

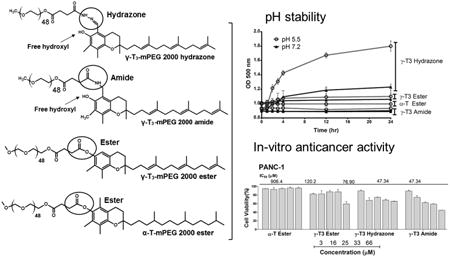
Abstract
The anticancer activity of water soluble methoxy polyethylene glycol (mPEG) derivatives of tocotrienol (T3) isomers of vitamin E was previously found to be reduced when compared to the parent free isomers. This could be due to the ester bond formation between the mPEG and the 6-OH group on the chroman moiety of the T3 isomer. To further investigate, the objectives of the current study were to (1) synthesize and characterize stable amide and cleavable hydrazone conjugates between mPEG and carbon-5 on the chroman moiety of T3, and (2) examine the cytotoxicity of the newly synthesized mPEG conjugates against breast (MCF-7 and MDA-MB-231) and pancreatic (BxPC-3 and PANC-1) cancer cells. Conjugates were synthesized by direct conjugation of succinyl chloride derivatives of mPEG to the α-tocopherol and γ-tocotrienol isomers of vitamin E, and were characterized by 1H-NMR, FT-IR, and mass spectrometry. The micelles of the amide and hydrazone self-assembled conjugates were characterized for size, zeta, CMC, and stability at different pH media. The hydrolysis of the hydrazone conjugate was pH dependent with highest release at acidic (pH 5.5) conditions, whereas the amide conjugate was stable in all tested media. The amide conjugate nonetheless showed greater cytotoxicity than the hydrazone conjugate, which suggested that maintaining solubility and the presence of free 6-OH group are important for γ-T3 to exert anticancer activity in vitro. The results from the current study demonstrated the importance of considering the nature of the chemical bond between T3 and mPEG when designing functional ingredients for use in drug delivery.
Keywords: vitamin E, Tocopherol, Tocotrienol, PEGylation, Acid-degradation, Hydrazone, Drug delivery, Cancer Chemotherapy, Nanotechnology
Graphical abstract
1. Introduction
Vitamin E refers to a group of eight isomers that include both tocopherols and tocotrienols [1]. While both possess antioxidant activity, the tocotrienol isomers were shown to exhibit potent anticancer activity at significantly lower concentrations than tocopherols [2, 3]. Tocotrienols, however, are poorly soluble in aqueous media [4]. To enhance their solubility, we synthesized mPEG derivatives of the tocotrienol isomers of vitamin E [1, 5]. Despite the enhancement in aqueous solubility, conjugating tocotrienols to a mPEG moiety was found to reduce their anticancer activity [1, 5]. While the exact reason is not known, a reduction in activity could be due to the conjugation of the mPEG moiety to the 6-OH group on the chroman ring of tocotrienols. One of the objectives of the present study was therefore to investigate this possibility. We synthesized a conjugate where the mPEG moiety is linked via an amide bond to carbon-5 of the chroman ring thereby leaving the 6-OH group intact. While the 6-OH group might be essential for activity, it was also reported that tocotrienols exert their effect by interfering with the integrity of the lipid rafts of the cell membrane [6]. Therefore, the self-assembly of the amphiphilic mPEG conjugates into micelles may hinder the docking of the polyunsaturated phytyl side chain of the tocotrienol isomers to the cellular membranes. To address this possibility, we also synthesized a mPEG tocotrienol conjugate where the mPEG moiety is linked to carbon-5 on the chroman ring via hydrazone linkage. A hydrazone linkage is an acid sensitive moiety that has been used in targeted drug delivery [7] because of its capacity to hydrolyze in the extracellular acidic environment of the tumor or the lysosomes [8, 9]. While the cleavage of the hydrazone bond in the acidic microenvironment of tumor tissues is expected if tested in animal models, the focus of the present study was to investigate whether the hydrolyzable conjugate of γ-tocotrienol will maintain or reduce the anticancer activity of γ-tocotrienol. A similar approach has been used to test gemcitabine lipid conjugates [7]. Since it is not known whether a mPEG tocotrienol conjugate is internalized into the cell or if it acts on the cell membrane we hypothesized that any differences in activity between the ester, amide, and hydrazone mPEG tocotrienol conjugates might be used to indirectly indicate whether the molecule is internalized or not. Therefore, the primary objective of the present manuscript was to detail the synthesis scheme and characterization of the hydrazone, amide, and ester PEG-tocotrienol conjugates, and to assess their pH stability and the ability of these molecules to self-assemble into micelles by measuring their critical micelles concentration (CMC). Preliminary in vitro data against breast and pancreatic tumor cells were provided to address the effect of the hydrolyzable and non-hydrolyzable γ-T3-mPEG on the anticancer activity of the conjugates. A secondary objective was to present an alternative synthesis scheme for the conjugation of mPEG to the γ-tocotrienol and α-tocopherol isomers of vitamin E. In our previous study [5], a two-step reaction procedure was utilized that involved succinate formation followed by PEGylation. In the current study, PEGylated vitamin E isomers were prepared by direct conjugation of succinyl chloride derivatives of mPEG to the α-tocopherol and γ-tocotrienol isomers of vitamin E.
2. Materials and Methods
2.1. Materials
mPEG 2000 succinyl chloride was purchased from Biochempeg Scientific Inc. (Watertown, MA). γ-T3 and α-T vitamin E isomers were isolated from Tocotrol™ L50P, a viscous Tocotrienol-rich fraction of palm fruit oil (Fuji Health Science Inc., Burlington, NJ). Paclitaxel was from LC Laboratories (Woburn, MA). Formaldehyde (37%) solution was from VWR life science (Raleigh, NC). Ammonia hydroxide solution (NH4OH) was from Allied Chemical (Morristown, NJ). Triethylamine and 1,4-dioxane anhydrous were from Alfa Aesar (Ward Hill, MA). Hydrazine hydrate (85%) was from Sigma-Aldrich (St. Louis, MO), Chloroform-d (CDCL3) was from Acros (Bridgewater, NJ). Methanol and ethyl acetate (EtOAc) were from Pharmco-AAPER (Shelbyville, KY). Sodium sulfate anhydrous (Na2SO4) was from Avantor (Center Valley, PA). Acetonitrile and dichloromethane (DCM) were from EMD Chemicals Inc. (Cibbstown, NJ). All chemicals and solvents were of reagent grade or higher and were used as supplied without further modification.
2.2. Synthesis of the hydrazone, amide, and ester mPEG-vitamin E conjugates
Conjugation of the mPEG moiety to the γ-T3 isomer via hydrazone and amide linkages is outlined in Figs. 1 and 2. For hydrazone derivative synthesis, 5-formy-γ-T3 was initially synthesized using Reimer-Tiemann Formylation reaction and was modified from Jung et al.[10]. Briefly, to a suspension of 500 mg γ-T3 and 60 ml water in 10 ml CHCl3, 400 mg NaOH was added. The mixture was then refluxed for 4 h and then diluted with water and EtOAc, and the aqueous layer was acidified to pH 1 with 1 N HCl and back extracted with EtOAc. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated using a Heidolph Laborota 4000 rotary evaporator (Elk Grove Village, IL). The reaction mixture was then purified using chromatography using CHCl3/MeOH (12:1) to afford 5-formy-γ-T3 (∼ 150 mg, Fig. 1). The 5-aminomethyl γ-T3 derivative was synthesized using a modified procedure from Nakamura et al. [7, 11]. Briefly, a mixture of 600 mg (1.5 mmol) of γ-T3, 1 mL of 30% ammonia solution (NH4OH) and 15 mL dioxane was cooled by ice water and stirred. 360 mg of 37% formaldehyde was then added dropwise to the reaction mixture. The reaction mixture was stirred for 1 h at room temperature and then refluxed for another 4 h. The reaction mixture was concentrated using a Heidolph Laborota 4000 rotary evaporator (Elk Grove Village, IL). 50 mL of EtOAc was then added to the residue and washed three times with brine solution; dried over anhydrous Na2SO4, and then concentrated by rotary evaporation to give a yellow to orange 5-aminomethyl γ-T3 derivative (Fig. 2). mPEG succinyl 2000 hydrazide (Fig. 1) was prepared by adding 400 μL triethylamine to a stirring mixture of 1000 mg of mPEG 2000 succinyl chloride and 16 mg of hydrazine at room temperature. The hydrazide derivative was then reacted with oxalyl chloride to form the reactive acyl chloride. To obtain the acid-sensitive PEGylated γ-T3 hydrazone, approximately 500 mg mPEG 2000 hydrazide chloride was reacted with 100 mg 5-formy-γ-T3 in 10 mL DCM at 50°C for 24 h. The mixture was concentrated with a rotary evaporator and then applied to a silica gel column. DCM:MeOH (96:04) was used as the mobile phase to collect acid-sensitive PEGylated γ-T3 hydrazone. PEGylated γ-T3 amide (Fig. 2) was synthesized by adding 500 mg of mPEG 2000 succinyl chloride to 5 mL DCM containing 300 mg 5-aminomethyl γ-T3, followed by dropwise addition of 300 μL triethylamine. The reaction mixture was stirred at room temperature for 6 h. The solvent was evaporated with a rotary evaporator, and the residue was purified by silica gel column using DCM:MeOH (96:04) as the mobile phase to afford the off-white solid γ-T3-mPEG 2000 amide conjugate (Fig. 2). For conjugating mPEG 2000 to γ-T3 and α-T via an ester linkage (Fig. 3); a mixture of 300 mg of α-T or γ-T3 and mPEG 2000 succinyl chloride (∼ 1.75 gm) in DCM were stirred at room temperature followed by dropwise addition of 300 μL triethylamine. The reaction mixture was then stirred for an additional hour. After evaporating the DCM, the residue was purified by silica gel column using DCM:MeOH (96:04) as the mobile phase to afford the off-white solid α-T and γ-T3 mPEG 2000 ester conjugates (Fig. 3).
Fig. 1.
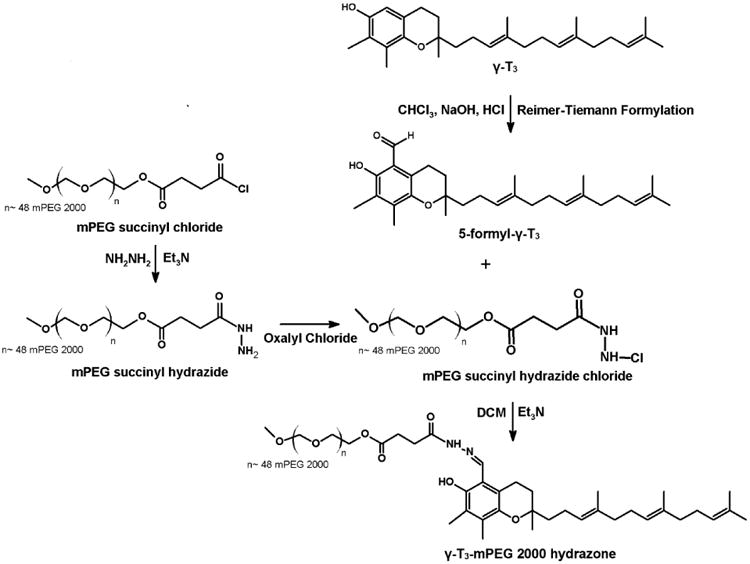
Synthesis scheme of γ-tocotrienol-mPEG 2000 hydrazone conjugate.
Fig. 2.
Synthesis scheme of γ-tocotrienol-mPEG 2000 amide conjugate.
Fig. 3.
Synthesis scheme of α-tocopherol-m-PEG 2000 ester conjugate.
2.3. 1H-NMR and FT-IR analysis of the conjugates
Proton-NMR studies were carried out to confirm the PEGylation of the isomers. Samples were prepared in CDCL3 and analyzed by a JEOL Eclipse NMR spectrometer (JEOL USA, Inc., MA) operating at 400 MHz and 20°C. Delta™ NMR Data Processing Software (JEOL USA, Inc., MA) was used for data acquisition and spectral processing with chemical shifts reported in ppm (d). One-dimensional spectra were collected with 64 scans of 16 K points over 20 ppm and a recycle delay of 5 s. Fourier transform infrared spectroscopy (FT-IR) analysis was employed to investigate the molecular structure of the conjugates using a PerkinElmer Spectrum Two™ spectrometer (Waltham, MA) attached to an attenuated total reflectance (ATR) accessory. Samples were directly placed on the diamond disk and scanned for absorbance over the range from 4000 to 500 wavenumbers (cm−1) at a resolution of 1 cm−1.
2.4. Mass spectrometry analysis of the conjugates
JEOL AccuTOF™ time-of-flight mass spectrometer (JEOL Ltd., Tokyo, Japan) equipped with orthogonal spray electrospray ionization (ESI) ion source was used to analyze the PEGylated products. HPLC grade methanol was used to dissolve the samples. 50 μL was injected through Rheodyne 6-port valve injector. The mass spectrometer was operated in positive-ion mode (ESI + ve) with needle voltage (2000 V). The atmospheric pressure interface potentials were set to the following values: orifice 1 = 55 V, ring lens voltage = 5 V and orifice 2 = 6 V. The detector voltage was set to 1900 V. Orifice 1 temperature was adjusted to 80°C with dissolving temperature at 250°C. Nebulizing and desolvation gas (N2) were adjusted to 2 and 5 L/min flow rate, respectively.
2.5. Thermal analysis of the conjugates
Thermal analysis was performed to examine the physical state of the PEGylated γ-T3 and α-T isomers using a TA 2920 modulated differential scanning calorimeter (DSC, TA Instruments-Waters LLC, New Castle, DE). Accurately weighed samples (3 to 5 mg) were hermetically sealed in aluminum pans. Sealed pans were then heated from 0°C to 80°C at a rate of 10°C/min. Melting endotherms were analyzed from the generated data using universal analysis 2000 version 4.2 software (TA Instruments-Waters LLC, New Castle, DE).
2.6. Determination of the critical micellar concentration (CMC) of the conjugates
The CMC of the PEGylated isomers in water was determined using pyrene as a fluorescence probe as previously reported [1]. Before analysis, 0.001 to 1mg/mL stock solutions of each the PEGylated derivatives were prepared by dissolving the conjugates in water. Before testing, 250 μL of pyrene solubilized in acetone at a concentration of 4.69 μg/mL was added to glass vials and flushed with N2 gas then dried under vacuum. A 2 mL sample of each stock solution of the PEGylated isomers was then added to the pyrene vials. The glass vials were then capped and incubated overnight at room temperature while shaking at 200 rpm. Samples from each vial were then examined by fluorescence spectroscopy using a K2 multifrequency cross-correlation phase and modulation fluorometer (ISS, Inc., Champaign, IL). The emission spectra of pyrene were recorded between 360 and 410 nm (Ex = 340 nm). CMC values were estimated from the plot of the peak height at 372 nm, which is inversely related to the fluorescence intensity of the samples, versus the logarithmic concentration of the PEGylated isomers.
2.7. Particle size and Zeta potential analysis of the conjugates
The mean particle size of the self-assembled PEGylated isomers in water was measured by photon correlation spectroscopy (PCS) using a Nicomp™380 ZLS submicron particle size analyzer (Particle Sizing System, Port Richey, FL) at 25°C and 90° lasers light scattering. Samples were diluted with deionized water in order to avoid side scattering and to achieve a scattering intensity of 300 kHz. The number-weighted mean diameter of the particles was calculated based on Stokes–Einstein law by curve fitting of the correlation function. Zeta-potential of the samples in deionized water was measured using the same instrument under the zeta mode.
2.8. pH stability of the hydrazone and amide mPEG conjugates of the γ-T3 isomer
pH stability of the ester, hydrazone and amide mPEG conjugates of the γ-T3 isomer was determined as previously reported [7]. Briefly, the esters, and the PEGylated γ-T3 hydrazone and amide conjugates were dissolved in acetate buffer (pH 5.5), and phosphate buffered saline (pH 6.8 and 7.4) to attain a final concentration of 2 mg/mL. A 0.5 mL sample of each solution was then transferred to a 24-well plate and incubated at 37°C while shaking at 100 RPM. At 0, 1, 2 3, 4, 12 and 24 h, the absorbance of the solutions in the wells was measured at 500 nm using a BioTek Synergy HT Multi-Mode Microplate Reader (BioTek Instruments, Inc. Winooski, VT).
2.9. In vitro cytotoxicity of the conjugates
The cytotoxicity of the PEGylated isomers was evaluated against MCF-7 and MDA-MB-231 breast and BxPC-3 and PANC-1 pancreatic cancer cell lines, which were obtained from ATCC™ (Manassas, VA). MCF-7, MDA-MB-231, and PANC-1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) whereas the BxPC-3 cells were maintained in RPMI-1640 medium (Invitrogen, Carlsbad, CA). All media were supplemented with 10% fetal bovine serum, 1% insulin, and 1% penicillin–streptomycin, all from Invitrogen (Carlsbad, CA). Cells at a density of 5000 cells/well were seeded into 96-well plates in 50 μL volume and incubated at 37°C with 5% CO2. After overnight incubation, an additional 50 μL of fresh medium containing the PEGylated isomers was added to the wells. After 72 h of incubation, media from the wells were replaced with 20 μL of CellTiter-Blue reagent (Promega Corporation, Madison, WI). The plates were allowed to incubate at 37°C for 1 h, and the fluorescence was measured using a BioTek Synergy HT multi-mode microplate reader (BioTek, Winooski, VT). Cell viability was calculated as the percentage of cells remaining viable compared to the untreated cells. The 50% inhibitory concentration (IC50) values were estimated using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA).
2.10. Statistical analysis
The statistical significance of the in-vitro anticancer activity of the PEGylated isomers against breast and pancreatic cell lines was analyzed by one-way analysis of variance with Tukey post-test calculation. A difference of P < 0.05 was considered to be statistically significant.
3. Results and Discussion
3.1. Synthesis and characterization of the PEGylated isomers
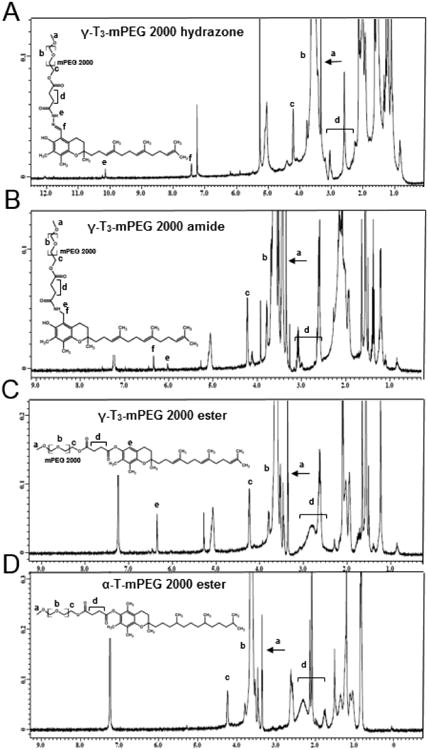
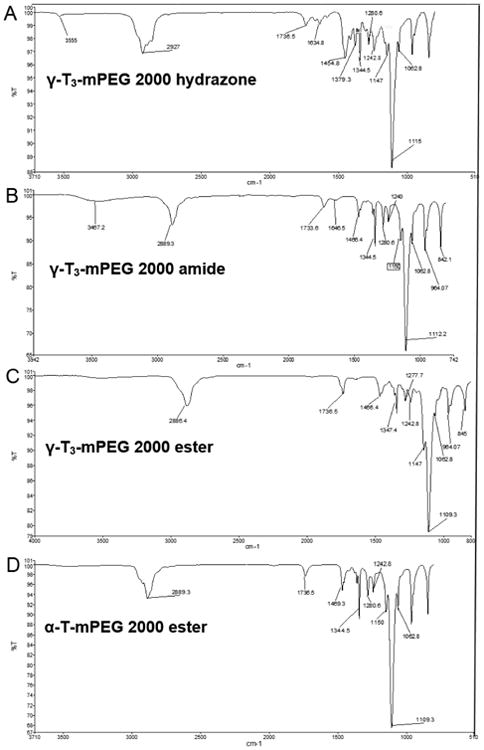
The synthesis schemes of the PEGylated isomers are outlined in Figs. 1-3. For the preparation of the hydrazone and amide derivatives (Fig. 1 and 2), a 5-aminomethyl group was introduced on the chroman ring of the γ-T3 isomer in order to provide a functional moiety to conjugate mPEG succinyl chloride. To create a cleavable linkage, mPEG succinyl chloride was reacted with hydrazine to substitute the chlorine atom with hydrazide functionality (Fig. 1). mPEG succinyl hydrazide, however, cannot react with the 5-aminomethyl group of the γ-T3 unless activated. mPEG succinyl hydrazide was, therefore, chlorinated with oxalyl chloride to generate mPEG succinyl hydrazide chloride, which easily reacted with 5-aminomethyl γ-T3 to yield the γ-T3-mPEG hydrazone derivative (Fig. 1). An amide conjugate was formed when mPEG succinyl chloride was reacted with the 5-aminomethyl group (Fig. 2). Ester derivatives of γ-T3 and α-T were prepared by direct conjugation of mPEG succinyl chloride with γ-T3 and α-T using triethylamine-assisted reaction (Fig. 3). PEGylated γ-T3 and α-T isomers were characterized by 1H-NMR. As shown in Fig. 4A, B, C and D, the appearance of ethanyl proton peaks of succinate at 2.75–2.86 ppm (m, 4H, COCH2CH2CO) in the 1H NMR spectrum and the disappearance of the chroman hydroxyl proton at 4.7 ppm (s, 1H) indicated successful PEGylation reaction. The PEGylation was further confirmed by the appearance of the peak at 4.23 ppm (m, 2H), which corresponded to the protons of the first carbon atom of the mPEG chain directly linked to the succinate. The chemical shift at 3.5–3.7 was due to mPEG chain [m, 192H, (CH2CH2O)48, Fig. 4]. The chemical shift at 3.36 was due to the terminal methoxy group on the PEG molecule (s,3H, OCH3, Fig. 4). The molecular structure of the conjugates was also investigated by FT-IR analysis (Fig. 5). For all conjugates, the carbonyl group bands appeared at 1736 cm−1. The bands at 2889-2927 cm−1 were attributed to −CH2 stretching and the absorption bands from 1062 to 1283 cm−1 were due to C-O stretching, all of which correspond to the PEGylation of the isomers. For the hydrazine conjugate, the bands at 3355 cm−1 and 1634 cm−1 (Fig. 5A) were due to -N-H- and -NH-N= stretching, respectively. The band at 1646 cm−1 was due to the -N-H- bending of the amide conjugate. The PEGylation of γ-T3 and α-T isomers was further confirmed by time-of-flight mass spectrometry (Fig. 6A, B, C and D). The average molecular weights of the hydrazone, amide, and ester γ-T3 conjugates; and the ester α-T conjugate were observed with peaks at m/z of 2377, 2365, 2350 and 2353, respectively (Na+ adducts), which were in agreement with the theoretical MWs of the conjugates.
Fig. 4.
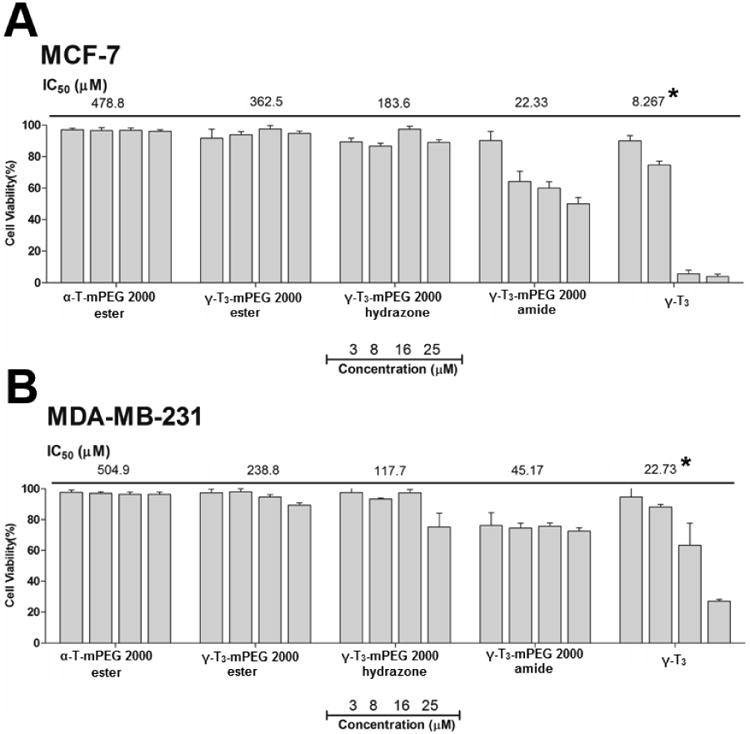
1H-NMR in CDCl3 spectra of (A) γ-T3-mPEG 2000 hydrazone, (B) γ-T3-mPEG 2000 amide, (C) γ-T3-mPEG 2000 ester and (D) α-T-mPEG 2000 ester.
Fig. 5.

FT-IR analysis of (A) γ-T3-mPEG 2000 hydrazone, (B) γ-T3-mPEG 2000 amide, (C) γ-T3-mPEG 2000 ester and (D) α-T-mPEG 2000 ester.
Fig. 6.

ESI mass spectra of (A) γ-T3-mPEG 2000 hydrazone, (B) γ-T3-mPEG 2000 amide, (C) γ-T3-mPEG 2000 ester and (D) α-T-mPEG 2000 ester.
3.2. Physicochemical characterization and pH stability of the conjugates
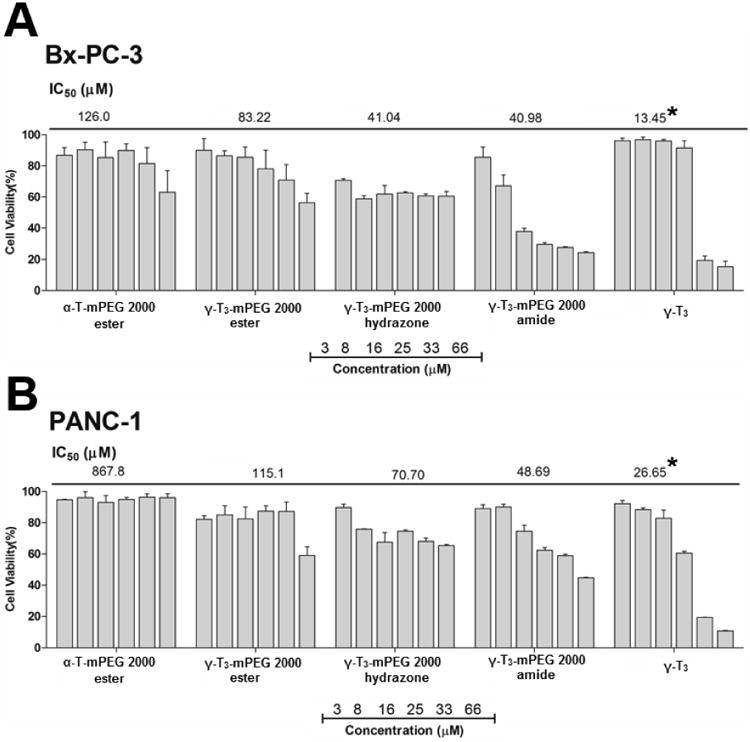
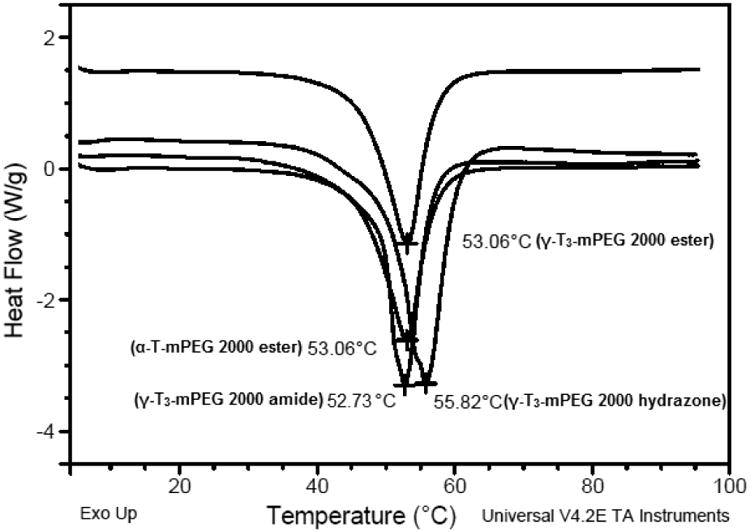
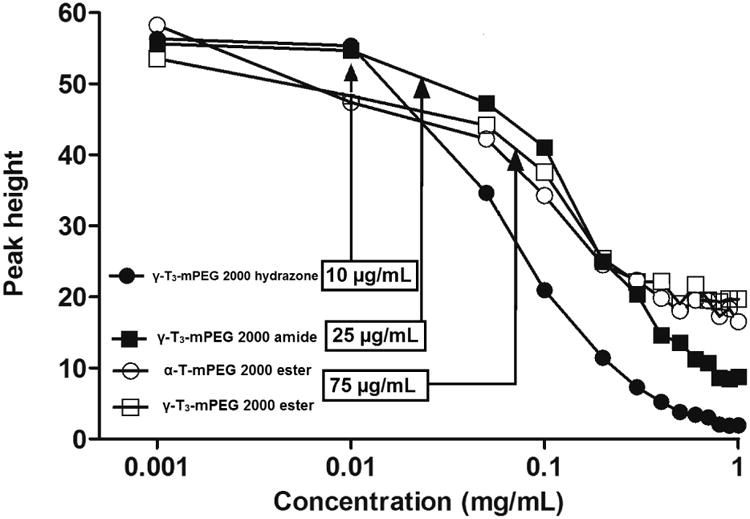
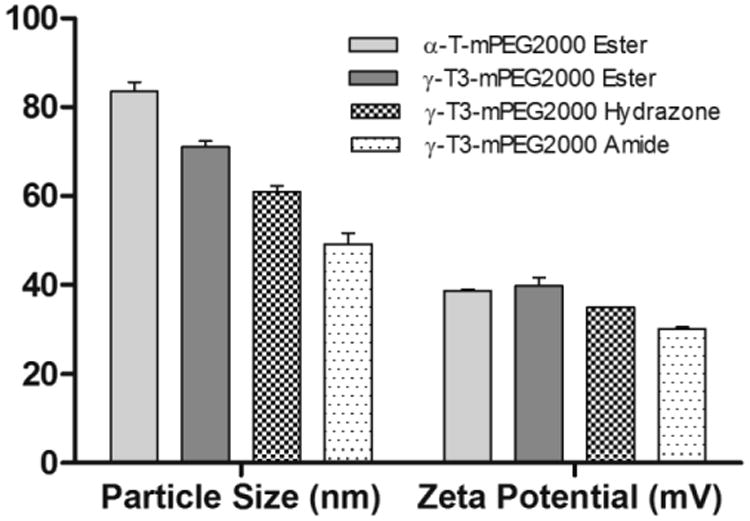
The physicochemical characterization of the PEGylated isomers was carried out to investigate their thermal properties, CMC, particle size, zeta potential, and stability in media with different pH values. As shown in Fig. 7, the melting point of the PEGylated conjugates was in the range of 52.7°C to 55.8°C, which is higher than the reported melting point of vitamin E TPGS; 36°C [1]. The Higher melting point is expected due to the higher molecular weight of the PEG moiety (2000) used in the present study as opposed to the PEG 1000 in vitamin E TPGS. An increase in the molecular weight of the mPEG increases its melting point [12]. Since the PEGylated isomers are amphiphilic, they self-assemble into micelles when introduced into an aqueous solution. The hydrazone conjugate had the least CMC value (10 μg/mL) followed by the amide conjugate with an observed CMC of approximately 25 μg/mL. Ester conjugates of the α-T and γ-T3 isomers had CMCs values of approximately 65 and 75 μg/ml, respectively (Fig. 8). Although different conclusions were reported on the effect of PEG molecular weight on CMC [13], for vitamin E/PEG conjugates, an increase in the molecular weight of the PEG chain was reported to lead to smaller CMC values [14]. This may explain the lower CMC values observed in the current study when compared to vitamin E TPGS, which was reported to have a CMC value of 200 μg/mL [1]. The lower CMC values of the hydrazone and amide conjugates could be attributed to their higher hydrophilicity due to the presence of -N- atom, which imparts basicity to the molecules due to the availability of a lone pair of electrons for proton acceptance. Surfactants with higher polarity partition more efficiently into the water interface leading to a decrease in surface free energy at lower surfactant concentrations and thereby surfactant aggregation and micelles formation at lower concentrations [15]. Micelles formed by the amide conjugate had the smallest particle size (∼50 nm, Fig. 9) followed by hydrazone (∼60 nm) and then the γ-T3 ester (∼70 nm). The largest particle size was observed with the α-T ester conjugate (∼80 nm). The zeta potential of the amide conjugate (30 mV) was also less than the other conjugates, which had a zeta potential in the range of 35-41 mV (Fig. 9). When tested for acid sensitivity, (Fig. 10) the hydrazone conjugate was found to hydrolyze in a pH-dependent manner. As expected, maximum hydrolysis was observed at pH 5.5 followed by pH 6.8 and 7.2 with approximately 70%, 30% and 20% being hydrolyzed at these pH values, respectively. No hydrolysis was observed for the ester and amide conjugate at the three pH conditions (Fig. 10). The degree of hydrolysis was quantified by measuring the optical density of the solutions, where a change in solution turbidity is caused by the water-insoluble γ-T3 isomer after the hydrolysis of the conjugates.
Fig. 7.
Differential scanning calorimetry (DSC) thermal profile, of α-tocopherol-m-PEG 2000 ester and γ-tocotrienol-mPEG 2000 ester, hydrazone, and amide conjugates.
Fig. 8.
Critical micelles concentration (CMC) of α-tocopherol-m-PEG 2000 ester and γ-tocotrienol-mPEG 2000 ester, hydrazone, and amide conjugates.
Fig. 9.
Particle size and zeta potential of α-tocopherol-m-PEG 2000 ester and γ-tocotrienol-mPEG 2000 ester, hydrazone, and amide conjugates. Each value represents the mean ± SD of triplicate samples.
Fig. 10.
pH stability profile of α-tocopherol-m-PEG 2000 ester and γ-tocotrienol-mPEG 2000 ester, hydrazone, and amide conjugates. Each value represents the mean ± SD of triplicate samples.
3.3. In-vitro cytotoxicity of the conjugates
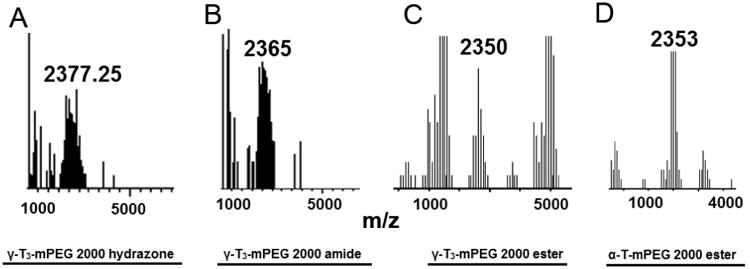
The in-vitro anticancer activity of the PEGylated α-T and γ-T3 isomers of vitamin E were carried out against pancreatic (Bx-PC-3 and PANC-1) and breast (MCF-7 and MDA-MB-231) cancer cells. Bx-PC-3 cells were derived from the body of the pancreas with no evidence of metastasis, whereas PANC-1 cells were derived from the head of the pancreas that has invaded the duodenal wall and metastasized to the lymph node [16]. PANC-1 is also more resistant to chemotherapy than BxPC-3 cells [17], which makes them suitable for preliminary screening of activity against pancreatic cancer. MCF-7 and MDA-MB-231 cells have also been used for preliminary screening of the activity of the conjugates against breast cancer. MCF-7 cells are estrogen and progesterone receptors positive and HER-2 negative, whereas MDA-MB-231 cells are triple negative.
As discussed earlier, the ester, hydrazone, and amide conjugates of γ-T3 were synthesized to investigate (1) whether the anticancer activity of the conjugates will be affected by the conjugation of the molecule to a PEG moiety, and (2) to observe the role of the free 6-OH group of the γ-T3 on the anticancer activity of the conjugates. Theoretically, as the hydrazone conjugate is prone to hydrolysis, the hydrolyzed conjugate was expected to have similar cytotoxic effect as the free γ-T3 sisomer. It was also assumed that a reduction in the anticancer activity of the γ-T3-mPEG ester conjugate might be a result of the esterification and masking of the 6-OH group on the chroman ring of the γ-T3 isomer. It was reported that the hydroxyl group on the chroman must be present for vitamin E activity, and that optimal activity occurs when there are three methyl groups on the ring [18, 19]It was also reported that the 6-OH group is responsible for the antioxidant activity of vitamin E isomers, especially in conditions associated with mitochondrial dysfunction, such as neoplastic diseases [18, 19]. Esterification of 6-OH group on the chroman ring when synthesizing redox-silent analogues was found to prevent vitamin E molecules from exerting biological activity [18].
As shown in Figs. 11 and 12, the free γ-T3 isomer was significantly more active on all cell lines than the conjugates. The activity of the free isomers was in agreement with several reports on the cytotoxicity of tocotrienols against breast and pancreatic cancer cells [20-22]. Although conjugating a PEG moiety to the tocotrienol isomers reduced their overall cytotoxic activity, the amide and hydrazone conjugates with a free unmasked 6-OH group on their chroman ring were significantly more active than the ester conjugates. This indicates that the 6-OH group is essential for tocotrienol activity as stipulated earlier. It is also probable that the toxic effect observed by the amide and hydrazone conjugates might be due to increased cellular uptake or due to their hydrophobic/hydrophilic balance and stronger detergency as indicated by their lower CMC and particle size than the ester conjugates. Surprisingly, the amide conjugate had significantly higher anticancer activity than the hydrazone conjugate. This observation might be explained by the fact that the amide bond is more stable in the experimental in vitro conditions than the hydrazone bond (Fig. 10). Since the mPEG chain would remain attached in the amide conjugate to the γ-T3 isomer, the molecule retains its solubility in culture media allowing for the γ-T3 to exert its anticancer activity. However, unlike the free isomers, which were stabilized in culture media with the aid of albumin, the cleavage of the PEG chain from γ-T3 in the unstable hydrazone conjugate would lead to the phase separation of the insoluble γ-T3 isomer and thereby loss of its anticancer activity. During the 72 h treatment phase, it is expected that the treated cancer cells will produce acidic by-products metabolites to the media, which would inadvertently affect the conjugates [23]. While the ester conjugates were least cytotoxic, perhaps due to the masking of the 6-OH group, the γ-T3 ester conjugate was more potent than the α-T ester conjugate as deduced from their IC50 values. This indicates that the polyunsaturated phytyl side is also essential for tocotrienol activity. As has been reported [24], the main criteria that differentiates tocotrienols from tocopherols is their polyunsaturated tail that has been shown to contribute to their cytotoxic activity. The authors recognize that the in-vitro cell culture conditions do not reflect the real acidic conditions of the tumor microenvironment. In vivo testing in animal models is warranted to confirm the observed in vitro data. Although critical, in vivo testing in animal models was beyond the scope of the present preliminary work and is the subject of future studies.
Fig. 11.
In-vitro anticancer activity of α-tocopherol and γ-tocotrienol-m-PEG conjugates against pancreatic cancer cell lines; (A) Bx-PC-3 and (B) PANC-1 cancer cell lines. Cells were treated for 72 hr. Values reported are the mean ± SD for triplicate samples. * P value < 0.05: indicates that the free unmodified γ-T3 isomer had significantly higher anticancer activity than the conjugates against both pancreatic cancer cell lines
Fig. 12.
In-vitro anticancer activity of α-tocopherol and γ-tocotrienol-m-PEG conjugates against pancreatic cancer cell lines (A) MCF-7 and (B) MDA-MB-231 breast cancer cell lines. Cells were treated for 72 hr. Values reported are the mean ± SD for triplicate samples. * P value < 0.05: indicates that the free unmodified γ-T3 isomer had significantly higher anticancer activity than the conjugates against both breast cancer cell lines.
4. Conclusion
In the current report, hydrazone, amide, and ester PEG derivatives of γ-T3 and α-T isomers of vitamin E were successfully synthesized, which demonstrated the feasibility of conjugating mPEG chain directly to vitamin E isomers using succinyl chloride derivative of the mPEG, instead of using a two-step reaction procedure as commonly cited in published literature. The chemical identify of the conjugates was confirmed by 1H-NMR. Conjugating the isomers to the higher molecular weight PEG (2000) increased their hydrophilicity when compared to the commercial vitamin E TPGS (1000) as indicated by their lower CMC values and the smaller size of their micellar assemblies in water. The nature of the link between the PEG moiety and the isomers, nonetheless, dictated their performance in aqueous media. Conjugate made with the amide bond was physically stable, whereas the hydrazone bond afforded an acid sensitive conjugate. Although this was confirmed experimentally, the therapeutic benefits of the hydrazone conjugate could not be deduced from the in-vitro cell culture data. The lower activity of the hydrazone conjugate when compared to the amide conjugate could be due to its instability and hydrolysis in culture media leading to the phase separation of the insoluble tocotrienol oily isomer. Nonetheless, the higher activity that was observed for the amide conjugate indicated that the 6-OH group on the chroman ring of the tocotrienol isomer is important for its activity. Masking the OH, however, may not be the only factor contributing to the observed differences in activity between the conjugates. It is also possible that differences in physical stability, cellular uptake, and hydrophilicity may have contributed to the observed cytotoxic effects. The results from this study highlighted the potential application of this new class of functional ingredients. PEG-tocotrienol conjugates were shown to possess dual functionality as antitumor agents and solubilizers. Special attention should nonetheless be paid to the location of the chemical bond between the PEG moiety and vitamin E isomers since it will dictate which of the two functional properties will predominate.
Acknowledgments
This work was supported in part by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103424. Drs. Jennifer L. Carroll and Ana-Maria Dragoi from the Feist-Weiller Cancer Center, Innovative Northwest Louisiana Experimental Therapeutics (INLET) at Louisiana State University Health Sciences Center, Shreveport, Louisiana, USA, are acknowledged for their assistance with the in-vitro cell culture studies. Acknowledgment is also extended to Dr. Robert Cody from JEOL USA, Inc. for his assistance with mass spectrometry analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abu-Fayyad A, et al. PEGylated γ-tocotrienol isomer of vitamin E: Synthesis, characterization, in vitro cytotoxicity, and oral bioavailability. Eur J Pharm Biopharm. 2015;96:185–95. doi: 10.1016/j.ejpb.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 2.Sen CK, Khanna S, Roy S. Tocotrienols: Vitamin E beyond tocopherols. Life Sci. 2006;78(18):2088–98. doi: 10.1016/j.lfs.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal B, Nesaretnam K. Vitamin E tocotrienols: life beyond tocopherols. Genes Nutr. 2012 Jan;7(1):1. doi: 10.1007/s12263-011-0234-x. Epub 2011 May 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yap SP, Yuen KH, Lim AB. Influence of route of administration on the absorption and disposition of alpha-, gamma- and delta-tocotrienols in rats. J Pharm Pharmacol. 2003;55(1):53–8. doi: 10.1111/j.2042-7158.2003.tb02433.x. [DOI] [PubMed] [Google Scholar]
- 5.Abu-Fayyad A, Nazzal S. Synthesis, characterization, and in-vitro antitumor activity of the polyethylene glycol (350 and 1000) succinate derivatives of the tocopherol and tocotrienol isomers of Vitamin E. Int J Pharm. 2017;519(1-2):145–156. doi: 10.1016/j.ijpharm.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alawin OA, et al. Antiproliferative effects of gamma-tocotrienol are associated with lipid raft disruption in HER2-positive human breast cancer cells. J Nutr Biochem. 2016;27:266–77. doi: 10.1016/j.jnutbio.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Zhu S, et al. Lysosomal Delivery of a Lipophilic Gemcitabine Prodrug Using Novel Acid-Sensitive Micelles Improved Its Antitumor Activity. Bioconjugate Chemistry. 2012;23(5):966–980. doi: 10.1021/bc2005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rijcken CJF, et al. Triggered destabilisation of polymeric micelles and vesicles by changing polymers polarity: An attractive tool for drug delivery. Journal of Controlled Release. 2007;120(3):131–148. doi: 10.1016/j.jconrel.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 9.Lee ES, Na K, Bae YH. Polymeric micelle for tumor pH and folate-mediated targeting. J Control Release. 2003;91(1-2):103–13. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 10.Jung ME, Lazarova TI. Efficient Synthesis of Selectively Protected l-Dopa Derivatives from l-Tyrosine via Reimer-Tiemann and Dakin Reactions. The Journal of Organic Chemistry. 1997;62(5):1553–1555. [Google Scholar]
- 11.Nakamura T, Kijima S. Studies on Tocopherol Derivatives. I. Conversion of β-, γ-, and δ-Tocopherol to α-Tocopherol. Chemical & Pharmaceutical Bulletin. 1971;19(11):2318–2324. [Google Scholar]
- 12.Andersson R, et al. Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions. Scand J Gastroenterol. 2009;44(7):782–6. doi: 10.1080/00365520902745039. [DOI] [PubMed] [Google Scholar]
- 13.Salmaso S, et al. Self-assembling nanocomposites for protein delivery: supramolecular interactions between PEG-cholane and rh-G-CSF. J Control Release. 2012;162(1):176–84. doi: 10.1016/j.jconrel.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Lu J, et al. Design and characterization of PEG-derivatized vitamin E as a nanomicellar formulation for delivery of paclitaxel. Mol Pharm. 2013;10(8):2880–90. doi: 10.1021/mp300729y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakiki F, Maharsi DA, Marhaendrajana T. Surfactant-Polymer Coreflood Simulation and Uncertainty Analysis Derived from Laboratory Study. 2015;47:2015. [Google Scholar]
- 16.Deer EL, et al. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas. 2010;39(4):425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Réjiba S, et al. Neoplasia. 7. Vol. 11. New York, N.Y.: 2009. Gemcitabine-Based Chemogene Therapy for Pancreatic Cancer Using Ad-dCK∷UMK GDEPT and TS/RR siRNA Strategies; pp. 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koufaki M. Vitamin E derivatives: a patent review (2010 - 2015) Expert Opin Ther Pat. 2016;26(1):35–47. doi: 10.1517/13543776.2016.1106476. [DOI] [PubMed] [Google Scholar]
- 19.Aurand LW. Springer, Editor. Springer; 1987. The Vitamines in Food Composition and Analysis. [Google Scholar]
- 20.Kunnumakkara AB, et al. {Gamma}-tocotrienol inhibits pancreatic tumors and sensitizes them to gemcitabine treatment by modulating the inflammatory microenvironment. Cancer Res. 2010;70(21):8695–705. doi: 10.1158/0008-5472.CAN-10-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hussein D, Mo H. d-delta-Tocotrienol-mediated suppression of the proliferation of human PANC-1, MIA PaCa-2, and BxPC-3 pancreatic carcinoma cells. Pancreas. 2009;38(4) doi: 10.1097/MPA.0b013e3181a20f9c. [DOI] [PubMed] [Google Scholar]
- 22.Kannappan R, et al. Tocotrienols fight cancer by targeting multiple cell signaling pathways. Genes & Nutrition. 2012;7(1):43–52. doi: 10.1007/s12263-011-0220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher T. Guidelines for Maintaining Cultured Cells. 2/2017]; Available from: https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/maintaining-cultured-cells.html.
- 24.Aggarwal BB, et al. Tocotrienols, the vitamin E of the 21st century: its potential against cancer and other chronic diseases. Biochem Pharmacol. 2010;80(11):1613–31. doi: 10.1016/j.bcp.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]