

Abstract
Due to China’s rapidly increasing population, the total arable land area has dramatically decreased; as a consequence, the competition for farming land allocated for grain and cotton production has become fierce. Therefore, to overcome the existing contradiction between cotton grain and fiber production and the limited farming land, development of early-maturing cultivars is necessary. In this research, a high-density linkage map of upland cotton was constructed using genotyping by sequencing (GBS) to discover single nucleotide polymorphism (SNP) markers associated with early maturity in 170 F2 individuals derived from a cross between LU28 and ZHONG213. The high-density genetic map, which was composed of 3978 SNP markers across the 26 cotton chromosomes, spanned 2480 cM with an average genetic distance of 0.62 cM. Collinearity analysis showed that the genetic map was of high quality and accurate and agreed well with the Gossypium hirsutum reference genome. Based on this high-density linkage map, QTL analysis was performed on cotton early-maturity traits, including FT, FBP, WGP, NFFB, HNFFB and PH. A total 47 QTLs for the six traits were detected; each of these QTLs explained between 2.61% and 32.57% of the observed phenotypic variation. A major region controlling early-maturity traits in Gossypium hirsutum was identified for FT, FBP, WGP, NFFB and HNFFB on chromosome D03. QTL analyses revealed that phenotypic variation explained (PVE) ranged from 10.42% to 32.57%. Two potential candidate genes, Gh_D03G0885 and Gh_D03G0922, were predicted in a stable QTL region and had higher expression levels in the early-maturity variety ZHONG213 than in the late-maturity variety LU28. However, further evidence is required for functional validation. This study could provide useful information for the dissection of early-maturity traits and guide valuable genetic loci for molecular-assisted selection (MAS) in cotton breeding.
Introduction
Upland cotton (Gossypium hirsutum L. AADD, 2n = 52), the most widely planted economic crop and the leading source of natural fiber worldwide, accounts for 95% of global cotton production [1]. Early maturity is an important breeding target in cotton cultivars; it can increase multiple crop indexes, reduce disaster losses and ensure stable cotton production. Short-season cotton, also called early-maturity cotton, generally exhibits a dwarf, compact plant architecture; shorter height of the node of the first fruiting branch (HNFFB); and shorter whole growth period (WGP) than those of middle- to late-maturity cotton. In recent years, with the limited arable land in China, there has been a competition between grain and cotton fiber production, which limits crop productivity. To address this and to efficiently utilize the limited farming land throughout the crop growing season, development of early-maturity cultivars with high yields and good fiber quality traits, coupled with resistance to major diseases, is needed. Flowering time (FT), the period from the first flower blooming to the first boll opening (FBP), WGP, node of the first fruiting branch (NFFB), HNFFB and plant height (PH) are important early-maturity-related traits of cotton [2–5]. WGP represents the entire duration of growth and development and consists of two periods: FT and FBP. All six traits are quantitatively inherited in cotton.
In cotton, the first genetic linkage map was constructed in 1994 using restriction fragment length polymorphism (RFLP) molecular markers from an interspecific F2 population of G. hirsutum and G. barbadense that comprised 705 loci and 57 lines. Even though single sequence repeat (SSR) markers are the most popular molecular markers in genetic map construction because of their specificity and simplicity, SSR markers cannot reach enough resolution for fine quantitative trait locus (QTL) mapping or map-based cloning. With the advancement of genome sequencing in cotton [6–9], several SNP markers have been identified and further assayed for polymorphisms, depending on the high-throughput platform [3, 10–16]. Among the different types of molecular markers, single nucleotide polymorphism (SNP) markers are currently the first choice for constructing genetic maps due to their high rate of polymorphism and various high-throughput automated platforms, such as Infinium, next-generation sequencing (NGS) and GoldenGate [17]. By using the high-throughput sequencing technologies, several thousand markers in parallel on automated platforms could be produced that are suitable for assaying. However, the utility of these technologies in high-density map construction and identification of qualitative trait loci (QTLs) in cotton using genome-wide linkage analysis needs to be explored. Linkage mapping and association mapping are powerful methods for detecting genetic loci underlying quantitative traits. Over the last two decades, many different quantitative traits in cotton have been reported, including fiber quality traits, yield and yield component traits, disease resistance traits, and drought tolerance-related traits [18–25]. However, early-maturity traits in cotton have received little attention, and to date, few QTLs related to early-maturity traits have been identified [3–5, 26–29]. For example, Fan et al. identified several QTLs related to FT and WGP in an F2 population derived from an upland cotton intraspecific cross [5]. Li et al developed two F2:3 populations and detected 4 common QTLs for early-maturity traits, including WGP, HNFFB, yield percentage before frost (YPBF) and the period from flower bud emergence to flowering (BP) on chromosome D03 [4]. Jia et al. constructed a high-density genetic map containing 6,295 SNPs and 139 SSR markers with an average interval of 0.63 cM and anchored one stable early-maturity-related QTL that spanned a 2-Mb region in 4 environments [10]. Su et al. employed association mapping techniques, which are different from bi-parental linkage mapping, using 81,675 high-density SNP markers in a set of 185 upland cotton accessions and identified 11 highly favorable SNP alleles for five early-maturity traits [3]. These results obtained through the study of early maturity in cotton may be valuable for improving cotton MAS breeding programs.
To better understand the genetic architecture of early-maturity traits in cotton, we present a linkage map generated from 3,978 SNPs that were developed through genotyping by sequencing (GBS). One major QTL was identified and explained 10.42–32.57% of the phenotypic variation (PV) in FT, FBP, WGP, NFFB and HNFFB. In addition, this QTL was also delimited to a 3.36-Mb interval on chromosome D03, a region spanning 112 genes. Candidate genes with known functions or Arabidopsis orthologs are also proposed. This study enriches our knowledge of the genetic bases of FT, FBP, WGP, NFFB, HNFFB and PH in cotton and provides valuable information for MAS breeding in the future.
Materials and methods
Parents and mapping population
ZHONG213 and LU28 were used as parents. ZHONG213, a short-season upland cotton variety, was developed by the Cotton Research Institute of Chinese Academy of Agricultural Sciences (CRICAAS) from the cross (Mei-R1 × CRI27) × (Mei-R1 × Texas29-047). It is an excellent early-maturing cultivar and harbors strong early-maturity genes. ZHONG213’s pedigree was summarized in S1 Fig. In contrast, upland cotton strain LU28, a Chinese commercial multiple-hybrid line, was bred at the Shandong Cotton Research Center and exhibits late-maturity phenotypic traits and wider adaptability (Fig 1). In the summer of 2013, ZHONG213 was crossed with LU28 to obtain F1 seeds at CRICAAS, Anyang, Henan, China (36°08′N, 114°48′E). The F1 seeds were planted during winter in Sanya, Hainan, China (18°29′N, 109°52′E), and self-pollinated to produce F2 generation. The F2 seeds were planted in 10 rows (each 8 m long and 0.8 m apart) and self-pollinated to produce F2:3 seeds at Anyang in 2014. In 2015, we selected 170 F2:3 families and planted them in Anyang, Henan, China. The 170 F2:3 plants were grown in single-row plots with the same row length and width. Pesticides were used to control insects and diseases. The parental accessions used in the cross were obtained from the CRICAAS.
Fig 1. Flowering time (FT) and whole growth period (WGP) performance of two parents.

A Phenotypes of LU28 and ZHONG213 with different FT and WGP. B Phenotypic effect values of FT for two parents. C Phenotypic effect values of WGP for two parents.
Field experiments and phenotyping
The F2 and the F2:3 families were planted at Anyang in 2014–2015, and the experimental protocol was approved by the CRICAAS. The following six traits related to early maturity were investigated in this study: FT, FBP, WGP, NFFB, HNFFB and PH. F2 families were investigated by single plant. In addition, for F2:3 families, all the field experiments followed a randomized complete block design with three replications. Ten plants in the middle of each F2:3 family row were investigated for NFFB, HNFFB and PH. The average was calculated as the last phenotypic value. The phenotypic data were analyzed using R software.
DNA extraction and SNP genotyping
Genomic DNA was isolated from the two parents and 170 F2 individuals using the modified CTAB method [30]. The GBS library was constructed based on the predesigned scheme. Genomic DNA was incubated at 37°C with Mse I (New England Biolabs, NEB), T4 DNA ligase (NEB), ATP (NEB), and MseI Y adapter N containing a barcode for the F2 population. The details of the GBS strategy are described by Zhang et al. [31] and Zhou et al. [32]. The sequences of each sample were sorted according to the barcodes. To ensure that the reads were reliable and without artificial bias (low-quality paired reads, which mainly resulted from base-calling duplicates and adapter contamination) in the subsequent analyses, raw data (raw reads) in fastq format were first processed through a series of quality control (QC) procedures by C scripts. QC standards were as follows: (1) Removing reads with ≥ 10% unidentified nucleotides (N); (2) Removing reads with > 50% bases having phred quality < 5; (3) Removing reads with > 10 nt aligned to the adapter, allowing ≤ 10% mismatches; (4) Removing reads containing Hae III or EcoR I. BWA (Burrows-Wheeler Aligner) was used to align the clean reads of each sample against the reference genome (settings: mem -t 4 -k 32 –M -R) [33]. Alignment files were converted to BAM files using SAMTools software (settings:–bS–t) [34]. If multiple read pairs had identical external coordinates, only the pair with the highest mapping quality was retained. Variant calling was performed for all samples using the GATK software [35]. SNPs were filtered by the PYTHON script. ANNOVAR, an efficient software tool, was used to annotate SNPs based on the GFF3 files for the Gossypium hirsutum genome [36]. Polymorphic markers were classified into eight segregation patterns (ab×cd, ef×eg, hk×hk, lm×ll, nn×np, aa×bb, ab×cc and cc×ab). For the F2 population, segregation patterns were chosen for the genetic map. Prior to map construction, the markers with segregation distortion (p < 0.001) or integrity (> 60%) or containing abnormal bases were filtered.
Genetic map construction
According to the position on the G. hirsutum genome, the linkage group was divided, and then we used PYTHON script to sort the markers in every linkage group. The linkage map was constructed by JoinMap version 4.0 software using a regression approach with a logarithm of odds (LOD) threshold of 6. We used jump threshold at 0.5 to assign segregating markers to linkage groups [37].
QTL mapping
A total of 3978 SNPs were used for QTL mapping. The QTL IciMapping software was used to identify QTLs [38]. The composite interval mapping (CIM) method was utilized to detect any significant association between phenotypic traits and marker loci in the datasets. A LOD threshold of 2.5 was used to declare presence of QTLs.
Candidate gene identification
Candidate genes that were located on chromosome D03 within the 29.51–32.88 Mb QTL region were compared between the parents. To identify candidate genes, all genes in these genomic regions were searched by PYTHON script for gene function in COTTONGENE (https://www.cottongen.org). The genes were functionally annotated using BLASTp [39] and BLAST2GO [40].
Quantitative real-time PCR
Total RNA was extracted from the samples using the Plant RNA Prep Pure Plant kit (Tiangen, Beijing, China). Then, purified total RNA was reverse-transcribed using the SuperScript III First-Stand Synthesis System to obtain cDNA for qRT-PCR (PrimeScript, Takara, Dalian, China). Real-time PCR was performed on the ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The detailed primer information used for PCR amplification is listed in S1 Table. The gene expression levels were calculated using the 2−ΔΔCT method, and each sample used three biological replicates.
Results
Phenotypic characteristics of traits related to early maturity in F2, F2:3 populations and two parents
Phenotypic analysis of the F2 and F2:3 populations (Table 1) revealed significant variation in all six early-maturity traits. The mean value of each early-maturity trait fell between or outside the values of the two parents. All traits showed transgressive segregation, and the absolute values of skewness and kurtosis for each trait were less than 1, which indicated that all the traits were normally distributed and deemed suitable for QTL mapping. Pearson’s correlation coefficient analysis of the six cotton early-maturity traits in the F2 and F2:3 populations are presented in Table 2. All the traits measured were positively correlated with each other (Table 2).
Table 1. Performance and analysis of early-maturity traits in F2, F2:3 populations and two parents.
| Parent | F2: | F2:3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Traits | P1 | P2 | Range | Mean | SD | Kurtosis | Skewness | Range | Mean | SD | Kurtosis | Skewness |
| LU28 | ZHONG213 | |||||||||||
| FT (d) | 56 | 42 | 42.50–62.00 | 58.74 | 3.50 | 0.39 | 0.31 | 41.00–62.00 | 57.61 | 1.94 | -0.32 | -0.18 |
| FBP (d) | 72 | 59 | 37.50–68.50 | 49.64 | 5.68 | 0.78 | 0.88 | 39.50–72.50 | 50.47 | 4.28 | -0.36 | 0.18 |
| WGP (d) | 128 | 101 | 96.00–126.00 | 108.4 | 7.50 | 0.59 | -0.53 | 98.00–128.00 | 108.1 | 5.19 | -0.71 | 0.13 |
| PH (cm) | 81.8 | 55.75 | 43.00–80.00 | 69.65 | 7.46 | 0.25 | 0.03 | 59.38–87.13 | 75.78 | 5.46 | -0.16 | -0.18 |
| HNFFB (cm) | 32.1 | 12 | 12.21–31.25 | 18.71 | 3.33 | 0.97 | 0.94 | 13.38–25.67 | 19.28 | 2.36 | 0.19 | 0.14 |
| NFFB | 7.1 | 4.4 | 4.51–9.52 | 5.86 | 0.79 | 0.32 | 0.13 | 4.49–7.49 | 5.69 | 0.59 | 0.08 | 0.40 |
Table 2. Correlation analysis of six cotton early-maturity traits in F2, F2:3 populations and two parents.
| Traits | FT-F2 | FBP-F2 | WGP-F2 | NFFB-F2 | HNFFB-F2 | PH-F2 |
| FT-F2 | 1 | |||||
| FBP-F2 | 0.294** | 1 | ||||
| WGP-F2 | 0.689** | 0.894** | 1 | |||
| NFFB-F2 | 0.305** | 0.334** | 0.396** | 1 | ||
| HNFFB-F2 | 0.095 | 0.181 | 0.182 | 0.557** | 1 | |
| PH-F2 | 0.017 | 0.020 | 0.066 | 0.293** | 0.425** | 1 |
| FT-F2:3 | FBP-F2:3 | WGP-F2:3 | NFFB-F2:3 | HNFFB-F2:3 | PH-F2:3 | |
| FT-F2:3 | 1 | |||||
| FBP-F2:3 | 0.296** | 1 | ||||
| WGP-F2:3 | 0.615** | 0.935** | 1 | |||
| NFFB-F2:3 | 0.550** | 0.437** | 0.565** | 1 | ||
| HNFFB-F2:3 | 0.496** | 0.389** | 0.505** | 0.561** | 1 | |
| PH-F2:3 | 0.022 | 0.081 | 0.075 | 0.212** | 0.411** | 1 |
**Indicates the correlation reaches significance at 0.01
Genotyping by sequencing (GBS)
In this study, GBS libraries were constructed using the Illumina HiSeq 2500 platform on 170 F2 populations and their parents. A total of 116.23 GB of data containing 650.41 Mb of reads was obtained; each read contained 150 bp (×2) (S2 Table). Among these data, 93.56% bases were high quality with Q30, and the average guanine cytosine (GC) content was 36.94%. The statistical sequencing depth corresponded to 23.90-fold in the LU28, 16.78-fold in the ZHONG213 and 11.68-fold in the 170 progenies (Table 3). The raw data are archived at the NCBI Sequence Read Archive (SRA) under Accession Number PRJNA381273.
Table 3. Summary of sequence data in the parents and F2 families.
| Average Q30 (%) | Average GC content (%) | Average depth (×) | ||
|---|---|---|---|---|
| LU28 | 92.58 | 36.28 | 23.90 | |
| ZHONG213 | 93.04 | 35.01 | 16.78 | |
| F2 | 93.56 | 36.94 | 11.68 | |
A total of 23,576 polymorphic SNP markers were identified from the two parents. All the polymorphic SNP markers contained four genotypes: aa×bb, hk×hk, lm×ll and nn×np (Table 4); however, only the aa×bb genotype was found to be homozygous between the two parents. Therefore, we used this genotype to construct the high-density genetic linkage map. In total, 7,258 SNP markers fell into this type, of which the At and Dt sub-genomes contained 3,645 and 3,613 SNPs, respectively. The percentage of the aa×bb marker on each chromosome varied from 0.85% on chromosome D06 to 9.62% on chromosome A09 (Fig 2).
Table 4. Statistics of marker type in the two parents.
| Marker Type | Number |
|---|---|
| nn×np | 2,707 |
| aa×bb | 7,258 |
| lm×ll | 8,625 |
| hk×hk | 4,986 |
| Total markers | 23,576 |
Fig 2. Genome-wide distribution of SNPs throughout the LU28 and ZHONG213 genomes.
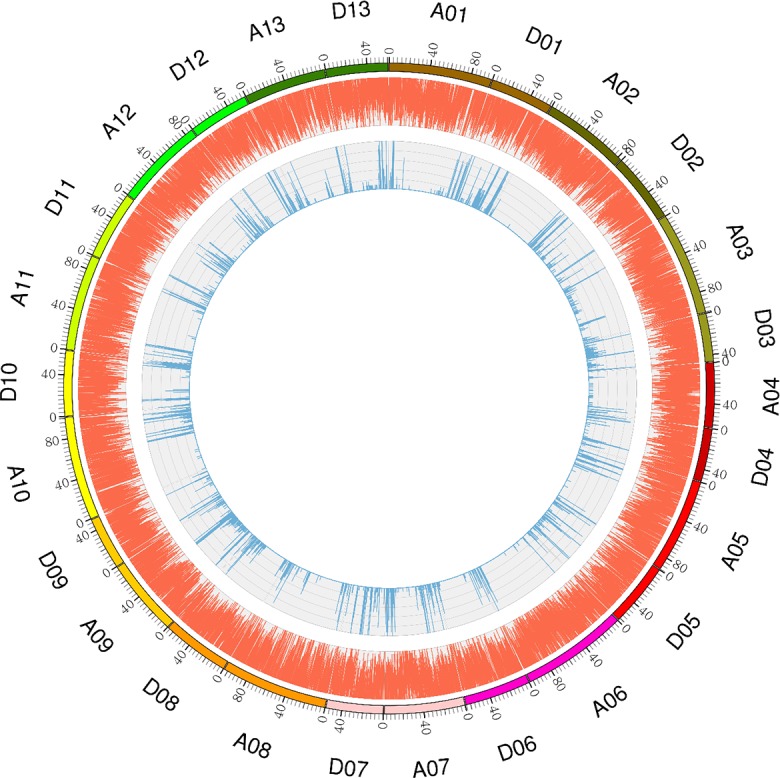
The outermost box with scale represents the 26 cotton chromosomes. The orange histogram represents the density of SNPs that are polymorphic between LU28 and ZHONG213; the blue histogram indicates the density of aa×bb markers between LU28 and ZHONG213.
Construction of the genetic map
As mentioned above, we have 23,576 SNPs in all. However, only the type of aa×bb marker could be used in the linkage analysis. After more than 40% missing SNPs were filtered, 3,978 SNP markers were left and mapped on the genetic map. The genetic map of the F2 population was distributed over 26 linkage groups (Fig 3, Table 5), which spanned a cumulative distance of 2480 cM, 2,117 loci in the At sub-genome (A01–A13) and 1,861 loci in the Dt sub-genome (D01–D13). Each linkage group ranged from 30.60 cM (A03) to 218.23 cM (A05). The number of SNP markers mapped in each linkage group varied from 34 markers on chromosome D06 to 383 markers on chromosome A09, with an average of 153 SNPs per linkage group (Table 5). The average distance between markers across the 26 linkage groups was 95.39 cM.
Fig 3. The genetic map constructed by SNP markers.

Table 5. Summary of the high-density SNP Map based on the F2 families.
| Chromosome | SNP number | Length (cM) | Average interval (cM) |
Largest gap (cM) |
|---|---|---|---|---|
| A01 | 79 | 50.75 | 0.64 | 12.5 |
| A02 | 67 | 44.21 | 0.66 | 8.67 |
| A03 | 145 | 30.6 | 0.21 | 12.44 |
| A04 | 49 | 65.95 | 1.35 | 16.62 |
| A05 | 221 | 218.23 | 0.99 | 24.4 |
| A06 | 117 | 122.71 | 1.05 | 16.85 |
| A07 | 358 | 149.19 | 0.42 | 10.99 |
| A08 | 84 | 74.73 | 0.89 | 14.36 |
| A09 | 383 | 79.75 | 0.21 | 6.45 |
| A10 | 340 | 130.66 | 0.38 | 15.73 |
| A11 | 126 | 74.06 | 0.59 | 14.91 |
| A12 | 94 | 67.74 | 0.72 | 9.67 |
| A13 | 54 | 44.66 | 0.83 | 10.09 |
| At | 2117 | 1153.24 | 0.54 | 24.4 |
| D01 | 175 | 142.55 | 0.81 | 12.78 |
| D02 | 185 | 102.85 | 0.56 | 13.19 |
| D03 | 242 | 101.4 | 0.42 | 18.05 |
| D04 | 128 | 119.72 | 0.94 | 12.61 |
| D05 | 147 | 57.16 | 0.39 | 19.03 |
| D06 | 34 | 103.85 | 3.05 | 13.42 |
| D07 | 233 | 117.5 | 0.5 | 11.83 |
| D08 | 81 | 86.57 | 1.07 | 18.66 |
| D09 | 103 | 86.63 | 0.84 | 18.28 |
| D10 | 162 | 47.71 | 0.29 | 6.25 |
| D11 | 48 | 118.8 | 2.48 | 14.22 |
| D12 | 197 | 109.37 | 0.56 | 14.12 |
| D13 | 126 | 133.01 | 1.06 | 21.9 |
| Dt | 1861 | 1327.12 | 0.71 | 21.9 |
| Total | 3978 | 2480.36 | 0.62 | 24.4 |
At: A sub-genome
Dt: D sub-genome
Collinearity analysis
Collinearity analysis between the linkage and physical maps indicated that the linkage map constructed in the present study had good collinearity with the G. hirsutum reference genome sequence (Fig 4), which suggests the high quality of the F2 genetic map. However, several inconsistencies on chromosomes A11, D09 and D10 were also detected. The At and Dt sub-genomes showed good coverage of the reference G. hirsutum genome, representing 87.37% and 91.02% of the genome assembly length (S3 Table; Fig 4).
Fig 4. Collinearity between the genetic map (LG01-LG26) and the physical map (A01-A13, D01-D13).

QTL mapping of early-maturity traits in the F2 and F2:3 families
A total of 47 QTLs for the six early-maturity traits were detected on the 26 chromosomes and explained 2.61–32.57% of the PV. Among the 47 QTLS, there were 12 QTLs for F2 generation and 35 QTLs for F2:3 generation. Eight QTLs, including two for WGP and NFFB and one each for HNFFB, WGP, FT, PBP and PH, accounted for more than 10% of the PV, whereas the remaining 39 QTLs accounted for less than 10% of the PV (Table 6). Of the total QTLs, only 18 (38%) were located on the At sub-genome, and 29 (62%) were located on the Dt sub-genome. Among these QTLs, 8 (17%) were located on chromosome D03. For FT, the ten QTLs identified were as follows: qFT-A02-1, qFT-A05-1, qFT-A07-1, qFT-A07-2, qFT-A08-1, qFT-A12-1, qFT-A12-2, qFT-D03-1, qFT-D03-2 and qFT-D06-1, which explained 3.57–30.07% of the observed PV, with LOD scores of 2.53–13.80. Ten QTLs for FBP were detected on eight chromosomes. Notably, only 2 (20%) were located on the At sub-genome, whereas 8 (80%) were located on the Dt sub-genome, explaining 3.52–16.96% of the PV. Nine QTLs for WGP were detected on six chromosomes. Here, we note that two significant QTLs, qWGP-D03-1 and qWGP-D03-2, not only had an LOD score > 3 but also explained 30.09% and 10.42% of the PV. A total of five QTLs were detected for PH in this study, and only 1 major QTL explained 10.75% of the PV on chromosome D06 at 20 cM. Four QTLs for HNFFB were identified on the Dt sub-genome and explained 4.53–22.03% of the observed PV, with LOD scores of 2.52–9.44. A total of 9 QTLs were detected for NFFB in this study, of which 2 major QTLs explained 10.88–32.57% of the PV. Three QTLs were on the At sub-genome, and six QTLs were on the Dt sub-genome; these QTLs explained 3.07–32.57% of the observed PV, with LOD scores of 2.52–17.12. Interestingly, a stable QTL region was revealed on chromosome D03 from 43–50 cM and was flanked by FT, FBP, WGP, NFFB and HNFFB. The proportion of PV explained by the QTL ranged from 10.42–32.57%, with LOD scores of 6.66–18.50. Thus, this QTL could be treated as a major QTL for further dissection.
Table 6. Stable QTLs for early maturity identified in F2 and F2:3 populations.
| Trait | QTL | Generation | Chromosome | Position (cM) | LOD | R2 (%) |
|---|---|---|---|---|---|---|
| FT | qFT-A02-1 | F2:3 | A02 | 22.00 | 2.58 | 3.72 |
| qFT-A05-1 | F2:3 | A05 | 179.00 | 2.53 | 3.57 | |
| qFT-A07-1 | F2:3 | A07 | 20.00 | 2.69 | 3.99 | |
| qFT-A07-2 | F2 | A07 | 149.00 | 2.59 | 4.21 | |
| qFT-A08-1 | F2:3 | A08 | 71.00 | 2.75 | 4.65 | |
| qFT-A12-1 | F2 | A12 | 26.00 | 3.11 | 5.27 | |
| qFT-A12-2 | F2 | A12 | 39.00 | 2.59 | 4.75 | |
| qFT-D03-1 | F2:3 | D03 | 0.00 | 2.61 | 4.07 | |
| qFT-D03-2 | F2:3 | D03 | 48.00 | 13.80 | 30.07 | |
| qFT-D06-1 | F2:3 | D06 | 22.00 | 2.54 | 4.77 | |
| FBP | qFBP-A08-1 | F2:3 | A08 | 36.00 | 4.65 | 8.33 |
| qFBP-A10-1 | F2:3 | A10 | 128.00 | 2.65 | 4.00 | |
| qFBP-D03-1 | F2:3 | D03 | 43.00 | 8.70 | 16.96 | |
| qFBP-D03-2 | F2:3 | D03 | 91.00 | 2.52 | 3.70 | |
| qFBP-D06-1 | F2:3 | D06 | 53.00 | 2.54 | 3.78 | |
| qFBP-D08-1 | F2:3 | D08 | 0.00 | 3.30 | 5.82 | |
| qFBP-D10-1 | F2:3 | D10 | 78.00 | 2.55 | 4.16 | |
| qFBP-D10-2 | F2 | D10 | 2.00 | 2.66 | 6.93 | |
| qFBP-D12-1 | F2:3 | D12 | 83.00 | 2.57 | 3.52 | |
| qFBP-D13-1 | F2 | D13 | 88.00 | 2.55 | 5.52 | |
| WGP | qWGP-A08-1 | F2:3 | A08 | 0.00 | 2.60 | 2.61 |
| qWGP-A08-2 | F2:3 | A08 | 36.00 | 5.89 | 7.79 | |
| qWGP-A12-1 | F2:3 | A12 | 34.00 | 3.54 | 4.60 | |
| qWGP-A12-2 | F2 | A12 | 45.00 | 2.60 | 2.91 | |
| qWGP-D01-1 | F2:3 | D01 | 81.00 | 3.04 | 3.90 | |
| qWGP-D03-1 | F2:3 | D03 | 49.00 | 18.50 | 30.09 | |
| qWGP-D03-2 | F2 | D03 | 50.00 | 6.66 | 10.42 | |
| qWGP-D08-1 | F2:3 | D08 | 0.00 | 5.43 | 7.16 | |
| qWGP-D10-1 | F2:3 | D10 | 74.00 | 4.07 | 5.40 | |
| PH | qPH-A01-1 | F2:3 | A10 | 15.00 | 2.73 | 5.98 |
| qPH-A08-1 | F2 | A08 | 35.00 | 3.25 | 4.92 | |
| qPH-D08-1 | F2 | D08 | 78.00 | 3.53 | 5.67 | |
| qPH-D06-1 | F2:3 | D06 | 20.00 | 3.48 | 10.75 | |
| qPH-D08-1 | F2:3 | D08 | 111.00 | 2.57 | 5.18 | |
| HNFFB | qHNFFB-D03-1 | F2:3 | D03 | 48.00 | 9.44 | 22.03 |
| qHNFFB-D06-1 | F2:3 | D06 | 25.00 | 3.90 | 8.31 | |
| qHNFFB-D09-2 | F2 | D09 | 29.00 | 2.52 | 6.06 | |
| qHNFFB-D12-1 | F2:3 | D12 | 34.00 | 2.52 | 4.53 | |
| NFFB | qNFFB-A05-1 | F2:3 | A05 | 46.00 | 2.54 | 3.11 |
| qNFFB-A07-1 | F2:3 | A07 | 128.00 | 2.90 | 4.45 | |
| qNFFB-A12-1 | F2:3 | A12 | 1.00 | 6.66 | 10.88 | |
| qNFFB-D02-1 | F2:3 | D02 | 70.00 | 2.69 | 3.87 | |
| qNFFB-D03-1 | F2:3 | D03 | 49.00 | 17.12 | 32.57 | |
| qNFFB-D05-1 | F2 | D05 | 104.00 | 2.53 | 3.91 | |
| qNFFB-D06-1 | F2:3 | D06 | 56.00 | 2.52 | 3.07 | |
| qNFFB-D10-1 | F2:3 | D10 | 49.00 | 2.60 | 3.60 | |
| qNFFB-D13-1 | F2 | D13 | 109.00 | 2.62 | 4.57 |
Identification of a candidate gene for traits related to early maturity on chromosome D03
As mentioned above, qFT-D03-2, qFBP-D03-1, qWGP-D03-1, qWGP-D03-2 qHNFFB-D03-1 and qNFFB-D03-1 were the six steady QTLs that showed significant effects on FT, FBP, WGP, HNFFB and NFFB from 43–50 cM (Table 6) and occupied a physical region of 3.36 Mb on chromosome D03. Based on the comparative mapping of the G. hirsutum reference genome [9], 112 genes were predicted (S4 Table). Among these genes, 14 candidate genes had no annotation information. Data analysis of qRT-PCR revealed that the expression levels of three genes, Gh_D03G0885, Gh_D03G0922 and Gh_D03G0961, were significantly lower in LU28 than in ZHONG213, whereas the expression levels of Gh_D03G0924, Gh_D03G0929 and Gh_D03G0949 were higher in LU28 than in ZHONG213. The expression levels of the remaining genes in LU28 were not significantly different from those of the genes in ZHONG213 (Fig 5).
Fig 5. Expression of the genes in the two-leaf stage determined by qRT-PCR.

Discussion
With the progress or completion of genome sequencing of many important crops, the molecular genetics and breeding of crops have entered into a high-throughput, large-scale whole-genome level molecular design platform. SNP molecular markers have gained great attention in molecular marker-assisted breeding programs, population and evolutionary genetics, bi-parental QTL mapping, and association mapping studies [10, 11, 41] because they are widely distributed, highly polymorphic and large in quantity [42–44].
The characteristics of markers determine the distribution density of a marker on a genetic linkage map, which further affects the accuracy of the QTLs. From the first-generation marker to the third-generation marker, the distribution of markers in the whole genome of cotton is becoming increasingly abundant, which greatly improves the density of the marker. In this study, we employed GBS to identify a major QTL related to early maturity in cotton using an F2 mapping population. This technique is rapid, cost-effective and widely used in many crops [14, 45–48]. Our map contained 3978 polymorphic SNP markers, and the average distance between markers was 0.62 cM. Compared with previous studies, regardless of whether RFLP, SSR or SNP markers constructed traditional linkage maps generated from the same cross, the total length of the map in our study is shorter [10, 36, 49, 50]. The main reasons for these differences are caused by the marker types, the size of the population and the type of mapping population. Moreover, in our 26 chromosomes, the distribution of some chromosome markers is nonuniform and the number of markers is low, which can decrease the genetic distance of genetic map. The shortest A03 chromosome, although having 145 markers, is unevenly distributed on chromosomes. The three short chromosomes (chromosomes A01 A02 and A13) only spanned 50.75 cM, 44.21 cM and 44.66 cM, harboring 79, 54 and 67 markers, respectively. However, the marker density of cotton has obviously improved, which is mainly attributed to the development and application of high-throughput sequencing. Wang et al. used 4,999,048 SNPs to construct an ultra-dense genetic map extended over 4,042 cM [50]. This map provides a meaningful reference for other scientists who are engaged in G. hirsutum genome assembly [9]. By taking advantage of the RAD-seq technique in upland cotton, the construction of more comprehensive genetic maps using SSR and RAD is becoming increasingly popular [10, 51]. Hulse-Kemp et al. constructed a genetic map that had 19,191 SNPs and 0.21-cM distribution density [52]. Zhang et al. used SALF-seq to construct a high-density genetic map comprised of 5,521 SNPs with 0.78-cM marker spacing [16]. These studies demonstrate that the number of high-throughput sequencing techniques is rapidly growing. The number of markers employed in this study is much higher than that in previous studies and greatly improved the marker density of the linkage map [10, 16]. Moreover, in our study, the number of large interval segments of the map is much lower than that of previous studies [16, 36, 49, 53, 54], which suggests the higher accuracy of the detected QTLs and the possibility of detecting large QTL interval regions in other molecular markers.
Collinearity is an important factor in determining the quality of a genetic map. Collinearity analysis showed that the constructed map had good collinearity with the G. hirsutum reference genome [9], which indicates the high quality and accuracy of the map. The coverage of genomes by the At sub-genome and Dt sub-genome is 87.37% and 91.02%, respectively. One main reason for the lower coverage by the At sub-genome than the Dt sub-genome is that two linkage groups, A03 and A13, represent only 70.27% and 78.80% of the corresponding chromosomes, respectively, while others chromosome range from 83.23% to 98.49% (S4 Table).
Early maturity is an important cotton trait, and short-season cotton is an important cotton resource in China. Given the need to ensure food security, it is necessary to increase the production level and improve yield and quality in cotton-growing regions. Early maturity of cotton is a comprehensive trait that includes WGP, FT, NFFB and other traits, which are important indicators of earliness [3, 4, 10]. These traits are quantitative traits controlled by multiple genes [55, 56].
In cotton, a few studies have mapped genomic regions associated with early-maturity traits. These studies included intra-specific crosses of G. hirsutum TM1 × CRI36 [5], Baimian2 × TM-1 [4], Baimian2 × CRI12 [4] and CRI36 × G2005 [10]. As shown in the pedigree (S1 Fig), ZHONG213, CRI36 and Baimian2 were all derived from the King, which is the founder of the most of China’s short-season cultivars. CRI36 and Baimian2 is a short-season cotton bred in 1999 and 2008, and the WGP was approximately 115 and 110 days respectively. However, with the improvement of breeding technology, ZHONG213 as a new line, with a WGP of 105 days; compared to CRI36 and Baimian2, it has a shorter WGP, better quality, good disease resistance and wide adaptability. It is worth mentioning that none of the parents involved in these studies were insect resistant. Thus, the accuracy of FT and WGP explained in these studies might be affected by insect infestation, such as cotton bollworm, which feeds on buds. To overcome this limitation and ensure the accuracy of the phenotypic data investigated in our study, we used BT cotton parents that have high resistance to the field environment.
In the present study, we uncovered a total of 47 QTLs, including ten for FBP, five for PH, four for HNFFB, ten for FT, seven for WGP, and nine for NFFB (Table 6). Most importantly, the main QTLs for FT (qFT-D03-2), FBP (qFBP-D03-1), WGP (qWGP-D03-1/ qWGP-D03-2), HNFFB (qHNFFB-D03-1) and NFFB (qNFFB-D03-1) were overlapped on chromosome D03 from 43–50 cM and explained 10.42–32.57% of the PV explained (PVE). In particular, one common QTL qWGP-D03-1/ qWGP-D03-2 for WGP was detected in both generations. Common QTLs should be reliable and could be used in maker-assisted selection to increase cotton early maturity. From previous studies, early-maturity QTLs were detected on almost all 26 chromosomes. However, in recent years, many researchers have demonstrated that chromosome D03 contains important segments for early-maturity traits, which indicates that chromosome D03 is responsible for early maturity [3, 4, 10]. When comparing our results with those previously reported by studies of early maturity, the main QTLs on the D03 chromosome could be validated based on previous linkage mapping. The six steady QTLs were positioned between DPL0041 and CIR347 [4] and had an overlap with five early-maturity QTLs that have been reported between Marker25958 and Marker25963 on chromosome D03 [10] (Fig 6). Subsequently, Su et al. found that these SSR markers mapped to the genome sequence by electronic PCR (e-PCR) and, using association mapping, found one peak SNP locus mapped to D03 at 31.98 Mb, positioned between DPL0200 and CIR347 [3]. Interestingly, we also discovered the same SNP located across the same markers and further narrowed the candidate region to 29.51–32.88 Mb on chromosome D03. These findings validate the QTL results and increase cotton breeders’ confidence in the identity of the main QTL.
Fig 6. Physical maps and linkage relationships among quantitative trait loci (QTLs) in previous and present studies.

One hundred and twelve genes were predicted and annotated in this interval. Among them, Gh_D03G0885 and Gh_D03G0922 caught our attention because their expression levels were higher in the early-maturity variety ZHONG213 than in the late-maturity variety LU28. In particular, Gh_D03G0922 was the only AGL8 homolog in the cotton genome, and AGL8 was the best match to Gh_D03G0922 in the Arabidopsis genome. AGL8 is a member of MADS-box gene family, which plays significant roles in regulating FT and flower initiation and participates in plant growth and development [57, 58]. Our previous association mapping study also showed that Gh_D03G0922 is a potential candidate gene for early maturity [3]. Therefore, we speculate that Gh_D03G0922 plays a major role in cotton flowering similar to AGL8 in Arabidopsis. The results of BLAST alignment showed that the CDS identity of Gh_D03G0885 with the Arabidopsis TOC1 gene was as high as 58%. Furthermore, Gh_D03G0885 encoded a protein sharing 49% sequence identity with the Arabidopsis TOC1 protein. Hence, we regard Gh_D03G0885 as the TOC1 homolog in cotton. TOC1 contributes to plant fitness by influencing the circadian clock period, and the expression of TOC1 is correlated with rhythmic changes in chromatin organization [59, 60]. Therefore, it is reasonable to postulate that Gh_D03G0885 and Gh_D03G0922 are candidate genes for early maturity in cotton. Of the remaining 110 genes in this region, 14 annotated genes were found to be unknown genes, and of the remaining 94 genes, none were identified as homologous to Arabidopsis genes that are involved in the FT pathway, such as CO, SOC1, FD, and TFL1. Therefore, to prove the hypothesis, further experiments are needed.
Supporting information
(TIF)
(XLS)
(XLS)
(XLS)
(XLS)
Acknowledgments
We thank the Novogene Company for the Illumina HiSeq sequencing service. This research was supported by the Chinese National Natural Science Foundation (31660409) and the Shandong TAISHAN industry leading talent program (LJNY201608).
Data Availability
Sequences are available on the Sequence Read Archive, http://www.ncbi.nlm.nih.gov/Traces/sra/ under accession number PRJNA381273.
Funding Statement
This work was supported by: Chinese National Natural Science Foundation to Junji Su, grant number: 31660409, https://isisn.nsfc.gov.cn/egrantindex/funcindex/prjsearch-list; Shandong TAISHAN industry leading talent program to Shuxun Yu, grant number: LJNY201608, http://sdgb.shandong.gov.cn/art/2016/12/16/art_4563_5473.html.
References
- 1.Chen ZJ, Scheffler BE, Dennis E, Triplett BA, Zhang T, Guo W, et al. (2007) Toward sequencing cotton (Gossypium) genomes. Plant Physiology 145: 1303–1310. doi: 10.1104/pp.107.107672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Godoy AS, Palomo GA (1999) Genetic analysis of earliness in upland cotton (Gossypium hirsutum L.). I. Morphological and phenological variables. Euphytica 105: 155–160. [Google Scholar]
- 3.Su J, Pang C, Wei H, Li L, Liang B, Wang C, et al. (2016) Identification of favorable SNP alleles and candidate genes for traits related to early maturity via GWAS in upland cotton. BMC Genomics 17: 687 doi: 10.1186/s12864-016-2875-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li C, Wang X, Na D, Zhao H, Zhe X, Rui W, et al. (2013) QTL analysis for early-maturing traits in cotton using two upland cotton (Gossypium hirsutum L.) crosses. Breeding Science 63: 154–163. doi: 10.1270/jsbbs.63.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan SL, Shu-Xun YU, Song MZ, Yuan RH (2006) Construction of molecular linkage map and QTL mapping for earliness in short-season cotton. Cotton Science 18: 135–139. [Google Scholar]
- 6.Li F, Fan G, Lu C, Xiao G, Zou C, Kohel RJ, et al. (2015) Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat Biotechnol 33: 524 doi: 10.1038/nbt.3208 [DOI] [PubMed] [Google Scholar]
- 7.Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, et al. (2014) Genome sequence of the cultivated cotton Gossypium arboreum. Nat Genet 46: 567–572. doi: 10.1038/ng.2987 [DOI] [PubMed] [Google Scholar]
- 8.Wang K, Wang Z, Li F, Ye W, Wang J, Song G, et al. (2012) The draft genome of a diploid cotton Gossypium raimondii. Nat Genet 44: 1098–1103. doi: 10.1038/ng.2371 [DOI] [PubMed] [Google Scholar]
- 9.Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, et al. (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol 33: 531–537. doi: 10.1038/nbt.3207 [DOI] [PubMed] [Google Scholar]
- 10.Jia X, Pang C, Wei H, Wang H, Ma Q, Yang J, et al. (2016) High-density linkage map construction and QTL analysis for earliness-related traits in Gossypium hirsutum L. BMC Genomics 17: 909 doi: 10.1186/s12864-016-3269-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su J, Fan S, Li L, Wei H, Wang C, Wang H, et al. (2016) Detection of favorable QTL alleles and candidate genes for lint percentage by GWAS in Chinese upland cotton. Front Plant Sci 7: 1576 doi: 10.3389/fpls.2016.01576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su J, Li L, Pang C, Wei H, Wang C, Song M, et al. (2016) Two genomic regions associated with fiber quality traits in Chinese upland cotton under apparent breeding selection. Sci Rep 6: 38496 doi: 10.1038/srep38496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hulsekemp A (2015) Development of a 63K SNP array for Gossypium and high-density mapping of intra- and inter-specific populations of cotton (G. hirsutum L.). Plasmid 45: 171–183. [Google Scholar]
- 14.Islam MS, Thyssen GN, Jenkins JN, Zeng L, Delhom CD, Mccarty JC, et al. (2016) A MAGIC population-based genome-wide association study reveals functional association of GhRBB1_A07 gene with superior fiber quality in cotton. Bmc Genomics 17: 903 doi: 10.1186/s12864-016-3249-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cong L, Dong Y, Zhao T, Ling L, Cheng L, Yu E, et al. (2016) Genome-wide SNP linkage mapping and QTL analysis for fiber quality and yield traits in the upland cotton recombinant inbred lines population. Front Plant Sci 7: 1356 doi: 10.3389/fpls.2016.01356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhen Z, Shang H, Shi Y, Long H, Li J, Ge Q, et al. (2016) Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to Quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum L.). BMC Plant Biology 16: 79 doi: 10.1186/s12870-016-0741-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varshney RK, Kudapa H, Roorkiwal M, Thudi M, Pandey MK, Saxena RK, et al. (2012) Advances in genetics and molecular breeding of three legume crops of semi-arid tropics using next-generation sequencing and high-throughput genotyping technologies. J Biosci 37: 811–820. [DOI] [PubMed] [Google Scholar]
- 18.Shen X, Guo W, Zhu X, Yuan Y, Yu JZ, Kohel RJ, et al. (2005) Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol Breed 15: 169–181. [Google Scholar]
- 19.Fang DD, Jenkins JN, Deng DD, Mccarty JC, Li P, Wu J (2014) Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (Gossypium hirsutum L.). BMC Genomics 15: 397 doi: 10.1186/1471-2164-15-397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan Z, Fang X, Tang S, Zhang J, Liu D, Teng Z, et al. (2015) Genetic map and QTL controlling fiber quality traits in upland cotton (Gossypium hirsutum L.). Euphytica 203: 1–14. [Google Scholar]
- 21.Xia Z, Zhang X, Liu YY, Jia ZF, Zhao HH, Cheng-Qi LI, et al. (2014) Major gene identification and quantitative trait locus mapping for yield related traits in upland cotton (Gossypium hirsutum L.). J Integr Agric 13: 299–309. [Google Scholar]
- 22.Feng J, Zhao J, Lei Z, Guo WZ, Zhang TZ (2009) Molecular mapping of Verticillium wilt resistance QTL clustered on chromosomes D7 and D9 in upland cotton. Sci China Life Sci 52: 872–884. [DOI] [PubMed] [Google Scholar]
- 23.Ulloa M, Hutmacher RB, Roberts PA, Wright SD, Nichols RL, Michael Davis R (2013) Inheritance and QTL mapping of Fusarium wilt race 4 resistance in cotton. Theor Appl Genet 126: 1405–1418. doi: 10.1007/s00122-013-2061-5 [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Wang H, Chen W, Li Y (2014) Genetic structure, linkage disequilibrium and association mapping of Verticillium wilt resistance in elite cotton (Gossypium hirsutum L.) germplasm population. Plos One 9: e86308 doi: 10.1371/journal.pone.0086308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levi A, Paterson AH, Cakmak I, Saranga Y (2011) Metabolite and mineral analyses of cotton near-isogenic lines introgressed with QTLs for productivity and drought-related traits. Physiol Plant 141: 265–275. doi: 10.1111/j.1399-3054.2010.01438.x [DOI] [PubMed] [Google Scholar]
- 26.Li CQ, Xu XJ, Dong N, Ai NJ, Wang QL (2016) Association mapping identifies markers related to major early-maturating traits in upland cotton (Gossypium hirsutum L.). Plant Breeding 135: 483–491. [Google Scholar]
- 27.LI C., Song L, Zhao H, Xia Z, Jia Z, Wang X, et al. (2014) Quantitative trait loci mapping for plant architecture traits across two upland cotton populations using SSR markers. J Agric Sci 152: 275–287. [Google Scholar]
- 28.Guo Y, Jackc MC, Johnien J, An C, Sukumar S (2009) Genetic detection of node of first fruiting branch in crosses of a cultivar with two exotic accessions of upland cotton. Euphytica 166: 317–329. [Google Scholar]
- 29.Li C, Wang C, Dong N, Wang X, Zhao H, Converse R, et al. (2012) QTL detection for node of first fruiting branch and its height in upland cotton (Gossypium hirsutum L.). Euphytica 188: 441–451. [Google Scholar]
- 30.Paterson AH, Brubaker CL, Wendwl JF (1993) A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RELP or PCR analysis. Plant Mol Biol Rep 11: 122–127. [Google Scholar]
- 31.Zhang Z, Wei T, Zhong Y, Li X, Huang J (2016) Construction of a high-density genetic map of Ziziphus jujuba Mill. using genotyping by sequencing technology. Tree Genet Genomes 12: 76. [Google Scholar]
- 32.Zhou Z, Zhang C, Zhou Y, Hao Z, Wang Z, Zeng X, et al. (2016) Genetic dissection of maize plant architecture with an ultra-high density bin map based on recombinant inbred lines. BMC Genomics 17: 178 doi: 10.1186/s12864-016-2555-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jun G, Wing MK, Abecasis GR, Kang HM (2015) An efficient and scalable analysis framework for variant extraction and refinement from population-scale DNA sequence data. Genome Res 25: 918–925. doi: 10.1101/gr.176552.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu D, Liu F, Shan X, Zhang J, Tang S, Fang X, et al. (2015) Construction of a high-density genetic map and lint percentage and cottonseed nutrient trait QTL identification in upland cotton (Gossypium hirsutum L.). Mol Genet Genomics 290: 1683–1700. doi: 10.1007/s00438-015-1027-5 [DOI] [PubMed] [Google Scholar]
- 37.Ooijen J, Jw VTV (2006) JoinMap 4, software for the calculation of genetic linkage maps in experimental populations.
- 38.Meng L, Li H, Zhang L, Wang J (2015) QTL IciMapping: Integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop Journal 3: 269–283. [Google Scholar]
- 39.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421 doi: 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conesa A, Gotz S, Garciagomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21: 3674–3676. doi: 10.1093/bioinformatics/bti610 [DOI] [PubMed] [Google Scholar]
- 41.Lin T, Zhu G, Zhang J, Xu X, Yu Q, Zheng Z, et al. (2014) Genomic analyses provide insights into the history of tomato breeding. Nat Genet 46: 1220–1226. doi: 10.1038/ng.3117 [DOI] [PubMed] [Google Scholar]
- 42.Ganal MW, Altmann T, Roder MS (2009) SNP identification in crop plants. Curr Opin Plant Biol 12: 211–217. doi: 10.1016/j.pbi.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 43.Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol 5: 94–100. [DOI] [PubMed] [Google Scholar]
- 44.Mcnally KL, Childs KL, Bohnert R, Davidson RM, Zhao K, Ulat VJ, et al. (2009) Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc Natl Acad Sci U S A 106: 12273–12278. doi: 10.1073/pnas.0900992106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bajgain P, Rouse MN, Tsilo TJ, Macharia GK, Bhavani S, Jin Y, et al. (2016) Nested association mapping of stem rust resistance in wheat using genotyping by sequencing. Plos One 11: e0155760 doi: 10.1371/journal.pone.0155760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furuta T, Ashikari M, Jena KK, Doi K, Reuscher S (2017) Adapting genotyping-by-sequencing for rice F2 populations. G3 (Bethesda) 7: 881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maschietto V, Colombi C, Pirona R, Strozzi F, Marocco A, Rossini L, et al. (2017) QTL mapping and candidate genes for resistance toFusariumear rot and fumonisin contamination in maize. BMC Plant Biology 17: 20 doi: 10.1186/s12870-017-0970-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monteropau J, Blanca J, Esteras C, Martínezpérez EM, Gómez P, Monforte AJ, et al. (2017) An SNP-based saturated genetic map and QTL analysis of fruit-related traits in Zucchini using Genotyping-by-sequencing. BMC Genomics 18: 94 doi: 10.1186/s12864-016-3439-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun FD, Zhang JH, Wang SF, Gong WK, Shi YZ, Liu AY, et al. (2012) QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol Breed 30: 569–582. [Google Scholar]
- 50.Wang S, Chen J, Zhang W, Hu Y, Chang L, Fang L, et al. (2015) Sequence-based ultra-dense genetic and physical maps reveal structural variations of allopolyploid cotton genomes. Genome Biol 16: 108 doi: 10.1186/s13059-015-0678-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Ning Z, Yan H, Chen J, Rui Z, Hong C, et al. (2015) Molecular mapping of restriction-site associated DNA markers in allotetraploid upland cotton. Plos One 10: e0124781 doi: 10.1371/journal.pone.0124781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hulse-Kemp AM, Lemm J, Plieske J, Ashrafi H, Buyyarapu R, Fang DD, et al. (2015) Development of a 63K SNP array for cotton and high-density mapping of intraspecific and interspecific populations of Gossypium spp. G3 (Bethesda) 5: 1187–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen H, Khan MKR, Zhou Z, Wang X, Cai X, Ilyas MK, et al. (2015) A high-density SSR genetic map constructed from a F2 population of Gossypium hirsutum and Gossypium darwinii. Gene 574: 273–286. doi: 10.1016/j.gene.2015.08.022 [DOI] [PubMed] [Google Scholar]
- 54.Zhao L, Lv Y, Cai C, Tong X, Chen X, Wei Z, et al. (2012) Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information. BMC Genomics 13: 539 doi: 10.1186/1471-2164-13-539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.White TG (1966) Diallel analyses of quantitatively inherited characters in Gossypium hirsutum L. Crop Sci 6: 253–255. [Google Scholar]
- 56.Godoy AS, Palomo GA (1999) Genetic analysis of earliness in upland cotton (Gossypium hirsutum L.). II. Yield and lint percentage. Euphytica 105: 161–166. [Google Scholar]
- 57.Theißen G (2001) Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol 4: 75–85. [DOI] [PubMed] [Google Scholar]
- 58.Becker A, Theißen G (2003) The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol 29: 464–489. [DOI] [PubMed] [Google Scholar]
- 59.Graf A, Coman D, Uhrig RG, Walsh S, Flis A, Stitt M, et al. (2017) Parallel analysis of Arabidopsis circadian clock mutants reveals different scales of transcriptome and proteome regulation. Open Biol 7: 160333 doi: 10.1098/rsob.160333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li X, Ma D, Lu SX, Hu X, Huang R, Liang T, et al. (2016) Blue light- and low temperature-regulated COR27 and COR28 play roles in the arabidopsis circadian clock. Plant Cell 28: 2755–2769. doi: 10.1105/tpc.16.00354 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(XLS)
(XLS)
(XLS)
(XLS)
Data Availability Statement
Sequences are available on the Sequence Read Archive, http://www.ncbi.nlm.nih.gov/Traces/sra/ under accession number PRJNA381273.