found several recurrent CNVs associated with CHD, including 1q21.1, 3p25.1, 16p13.11, 15q11.2 and 2p13.331, 41.
Beyond specific syndromes associated with CNVs, their global contribution to CHD has been investigated in several large cohorts of patients with specific CHD: Tetralogy of Fallot41, Heterotaxy42 and Hypoplastic Left Heart43–45, all of which show an overrepresentation of rare CNVs, and de-novo CNVs, in patients with CHD compared to controls46. An increased burden of CNVs was also detected in non-syndromic patients with mild-to-moderate severity CHD47. The availability of chromosomal microarray testing as a standard clinical test has increased the awareness of the contribution of CNVs to congenital heart disease, and clinical and research-based testing suggests that CNVs contribute to 10–15% of CHD48. As previously noted, given the inherent limitations of most commonly used platforms for CNV detection for optimal sensitivity, the contribution of CNVs to disease phenotypes may underestimate their contribution31.
Inherited point mutations: Mendelian and inherited CHD
Remarkable insights into Mendelian and inherited forms of CHD have emerged from classic linkage analyses, positional cloning and targeted sequencing of CHD candidate genes. Many of the genes first implicated in inherited CHD are members of a core group of cardiac transcription factors that includes NKX2.5, the GATA family of zinc-finger proteins, T-box factors including TBX5 and TBX1 and MEF2 factors (reviewed in49–51). Mutations in NKX2.5 were one of the first inherited point mutations clearly shown to cause human CHD. NKX2.5 is a transcriptional regulator that interacts with GATA4 to specify cardiac mesoderm, first identified in Drosophila mutants that had complete failure to form a heart tube52. Evaluation of large pedigrees that included individuals with isolated ASDs, and individuals with ASDs along with abnormalities of the conduction system subsequently identified NKX2.5 mutation underlying both the ASD and conduction system defects. Notably, some affected individuals had the ASD alone, others had the conduction defect alone, and some had both the ASD and the conduction defect53. The phenotypic heterogeneity associated with NKX2.5 mutations is remarkable, encompassing a wide range of CHD beyond ASDs, including HTX and Tetralogy of Fallot54. It is interesting to speculate whether the wide spectrum of CHD results from differences in genetic background, or interaction between an at-risk genotype and environmental influences that may include subtle variation in hemodynamics in-utero during critical times in cardiac development. GATA4 is a zinc-finger transcription factor essential for cardiogenesis that directly associates with NKX2.555. GATA4 mutations were implicated in two families with CHD with cardiac septal defects56.
Mutations in TBX5, a T-box protein, were likewise implicated in two families with Holt-Oram Syndrome, a disease characterized by upper limb malformations and cardiac abnormalities (septation and conduction defects)57, 58. TBX5 is notably expressed in both the developing forelimb buds and the heart, and similar to other T-box proteins, regulates cell fate and crucial developmental processes. Further evidence of causality has emerged from heterozygous Tbx5 null mice displaying limb abnormalities, septal defects, deformed hearts, and other complex cardiac malformations59.
X-linked ZIC3 mutations were identified in several multi-generational pedigrees, and were carried by family members with the mutation, some of whom by chance ended up with either complete situs solitus or complete situs inversus and functionally normal hearts19, 60, 61. ZIC3 is a zinc-finger transcription factor that is required to form a functional left-right organizer62 and is required to direct the directionality of heart looping. Since absence of left-right organizer function leads to random heart looping, these pedigrees show striking incomplete penetrance: some affected family members will have normal heart looping and appear phenotypically normal and transmit the disease allele, while others will have situs inversus or heterotaxy and complex CHD.
In addition to cardiac transcriptional regulators, genes coding for a variety of signaling molecules and cellular structural components have been identified in Mendelian inherited CHD. Dominantly inherited NOTCH1 mutations were first described in 2 multigeneration pedigrees that included family members with bicuspid aortic valve with only hemodynamic impairment, while other family members have complex CHD including hypoplastic left heart syndrome (HLHS)22. Since then, NOTCH1 mutations have been found in additional CHD pedigrees63, and in ~5% of cases of one of the most common cardiac defects, bicuspid aortic valve, which is found in up to 2% of adults64. Similarly, mutations in JAG1 were mapped to affected family members with Alagille syndrome, a multi-system disorder with diverse cardiac phenotypes. JAG1 is one of the five ligands for receptors in the Notch signaling pathway, which results in localization of Notch to the nucleus and downstream activation of target genes. In addition, genes coding for the focal adhesion protein Tns1 (tensin 1) and the planar cell polarity protein Dchs1 (dachsous1) were identified from several large pedigrees of severe mitral valve prolapse20, 21.
Less is known about the impact of recessive inheritance on CHD, although several lines of evidence support a recessive model contributing to some types of CHD. CHD is more prevalent in populations with a high degree of consanguinity23. Although there is an increase in all types of CHD in consanguineous populations, HTX and the associated complex cardiac malformations are observed at a higher frequency in consanguineous populations65. Pedigrees of consanguineous families with HTX identified recessively inherited mutations in genes including SHROOM3 66(cytoskeletal protein), WDR16 67(cilia-associated WD40 repeat-protein), and MMP21 68(matrix metalloproteinase 21), and NPHP4 69 nephronopthisis 4).
A different approach to understanding the recessive contribution to severe CHD was undertaken through an unbiased recessive mutagenesis screen in mouse70. Here, the offspring of ENU-mutagenized mice were intercrossed, and the pregnancies studied for any severe congenital heart disease by fetal ultrasound. This identified 61 genes contributing to recessively-inherited CHD. Several notable findings from this landmark study were that, of the 61 genes identified, 34 were cilia-related genes, several of which had previously been identified in human CHD. Further, cilia-related genes contributed to both HTX-type CHD, and CHD not associated with laterality defects. Together, the observations in a limited number of human pedigrees and in model organism suggest that recessive inheritance contributes to CHD, in particular to HTX-type (laterality) CHD.
Beyond large structural variation and Mendelian CHD: the impact of next generation sequencing on CHD genetics
The explosion of technological approaches and analysis tools for next generation sequencing, which has occurred over almost a decade has opened the door for understanding the genetics of complex disease such as CHD. In particular, whole exome sequencing has allowed identification of mutations that were undefinable through traditional genomic methods, such as de novo variation, variants without clear Mendelian inheritance patterns, variants with marked reduced penetrance, and somatic alterations, among others.
Whole-exome sequencing
The development of robust methods of whole-exome sequencing (WES) has created new opportunities for genomic discovery71, 72. The complete coding regions of the ~20,000 genes in the human genome plus their flanking splice sites comprises only ~33 Mb of DNA, about 1% of the human genome sequence. Unbiased genetic discovery by positional cloning in humans, mice and fruit flies has demonstrated that the overwhelming majority of phenotypes caused by large-effect mutations are caused by coding sequence mutations. This has identified mutations in ~3,500 genes underlying known Mendelian phenotypes. The recognition that the 20,000 human genes are largely conserved across vertebrate phylogeny strongly suggests that mutation of most will lead to phenotypic consequences, although how many of these phenotypes will manifest as disease remains unknown. These observations motivated the development of methods for selective sequencing of this 1% of the genome, which now can be completed at about 20% of the cost of sequencing complete genomes, affording a very significant cost advantage and allowing large cohorts of patients with unexplained phenotypes to be sequenced. Sequential improvements have resulted in virtually complete detection of point mutations in the full coding region; challenges nonetheless remain in detecting certain CNVs and chromosomal translocations. WES can be used to identify genes that are mutated more often than expected by chance after accounting for sequencing 20,000 genes. This has allowed for the identification of novel disease genes for a range of disease phenotypes ranging from autism to congenital malformations to cancer through the analysis of transmitted, de novo and somatic mutations.
Whole exome sequencing identifies de novo mutations in CHD
The first advance derived from applying next generation sequencing to the study of CHD was the discovery of the role of de novo mutation in CHD. Most CHD is sporadic: only 2.2% patients with CHD have affected first-degree relatives16. That sporadic disease such as CHD has stable incidence despite low reproductive potential suggests that new mutation occurs as existing mutations are lost due to impaired reproductive fitness, and suggests that de novo mutations underlie some CHD73. De novo mutations are on average more deleterious than inherited mutations, as there has been less evolutionary selection. These mutations occur at approximately 1.8 × 10−8/nucleotide/generation, resulting in ~1 de novo mutations per coding-region (exome), and ~100–115 de novo mutations per genome74, 75. As spermatogenesis has many more germline cell divisions than oogenesis, this results in a 3.9:1 ratio of de novo mutations when comparing the paternal allele to the maternal allele76, 77. De novo mutations occur throughout the genome, but are not entirely randomly distributed. Factors that influence DNA mutation rate include high CpG density, segmental duplications, paternal age, and mutations conferring advantages during spermatogenesis.78
Since there is strong evidence for impaired reproductive fitness in a majority of CHD subjects, it is likely that de novo mutations confer a major contribution. This hypothesis has been tested in several studies that employed whole-exome sequencing of large cohorts of patient-parent trios affected by CHD. The first of these studies analyzed 362 trios with the patient affected by severe CHD. While the overall rate of de novo mutations was not significantly different between CHD cases and controls, there was a marked enrichment when stratifying for protein-altering de novo mutations in genes highly expressed in the developing heart (top quartile of gene expression in murine hearts at E9.5 and E14.5) in CHD cases. Further stratification by mutation type graded by stringency, from all protein-altering mutations to highly conserved missense and LOF mutations to LOF mutations alone, produced a significant rise in odds ratio. De novo mutations were found to collectively contribute to 10% of severe CHD79. Moreover, an excess of de novo mutations was identified in chromatin remodeling genes that affected the reading, writing, and removal of two bivalent marks, H3K4 and H3K27 methylation, found at the promoters and enhancers of key developmental genes posed for activation79. Expanding the cohort size from 362 to 1,213 CHD trios comprising patients with the complete spectrum of CHD including less complicated CHD such as isolated atrial septal defects, in addition to moderate and severe CHD, reinforced the contribution of de novo mutations to ~10% of CHD. This cohort included patients with isolated CHD, CHD associated with known syndromes, and CHD with extracardiac malformations and/or neurodevelopmental abnormalities. More extensive phenotyping coupled with a larger patient cohort demonstrated that de novo mutations disproportionately contributed to CHD in patients with associated syndromes, extracardiac malformations and/or neurodevelopmental abnormalities. Notably, de novo mutations accounted for at least 20% of CHD with associated extracardiac and neurodevelopmental abnormalities75. While these mutations are more prominently associated with syndromic rather than non-syndromic CHD80, there is a small but measurable contribution to isolated CHD (CHD not associated with a known syndrome, and without any extracardiac malformations or neurodevelopmental abnormalities)81, which may become more clearly delineated when larger cohorts are analyzed82,75.
Biological pathways in CHD
The genetics underlying CHD have identified critical biological pathways involved in CHD including chromatin remodeling, Notch signaling, cilia function, sarcomere structure and function, and RAS signaling. These pathways are anticipated to provide direct insights into the mechanism of heart development, and provide insights into potential CHD co-morbidities such as ventricular dysfunction observed in the setting of sarcomere and RAS pathway mutations. Furthermore, identification of common developmental pathways shared between cardiac development and other systems, such as the nervous system in the setting of chromatin modifier mutations, and the respiratory system in the setting of cilia mutations, are anticipated to directly inform outcomes and prognosis for CHD patients. We will outline studies linking three of these pathways to CHD: Chromatin remodeling, Notch signaling and cilia genes.
Chromatin modifiers and overlapping biology
As noted above, one of the most significant findings arising from the recent analysis of large cohorts of patients with CHD by whole-exome sequencing is the important role of mutations affecting chromatin regulating genes in CHD75, 79, 80. The first of these studies showed that de-novo mutations affecting chromatin regulating genes contribute to ~3% of CHD. This observation was reinforced by a larger analysis of 1,213 trios, noting LOF mutations in chromatin regulating genes in 25/1213 CHD cases, while only 3/900 controls (p=5.7×10−11). All mutated genes with damaging mutations in cases and their affected chromatin marks are shown in Fig. 2a. Genes identified are involved in production, removal or reading of H3K4 methylation (H3K4me), H3K9 methylation (H3K9me), H3K27 methylation (H3K27me), H4K20 methylation (H4K20me), and ubiquitylation of H2BK120, which is required for H3K4 methylation79.
Figure 2.
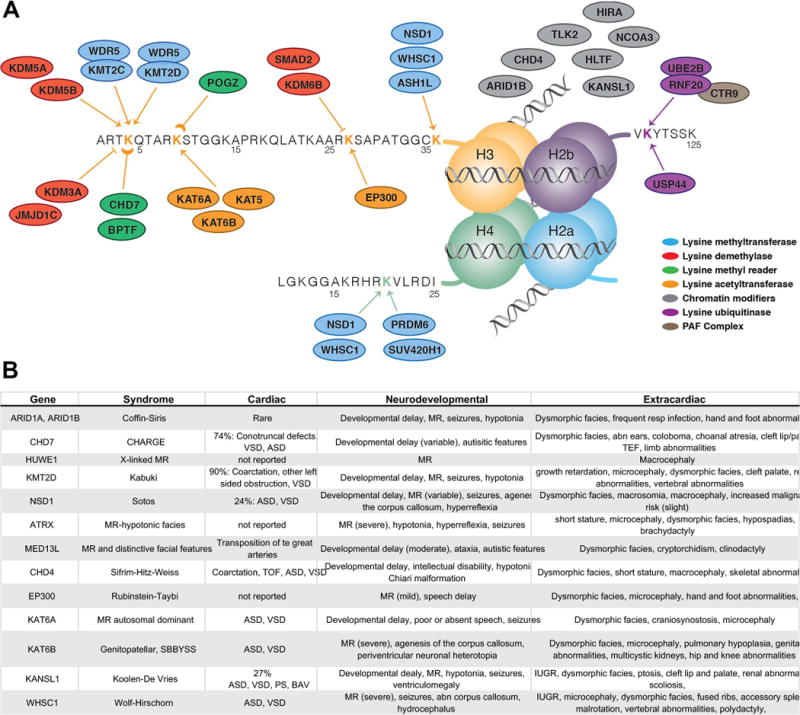
Chromatin remodeling genes in CHD. A Chromatin remodeling genes with mutations identified in CHD patients to date, highlighting the overlap between CHD and NDD genes. Nucleosomes with H3K4, H3K9, H3K27, H3K36 and H4K20 methylation and/or acetylation, and H2BK120 ubiquitylation are shown (adapted from Homsy et al, Science 2015). B Syndromes associated with chromatin remodeling gene mutations and their associated CHD and neurodevelopmental abnormalities.
Chromatin regulating genes encompass ~ 600 genes that orchestrate dynamic gene expression during development by addition or removal of chemical marks on chromatin or by catalyzing changes in chromatin structure. The biological state of chromatin is controlled by ATP-dependent chromatin modifiers, including the Baf complex and Chd8, and by histone modifiers. Both have been linked to heart development; Baf60c regulates early heart development through cooperation with the GATA4 transcription factor83. H3K4me and H3K27me constitute ‘bivalent’ marks that are found on the promoters and enhancers of key cardiac developmental genes poised for activation84. A significant burden of haploinsufficient (dominant) de novo mutations within these elements therefore indicate dosage sensitivity of the chromatin pathway in heart development. Although relatively rare, chromatin modifiers have been linked to isolated CHD such as the histone methyl transferase PRDM6 which has been associated with non-syndromic Patent Ductus Arteriosus85.
The same control of gene expression required for normal cardiac development is also essential for brain development, and many chromatin regulating genes have been directly implicated in brain development, including members of the BAF complex, CHD8, HDAC4 and polycomb group protein EZH286. The overlap between specific chromatin regulators required for both heart and brain development remains unclear. Chromatin regulators are widely expressed, and mouse knockout frequently leads to very early lethality, precluding analysis of specific brain or heart phenotypes87. Heart-specific knockout of Kmtd2, a H3K4 methylase that is associated with Kabuki syndrome, results in very abnormal hearts with outflow tract septation defects in mice88. Mutations in chromatin modifying genes have been identified in patients with CHD have been associated with a range of syndromes, including Sotos Syndrome, Kabuki Syndrome, CHARGE and others (Fig. 2b). At the same time, genome sequencing studies in human neurodevelopmental and psychiatric disorders have identified mutations in chromatin modifying genes in Kleefstra, Schinzel-Giedion, Claes-Jensen, Weaver, Sotos, and Coffin-Siris syndromes among others. Although neurodevelopmental abnormalities are the most prominent feature, up to 50% of affected patients also have a CHD. These observations indicate that chromatin regulating mutations result in both cardiac and neurodevelopmental sequelae, and begin to shed light on potential developmental-genetic causes of the NDD associated with a subset of CHD patients.
Notch pathway genes
Notch signaling is a highly conserved pathway mediating local intercellular communication that has important roles in a host of developmental processes that are very relevant to heart development, including formation of the left-right organizer89, blood vessel development90 and ventricular chamber development91. Notch signaling provides a way for a Notch ligand from one cell to influence a directly neighboring cell via its Notch receptor (Fig. 3a) and determine cell fate. Upon binding of Notch ligand and receptor, signaling requires release of the Notch intracellular domain (NICD) via a coordinated series of tightly regulated steps including glycosylation of the receptor, ubiquitylation of the ligands by Mib1 and cleavage of the receptor triggered by Adam17/Tace. NICD can then function as a transcription factor regulating a large number of targets, including SNAIL1, HES, HEY and NRARP. Notch pathway genes implicated in CHD are outlined in Fig. 3b. This pathway was initially implicated in CHD when NOTCH1 mutations were identified in families with dominantly inherited bicuspid aortic valve and other left-ventricular outflow tact obstructive lesions22. Within a single family, NOTCH mutation associated with a range of CHD ranging from BAV to CoA to HLHS. Mutations affecting multiple components of Notch signaling (NOTCH1, MAML1, JAG1) were significantly enriched in 51 families with multiple family members affected by a range of LVO-type CHD63. NOTCH1 function does not appear to be restricted to left-sided heart development, as additional family studies identified high-impact NOTCH1 mutations in families with both LVO-type CHD and TOF63.
Figure 3.
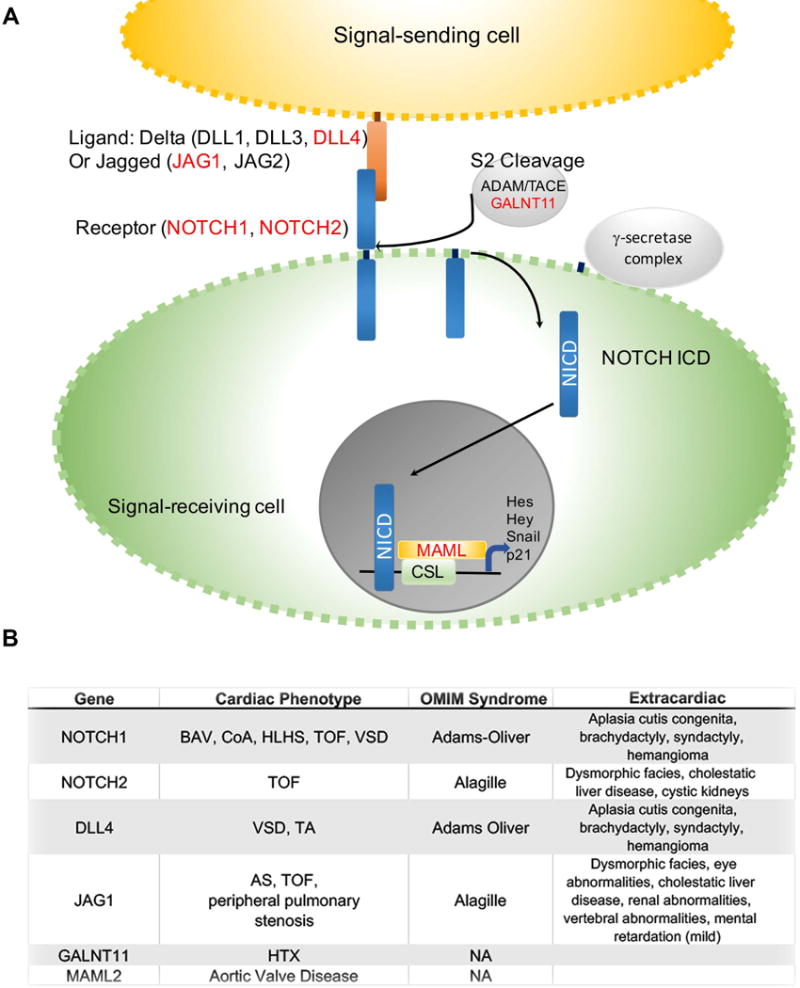
NOTCH signaling in CHD A outline of NOTCH signaling pathway showing signal-sending cell in yellow, and signal receiving cell in green. B Syndromes and CHD associated with NOTCH pathway gene mutations.
Further, NOTCH1 and Notch ligand DLL4 mutations are the most common cause of Adams-Oliver Syndrome, a rare syndrome comprising CHD, aplasia cutis of the scalp and limb defects. Alagille syndrome (Arteriohepatic Dysplasia) is an autosomal dominant inherited syndrome characterized by cholestatic liver disease, variable degrees of kidney involvement and CHD that is most commonly TOF. Mutations in the Notch ligand JAG1 are found in ~90% of patients with Alagille Syndrome92, 93, and mutations in the NOTCH2 receptor are found in another 2% of individuals with Alagille Syndrome94. Similar to mutations in other Notch pathway members, JAG1 mutations underlie both syndromic and isolated CHD, most notably TOF95. Finally, GALNT11, which is required for the S2 cleavage step of Notch receptor processing, has been linked to human HTX by affecting Notch-mediated specification of cilia function at the left-right organizer42, 89.
Cilia genes
Mutations affecting cilia structure and function have been identified in patients with CHD, and notably, cilia mutations were the major class of mutations found in a recessive mouse screen for severe CHD70. Cilia are hair-like organelles found on the surface of most vertebrate cell types and serve a multitude of functions, including signaling, extracellular fluid propulsion and cell cycle control (Fig. 4a). Defects affecting cilia structure and/or function have been intimately linked to a group of diverse human disorders characterized by pleiotropic phenotypes including renal, neurological, sensory and laterality defects coined ‘ciliopathies’. In heart development, the best understood role for cilia is establishing left-right (LR) asymmetry and determining the direction of heart looping. Here, a highly conserved ciliated left right organizer (LRO) utilizes cilia to generate and sense directional flow of extraembryonic fluid and transduce it in a polycystin-dependent manner to a calcium signal96–99. This triggers asymmetric gene expression in the lateral plate mesoderm, eventually leading to asymmetric heart looping. Due to the role of cilia in determining LR patterning, mutations affecting ciliary motility100 and sensing101 machinery result in HTX and CHD.
Figure 4.
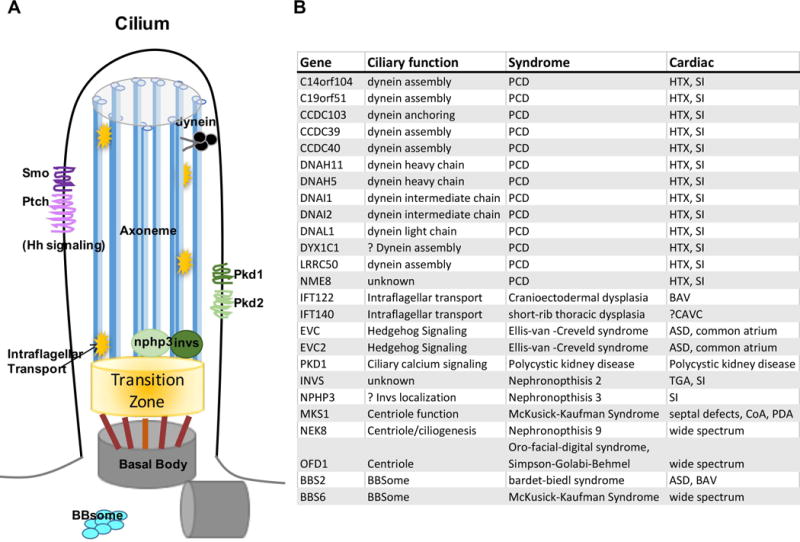
Cilia in CHD A Diagram of a cilium, showing the ciliary axoneme (blue) based on the mother centriole (gray) and linked via the transition zone (orange). B Syndromes and CHD linked to human cilia mutations.
In mice, mutations in components of the dynein motor complex, such as left-right dynein (Dnah11/Lrd) and dynein heavy chain 5 (Dnah5), result in cardiac and visceral LR abnormalities102, 103. Not surprisingly, 6.5% of patients with PCD (primary ciliary dyskinesia), a disorder defined by abnormal ciliary motility in the airway epithelia, also display heterotaxy100. PCD is genetically highly heterogeneous, and there are currently 35 genes that have been linked to PCD. It remains unknown how many of the PCD genes cause CHD, since a diagnosis of PCD in patients with CHD is made more challenging, due to the difficulty differentiating whether respiratory symptoms are primary, or secondary to the underlying cardiac pathology and the medical and surgical interventions required to manage the CHD. Other cilia genes that are not required for cilia motility and left-right development, but instead are involved in ciliogenesis or cilia-mediated sensation are also associated with CHD (Fig. 4b). It is interesting to speculate that cilia found in the developing heart and vasculature have a function in cardiac morphogenesis extending beyond their role in LR development104, 105, and that similar to mouse, cilia defects may underlie a broader range of human CHD than suspected to date.
Future Efforts in CHD Genomics
State of the art understanding of CHD genetics
The genetic basis of CHD has now been established in 1 out of 3 affected cases (Fig. 1 C). These comprise a broad array of genetic alterations in a large, heterogeneous group of genes. Earliest insights arose from aneuploidies in CHD. Likewise, there is a growing catalogue of CNVs in CHD, typified by well characterized deletions, such as del22q11. Given the large number of genes involved in aneuploidies and CNVs, identification of specific disease associated genes is challenging. In addition, inherited forms of CHD have been identified through traditional genetic tools, such as linkage. These linkage studies have ranged from mutations in cardiac transcription factors, such as NKX2.5, and GATA4 to signaling molecules and cellular structural components, such as NOTCH1 and JAG1. Through critical advances in next generation sequencing, our understanding of CHD biology has expanded rapidly over the past decade. Seminal studies have found that 10% of CHD is due to de novo mutations, which increases to greater than 20% when stratifying for CHD with associated EM or NDD. Indeed, certain biological pathways, including chromatin modification genes, cilia genes and the Notch signaling pathway, have been implicated in CHD, raising the possibility that environmental perturbations might phenocopy the effects of these mutations. Despite these significant advances, the genetic underpinnings of over 50% of CHD remain unknown. Barriers to a complete understanding of CHD genetics include the extreme genetic heterogeneity coupled with limited genotype-phenotype correlation. Some of the “unexplained CHD cases” could be due to mutations affecting the as of yet underexplored non-coding DNA, somatic mutations and gene-environment interactions as discussed below. In addition, CHD due to biallelic mutations has been underexplored, and thus far has largely focused on candidate gene analysis and familial CHD101. As new statistical approaches are coupled with larger CHD cohorts, the inherent challenges in an unbiased analysis of recessive mutations in sporadic CHD can be surmounted.
Most CHD is sporadic, with no affected family members. Beyond the 20% of sporadic CHD caused by de-novo CNVs and SNVs, it is likely that some CHD will be secondary to complex inheritance, wherein for example, a heterozygous mutation requires a modifier mutation, or the absence of a protective variant, in order to manifest as disease. This is supported by incomplete or non-penetrance in extended families carrying mutations in well-characterized CHD genes including ZIC3 and NOTCH119, 61, 106. Animal models provide an avenue for testing the complex trait hypothesis in a more controlled genetic environment, and for example the susceptibility to VSDs in mice heterozygous for mutation in the cardiac transcription factor Nkx2.5 was modified by loci on mouse chromosomes 6, 8 and 10107. Another example is provided by the observation that introduction of a null allele for the VEGF-A pathway gene CRELD1 into a mouse model of Down Syndrome raises the incidence of AVSDs108, which supports prior observations on human Down Syndrome cohorts109. Correspondingly, several large GWAS studies in patients with CHD also identified possible loci influencing the susceptibility to CHD110, 111. In addition, it is also quite likely that since heart development appears to be highly dosage sensitive, some CHD may result from convergence of hypomorphic mutations in several components of a single pathway to exceed a threshold and manifest as disease. This model has previously been shown in a mouse model of congenital heart disease where the penetrance of the CHD phenotype is increased in mice compound heterozygous for Zic3(+/−); Nodal (+/−) 112. As available gene- and protein-level interactome databases become more robust and comprehensive, it may very well become possible to link CHD genomics data with genetic interactome databases to better explore multigenic CHD.
Exhausting the Coding Region: Number of Genes Contributing to CHD
Analysis of de novo mutations has illuminated the immense genetic heterogeneity underlying CHD pathogenesis. Recent work analyzing de novo mutations in 1,213 CHD subjects showed that ~392 genes, albeit with wide confidence intervals, collectively contribute to CHD. This estimation of the number of risk genes was performed using a maximum likelihood function, details of the simulation and derivation are noted in Homsy et al. and Iossifov et al. 75, 113. Based on this function in 1,213 CHD cases, 392 genes were estimated to contribute to CHD 75. An approach to identify a greater fraction of the CHD risk genes is to identify additional genes with more than one de-novo mutation in CHD patients. In a cohort double the size (2,426 trios), power simulations approximate 61 genes with more than one damaging mutation (~40 new genes in the additional 1,213 trios and 21 previously identified in the original 1,213 trios). To identify all 392 genes would require a significant increase in the number of CHD trios analyzed. Furthermore, recent work suggests that WES of 10,000 trios would permit ~80% saturation for detecting genes contributing to haploinsufficient syndromic CHD alone 80; it is likely that a significantly larger number of trios will have to be analyzed to approach a complete gene set for all CHD. As sequencing continues to become faster and less expensive, it is anticipated that large-scale collaboration of CHD genetics programs such as the Pediatric Cardiac Genomics Consortium (PCGC) 114, Pediatric Heart Network (PHN) 115, and the UK10K consortium 116 could allow for capture of the estimated ~392 CHD risk genes, and make a previously daunting task achievable.
Contribution of Somatic Mosaicism in CHD
The wealth of sequencing data at high coverage has prompted the search for genetic mosaicism. Genetic mosaicism is defined as the presence of having multiple populations of genetically distinct cells within an individual. Mosaic de novo variants have been shown to contribute up to 20% of sporadic cases in a number of developmental disorders, including Sturge Weber syndrome 117, facioscapulohumeral muscular dystrophy 118, and segmental neurofibromatosis 119. There have also been clinical reports suggesting pathogenic mosaic CNVs in patients with CHD 120. Small studies using array comparative genomic hybridization (aCGH) have not identified any CNVs with differential presence between cardiac tissue and peripheral whole blood 121. A recent study identified an excess of extreme allele-specific expression events in cardiac tissue from CHD patients compared to controls, and since only 15% of the ASE events were explained by genomic variants, it is possible that some of these were secondary to mosaicism 122. However, such studies are limited due to sample size, lack of developmentally relevant cardiac tissues, and imperfect statistical tools to detect mosaic variation. Larger cohorts of sequencing data, continued developmental of analysis tools, and ascertainment of cardiac tissues could help in identification of mosaic mutations, such as (1) de novo mutations with mosaic tissue distributions with involvement of cardiac tissues or precursors that would directly influence heart development, and (2) parental mosaicism where the unaffected parents of an affected offspring with CHD harbors mutation in the germline and any somatic tissue not involved in cardiac developmental, such that the mutation is constitutively transmitted to the affected offspring.
Environmental Phenocopies
Considering the overrepresentation of mutations in certain pathways, such as chromatin modifying genes, in which dosage sensitive mutations confer CHD, it is possible that environmental triggers phenocopy the effects of these mutations. Many environmental exposures have been studied through observational and epidemiological studies in CHD 123. A large Canadian population study showed a modest association between folic acid supplementation and reduction in several subtypes of CHD 124; of note is that a maternal MTHFR polymorphism contributes to the CHD risk pointing to gene-environment interaction in the development of CHD 125. The best documented environmental exposure that contributes to CHD is maternal pre-pregnancy diabetes which leads to an adjusted relative risk for CHD of 4.0 126. The effect is independent of whether the mother is affected by type 1 or type 2 diabetes, and studies of the NOD (non-obese diabetic) mouse show that CHD in offspring correlates with elevated glucose during embryogenesis 127. Especially in view of the increasing rates of type 2 diabetes in the younger population, this represents an important contributor to CHD; for example, maternal diabetes is thought to contribute to 6–8% of HLHS and TOF 128. The potential for gene-environment interactions highlight the continued need to catalogue environmental exposures within a cohort which also has corresponding DNA sequencing data. This should further expand our understanding of the genetic and non-genetic mechanisms of CHD, and moreover tell us how these two etiologies may converge.
Contribution of Non-Coding Mutations
The prominent role of transcriptional regulation in CHD predicts that mutations affecting regulatory elements will contribute to CHD. For example, homozygous variation in a TBX5 enhancer was found in a patient with isolated septal defects 129. An important obstacle of detecting non-coding mutations in CHD is to delineate cardiac-specific regulatory elements and promoters at appropriate developmental time-points. Projects, such as ENCODE and the Cardiovascular Genomic Consortium, continue to build these datasets, and thus may be helpful in identifying rare de novo events in these non-coding elements. Other sources of WGS discovery could focus on cis-acting regulatory sequences, allelic selective gene expression in regulatory elements, and identification of epistatic and modifying mutations in diseases with known coding mutations, but with poor penetrance. An example of the latter has been shown in another developmental disorder, craniosynostosis, where rare SMAD6 LOF mutations modified by a common variant in BMP2 resulted in complete penetrance of this disease 130. Exploration of the non-coding DNA will require whole genome sequencing (WGS), which provides the most comprehensive view of the genome. Beyond complete determination of mutations outside the coding region, WGS provides more complete coverage of the exome and leads to improved detection of exonic CNVs and translocations. The potential challenges of WGS include greater expense, larger amounts of acquired and stored data, and the greater challenge of interpreting sequence variation in non-coding DNA. At present, evidence that WGS comes close to WES in efficiency of discovery of rare mutations with large effect remains limited 131. Moreover, the 10-fold lower conservation of enhancer sequences indicates a much lower power to find disease-related mutations and adds to the challenge. One study of patients with severe intellectual disability, however, identified a conclusive cause in 42% of patients by WGS, compared to 27% by WES; it is notable that many of the mutations identified by WGS in this study actually affected the exome 132. This limitation of WES is progressively being overcome by improved capture technologies that generate progressively more complete coverage of all exonic sequences 133. The genomic technologies applied to CHD gene discovery and CHD patient diagnosis are rapidly evolving. Currently, WGS is likely contribute to understanding the genetic cause of CHD, but will is most effective when applied in patients without WES evidence of damaging de novo mutations or likely pathogenic dominant and recessive mutations.
Clinical impact of CHD genetics
For CHD genetics to become part of standard care for CHD patients, there are a few essential considerations. First, testing should be broadly available, and specific testing (chromosomal microarray, karyotype, targeted sequencing, exome sequencing or genome sequencing) should be tailored to the specific patient’s case with regard to type of CHD and presence or absence of extracardiac abnormalities. Second, the results of genetic testing for CHD should be actionable, i.e. impact management and hopefully contribute to improved outcome. Third, when a genetic cause for CHD can be identified, it becomes possible to provide much more specific information regarding recurrence risk in other family members; this will become increasingly important as many more patients with CHD reach reproductive age in the context of the growing population of adults with CHD. The extreme heterogeneity and variable expressivity of CHD make it difficult to directly link specific genes to specific outcomes. However, evidence is mounting that defects affecting specific gene ontologies and pathways predispose patients to defined groups of potential complications. These risks may be defined by the genetic defect, in addition to the specific anatomic-physiologic cardiac defect. We will focus on two outcomes associated with CHD for which there is mounting evidence that the genetic cause of the CHD is a major contributor: neurodevelopmental abnormalities, and surgical outcome focusing on respiratory complications. Other co-morbidities such as renal and myocardial dysfunction may also be influenced by genetic findings contributing to the CHD 134.
Neurodevelopmental outcomes
One of the most impactful associations with CHD are neurodevelopmental abnormalities. They affect 10% of all patients with CHD, and 50% of patients with severe CHD 135. The spectrum of associated neurodevelopment abnormalities includes intellectual disability, language deficits, autism spectrum, executive function deficits, deficits in non-verbal skills including motor skills and social cognition. Attention-deficit hyperactivity disorder is also observed at a prevalence of up to 3–4 times higher than the general population 135, 136. Although the incidence of NDD is increased in the setting of complex CHD and CHD in the setting of known genetic syndromes, the underlying causes remain poorly defined. Investigation of risk factors for CHD-associated NDD has focused on the role of complications of cardiopulmonary bypass, effects of abnormal physiology preceding repair (including during fetal life), and complications of hospitalization including prolonged requirement for intensive care; it is likely that these factors interrelate with the genetic substrate 137. Thus far, no dominant major contributor to the NDD associated with CHD has been identified, and it remains difficult to identify at-risk children prospectively. While some studies show a modest correlation with type of CHD and length of deep hypothermic arrest 138, the most striking predictor of poor neurodevelopmental outcome at age two was preoperative microcephaly. Similarly, preoperative brain MRI has demonstrated white-matter abnormalities in 32% of newborns with D-Transposition of the Great Arteries or single ventricle, compared to none of the control infants 139; both of these observations support the association of CHD and abnormal brain development independent of surgical management.
Although recent recommendations include developmental evaluation in a subset of high-risk patients with CHD, ND risk-stratification for CHD remains difficult. Poor neurodevelopmental outcomes are much more prevalent in CHD patients with a diagnosed genetic syndrome, and a link between genetic factors and weight growth and head circumference has been identified 140. Genetic analysis of patients of 1,213 CHD patients revealed that de novo risk increases when stratifying for CHD cases with NDD and/or extracardiac manifestations (EM). Specifically, 10% of patients with CHD and NDD were found to be attributable to damaging de novo mutations, which increased to 20% when looking at patients with both NDD and EM. These mutations tended to occur in genes that were highly expressed in both the heart and the brain. Furthermore, within this set of cases, there were 66 genes, which were mutated in both CHD probands and seven published cohorts ascertained for neurodevelopmental phenotypes. These findings were suggestive of common genetic etiologies. Common pathways converged on chromatin modification, transcriptional regulations, Notch and Wnt Signaling, among other pathways in cardiac development.75 These findings open an avenue to identification of CHD patients who are at risk for neurodevelopmental difficulties early during their clinical course. Several studies show that early interventions impact neurodevelopmental outcomes such as executive function in at-risk children 141. Whether these or other interventions could also benefit children with CHD who are at high risk for neurodevelopmental sequelae still needs to be rigorously tested.
Postoperative and Respiratory Outcomes
Surgical correction or palliation of even the most complex CHD has been one of the main drivers of the remarkable increase in survival for patients with CHD. One of the challenging aspects of caring for these patients is the variable outcome resulting from surgery for anatomically and physiologically identical CHD. Two of the most significant modulators of post-operative outcome that may be influenced by the genetic underpinnings of the CHD are respiratory complications and myocardial dysfunction, and the ability to identify at-risk patients preoperatively may allow improved surgical and post-operative care that is better tailored to the individual patient. Mutations in cilia genes are known to cause heterotaxy, and are also likely contributing to some types of non-heterotaxy CHD 70, 100. In addition to CHD, cilia mutations cause PCD (Primary Ciliary Dyskinesia), a genetically heterogeneous disorder leading to pulmonary dysfunction, male infertility, and organ laterality defects 142, 143. In the respiratory tract, PCD can result in immotile or dyskinetic cilia that fail to coordinate mucociliary clearance of pathogens and debris from the respiratory tract. Poor mucociliary clearance leads to infection and inflammation that damage the airway, and it is especially important to note that patients with ciliary dysfunction depend entirely on cough for mucociliary clearance, a function that is compromised in patients on mechanical ventilatory support such as post-operative CHD patients. With this in mind, it is not surprising that patients with airway ciliary dysfunction and heterotaxy with CHD have a higher rate of respiratory complication post-operatively compared with patients without airway ciliary dysfunction 144; these findings suggest that prospective knowledge of which patients have airway ciliary dysfunction could improve postoperative outcome by tailored modifications to their respiratory care.
Genetic testing in CHD
The utility of genetic information in tailoring care for the CHD patient, risk stratification, establishing prognosis, and counseling families affected by CHD is continually expanding as more is learned about the genetic contribution to CHD. Genetic testing is being offered to an increasing portion of CHD families, however, at this time there is little consensus on the type of testing, the specific clinical indication for testing, and the interpretation of testing results. Increasingly, specialty clinics in CHD genetics where testing can be ordered, and patient counseling provided by geneticists and genetic counselors working together with pediatric cardiologists are being offered to patients 145. Below, we outline CHD genetic testing available on a clinical basis at the time of this writing; notably, the technology for genetic testing along with the interpretation of results is evolving at an extremely rapid pace, and it is distinctly possible that whole-exome or whole-genome sequencing will become more universally employed in the diagnosis and management of CHD in the very near future.
The most common clinical genetic tests utilized in CHD are karyotypes, chromosomal microarray, targeted FISH (Fluorescence in-situ hybridization), directed panel sequencing, and more recently whole-exome sequencing. The decision on which type of testing is appropriate depends on the clinical presentation. Karyotyping is commonly the first line of testing used. In studies of CHD patients admitted to a cardiac intensive care unit, or who had surgery in the first year of life, karyotyping yielded a diagnosis in 10.5%–23% of patients 146, 147. The majority of the positive results from karyotyping were either Down Syndrome or Turner Syndrome, both of which are often clinically recognized. Genome-wide microarray provides information on deletions and duplications including common CNVs associated with CHD such as del22q11 (DiGeorge Syndrome) and del7q11 (William Syndrome). Microarray has yielded a diagnostic result in 10–25% of patients tested, with an additional 8% of patients carrying a variant of unknown significance. Targeted FISH (fluorescent in-situ hypbridization), most commonly focusing on the 22q11 deletion, identifies the presence or absence of the specific target at lower cost and slightly more rapid turnover than genome-wide microarray, and had a positive diagnostic rate of 12%. Whether targeted FISH is positive correlated most directly with the specific cardiac lesion being tested, with 25–50% of patients with interrupted aortic arch, pulmonary atresia with ventricular septal defect or truncus arteriosus having a positive 22q11 FISH 147. It is important to note that the abnormalities detected by targeted FISH will also be detected by genome-wide microarray, and at least one study suggests that except in specific clinical scenarios, microarray testing is more cost effective 146.
Finally, sequencing is increasingly being utilized to identify genetic causes of CHD at the clinical level. Targeted sequencing is being offered for panels of CHD candidate genes. Two studies using similar sets of 57 genes previously implicated in CHD to test a group of patients from non-syndromic CHD families with probable dominantly inherited CHD identified likely causative mutations in 25–46% of the families 148, 149. This approach relies on variants segregating with disease within a family, and thus becomes more difficult in non-familial CHD, first because the composition of the currently utilized CHD gene sets in the targeted sequencing approach are biased towards inherited CHD, and second because variant interpretation in an isolated case is more challenging. When there is strong clinical suspicion for CHD associated with a syndrome that has known genetic cause, targeted sequencing of a gene or group of genes associated with that syndrome is indicated, and one study finds that targeted sequencing that is driven by clinical evaluation identified the etiology of CHD in 17% of the cases 147. In specific syndromes that have been well characterized at the molecular level, such as Noonan Syndrome or Marfan Syndrome, the diagnostic yield can be as high as 80% (Noonan Syndrome) to 90% (Marfan Syndrome). Finally, whole-exome or even whole-genome sequencing, which have been extensively utilized in management of oncology patients, are increasingly being offered as a clinical tool in the care of CHD patients. Although these methods are more expensive and time-consuming than targeted sequencing, they are unbiased and allow re-analysis when additional clinical or genetic information come to light. Whole-exome sequencing has been successfully utilized clinically for almost a decade 72, and is now becoming part of genetic testing for CHD 150, 151. It provides information for the entire coding region of the genome at a cost of only 2–3 times the cost of targeted panels. The current clinical approach to WES in CHD is to obtain the data for the entire exome, and then interrogate gene sets that are driven by the clinical phenotype. If there are new pieces of clinical information obtained during the patient’s course, it is possible to re-interrogate and look for mutations in a different set of genes. Further, if new genes are identified as possible etiologies for the patient’s phenotype, it is possible to obtain patient data for those genes without reobtaining a sample and repeating sequencing. Because of the large amounts of data obtained by WES, the most challenging aspect of this methodology is identifying whether rare variants that are biologically plausible, but have not previously been linked to disease (variants of unknown significance, VUS), are actually causal. Since de-novo mutations have been associated with CHD, interpretation of clinical WES results is greatly helped by obtaining parental samples; if a variant is not identified in either phenotypically normal parent, the likelihood that it is contributing to the patient’s CHD is much more likely.
Genetic testing for CHD is increasingly becoming part of standard care, especially for severe CHD requiring intervention, and for CHD associated with extracardiac abnormalities. Specific testing should be strongly guided by the cardiac and extracardiac phenotype; a proposed strategy for clinical genetic testing in CHD is outlined in Figure 5. Phenotyping and family history are exceptionally important in this scenario, as they inform the type of testing, and will help guide the genetic testing laboratory. Equally important will be careful adjudication of those variants that are not known disease-causing variants, but variants of unknown significance (VUS). The first step here is to test whether the variant is de-novo, which greatly raises the likelihood that the variant is disease causing. Additional information on the likelihood that a given variant is disease-causing is provided by measures of evolutionary conservation of the position at which the variant is occurring, and by the biological impact of the resulting amino acid substitution. Computational algorithms that integrate sequence and functional parameters provide indices of whether a given missense variant is predicted to be damaging or benign are also able to add information on the likely pathogenicity of a sequence variant (Meta-SVM, Polyphen-2, SIFT, MutationTaster, among others) 152. As more and increasingly complex technologies reveal an expanding array of genomic variation, criteria for interpretation of clinical genetic testing are being developed and standardized by workgroups including the American College of Medical Genetics and Genomics 153. Finally, the implications of genetic testing for CHD patients and their families are significant, ranging from prognostic risk factors for neurodevelopmental outcome, to estimates of recurrence risk in siblings, and increasingly as CHD patients reach reproductive age, to recurrence risk in their own offspring. The increasing awareness of the major genetic contribution to CHD provides a strong argument for providing broad access for CHD patients and families to specialized cardiac genetics clinics that can provide high-quality genetic counseling, along with training of pediatric and adult cardiologists, and genetic counselors, in CHD genetics 150.
Figure 5.
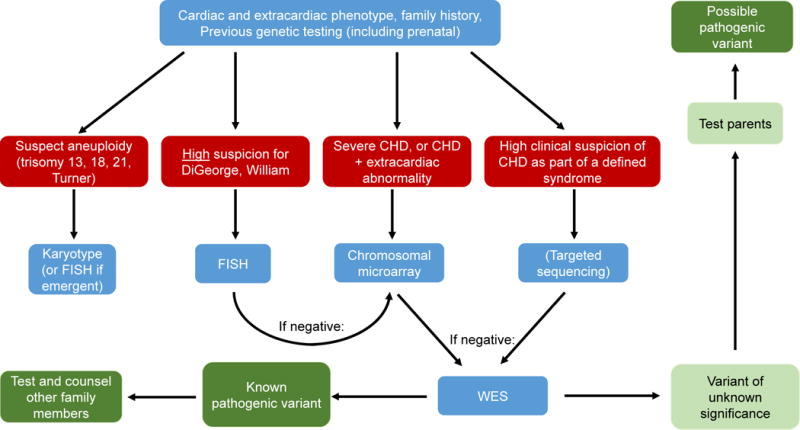
Outline of proposed clinical genetic testing for patients with CHD
Summary
Genetics of CHD has made giant leaps forward in parallel with the evolution of genome analysis technologies. The suspicion that CHD is extremely heterogenic has been validated, and the anticipated complexity of CHD genetics further increased by relatively limited observed genotype-phenotype correlations. Even syndromes that were thought to be well-defined clinically, such as CHARGE and Kabuki, are showing tremendous variation in phenotype when they are defined on the basis of the molecular finding. It is distinctly possible that some of the outcome in CHD is substantially influenced by the underlying genetic cause, in addition to the morphology and hemodynamics that underpin the impressively successful medical and surgical management of CHD to date 3. This observation drives the hope that early identification of genetic causes of CHD will allow more tailored management of CHD, and will hopefully improve the outcome especially with respect to the many co-morbidities of CHD that have a profound impact in quality of life for patients living with CHD. For example, identification of neurodevelopmental risk genes can identify patients who can benefit from early intervention programs long before any clinical signs of NDD such as learning disabilities become apparent. In addition to the clinical implications of a more complete understanding of CHD genetics, the genes uncovered in human patients have already provided tremendous insights into the basic mechanisms underlying cardiac development.
Going forward, there is still much work to be done. Studies to date have at most defined the cause of 45–50% of CHD. Current analysis protocols are possibly underestimating the CNV and SNV contributions due to detection limitations and difficulty predicting whether identified variants are pathogenic or not. In addition, it is highly likely that some CHD is due to multi-locus inheritance, and that some is caused by mutations in non-coding DNA. As larger cohorts of CHD patients are being evaluated with progressively more comprehensive sequencing, it appears ever more likely that we will be able to identify the genetic underpinning of the majority of CHD, and to translate these findings into precision medicine for the care of CHD patients from infants to adults.
Supplementary Material
Acknowledgments
The authors are grateful for the invaluable contribution to the manuscript made by Dr. Richard P. Lifton, President of Rockefeller University. We also thank Dr. Robert W. Elder, Yale University Department of Pediatrics/Cardiology for critical review of the manuscript. MB is supported by NIH UM1HL098162-07 and NIH RO1HL125885-01
Non-standard abbreviations and acronyms
- CHD
Congenital Heart Disease
- CTD
Conotruncal Defects
- LVO
Left-ventricular outflow defects
- HTX
Heterotaxy-associated defects
- NDD
Neurodevelopmental Disabilities
- RV
Right Ventricle
- LV
Left Ventricle
- ASD
Atrial Septal Defect
- HLHS
Hypoplastic Left Heart Syndrome
- CoA
Coarctation of the Aorta
- BAV
Bicuspid Aortic Valve
- TOF
Tetralogy of Fallot
- AVSD
Atrio-ventricular Septal Defect
- CNV
Copy Number Variation
- WES
Whole-Exome Sequencing
- WGS
Whole Genome Sequencing
- LOF
Loss of Function
- ASE
Allele Specific Expression
- VUS
Variant of Unknown Significance
- PCD
Primary Ciliary Dyskinesia
Footnotes
Disclosures
None
References
- 1.Triedman JK, Newburger JW. Trends in Congenital Heart Disease: The Next Decade. Circulation. 2016;133:2716–33. doi: 10.1161/CIRCULATIONAHA.116.023544. [DOI] [PubMed] [Google Scholar]
- 2.van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–7. doi: 10.1016/j.jacc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 3.Gelb BD. History of Our Understanding of the Causes of Congenital Heart Disease. Circ Cardiovasc Genet. 2015;8:529–36. doi: 10.1161/CIRCGENETICS.115.001058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leirgul E, Fomina T, Brodwall K, Greve G, Holmstrom H, Vollset SE, Tell GS, Oyen N. Birth prevalence of congenital heart defects in Norway 1994-2009–a nationwide study. Am Heart J. 2014;168:956–64. doi: 10.1016/j.ahj.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 5.Kwan GF, Mayosi BM, Mocumbi AO, Miranda JJ, Ezzati M, Jain Y, Robles G, Benjamin EJ, Subramanian SV, Bukhman G. Endemic Cardiovascular Diseases of the Poorest Billion. Circulation. 2016;133:2561–75. doi: 10.1161/CIRCULATIONAHA.116.008731. [DOI] [PubMed] [Google Scholar]
- 6.Blalock A, Taussig HB. Landmark article May 19, 1945: The surgical treatment of malformations of the heart in which there is pulmonary stenosis or pulmonary atresia. By Alfred Blalock and Helen B. Taussig. Jama. 1984;251:2123–38. doi: 10.1001/jama.251.16.2123. [DOI] [PubMed] [Google Scholar]
- 7.Marelli AJ, Ionescu-Ittu R, Mackie AS, Guo L, Dendukuri N, Kaouache M. Lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation. 2014;130:749–56. doi: 10.1161/CIRCULATIONAHA.113.008396. [DOI] [PubMed] [Google Scholar]
- 8.Egbe A, Lee S, Ho D, Uppu S, Srivastava S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann Pediatr Cardiol. 2014;7:86–91. doi: 10.4103/0974-2069.132474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bjornard K, Riehle-Colarusso T, Gilboa SM, Correa A. Patterns in the prevalence of congenital heart defects, metropolitan Atlanta, 1978 to 2005. Birth Defects Res A Clin Mol Teratol. 2013;97:87–94. doi: 10.1002/bdra.23111. [DOI] [PubMed] [Google Scholar]
- 10.Oyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PK, Melbye M. National time trends in congenital heart defects, Denmark, 1977–2005. Am Heart J. 2009;157:467–473 e1. doi: 10.1016/j.ahj.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs EG, Leung MP, Karlberg J. Distribution of symptomatic congenital heart disease in Hong Kong. Pediatr Cardiol. 2000;21:148–57. doi: 10.1007/s002469910025. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Li P, Chen S, Xi L, Guo Y, Guo A, Sun K. Influence of genes and the environment in familial congenital heart defects. Mol Med Rep. 2014;9:695–700. doi: 10.3892/mmr.2013.1847. [DOI] [PubMed] [Google Scholar]
- 13.Nora JJ, Dodd PF, McNamara DG, Hattwick MA, Leachman RD, Cooley DA. Risk to offspring of parents with congenital heart defects. Jama. 1969;209:2052–3. [PubMed] [Google Scholar]
- 14.Hyatt BA, Lohr JL, Yost HJ. Initiation of vertebrate left-right axis formation by maternal Vg1. Nature. 1996;384:62–5. doi: 10.1038/384062a0. [DOI] [PubMed] [Google Scholar]
- 15.Herskind AM, Almind Pedersen D, Christensen K. Increased prevalence of congenital heart defects in monozygotic and dizygotic twins. Circulation. 2013;128:1182–8. doi: 10.1161/CIRCULATIONAHA.113.002453. [DOI] [PubMed] [Google Scholar]
- 16.Oyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PK, Melbye M. Recurrence of congenital heart defects in families. Circulation. 2009;120:295–301. doi: 10.1161/CIRCULATIONAHA.109.857987. [DOI] [PubMed] [Google Scholar]
- 17.Oyen N, Poulsen G, Wohlfahrt J, Boyd HA, Jensen PK, Melbye M. Recurrence of discordant congenital heart defects in families. Circ Cardiovasc Genet. 2010;3:122–8. doi: 10.1161/CIRCGENETICS.109.890103. [DOI] [PubMed] [Google Scholar]
- 18.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108–11. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 19.Gebbia M, Ferrero GB, Pilia G, Bassi MT, Aylsworth A, Penman-Splitt M, Bird LM, Bamforth JS, Burn J, Schlessinger D, Nelson DL, Casey B. X-linked situs abnormalities result from mutations in ZIC3. Nature genetics. 1997;17:305–8. doi: 10.1038/ng1197-305. [DOI] [PubMed] [Google Scholar]
- 20.Dina C, Bouatia-Naji N, Tucker N, Delling FN, Toomer K, Durst R, Perrocheau M, Fernandez-Friera L, Solis J, investigators P. Le Tourneau T, Chen MH, Probst V, Bosse Y, Pibarot P, Zelenika D, Lathrop M, Hercberg S, Roussel R, Benjamin EJ, Bonnet F, Lo SH, Dolmatova E, Simonet F, Lecointe S, Kyndt F, Redon R, Le Marec H, Froguel P, Ellinor PT, Vasan RS, Bruneval P, Markwald RR, Norris RA, Milan DJ, Slaugenhaupt SA, Levine RA, Schott JJ, Hagege AA, France MVP, Jeunemaitre X, Leducq Transatlantic MN. Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nat Genet. 2015;47:1206–11. doi: 10.1038/ng.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durst R, Sauls K, Peal DS, deVlaming A, Toomer K, Leyne M, Salani M, Talkowski ME, Brand H, Perrocheau M, Simpson C, Jett C, Stone MR, Charles F, Chiang C, Lynch SN, Bouatia-Naji N, Delling FN, Freed LA, Tribouilloy C, Le Tourneau T, LeMarec H, Fernandez-Friera L, Solis J, Trujillano D, Ossowski S, Estivill X, Dina C, Bruneval P, Chester A, Schott JJ, Irvine KD, Mao Y, Wessels A, Motiwala T, Puceat M, Tsukasaki Y, Menick DR, Kasiganesan H, Nie X, Broome AM, Williams K, Johnson A, Markwald RR, Jeunemaitre X, Hagege A, Levine RA, Milan DJ, Norris RA, Slaugenhaupt SA. Mutations in DCHS1 cause mitral valve prolapse. Nature. 2015;525:109–13. doi: 10.1038/nature14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 23.Shieh JT, Bittles AH, Hudgins L. Consanguinity and the risk of congenital heart disease. American journal of medical genetics Part A. 2012;158A:1236–41. doi: 10.1002/ajmg.a.35272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurvitz M, Ionescu-Ittu R, Guo L, Eisenberg MJ, Abrahamowicz M, Pilote L, Marelli AJ. Prevalence of Cancer in Adults With Congenital Heart Disease Compared With the General Population. Am J Cardiol. 2016;118:1742–1750. doi: 10.1016/j.amjcard.2016.08.057. [DOI] [PubMed] [Google Scholar]
- 25.Gilboa SM, Devine OJ, Kucik JE, Oster ME, Riehle-Colarusso T, Nembhard WN, Xu P, Correa A, Jenkins K, Marelli AJ. Congenital Heart Defects in the United States: Estimating the Magnitude of the Affected Population in 2010. Circulation. 2016;134:101–9. doi: 10.1161/CIRCULATIONAHA.115.019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragan JD, Shin M, Correa A. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatric cardiology. 2011;32:1147–57. doi: 10.1007/s00246-011-0034-5. [DOI] [PubMed] [Google Scholar]
- 27.Wimalasundera RC, Gardiner HM. Congenital heart disease and aneuploidy. Prenat Diagn. 2004;24:1116–22. doi: 10.1002/pd.1068. [DOI] [PubMed] [Google Scholar]
- 28.Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, Sobel EM, Barlow GM, Aylsworth AS, Carpenter NJ, Clark RD, Cohen MY, Doran E, Falik-Zaccai T, Lewin SO, Lott IT, McGillivray BC, Moeschler JB, Pettenati MJ, Pueschel SM, Rao KW, Shaffer LG, Shohat M, Van Riper AJ, Warburton D, Weissman S, Gerstein MB, Snyder M, Korenberg JR. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci U S A. 2009;106:12031–6. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grossman TR, Gamliel A, Wessells RJ, Taghli-Lamallem O, Jepsen K, Ocorr K, Korenberg JR, Peterson KL, Rosenfeld MG, Bodmer R, Bier E. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS genetics. 2011;7:e1002344. doi: 10.1371/journal.pgen.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nature reviews Genetics. 2016;17:224–38. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glessner JT, Bick AG, Ito K, Homsy JG, Rodriguez-Murillo L, Fromer M, Mazaika E, Vardarajan B, Italia M, Leipzig J, DePalma SR, Golhar R, Sanders SJ, Yamrom B, Ronemus M, Iossifov I, Willsey AJ, State MW, Kaltman JR, White PS, Shen Y, Warburton D, Brueckner M, Seidman C, Goldmuntz E, Gelb BD, Lifton R, Seidman J, Hakonarson H, Chung WK. Increased frequency of de novo copy number variants in congenital heart disease by integrative analysis of single nucleotide polymorphism array and exome sequence data. Circulation research. 2014;115:884–96. doi: 10.1161/CIRCRESAHA.115.304458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature genetics. 2001;27:286–91. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 33.Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 34.Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–29. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 35.Fulcoli FG, Franzese M, Liu X, Zhang Z, Angelini C, Baldini A. Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nature communications. 2016;7:11688. doi: 10.1038/ncomms11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, Wilson DB, Watson MS, Hing AV. GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am J Med Genet. 1999;83:201–6. [PubMed] [Google Scholar]
- 37.Nickerson E, Greenberg F, Keating MT, McCaskill C, Shaffer LG. Deletions of the elastin gene at 7q11.23 occur in approximately 90% of patients with Williams syndrome. American journal of human genetics. 1995;56:1156–61. [PMC free article] [PubMed] [Google Scholar]
- 38.Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Human molecular genetics. 1997;6:1021–8. doi: 10.1093/hmg/6.7.1021. [DOI] [PubMed] [Google Scholar]
- 39.Ye M, Coldren C, Liang X, Mattina T, Goldmuntz E, Benson DW, Ivy D, Perryman MB, Garrett-Sinha LA, Grossfeld P. Deletion of ETS-1, a gene in the Jacobsen syndrome critical region, causes ventricular septal defects and abnormal ventricular morphology in mice. Human molecular genetics. 2010;19:648–56. doi: 10.1093/hmg/ddp532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, Jones C. The 11q terminal deletion disorder: a prospective study of 110 cases. American journal of medical genetics Part A. 2004;129A:51–61. doi: 10.1002/ajmg.a.30090. [DOI] [PubMed] [Google Scholar]
- 41.Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, Quintanilla-Dieck Mde L, Artunduaga MA, Eavey RD, Plenge RM, Shadick NA, Weinblatt ME, De Jager PL, Hafler DA, Breitbart RE, Seidman JG, Seidman CE. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nature genetics. 2009;41:931–5. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fakhro KA, Choi M, Ware SM, Belmont JW, Towbin JA, Lifton RP, Khokha MK, Brueckner M. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci U S A. 2011;108:2915–20. doi: 10.1073/pnas.1019645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warburton D, Ronemus M, Kline J, Jobanputra V, Williams I, Anyane-Yeboa K, Chung W, Yu L, Wong N, Awad D, Yu CY, Leotta A, Kendall J, Yamrom B, Lee YH, Wigler M, Levy D. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Human genetics. 2014;133:11–27. doi: 10.1007/s00439-013-1353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hitz MP, Lemieux-Perreault LP, Marshall C, Feroz-Zada Y, Davies R, Yang SW, Lionel AC, D’Amours G, Lemyre E, Cullum R, Bigras JL, Thibeault M, Chetaille P, Montpetit A, Khairy P, Overduin B, Klaassen S, Hoodless P, Awadalla P, Hussin J, Idaghdour Y, Nemer M, Stewart AF, Boerkoel C, Scherer SW, Richter A, Dube MP, Andelfinger G. Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet. 2012;8:e1002903. doi: 10.1371/journal.pgen.1002903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Glidewell SC, Miyamoto SD, Grossfeld PD, Clouthier DE, Coldren CD, Stearman RS, Geraci MW. Transcriptional Impact of Rare and Private Copy Number Variants in Hypoplastic Left Heart Syndrome. Clin Transl Sci. 2015;8:682–9. doi: 10.1111/cts.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soemedi R, Wilson IJ, Bentham J, Darlay R, Topf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, Blue GM, Breckpot J, Hellens S, Zwolinkski S, Glen E, Mamasoula C, Rahman TJ, Hall D, Rauch A, Devriendt K, Gewillig M, OS J, Winlaw DS, Bu’Lock F, Brook JD, Bhattacharya S, Lathrop M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. American journal of human genetics. 2012;91:489–501. doi: 10.1016/j.ajhg.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao W, Niu G, Shen B, Zheng Y, Gong F, Wang X, Lee J, Mulvihill JJ, Chen X, Li S. High-resolution analysis of copy number variants in adults with simple-to-moderate congenital heart disease. American journal of medical genetics Part A. 2013;161A:3087–94. doi: 10.1002/ajmg.a.36177. [DOI] [PubMed] [Google Scholar]
- 48.Kim DS, Kim JH, Burt AA, Crosslin DR, Burnham N, Kim CE, McDonald-McGinn DM, Zackai EH, Nicolson SC, Spray TL, Stanaway IB, Nickerson DA, Heagerty PJ, Hakonarson H, Gaynor JW, Jarvik GP. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J Thorac Cardiovasc Surg. 2016;151:1147–51 e4. doi: 10.1016/j.jtcvs.2015.09.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Prendiville T, Jay PY, Pu WT. Insights into the genetic structure of congenital heart disease from human and murine studies on monogenic disorders. Cold Spring Harb Perspect Med. 2014;4 doi: 10.1101/cshperspect.a013946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253–77. doi: 10.1016/B978-0-12-387786-4.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruneau BG. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb Perspect Biol. 2013;5:a008292. doi: 10.1101/cshperspect.a008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bodmer R. The gene tinman is required for specification of the heart and visceral muscles in Drosophila. Development. 1993;118:719–29. doi: 10.1242/dev.118.3.719. [DOI] [PubMed] [Google Scholar]
- 53.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, Smalls O, Johnson MC, Watson MS, Seidman JG, Seidman CE, Plowden J, Kugler JD. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. The Journal of clinical investigation. 1999;104:1567–73. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–5. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 55.Lee Y, Shioi T, Kasahara H, Jobe SM, Wiese RJ, Markham BE, Izumo S. The cardiac tissue-restricted homeobox protein Csx/Nkx2.5 physically associates with the zinc finger protein GATA4 and cooperatively activates atrial natriuretic factor gene expression. Molecular and cellular biology. 1998;18:3120–9. doi: 10.1128/mcb.18.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–7. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 57.Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, Renault B, Kucherlapati R, Seidman JG, Seidman CE. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997;15:30–5. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- 58.Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, Bonnet D, Lyonnet S, Young ID, Raeburn JA, Buckler AJ, Law DJ, Brook JD. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet. 1997;15:21–9. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- 59.Takeuchi JK, Ohgi M, Koshiba-Takeuchi K, Shiratori H, Sakaki I, Ogura K, Saijoh Y, Ogura T. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development. 2003;130:5953–64. doi: 10.1242/dev.00797. [DOI] [PubMed] [Google Scholar]
- 60.D’Alessandro LC, Casey B, Siu VM. Situs inversus totalis and a novel ZIC3 mutation in a family with X-linked heterotaxy. Congenital heart disease. 2013;8:E36–40. doi: 10.1111/j.1747-0803.2011.00602.x. [DOI] [PubMed] [Google Scholar]
- 61.Megarbane A, Salem N, Stephan E, Ashoush R, Lenoir D, Delague V, Kassab R, Loiselet J, Bouvagnet P. X-linked transposition of the great arteries and incomplete penetrance among males with a nonsense mutation in ZIC3. European journal of human genetics : EJHG. 2000;8:704–8. doi: 10.1038/sj.ejhg.5200526. [DOI] [PubMed] [Google Scholar]
- 62.Sutherland MJ, Wang S, Quinn ME, Haaning A, Ware SM. Zic3 is required in the migrating primitive streak for node morphogenesis and left-right patterning. Human molecular genetics. 2013;22:1913–23. doi: 10.1093/hmg/ddt001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Preuss C, Capredon M, Wunnemann F, Chetaille P, Prince A, Godard B, Leclerc S, Sobreira N, Ling H, Awadalla P, Thibeault M, Khairy P, consortium ML. Samuels ME, Andelfinger G. Family Based Whole Exome Sequencing Reveals the Multifaceted Role of Notch Signaling in Congenital Heart Disease. PLoS genetics. 2016;12:e1006335. doi: 10.1371/journal.pgen.1006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andreassi MG, Della Corte A. Genetics of bicuspid aortic valve aortopathy. Curr Opin Cardiol. 2016;31:585–592. doi: 10.1097/HCO.0000000000000328. [DOI] [PubMed] [Google Scholar]
- 65.Gatrad AR, Read AP, Watson GH. Consanguinity and complex cardiac anomalies with situs ambiguus. Arch Dis Child. 1984;59:242–5. doi: 10.1136/adc.59.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tariq M, Belmont JW, Lalani S, Smolarek T, Ware SM. SHROOM3 is a novel candidate for heterotaxy identified by whole exome sequencing. Genome Biol. 2011;12:R91. doi: 10.1186/gb-2011-12-9-r91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ta-Shma A, Perles Z, Yaacov B, Werner M, Frumkin A, Rein AJ, Elpeleg O. A human laterality disorder associated with a homozygous WDR16 deletion. European journal of human genetics : EJHG. 2015;23:1262–5. doi: 10.1038/ejhg.2014.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perles Z, Moon S, Ta-Shma A, Yaacov B, Francescatto L, Edvardson S, Rein AJ, Elpeleg O, Katsanis N. A human laterality disorder caused by a homozygous deleterious mutation in MMP21. J Med Genet. 2015;52:840–7. doi: 10.1136/jmedgenet-2015-103336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.French VM, van de Laar IM, Wessels MW, Rohe C, Roos-Hesselink JW, Wang G, Frohn-Mulder IM, Severijnen LA, de Graaf BM, Schot R, Breedveld G, Mientjes E, van Tienhoven M, Jadot E, Jiang Z, Verkerk A, Swagemakers S, Venselaar H, Rahimi Z, Najmabadi H, Meijers-Heijboer H, de Graaff E, Helbing WA, Willemsen R, Devriendt K, Belmont JW, Oostra BA, Amack JD, Bertoli-Avella AM. NPHP4 variants are associated with pleiotropic heart malformations. Circulation research. 2012;110:1564–74. doi: 10.1161/CIRCRESAHA.112.269795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, Chang C, Yagi H, San Agustin JT, Thahir M, Anderton S, Lawhead C, Vescovi A, Pratt H, Morgan J, Haynes L, Smith CL, Eppig JT, Reinholdt L, Francis R, Leatherbury L, Ganapathiraju MK, Tobita K, Pazour GJ, CW Lo. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015 doi: 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, Mane S, Lifton RP. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Ligt J, Veltman JA, Vissers LE. Point mutations as a source of de novo genetic disease. Curr Opin Genet Dev. 2013;23:257–63. doi: 10.1016/j.gde.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 74.Campbell CD, Eichler EE. Properties and rates of germline mutations in humans. Trends in genetics : TIG. 2013;29:575–84. doi: 10.1016/j.tig.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, Jin SC, Deanfield J, Giardini A, Porter GA, Jr, Kim R, Bilguvar K, Lopez-Giraldez F, Tikhonova I, Mane S, Romano-Adesman A, Qi H, Vardarajan B, Ma L, Daly M, Roberts AE, Russell MW, Mital S, Newburger JW, Gaynor JW, Breitbart RE, Iossifov I, Ronemus M, Sanders SJ, Kaltman JR, Seidman JG, Brueckner M, Gelb BD, Goldmuntz E, Lifton RP, Seidman CE, Chung WK. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–6. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goldmann JM, Wong WS, Pinelli M, Farrah T, Bodian D, Stittrich AB, Glusman G, Vissers LE, Hoischen A, Roach JC, Vockley JG, Veltman JA, Solomon BD, Gilissen C, Niederhuber JE. Parent-of-origin-specific signatures of de novo mutations. Nat Genet. 2016;48:935–9. doi: 10.1038/ng.3597. [DOI] [PubMed] [Google Scholar]
- 77.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–5. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nature reviews Genetics. 2012;13:565–75. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 79.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, Carriero NJ, Cheung YH, Deanfield J, DePalma S, Fakhro KA, Glessner J, Hakonarson H, Italia MJ, Kaltman JR, Kaski J, Kim R, Kline JK, Lee T, Leipzig J, Lopez A, Mane SM, Mitchell LE, Newburger JW, Parfenov M, Pe’er I, Porter G, Roberts AE, Sachidanandam R, Sanders SJ, Seiden HS, State MW, Subramanian S, Tikhonova IR, Wang W, Warburton D, White PS, Williams IA, Zhao H, Seidman JG, Brueckner M, Chung WK, Gelb BD, Goldmuntz E, Seidman CE, Lifton RP. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–3. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, Prigmore E, Rajan D, Abdul-Khaliq H, Banka S, Bauer UM, Bentham J, Berger F, Bhattacharya S, Bu’Lock F, Canham N, Colgiu IG, Cosgrove C, Cox H, Daehnert I, Daly A, Danesh J, Fryer A, Gewillig M, Hobson E, Hoff K, Homfray T, Study I. Kahlert AK, Ketley A, Kramer HH, Lachlan K, Lampe AK, Louw JJ, Manickara AK, Manase D, McCarthy KP, Metcalfe K, Moore C, Newbury-Ecob R, Omer SO, Ouwehand WH, Park SM, Parker MJ, Pickardt T, Pollard MO, Robert L, Roberts DJ, Sambrook J, Setchfield K, Stiller B, Thornborough C, Toka O, Watkins H, Williams D, Wright M, Mital S, Daubeney PE, Keavney B, Goodship J, Consortium UK. Abu-Sulaiman RM, Klaassen S, Wright CF, Firth HV, Barrett JC, Devriendt K, FitzPatrick DR, Brook JD, Deciphering Developmental Disorders S. Hurles ME. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060–5. doi: 10.1038/ng.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz MP, Lindsay S, D’Alessandro LC, Swaminathan GJ, Bentham J, Arndt AK, Louw J, Breckpot J, Gewillig M, Thienpont B, Abdul-Khaliq H, Harnack C, Hoff K, Kramer HH, Schubert S, Siebert R, Toka O, Cosgrove C, Watkins H, Lucassen AM, O’Kelly IM, Salmon AP, Bu’lock FA, Granados-Riveron J, Setchfield K, Thornborough C, Brook JD, Mulder B, Klaassen S, Bhattacharya S, Devriendt K, Fitzpatrick DF, Consortium UK. Wilson DI, Mital S, Hurles ME. Rare variants in NR2F2 cause congenital heart defects in humans. American journal of human genetics. 2014;94:574–85. doi: 10.1016/j.ajhg.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott D, Haddad F, Reddy S, Bernstein D, Burns T, Steimle JD, Yang XH, Moskowitz IP, Hurles M, Lifton RP, Nickerson D, Bamshad M, Eichler EE, Mital S, Sheffield V, Quertermous T, Gelb BD, Portman M, Ashley EA. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS genetics. 2016;12:e1005963. doi: 10.1371/journal.pgen.1005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–11. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–20. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li N, Subrahmanyan L, Smith E, Yu X, Zaidi S, Choi M, Mane S, Nelson-Williams C, Bahjati M, Kazemi M, Hashemi M, Fathzadeh M, Narayanan A, Tian L, Montazeri F, Mani M, Begleiter ML, Coon BG, Lynch HT, Olson EN, Zhao H, Ruland J, Lifton RP, Mani A. Mutations in the Histone Modifier PRDM6 Are Associated with Isolated Nonsyndromic Patent Ductus Arteriosus. American journal of human genetics. 2016;98:1082–91. doi: 10.1016/j.ajhg.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nature reviews Genetics. 2013;14:347–59. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tarcic O, Pateras IS, Cooks T, Shema E, Kanterman J, Ashkenazi H, Boocholez H, Hubert A, Rotkopf R, Baniyash M, Pikarsky E, Gorgoulis VG, Oren M. RNF20 Links Histone H2B Ubiquitylation with Inflammation and Inflammation-Associated Cancer. Cell reports. 2016;14:1462–76. doi: 10.1016/j.celrep.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ang SY, Uebersohn A, Spencer CI, Huang Y, Lee JE, Ge K, Bruneau BG. KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development. 2016;143:810–21. doi: 10.1242/dev.132688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boskovski MT, Yuan S, Pedersen NB, Goth CK, Makova S, Clausen H, Brueckner M, Khokha MK. The heterotaxy gene GALNT11 glycosylates Notch to orchestrate cilia type and laterality. Nature. 2013;504:456–9. doi: 10.1038/nature12723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. 2007;445:781–4. doi: 10.1038/nature05577. [DOI] [PubMed] [Google Scholar]
- 91.Han P, Bloomekatz J, Ren J, Zhang R, Grinstein JD, Zhao L, Burns CG, Burns CE, Anderson RM, Chi NC. Coordinating cardiomyocyte interactions to direct ventricular chamber morphogenesis. Nature. 2016;534:700–4. doi: 10.1038/nature18310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 93.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–42. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 94.McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. American journal of human genetics. 2006;79:169–73. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, Dietz HC. Familial Tetralogy of Fallot caused by mutation in the jagged1 gene. Human molecular genetics. 2001;10:163–9. doi: 10.1093/hmg/10.2.163. [DOI] [PubMed] [Google Scholar]
- 96.Yoshiba S, Hamada H. Roles of cilia, fluid flow, and Ca signaling in breaking of left-right symmetry. Trends Genet. 2013 doi: 10.1016/j.tig.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 97.McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 98.Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 99.Yoshiba S, Shiratori H, Kuo IY, Kawasumi A, Shinohara K, Nonaka S, Asai Y, Sasaki G, Belo JA, Sasaki H, Nakai J, Dworniczak B, Ehrlich BE, Pennekamp P, Hamada H. Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science. 2012;338:226–31. doi: 10.1126/science.1222538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, Robinson BV, Minnix SL, Olbrich H, Severin T, Ahrens P, Lange L, Morillas HN, Noone PG, Zariwala MA, Knowles MR. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–21. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 101.Vetrini F, D’Alessandro LC, Akdemir ZC, Braxton A, Azamian MS, Eldomery MK, Miller K, Kois C, Sack V, Shur N, Rijhsinghani A, Chandarana J, Ding Y, Holtzman J, Jhangiani SN, Muzny DM, Gibbs RA, Eng CM, Hanchard NA, Harel T, Rosenfeld JA, Belmont JW, Lupski JR, Yang Y. Bi-allelic Mutations in PKD1L1 Are Associated with Laterality Defects in Humans. American journal of human genetics. 2016;99:886–893. doi: 10.1016/j.ajhg.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Olbrich H, Haffner K, Kispert A, Volkel A, Volz A, Sasmaz G, Reinhardt R, Hennig S, Lehrach H, Konietzko N, Zariwala M, Noone PG, Knowles M, Mitchison HM, Meeks M, Chung EM, Hildebrandt F, Sudbrak R, Omran H. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nature genetics. 2002;30:143–4. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 103.Supp DM, Witte DP, Potter SS, Brueckner M. Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature. 1997;389:963–6. doi: 10.1038/40140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Slough J, Cooney L, Brueckner M. Monocilia in the embryonic mouse heart suggest a direct role for cilia in cardiac morphogenesis. Dev Dyn. 2008;237:2304–14. doi: 10.1002/dvdy.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goetz JG, Steed E, Ferreira RR, Roth S, Ramspacher C, Boselli F, Charvin G, Liebling M, Wyart C, Schwab Y, Vermot J. Endothelial cilia mediate low flow sensing during zebrafish vascular development. Cell reports. 2014;6:799–808. doi: 10.1016/j.celrep.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 106.Kerstjens-Frederikse WS, van de Laar IM, Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, Klein Wassink-Ruiter JS, van der Zwaag PA, du Marchie Sarvaas GJ, Bergman KA, Bilardo CM, Roos-Hesselink JW, Janssen JH, Frohn-Mulder IM, van Spaendonck-Zwarts KY, van Melle JP, Hofstra RM, Wessels MW. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genet Med. 2016;18:914–23. doi: 10.1038/gim.2015.193. [DOI] [PubMed] [Google Scholar]
- 107.Winston JB, Schulkey CE, Chen IB, Regmi SD, Efimova M, Erlich JM, Green CA, Aluko A, Jay PY. Complex trait analysis of ventricular septal defects caused by Nkx2-5 mutation. Circ Cardiovasc Genet. 2012;5:293–300. doi: 10.1161/CIRCGENETICS.111.961136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li H, Edie S, Klinedinst D, Jeong JS, Blackshaw S, Maslen CL, Reeves RH. Penetrance of Congenital Heart Disease in a Mouse Model of Down Syndrome Depends on a Trisomic Potentiator of a Disomic Modifier. Genetics. 2016;203:763–70. doi: 10.1534/genetics.116.188045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ackerman C, Locke AE, Feingold E, Reshey B, Espana K, Thusberg J, Mooney S, Bean LJ, Dooley KJ, Cua CL, Reeves RH, Sherman SL, Maslen CL. An excess of deleterious variants in VEGF-A pathway genes in Down-syndrome-associated atrioventricular septal defects. American journal of human genetics. 2012;91:646–59. doi: 10.1016/j.ajhg.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cordell HJ, Bentham J, Topf A, Zelenika D, Heath S, Mamasoula C, Cosgrove C, Blue G, Granados-Riveron J, Setchfield K, Thornborough C, Breckpot J, Soemedi R, Martin R, Rahman TJ, Hall D, van Engelen K, Moorman AF, Zwinderman AH, Barnett P, Koopmann TT, Adriaens ME, Varro A, George AL, Jr, dos Remedios C, Bishopric NH, Bezzina CR, O’Sullivan J, Gewillig M, Bu’Lock FA, Winlaw D, Bhattacharya S, Devriendt K, Brook JD, Mulder BJ, Mital S, Postma AV, Lathrop GM, Farrall M, Goodship JA, Keavney BD. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat Genet. 2013;45:822–4. doi: 10.1038/ng.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hu Z, Shi Y, Mo X, Xu J, Zhao B, Lin Y, Yang S, Xu Z, Dai J, Pan S, Da M, Wang X, Qian B, Wen Y, Wen J, Xing J, Guo X, Xia Y, Ma H, Jin G, Yu S, Liu J, Zhou Z, Wang X, Chen Y, Sha J, Shen H. A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat Genet. 2013;45:818–21. doi: 10.1038/ng.2636. [DOI] [PubMed] [Google Scholar]
- 112.Ware SM, Harutyunyan KG, Belmont JW. Heart defects in X-linked heterotaxy: evidence for a genetic interaction of Zic3 with the nodal signaling pathway. Dev Dyn. 2006;235:1631–7. doi: 10.1002/dvdy.20719. [DOI] [PubMed] [Google Scholar]
- 113.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, Sullivan CA, Walker MF, Waqar Z, Wei L, Willsey AJ, Yamrom B, Lee YH, Grabowska E, Dalkic E, Wang Z, Marks S, Andrews P, Leotta A, Kendall J, Hakker I, Rosenbaum J, Ma B, Rodgers L, Troge J, Narzisi G, Yoon S, Schatz MC, Ye K, McCombie WR, Shendure J, Eichler EE, State MW, Wigler M. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pediatric Cardiac Genomics C. Gelb B, Brueckner M, Chung W, Goldmuntz E, Kaltman J, Kaski JP, Kim R, Kline J, Mercer-Rosa L, Porter G, Roberts A, Rosenberg E, Seiden H, Seidman C, Sleeper L, Tennstedt S, Kaltman J, Schramm C, Burns K, Pearson G, Rosenberg E. The Congenital Heart Disease Genetic Network Study: rationale, design, and early results. Circulation research. 2013;112:698–706. doi: 10.1161/CIRCRESAHA.111.300297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mahony L, Sleeper LA, Anderson PA, Gersony WM, McCrindle BW, Minich LL, Newburger JW, Saul JP, Vetter VL, Pearson GD, Pediatric Heart Network I The Pediatric Heart Network: a primer for the conduct of multicenter studies in children with congenital and acquired heart disease. Pediatric cardiology. 2006;27:191–8. doi: 10.1007/s00246-005-1151-9. [DOI] [PubMed] [Google Scholar]
- 116.Consortium UK. Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JR, Xu C, Futema M, Lawson D, Iotchkova V, Schiffels S, Hendricks AE, Danecek P, Li R, Floyd J, Wain LV, Barroso I, Humphries SE, Hurles ME, Zeggini E, Barrett JC, Plagnol V, Richards JB, Greenwood CM, Timpson NJ, Durbin R, Soranzo N. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. The New England journal of medicine. 2013;368:1971–9. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van der Maarel SM, Deidda G, Lemmers RJ, van Overveld PG, van der Wielen M, Hewitt JE, Sandkuijl L, Bakker B, van Ommen GJ, Padberg GW, Frants RR. De novo facioscapulohumeral muscular dystrophy: frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. American journal of human genetics. 2000;66:26–35. doi: 10.1086/302730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, Garcia-Linares C, Cooper DN, Lazaro C, Kehrer-Sawatzki H. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1) Hum Mutat. 2011;32:213–9. doi: 10.1002/humu.21418. [DOI] [PubMed] [Google Scholar]
- 120.Prabhu S, Jenny B, James H, Provenzano S. Mosaic 22q11.2 deletion and tetralogy of Fallot with absent pulmonary valve: an unreported association. World J Pediatr Congenit Heart Surg. 2015;6:342–5. doi: 10.1177/2150135114561686. [DOI] [PubMed] [Google Scholar]
- 121.Winberg J, Berggren H, Malm T, Johansson S, Johansson Ramgren J, Nilsson B, Lieden A, Nordenskjold A, Gustavsson P, Nordgren A. No evidence for mosaic pathogenic copy number variations in cardiac tissue from patients with congenital heart malformations. Eur J Med Genet. 2015;58:129–33. doi: 10.1016/j.ejmg.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 122.McKean DM, Homsy J, Wakimoto H, Patel N, Gorham J, DePalma SR, Ware JS, Zaidi S, Ma W, Patel N, Lifton RP, Chung WK, Kim R, Shen Y, Brueckner M, Goldmuntz E, Sharp AJ, Seidman CE, Gelb BD, Seidman JG. Loss of RNA expression and allele-specific expression associated with congenital heart disease. Nature communications. 2016;7:12824. doi: 10.1038/ncomms12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL, American Heart Association Council on Cardiovascular Disease in the Y Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:2995–3014. doi: 10.1161/CIRCULATIONAHA.106.183216. [DOI] [PubMed] [Google Scholar]
- 124.Liu S, Joseph KS, Luo W, Leon JA, Lisonkova S, Van den Hof M, Evans J, Lim K, Little J, Sauve R, Kramer MS, Canadian Perinatal Surveillance S Effect of Folic Acid Food Fortification in Canada on Congenital Heart Disease Subtypes. Circulation. 2016;134:647–55. doi: 10.1161/CIRCULATIONAHA.116.022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang W, Wang Y, Gong F, Zhu W, Fu S. MTHFR C677T polymorphism and risk of congenital heart defects: evidence from 29 case-control and TDT studies. PloS one. 2013;8:e58041. doi: 10.1371/journal.pone.0058041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oyen N, Diaz LJ, Leirgul E, Boyd HA, Priest J, Mathiesen ER, Quertermous T, Wohlfahrt J, Melbye M. Prepregnancy Diabetes and Offspring Risk of Congenital Heart Disease: A Nationwide Cohort Study. Circulation. 2016;133:2243–53. doi: 10.1161/CIRCULATIONAHA.115.017465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Morishima M, Yasui H, Ando M, Nakazawa M, Takao A. Influence of genetic and maternal diabetes in the pathogenesis of visceroatrial heterotaxy in mice. Teratology. 1996;54:183–90. doi: 10.1002/(SICI)1096-9926(199610)54:4<183::AID-TERA2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 128.Simeone RM, Tinker SC, Gilboa SM, Agopian AJ, Oster ME, Devine OJ, Honein MA, National Birth Defects Prevention S Proportion of selected congenital heart defects attributable to recognized risk factors. Ann Epidemiol. 2016;26:838–845. doi: 10.1016/j.annepidem.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Human molecular genetics. 2012;21:3255–63. doi: 10.1093/hmg/dds165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Timberlake AT, Choi J, Zaidi S, Lu Q, Nelson-Williams C, Brooks ED, Bilguvar K, Tikhonova I, Mane S, Yang JF, Sawh-Martinez R, Persing S, Zellner EG, Loring E, Chuang C, Galm A, Hashim PW, Steinbacher DM, DiLuna ML, Duncan CC, Pelphrey KA, Zhao H, Persing JA, Lifton RP. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife. 2016;5 doi: 10.7554/eLife.20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Deciphering Developmental Disorders S. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017 doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, Kwint M, Janssen IM, Hoischen A, Schenck A, Leach R, Klein R, Tearle R, Bo T, Pfundt R, Yntema HG, de Vries BB, Kleefstra T, Brunner HG, Vissers LE, Veltman JA. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 133.Shigemizu D, Momozawa Y, Abe T, Morizono T, Boroevich KA, Takata S, Ashikawa K, Kubo M, Tsunoda T. Performance comparison of four commercial human whole-exome capture platforms. Sci Rep. 2015;5:12742. doi: 10.1038/srep12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Theis JL, Zimmermann MT, Evans JM, Eckloff BW, Wieben ED, Qureshi MY, O’Leary PW, Olson TM. Recessive MYH6 Mutations in Hypoplastic Left Heart With Reduced Ejection Fraction. Circ Cardiovasc Genet. 2015;8:564–71. doi: 10.1161/CIRCGENETICS.115.001070. [DOI] [PubMed] [Google Scholar]
- 135.Marino BS, Lipkin PH, Newburger JW, Peacock G, Gerdes M, Gaynor JW, Mussatto KA, Uzark K, Goldberg CS, Johnson WH, Jr, Li J, Smith SE, Bellinger DC, Mahle WT, American Heart Association Congenital Heart Defects Committee CoCDitYCoCN and Stroke C Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126:1143–72. doi: 10.1161/CIR.0b013e318265ee8a. [DOI] [PubMed] [Google Scholar]
- 136.Calderon J. Executive Function in Patients with Congenital Heart Disease: Only the Tip of the Iceberg? The Journal of pediatrics. 2016;173:7–9. doi: 10.1016/j.jpeds.2016.02.066. [DOI] [PubMed] [Google Scholar]
- 137.Marelli A, Miller SP, Marino BS, Jefferson AL, Newburger JW. Brain in Congenital Heart Disease Across the Lifespan: The Cumulative Burden of Injury. Circulation. 2016;133:1951–62. doi: 10.1161/CIRCULATIONAHA.115.019881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Limperopoulos C, Majnemer A, Shevell MI, Rohlicek C, Rosenblatt B, Tchervenkov C, Darwish HZ. Predictors of developmental disabilities after open heart surgery in young children with congenital heart defects. The Journal of pediatrics. 2002;141:51–8. doi: 10.1067/mpd.2002.125227. [DOI] [PubMed] [Google Scholar]
- 139.Miller SP, McQuillen PS, Hamrick S, Xu D, Glidden DV, Charlton N, Karl T, Azakie A, Ferriero DM, Barkovich AJ, Vigneron DB. Abnormal brain development in newborns with congenital heart disease. The New England journal of medicine. 2007;357:1928–38. doi: 10.1056/NEJMoa067393. [DOI] [PubMed] [Google Scholar]
- 140.Burnham N, Ittenbach RF, Stallings VA, Gerdes M, Zackai E, Bernbaum J, Clancy RR, Gaynor JW. Genetic factors are important determinants of impaired growth after infant cardiac surgery. J Thorac Cardiovasc Surg. 2010;140:144–9. doi: 10.1016/j.jtcvs.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Diamond A, Barnett WS, Thomas J, Munro S. Preschool program improves cognitive control. Science. 2007;318:1387–8. doi: 10.1126/science.1151148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–9. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 143.Horani A, Ferkol TW, Dutcher SK, Brody SL. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev. 2016;18:18–24. doi: 10.1016/j.prrv.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harden B, Tian X, Giese R, Nakhleh N, Kureshi S, Francis R, Hanumanthaiah S, Li Y, Swisher M, Kuehl K, Sami I, Olivier K, Jonas R, Lo CW, Leatherbury L. Increased postoperative respiratory complications in heterotaxy congenital heart disease patients with respiratory ciliary dysfunction. J Thorac Cardiovasc Surg. 2014;147:1291–1298 e2. doi: 10.1016/j.jtcvs.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 145.Beausejour Ladouceur V. Training in cardiovascular genetics. J Am Coll Cardiol. 2015;65:856–8. doi: 10.1016/j.jacc.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 146.Geddes GC, Butterly M, Sajan I. FISH for 22q11.2 deletion not cost-effective for infants with congenital heart disease with microarray. Pediatric cardiology. 2015;36:531–6. doi: 10.1007/s00246-014-1045-9. [DOI] [PubMed] [Google Scholar]
- 147.Ahrens-Nicklas RC, Khan S, Garbarini J, Woyciechowski S, D’Alessandro L, Zackai EH, Deardorff MA, Goldmuntz E. Utility of genetic evaluation in infants with congenital heart defects admitted to the cardiac intensive care unit. American journal of medical genetics Part A. 2016 doi: 10.1002/ajmg.a.37891. [DOI] [PubMed] [Google Scholar]
- 148.Jia Y, Louw JJ, Breckpot J, Callewaert B, Barrea C, Sznajer Y, Gewillig M, Souche E, Dehaspe L, Vermeesch JR, Lambrechts D, Devriendt K, Corveleyn A. The diagnostic value of next generation sequencing in familial nonsyndromic congenital heart defects. American journal of medical genetics Part A. 2015;167A:1822–9. doi: 10.1002/ajmg.a.37108. [DOI] [PubMed] [Google Scholar]
- 149.Blue GM, Kirk EP, Giannoulatou E, Dunwoodie SL, Ho JW, Hilton DC, White SM, Sholler GF, Harvey RP, Winlaw DS. Targeted next-generation sequencing identifies pathogenic variants in familial congenital heart disease. J Am Coll Cardiol. 2014;64:2498–506. doi: 10.1016/j.jacc.2014.09.048. [DOI] [PubMed] [Google Scholar]
- 150.Landis BJ, Ware SM. The Current Landscape of Genetic Testing in Cardiovascular Malformations: Opportunities and Challenges. Front Cardiovasc Med. 2016;3:22. doi: 10.3389/fcvm.2016.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.LaHaye S, Corsmeier D, Basu M, Bowman JL, Fitzgerald-Butt S, Zender G, Bosse K, McBride KL, White P, Garg V. Utilization of Whole Exome Sequencing to Identify Causative Mutations in Familial Congenital Heart Disease. Circ Cardiovasc Genet. 2016;9:320–9. doi: 10.1161/CIRCGENETICS.115.001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human molecular genetics. 2015;24:2125–37. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.