

Abstract
The secretome, defined as a portion of proteins secreted by specific cells to the extracellular space, secures a proper microenvironmental niche not only to the donor cells, but equally to the neighboring cells, thus maintaining tissue homeostasis. Communication via secretory products exists between endothelial cells and fibroblasts and this local mechanism maintains the viability and density of each compartment. Endothelial dysfunction, apart from obvious cell-autonomous defects, leads to the aberrant secretome which predisposes fibroblasts to acquire myofibroblastic fibrogenic phenotype. In our recent profiling of the secretome of such dysfunctional pro-fibrogenic renal microvascular endothelial cells we identified unique pro-fibrogenic signatures among which we detected ligands of Notch and Wnt/beta-catenin pathways. Here, we stress the role of reprogramming cues in the immediate microenvironment of (myo)fibroblasts and the contribution of endothelial secretome to the panoply of instructive signals in the vicinity of fibroblasts. We hope that this brief overview of endothelial-fibroblast communication in health and disease will lead to eventual unbiased proteomic mapping of individual secretomes of glomerular and tubular epithelial cells, pericytes and podocytes through reductionist approaches to allow for the synthetic creation of a complex network of secretomic signals acting as reprogramming factors on individual cell types in the kidney. Knowledge of pro- and anti-fibrogenic signatures in the secretome may garner future therapeutic efforts.
Keywords: fibroblast, endothelial cell, reprogramming, microenvironment, secretome, proteomics
General concept of the secretome
Communication is paramount for homeostasis in biological systems. Impaired communication is implicated in initiation and progression of majority of chronic diseases, from genetic, neurologic, and immune to cardiovascular and metabolic. Communication is universal wherever exists a community of individual species. The science of allelopathy, study of plant-released chemicals that influence growth and survival of neighboring organisms, is an example of plants communication with each other or the microbiome of the soil. Positive and negative allelopathy occurs as judged on the basis of whether released chemicals are supportive or harmful to the neighbors. Similar principles operate in the field of communication in the animal kingdom and between various cells. Hierarchical regulation by the central nervous and endocrine systems describes the control mechanisms governing local functions through the central regulators. In parallel with those, there is growing awareness that the local regulators per se determine or modulate local functions. More recently, it has been realized that secretory products of individual cells within an organ affect their own behavior, as well as that of the neighboring cells and distant organs.
The cell secretome is defined as a portion of proteins secreted to the extracellular space, which in humans constitutes 13–20% of the entire proteome1. More recently, proteins present in microvesicles (100nm-1um in diameter) and exosomes (<100nm in diameter), both organelles secreted by cells and containing up to 42% of the secretome2 (see ExoCarta database of exosomal proteins in http://exocarta.ludwig.edu.au), have been incorporated into the original term. Obviously, the secretome is cell- and tissue-specific, and the secretomic signatures change in response to fluctuations in physiologic states or pathologic conditions. Notably, multitude of non-protein products released by the cells participate in cell-to-cell communication, but those are considered as part of broader angiocrine signaling, the concept closely related to that of endothelial secretome. Angiocrine signaling is defined as endothelium-produced instructive factors that influence adjacent cells “through secreted or membrane-bound inhibitory and stimulatory growth factors, trophogens, chemokines, cytokines, extracellular matrix components, exosome and other cellular products that are supplied by tissue-specific endothelial cells to help regulate homeostatic and regenerative processes in a paracrine or juxtacrine manner”3. Alas, the panoply of those signals is beyond the scope of this brief overview. Not surprisingly, studies of the secretome/angiocrine signaling are considered to be of paramount importance in discovery of diagnostic tools for cancer, infectious diseases, senescence-messaging, to name a few, and could serve as a guide toward therapeutic strategies or explanation for adaptive transfer of stem cell4. The field, however, remains immature.
Interstitial fibroblasts, interlocked between tubular epithelial cells and peritubular capillaries, receive secretory signals from both sources. This raises several questions: “Are instructive signals from those sources unidirectionally coherent or signals are reciprocal?”, and “How signals from both sources are coordinated?”, and “How does a fibroblast make a decision which source to prefer in every case?” Preciously little information exist to address such a complexity of the four-cellular compartment (endothelial and epithelial cells, pericytes, and fibroblasts). Here, we shall take a reductionist approach and intentionally simplify the task to describe a role of endothelial secretome in kidney maintenance, repair or fibrogenesis5 (Fig. 1).
Figure 1. Four-compartmental model of (myo)fibroblast cross-talk with endothelial and epithelial cells. Grey background brackets the main subject of the present discussion.
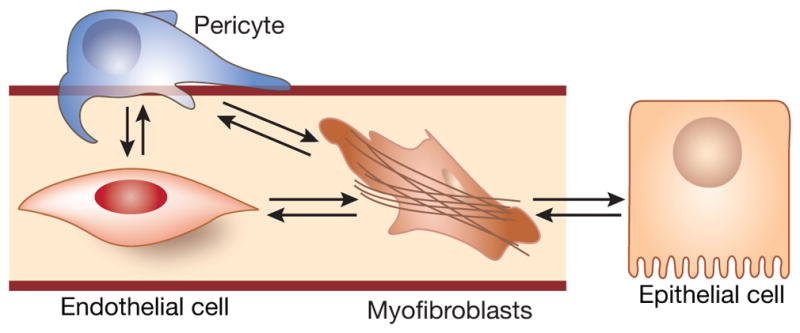
(Most recently, the fifth cell type, kidney-resident macrophages, closely associated with peritubular capillary bed, has been identified)5.
Ancestral uncertainty of myofibroblast
True to the Virchow’s theory of cell pathology, two main hypotheses currently ascribe progression of CKD to either the epithelial-mesenchymal transition (EMT) and its more recent partial EMT variation6,7,8, or to the pericyte-mesenchymal transition9. [Circulating fibrocytes, advocated by Asada et al10, may represent yet another source of fibroblasts.] While each of the proposed derivations has generated experimental evidence, at times conflicting, it is undisputed that fibrogenesis is a result of convergence of these pathways to generate myofibroblasts or to actuate the transition of quiescent fibroblasts to the matrix-synthesizing contractile myofibroblasts. Multiple reviews and explanatory comments11,12 detail these pathways, thus they will be out of scope of this essay. Instead, we shall focus on a fledgling data to suggest the existence of the third, endothelial, pathway of fibrogenesis.
In general, vascular endothelium, apart from playing a supportive role in the maintenance of vascular structure and tissue architecture3, participates in acute kidney injury, and in progression or regression of chronic kidney disease13,14 in part through deployment of diverse secretory products. What is the proof that endothelial cells possess the capacity to affect the fibrogenic behavior of the neighboring cells, such as (myo)fibroblasts? In our studies of mice with the catalytically disabled silent information regulation homolog 2 (SIRT1) exclusively in endothelial cells, that are nearly indistinguishable from control mice under basal conditions, we found an exaggerated fibrogenesis after diverse noxious stimuli15. On the other hand, mice heterozygote for transforming growth factor (TGF) receptor 2 exclusively in endothelial cells, while indistinct from controls under basal conditions, are significantly protected against fibrogenic stressful stimuli16. Such opposing responses to fibrogenic stimuli in mice, distinguishable exclusively by their endothelial cell metabolic and signaling properties, can be attributed to context-dependent contrasting signals emanating from the vascular endothelium and differentially instructing (myo)fibroblasts to attain pro- or anti-fibrogenic phenotype.
Here, we shall advance the thesis that endothelial cell is an independent modifier of fibrogenesis. It is accomplished mainly via its secretome acting on the (myo)fibroblast (paracrine regulation), as well as on endothelial cells (juxtacrine regulation). On the other hand, there may exist a counter-regulatory loop through a recently advocated putative (myo)fibroblast-to-endothelial cell transition supporting angiogenesis (see below) (Fig. 2). In either case, however, this is accomplished by the secretome-induced changes in the cellular microenvironment. Notably, the juxtacrine endothelial signaling or paracrine signals from neighboring cells participate in pruning of renal microvasculature leading to its rarefaction, a critical driver of fibrogenesis (the most recent analysis has confirmed this concept of Bohle’s group and Johnson’s group17,18,19.) The existing dichotomy of endothelial secretome is illustrated in Fig. 2. While normally the secretome maintains the stem/progenitor cell niche and supports the neighboring epithelia, under pathological conditions, endothelial secretome provides instructions towards regeneration and fibrosis – the balance between the two determines the outcome. Distinct mechanistic elements of such signaling are enumerated in the lower part of the Fig. 2.
Figure 2.

Endothelial secretome-dependent pathways for the maintenance and restoration of kidney architecture and pro-fibrogenic properties of aberrant endothelial secretome.
Myofibroblasts and tissue rigidity
Every researcher who has ever cultured primary epithelial or endothelial cells knows first-hand that, after several passages, these cells acquire a fibroblastic phenotype. Fibroblasts, not surprisingly, comprise a heterogeneous population. Indeed, they derive from distinct embryologic roots (for instance, dermal fibroblasts, depending on the site, derive from the dermomyotome or neural crest)20,21,22,23,24. Fetal fibroblasts derive from the undifferentiated mesenchyme and critically differ from their adult progeny by the ability of scar-free wound repair. A small proportion of fibroblasts found in the bone marrow and in the circulation is represented by either mesenchymal stem cells or CD45+/CD34+ fibrocytes. In the heart, Moore-Morris et al25 studied the origin of fibroblasts in pressure overload-induced fibrosis using fate tracing experiments and demonstrated that cardiac fibroblasts were not derived from hematopoietic cells, or via Endo-MT, or epicardial EMT, rather they originated from the proliferation of two populations of cells, epicardial and endothelial. In the kidney, cortical and medullary/papillary fibroblasts differ morphologically and functionally26. It is consistent with a broader view on heterogeneity of fibroblasts22,24. Fibroblasts of different organs, including kidneys, express collagen-1α121,27. Pulmonary and liver fibroblasts consist of at least two subsets – Thy-1 positive and negative, with Thy-1-negative population being more fibrogenic and Thy-1-positive cells confined to perivascular niche (quoted from 24). Most recent fate-mapping studies of dermal fibroblasts using Engrailed-1 lineage labeling28 disclosed a subpopulation of CD26/dipeptidyl peptidase-4 (DPP4) positive cells possessing intrinsic fibrogenic potential. Inhibition of CD26/DPP4 enzymatic activities using small-molecule inhibitors ameliorated scarring. Knowledge on the origins of fibroblasts in each particular organ is of critical importance for resolving the “identity crisis” in assigning the origins to myofibroblasts in fibrosis in general, and tubulointerstitial fibrosis in particular. These cells, characterized by the expression of α-smooth muscle actin (αSMA), stress fibers and contractile force, as well as variously expressing endosialin, P311 (a gene product overexpressed in hypertrophic scars), integrin α11β1, osteopontin and periostin29, though neither of these markers unique, are critically involved in pathogenesis of fibrosis. Contractile properties of myofibroblasts lacking myosin heavy chain, in contrast to smooth muscle cells, are characterized by the incremental generation of force using a lock-step mechanism dependent on RhoA/Rho-associated kinase30 and are responsible for micronewtons-strong and micrometers-ranging long-lasting contraction. This mechanism is a centerpiece of connective tissue remodeling.
Direct detection of mechanical properties of fibrotic tissues showed a several-fold higher rigidity than that of corresponding normal tissues31. High rigidity of connective tissue predisposes to tighter cell adhesion and spreading, a process termed “durotaxis”. Fibroblast-to-myofibroblast transition depends on convergence of two signals: the elevated levels of TGF-β and the stiff matrix with elastic modulus E>20 kPa. (Note that the rigid cell culture dish emulates the stiff matrix and creates a simplified view of the potency of TGF-β.) Moreover, liberation of TGF-β normally sequestered in the latent form by the extracellular matrix is mediated by contraction-induced unfolding of ECM proteins. Spreading and tight adhesion resulting in contraction of the matrix and deformation of tissue architectonics depend on the expression of non-muscle myosin, integrins, and Rac/Rho participation. Such a synergy between durotaxis and TGF-β pathways of fibrogenesis forms a vicious circle where one component triggers another. In accord with this, inhibition of Rho kinase effector, ROCK, which inactivates myosin, eliminates durotaxis effects of rigid substrates.
Possible dissociation between synthetic and contractile functions of myofibroblast
TGF-β signaling induces both the programs responsible for collagen and the programs responsible for α-SMA synthesis. Apart from this common denominator, the wiring of programs initiating each of these two arms of TGF-β effect is distinct. TGF-β induces collagen I synthesis by binding to the collagen alpha2(1) promoter32. α-SMA promoter region responsible for TGF-β signaling has been identified as a putative GATA-binding site. Known factors promoting α-SMA expression in fibroblasts include intracellular signals like Smad 3, KLF5, miRNAs -192, -132, -21, -129, -200 b/c, -216a; and extracellularly Wnt and Jagged 133. Factors suppressing myofibroblastic conversion include KLF4, PPARγ, interferon-γ, miRNA 200a, Let 7, and supple matrix, among others33. We encountered a possible dissociation between collagen I synthesis and α-SMA in our recent studies of effects of LIF on activated fibroblasts34. While LIF reduced the proportion of α-SMA-positive fibroblasts in vitro and in vivo in models of tubulointerstitial fibrosis, it did not reduce collagen I synthesis and the extent of renal fibrosis (see below). Collectively, the data allude to the possible existence of at least three subpopulations of activated fibroblasts: those predominantly expressing α-SMA, those that are mainly collagen I-synthetic, and those which have both up-regulated.
Microenvironment predisposing to cell reprogramming and its reversibility
The view that only cell-autonomous mechanisms define cell fate and state of differentiation have recently been challenged by the findings of cell-cell communication and the instructive role of the microenvironment in determining programs for cell differentiation35,36,37. Capillary endothelial cells fulfill instructive functions for adjacent cells by creating a supportive microenvironment (niche) for maintenance and renewal of various stem and mature differentiated cells3. For instance, bone marrow sinusoidal endothelial cells orchestrate differentiation of hematopoietic stem cells toward distinct lineages by secreting GM-CSF, IL-6, IL-8, G-CSF, IL-1, TNF, chemokines and metalloproteinases38. Hematopoietic and thrombopoietic regeneration after myeloablation critically depend on the secretome of sinusoidal endothelial cells39, and conditional endothelial deletion of Jagged-1 impairs this regeneration40. Regenerated mature megacaryocytes terminate stimulatory signaling from sinusoidal endothelial cells by secreting thrombospondin-1, thus closing the feedback loop between endothelial and hematopoietic cell balance. Compensatory growth of alveolar type 2 epithelial cells after unilateral pulmonectomy has been found to be driven by endothelial cell-induced up-regulation of MMP-14 which in turn activates EGF receptor on pulmonary epithelial cells41. These and other similar studies raise the question whether renal (glomerular and peritubular) capillary endothelium maintains the homeostasis with adjacent pericytes, podocytes and fibroblasts? The cross-talk between glomerular endothelial cells and podocytes has been described42,43.
Pro-fibrotic secretome of vascular endothelium
An observation that tissue fibrosis progresses pari passu with the site-specific rarefaction of the microvasculature, first made by Bohle’s laboratory17, still lacks a molecular explanation. One of the possible explanations posits that mutual communication, via secretory products, exists between fibroblasts and endothelial cells and this local mechanism maintains the viability and density of each compartment. In fact, the most detailed so far proteomic studies by M. Mayr’s group44 revealed that up to 248 distinct proteins are secreted by cardiac fibroblasts and that activation by TGFβ affects secretion of 148 proteins. Studies showed that cardiac fibroblasts, after myocardial infarction or activation by TGFβ, exhibit reduced expression of secreted microRNAs, like miR-29b, leading to cardiac fibrosis. Normally, miR-29b is responsible for up-regulation of collagens, matrix metalloproteinases, leukemia-inhibitory factor, insulin-like growth factor-1, and pentraxin-3 and attenuates responses to TGFβ, all having a protective effect44. In the case of liver sinusoidal endothelial cells, the balance between pro-fibrotic and pro-regenerative signaling is maintained by the expression of stromal-derived factor-1 receptors CXCR7 and CXCR445. After an acute injury, CXCR7 upregulation leads to Id-1-dependent induction of secretory angiocrine factors which stimulate regeneration. On the other hand, chronic liver injury augments CXCR4 signaling and production of pro-fibrotic secretome.
The role of microRNAs in promoting organ fibrosis (fibromiRs) is rapidly expanding46. A list of fibromiRs includes miR-15 family, miR-21, miR-34a, miR-192, miR-199b, miR-208; whereas other miRs exhibit antifibrotic signaling, such as miR-1, miR-24, miR-29b, miR-101, and miR-200b. We (SD and MSG, unpublished observations) have recently screened miRs of renal microvascular endothelial cells obtained from SIRT1endo−/− and control mice and, among other differentially displayed miRs, identified a unique miR-127 3p (known to promote cell senescence) highly enriched in senescence- and fibrosis-prone SIRT1endo−/− mice. We performed preliminary studies based on the assumption that miR-127-3p has pro-fibrogenic effects and employed anti-miR strategy using complementary locked nucleic acid (LNA) oligonucleotides and respective scrambled controls. Data presented in Fig. 3,A and B demonstrate that LNA targeting miR-127-3p effectively ameliorates fibrosis in UUO. Notably, Wnt and Notch pathway components, in addition to histone methyltransferase-8 gene (SETD8), are identified as targets of upregulated miR-127-3p in dysfunctional endothelial cells. All three are highly interconnected due to their regulation of Wnt and Notch pathways. Specific targets of SETD8 include Wnt (it regulates transcription of Wnt-activated genes), as depicted in Fig. 3, C.
Figure 3. Profibrogenic effect of miR-127-3p.

A and B: Masson’s trichrome staining of UUO kidneys in control mice, mice treated with the negative (scrambled) LNAs, and mice receiving complementary LNAs to miR-127 and miR-410. Ordinate shows the number of pixels corresponding to the detection of fibrotic areas. Horizontal lines above individual bars indicate p<0.05 between the corresponding groups. N=5 per each group of animals. Note that LNA to miR-127 mitigates fibrosis compared to both control groups, whereas LNA to miR-410 (another miR screen finding) is not effective, at least at the dose used.
C: Targets of miR-127-3p. TargetScan identifies these target gene promoters relevant to Wnt/β-catenin and Notch pathways. Note a remarkable overlap with proteomic findings. In red are components of main focus.
In a series of recent studies our group has found that the extract of endothelial progenitor cells prevents TGFβ-induced activation of fibroblasts and their conversion to myofibroblasts. In vivo it prevents and reverses renal fibrosis34. Multiplex analysis of cyto-/chemokines in the medium conditioned by endothelial progenitors revealed that it is highly enriched in the vascular endothelial growth factor and leukemia inhibitory factor (LIF). While LIF alone activated gp130/STAT3 pathway and mimicked anti-fibrotic effects of the extract of endothelial progenitor cells in cultured fibroblasts, the effect was not entirely reproducible in vivo, probably due to the variable expression of LIF receptor. Therefore, we next employed a receptor-independent agonist of gp130/STAT3 pathway, hyper-IL-6, which turned to produce a powerful anti-fibrotic effect in models of renal fibrogenic diseases. These findings not only reveal an anti-fibrogenic component of the secretome which consistently induces gp130/STAT3 pathway, but also propose a novel therapeutic strategy entirely based on secretomic findings. There is, however, a recent controversy related to the role of STAT3 pathway in fibrogenesis. One study reports on the protective effect of LIF, acting via STAT3 and miR-29c, against renal fibrosis47. As noted, our work34 introduced hypert-IL-6, a receptor-independent gp130/STAT3 activator, as a therapeutic that ameliorates renal fibrosis in two different models, unilateral ureteral obstruction and chronic phase of folic acid nephropathy. Yet, recent publication by Terzi’s group48 implicates STAT3 activation in the lesion-prone strain of mice, but not in C57BL/6 mice, in progression of fibrosis after nephron reduction. It is not excluded that differences in matrix rigidity and mouse background (our studies were performed using αSMA-GFP transgenic mice on C57BL/6J background) are responsible for disparate results.
Senescence-associated endothelial secretory products
It is remarkable that CKD predisposes to premature aging. Renal biopsies in young individuals with diverse CKD reveal senescent cells in all kidney compartments49, 50. One of the contributors to organismal aging is cell senescence. This thesis is confirmed by the findings that progeroid mice subjected to elimination of p16Ink4a-positive senescent cells exhibit a delay in age-related dysfunction51. One of the mechanistic scenarios explaining how a relatively few senescent cells can affect organ and organismal functions stems from the idea that these cells secrete substances which could be deleterious to neighboring cells by amplifying pro-senescence signaling – the concept of senescence-associated secretory phenotype/products (SASP). The main hitherto disclosed components of SASP include IL-6, IL-8, TNF-α, TGF-β, matrix metalloproteinases, insulin-like growth factor (IGF)-binding proteins, monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor (PAI)-1, among many other components which vary depending on the cell type52. SASP accounts for both the autocrine and the paracrine effects, which are important for diverse pathological processes from cancer progression to immunomodulation, changes in cell microenvironment, and propagation of cellular senescence53,54. SASP is responsible at least in part for the sterile systemic inflammation, one of the cardinal features of aging. Initially, activation of inflammasomes leads to activation of caspase-1 and amplification of IL-1 signaling55. It is followed by IL-1 activation of NF-kB and C/EBP-β transcription factors, which lead to the induction of IL-6 and IL-8. Kuilman et al showed that by depleting IL-6 it was possible to ameliorate inflammatory SASP network56. Pharmacological design of future senolytic therapies should target elements of SASP53. It is remarkable that the concept of senescence and SASP is not limited to aging, but is highly relevant to chronic kidney disease (CKD). This implies a much broader applicability of SASP findings to a range of CKD. Dysfunctional senescent endothelial cells produce excessive amounts of collagen XVIII and its C-terminal antiangiogenic fragment, endostatin57, in part responsible for endothelial-mesenchymal transition.
Endothelial-mesenchymal transition (EndoMT) and microvascular rarefaction
Endo-MT, a process of acquisition by endothelial cells of mesenchymal and stem cell-like phenotypic characteristics, is a normal developmental stage taking place during embryonic formation of heart valves and septa. In adulthood, Endo-MT is a contributor to vascular drop-out and development of fibrosis, as detected in multiple organs, including heart and kidney57,58,59. It has been demonstrated that glomerulosclerosis in tensin-2-deficient mice is associated with Endo-MT of glomerular endothelial cells60. Some studies show that 30–50% of interstitial myofibroblasts originate from the endothelium, although other investigations dramatically reduce this figure61. Dejana’s group has unequivocally demonstrated Endo-MT at the onset and progression of cerebral cavernous malformation62. These investigators pursued the mechanism of Endo-MT and showed that it is due to activation of Wnt-β-catenin pathway and inhibition of β-catenin prevents cerebral-cavernous malformation63,64. Xu et al64 demonstrated that aberrant Endo-MT causes endocardial fibroelastosis. In fibrodysplasia ossificans progressiva Endo-MT is responsible for excessive generation of osteoblasts and chondrocytes from endothelial cells, thus illustrating yet another end-result of this process65. TGF-β is a major mediator of Endo-MT, whereas BMP-7 counteracts Endo-MT as demonstrated using endothelial cell fate-tracing66. Actions of TGF-β are mediated via activin receptor-like kinases 1 and 5 (Alk1 and Alk5). Activation of Alk1 uniquely expressed on endothelial cells results in cell migration, proliferation and angiogenesis, whereas stimulation of Alk5 induces, via Smads 2/3 phosphorylation, the transcription of SM22α, fibronectin, and PAI-1, thus mediating differentiation along the smooth muscle/mesenchymal phenotype and leading to formation of myofibroblasts. Endoglin, an endothelium-specific TGF-β co-receptor, regulates balance between activation of Alk-1 and Alk-5 pathways. Noteworthy, prolonged activation with TGF-β results in the down-regulation of Alk1 signaling and predominant signaling via Alk 5, thus promoting Endo-MT65. Ubil et al67 has recently described the possibility of resolving cardiac fibrosis by reversing Endo-MT, and thus generating endothelial cells from mesenchymal cells.
Gene microarray analysis of cultured endothelial cells treated with an inhibitor of nitric oxide synthase to induce endothelial dysfunction, revealed upregulation of collagen XVIII and its anti-angiogenic fragment endostatin, a finding confirmed in vivo in mice chronically treated with NOS inhibitor57. Endostatin in these animals leads to the development of Endo-MT and eventual rarefaction of renal microvasculature, thus further compounding vascular and parenchymal pathology. In fact, we recently demonstrated that young mice genetically overexpressing endostatin or young mice chronically infused with endostatin via osmotic minipumps show signs of premature vascular senescence, microvascular rarefaction, and renal fibrosis68,69. Endo-MT can be induced not only by TGF-β or chronic activation of Wnt-β-catenin pathway, but also by activation of Notch which could serve as a regulator of partial or complete Endo-MT70,71,72. FGF and its downstream target let-7 miRNA suppress TGF-β-induced Endo-MT, thus preventing fibrosis71.
It is remarkable that cell-specific translational profiling of distinct kidney resident cells using CRE-recombinase-dependent activation of eGFP-L10a ribosomal protein subunit (this allows translating ribosome affinity purification of mRNAs in CRE-expressing cells) detected 384 endothelium-specific genes and documented that in acute kidney injury “the largest sets of unique responses were observed in the vascular endothelium …” and point to endothelium-dependent recruitment of other cell types73. Specific unique canonical pathways enriched 4–8-fold in endothelial cells after acute kidney injury include leukocyte extravasation, IL-8, high mobility group box 1 protein, Toll-like receptor, CXCR4, endothelin-1, chemokine, thrombopoietin, JAK/STAT, acute-phase response, and IL-10 signaling, thus emphasizing pro-inflammatory and anti-angiogenic state of damaged endothelium. This set of findings is in a good agreement with the fluorescence microangiography-detected loss of peritubular capillary density and caliber at 8 weeks, correlating with severity of initial renal ischemia74.
All these lines of evidence reinforce the concept of microvascular rarefaction occurring in parallel and pari passu with the tubulointerstitial fibrosis. We have recently found several fibrogenic components of the aberrant endothelial secretome. The ligand-receptor pair, semaphorin 7A (sema-7A) and neuropilin 1 (NP-1), is remarkable for its involvement in angiogenesis and fibrosis (shown for the lung75, where sema-7A knockout ameliorated pulmonary fibrosis). In fact, thrombospondin 1 and 2 also discovered in pro-fibrogenic endothelial secretome, have a similar anti-angiogenic effect. In our unpublished studies we found that Sema-7A–Fc (5 ug/ml) induces premature senescence in primary renal fibroblasts (Fig. 4). That would suggest that these cells become the source of SASP, which, in turn, may affect functional state of neighboring endothelial and epithelial cells. It is conceivable that Endo-MT is at the cross-section of several pathophysiological conditions and is an important contributor to microvascular rarefaction. The earlier stage of microvascular drop-out is characterized by the loss of vessel patency detected as dissociation between the near-normal number of CD-31-positive endothelial cells and reduced number of actually perfused capillaries after intravital injection of Lycopersicon esculentum.
Fig. 4. Example of a dialog between the dysfunctional endothelium secreting semaphorin 7A and fibroblasts.

A: Primary renal fibroblasts cultured in the presence of Sema-7A–Fc exhibit a 2-fold increase in prematurely senescent cells after only 48 h in culture.
B: A cartoon depicting the vicious circle between dysfunctional endothelial secretome containing, among others, semaphorin 7A, and fibroblasts, which develop premature senescence and release SASP in turn affecting endothelial cells.
We observed such dissociation between the vascularity and patency in murine kidneys undergoing fibrogenesis and in mice exposed to elevated levels of endostatin16,69. The next stage is characterized by the obliteration of the capillary lumen, which leaves behind the de-endothelialized basement membranes, variably referred to as a “string vessels” or “empty basement membrane tubes”, or “empty sleeves” - the remnants of preexisting vessels, as discussed in detail in our recent review14. This remaining scaffold of the basement membrane, rich in endothelial and pericyte growth factors, such as VEGF, bFGF, and PDGF, facilitates the repopulation of string vessels with endothelial cells. Restoration of endothelial lining of string vessels could be orchestrated by secretomic instructions and may occur by endothelial proliferation and be assisted by endothelial progenitor cells76.
Endothelial regulation of fibroblast acquisition of myofibroblastic phenotype
To study the endothelial secretomic pro- and anti-fibrogenic signatures, we examined proteomic signatures of endothelium-conditioned media obtained from primary cultures of CD31-magnetically isolated high-purity populations of renal microvascular endothelial cells from 1) control wild-type, 2) endothelial SIRT-1−/− or 3) endothelial deficient in TGFβRII+/− mice under control conditions and after TGFβ (unpublished data). Conditioned media were analyzed using unbiased, non-targeted MS/MS. We were able to detect 332 non-redundant proteins, which belong to diverse categories (Fig. 5). We argued that the differential signatures of proteins secreted uniquely by endothelial SIRT-1−/− vis-a-vis endothelial TGFβRII+/− could theoretically contain protein culprits for pro- and anti-fibrogenic phenotype of the respective mice, as depicted in Venn’s diagram (Fig. 5). Remarkably, among unique pro-fibrogenic signatures of endothelial cells we detected ligands of Notch and Wnt pathways, jagged 1 and Dickkopf 3, respectively. Work-in-progress now seeks to determine the precise action of these endothelial-secreted ligands on the phenotype of primary cultures of renal interstitial fibroblasts.
Figure 5.

The secretome of cultured murine renal microvascular endothelial cells. A – categories of proteins detected in the secretome; B – a Venn diagram comparing the protein components of the secretome of endothelial cells isolated from SIRT1−/−, TGFR2+/−, and wild type mice and stimulated with TGF-β1.
In addition to this, we (HD and GM) have initiated studies of epithelial and fibroblastic secretomes. To identify the secretome components from renal cells and to understand their role in progression of renal fibrosis we investigated the supernatant of renal fibroblasts and epithelial cell line subjected to TFG-β1 treatment using two-dimensional electrophoresis and mass spectrometry. The study yielded a panel of protein sets of 167, and 112 which were differently secreted upon TGF-β1 treatment by renal fibroblasts and renal epithelial cell lines respectively. Among the identified proteins, 50 were shared in both cell lines (Fig. 6), whereas 117 were identified only in renal fibroblasts secretome and 62 were identified only in epithelial cell secretome, thus highlighting the cell type-specific signatures. The differentially secreted proteins were classified using DAVID GO-biological process analysis tool. The resulting data show that the differentially secreted proteins are involved in several important biological processes. The major functions of these proteins are related to regulation, cellular and metabolic categories (Fig. 6). In addition, proteins involved in the regulation of catalytic activity, response to hypoxia, regulation of actin cytoskeleton organization, and regulation of macromolecule metabolic processes were particularly reinforced (Fig. 6).
Figure 6. Fibroblast and epithelial cells secretome.

Human kidney fibroblast cell line and human epithelial cell line were grown to subconfluency. Medium was removed, and after washing in phosphate-buffered saline (PBS) the cells were incubated in serum-free DMEM. The cells were treated for 48 h with purified human TGFβ1 (5 ng/ml), and the supernatants were collected. The secretome proteins were prepared from the supernatant and separated by two-dimensional gel electrophoresis. The protein mapcompared to the control one and the differentially secreted proteins were identified by mass spectrometry.
It is tempting to consider here a more realistic situation when a fibroblast receives input from both the epithelial and endothelial compartments. For instance, our finding of a putative Wnt ligand, Dickkopf-3, secreted by the dysfunctional endothelium concatenates with the finding that epithelium-secreted Wnt-1 is capable of inducing tubulointerstitial fibrosis72. Whether this is this an example of a coherent or reciprocal signaling inputs remains to be determined.
(Myo)fibroblast-to-endothelial cell transition – does it occur?
Reversal of fibrotic disease, in theory, is achievable by either shifting the balance between fibrogenesis and degradation of extracellular matrix proteins towards their degradation, or by reducing the burden of myofibroblasts in affected tissues, or both. Bechtel et al77 have previously demonstrated the role of hypermethylation of CpG island promoters in fibroblast activation and renal fibrosis. More recently, Tampe et al78 demonstrated that BMP7-induced reversal of renal fibrosis in UUO or diabetic nephropathy is associated with the restoration of Tet3 expression and normalization of Rasal1 promoter hypermethylation, thus preventing fibroblast activation. These studies outline one of the epigenetic paths toward reprogramming of myofibroblasts. Margarity et al79 employed ex vivo strategy to generate from fibroblasts partially induced pluripotent stem cells (PiPS) using reprogramming factors OCT4, SOX2, KLF4 and c-MYC. Treatment of PiPS with VEGF resulted in SET translocation (myeloid leukemia-associated) similar protein to the nucleus where it binds to VE-cadherin promoter and induces reprogramming toward endothelial cells. The role of p53 in (myo)fibroblast-endothelial transition has been demonstrated by Ubil et al67. These investigators showed that endothelial-to-mesenchymal transition is reversible. Using genetic fate mapping, they demonstrated that cardiac fibroblasts acquire an endothelial-cell-like phenotype after acute ischemic cardiac injury and that transcription factor p53 regulates this switch. Loss-of-function experiments showed that it decreases the formation of fibroblast-derived endothelial cells, reduces post-infarct vascular density and worsens cardiac function. Gain-of-function experiments with stimulation of the p53 pathway in cardiac fibroblasts resulted in augmentation of mesenchymal-to-endothelial transition, enhancement of vascular density and improvement of cardiac function. These studies established that mesenchymal-to-endothelial transition occurs in vivo and contributes to neovascularization of the injured heart.
In our studies of renal fibrosis34 we also initially set out to induce mesenchymal-to-endothelial transition using an extract of endothelial progenitor cells both in vitro and in vivo. Indeed, this therapy resulted in a significant inhibition and reversal of fibrosis, reduction of myofibroblasts (α-SMA-GFP-positive cells), and improvement of microvascular density, but we were unable to detect the appearance of markers of endothelial specification in treated myofibroblasts in vitro. The finding that administration of the EPC extract improved renal microvascular density, could be interpreted by the fibroblast-promoted angiogenesis, as previously demonstrated by others80,81. Our findings are more consistent with a reversal of myofibroblastic to fibroblastic phenotype, similar to what has been described previously82. In addition, there is a precedent to our findings: it has been shown that amniotic membrane stromal extract and atrial natriuretic peptide actuate myofibroblast reversal to fibroblastic phenotype83.
One of the possible reasons for the ability of microenvironmental cues to reprogram (myo)fibroblasts is that the cells subjected to pro-fibrotic stimuli, like TGF-β, acquire a more open chromatin configuration (J Chen and MSG, unpublished) that facilitates reprogramming signal transduction. In the case of LIF or, more significantly, hyper-IL-6, their presence in the interstitial microenvironment of the kidney induces acquisition by myofibroblasts of a state of transient pluripotency, as judged by the short-lived surge in pluripotency transcription factors c-Myc and Nanog with the later induction of Oct434. This makes myofibroblasts especially sensitized, within a narrow time-frame, to other instructive cues which could enhance either a pro-fibrogenic or anti-fibrogenic phenotype. Enhancing the latter while suppressing the former may constitute a rational therapeutic strategy.
In summary, this brief overview of endothelial secretome-induced modulation of (myo)fibroblast behavior provides a clarion call to explore in depth the wiring of local regulators of fibrogenesis. It stresses the role of reprogramming cues in the immediate microenvironment of (myo)fibroblasts and the contribution of endothelial secretome to the panoply of instructive signals in the vicinity of fibroblasts. By the same token, the secretomes of tubular and glomerular epithelial cells and pericytes, contributing as well to the overall signaling landscape are in need of further exploration. On the other hand, the feedback secretomic signatures of (myo)fibroblast and their effects on its neighbors remains poorly understood. There is also a need to better understand how senescence or noxious stimuli affect the secretome of endothelial, epithelial cells and pericytes to sensitize fibroblast activation to acquire myofibroblast phenotype. When the mapping of individual secretomes in health and disease has been accomplished through reductionist approaches, then will it become possible to synthesize a complex network of secretomic signals acting as reprogramming factors on individual cell types in the kidney. Knowledge of pro- and anti-fibrogenic signatures in the secretome may garner future therapeutic efforts.
Acknowledgments
These studies were supported in part by NIH grants DK54602, DK052783 and DK45462 (MSG); New York Trust Fund (MSG); Dr. Werner Jackstaedt Foundation (ML); the “ILJIN” Faculty Research Assistance Program of Yonsei University College of Medicine for 2013 (6-2013-0068) and Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (NRF-2013R1A1A1010863) (JWS); Showa University Research grant for Young Researchers from Showa University Research Fund (KM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907–20. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee P, Mani S. Methodologies to decipher the cell secretome. Biochim Biophys Acta. 2013;1834:2226–32. doi: 10.1016/j.bbapap.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529:316–25. doi: 10.1038/nature17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ranganath SH, Levy O, Inamdar MS, et al. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell. 2012;10:244–58. doi: 10.1016/j.stem.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stamatiades EG, Tremblay ME, Bohm M, et al. Immune Monitoring of Trans-endothelial Transport by Kidney-Resident Macrophages. Cell. 2016;166:991–1003. doi: 10.1016/j.cell.2016.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwano M, Plieth D, Danoff TM, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–50. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lovisa S, LeBleu VS, Tampe B, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens. 2011;20:297–305. doi: 10.1097/MNH.0b013e328344c3d4. [DOI] [PubMed] [Google Scholar]
- 10.Asada N, Takase M, Nakamura J, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121:3981–90. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kramann R, DiRocco DP, Humphreys BD. Understanding the origin, activation and regulation of matrix-producing myofibroblasts for treatment of fibrotic disease. J Pathol. 2013;231:273–89. doi: 10.1002/path.4253. [DOI] [PubMed] [Google Scholar]
- 12.Quaggin SE, Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int. 2011;80:41–50. doi: 10.1038/ki.2011.77. [DOI] [PubMed] [Google Scholar]
- 13.Brodsky SV, Goligorsky MS. Endothelium under stress: local and systemic messages. Semin Nephrol. 2012;32:192–8. doi: 10.1016/j.semnephrol.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goligorsky MS, Hirschi K. Stress-Induced Premature Senescence of Endothelial and Endothelial Progenitor Cells. Adv Pharmacol. 2016;77:281–306. doi: 10.1016/bs.apha.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vasko R, Xavier S, Chen J, et al. Endothelial sirtuin 1 deficiency perpetrates nephrosclerosis through downregulation of matrix metalloproteinase-14: relevance to fibrosis of vascular senescence. J Am Soc Nephrol. 2014;25:276–91. doi: 10.1681/ASN.2013010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xavier S, Vasko R, Matsumoto K, et al. Curtailing endothelial TGF-β signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol. 2015;26:817–29. doi: 10.1681/ASN.2013101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bohle A, Kressel G, Müller CA, et al. The pathogenesis of chronic renal failure. Pathol Res Pract. 1989;185:421–40. doi: 10.1016/S0344-0338(89)80058-5. [DOI] [PubMed] [Google Scholar]
- 18.Kang DH, Kanellis J, Hugo C, et al. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol. 2002;13:806–16. doi: 10.1681/ASN.V133806. [DOI] [PubMed] [Google Scholar]
- 19.Bábíčková J, Klinkhammer BM, Buhl EM, et al. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017;91:70–85. doi: 10.1016/j.kint.2016.07.038. [DOI] [PubMed] [Google Scholar]
- 20.Müller GA, Rodemann HP. Characterization of human renal fibroblasts in health and disease: I. Immunophenotyping of cultured tubular epithelial cells and fibroblasts derived from kidneys with histologically proven interstitial fibrosis. Am J Kidney Dis. 1991;17:680–3. doi: 10.1016/s0272-6386(12)80351-9. [DOI] [PubMed] [Google Scholar]
- 21.Rodemann HP, Müller GA. Characterization of human renal fibroblasts in health and disease: II. In vitro growth, differentiation, and collagen synthesis of fibroblasts from kidneys with interstitial fibrosis. Am J Kidney Dis. 1991;17:684–6. doi: 10.1016/s0272-6386(12)80352-0. [DOI] [PubMed] [Google Scholar]
- 22.Müller GA, Strutz FM. Phenotypic heterogeneity of renal fibroblasts. Kidney Int. 1995;48(Suppl):S33–S36. [PubMed] [Google Scholar]
- 23.Zeisberg M, Strutz F, Müller GA. Role of fibroblast activation in inducing interstitial fibrosis. J Nephrol. 2000;13(Suppl 3):111–120. [PubMed] [Google Scholar]
- 24.Sorrell JM, Caplan AI. Fibroblasts-a diverse population at the center of it all. Int Rev Cell Mol Biol. 2009;276:161–214. doi: 10.1016/S1937-6448(09)76004-6. [DOI] [PubMed] [Google Scholar]
- 25.Moore-Morris T, Guimarães-Camboa N, Banerjee I, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921–34. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grgic I, Krautzberger AM, Hofmeister A, et al. Translational profiles of medullary myofibroblasts during kidney fibrosis. J Am Soc Nephrol. 2014;25:1979–90. doi: 10.1681/ASN.2013101143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin SL, Kisseleva T, Brenner DA, et al. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–27. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rinkevich Y, Walmsley GG, Hu MS, et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015;348:aaa2151. doi: 10.1126/science.aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinz B, Phan SH, Thannickal VJ, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–55. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castella LF, Buscemi L, Godbout C, et al. A new lock-step mechanism of matrix remodelling based on subcellular contractile events. J Cell Sci. 2010;123:1751–60. doi: 10.1242/jcs.066795. [DOI] [PubMed] [Google Scholar]
- 31.Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324:1673–7. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nieto N, Cederbaum AI. S-adenosylmethionine blocks collagen I production by preventing transforming growth factor-beta induction of the COL1A2 promoter. J Biol Chem. 2005;280:30963–74. doi: 10.1074/jbc.M503569200. [DOI] [PubMed] [Google Scholar]
- 33.Ghosh AK, Quaggin SE, Vaughan DE. Molecular basis of organ fibrosis: potential therapeutic approaches. Exp Biol Med (Maywood) 2013;238:461–81. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto K, Xavier S, Chen J, et al. Instructive Role of the Microenvironment in Preventing Renal Fibrosis. Stem Cells Transl Med. 2016 doi: 10.5966/sctm.2016-0095. pii:sctm. 2016–0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rechavi O, Erlich Y, Amram H, et al. Cell contact-dependent acquisition of cellular and viral nonautonomously encoded small RNAs. Genes Dev. 2009;23:1971–9. doi: 10.1101/gad.1789609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rechavi O, Goldstein I, Kloog Y. Intercellular exchange of proteins: the immune cell habit of sharing. FEBS Lett. 2009;583:1792–9. doi: 10.1016/j.febslet.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 37.Roobrouck VD, Vanuytsel K, Verfaillie CM. Concise review: culture mediated changes in fate and/or potency of stem cells. Stem Cells. 2011;29:583–9. doi: 10.1002/stem.603. [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi H, Butler JM, O’Donnell R, et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol. 2010;12:1046–56. doi: 10.1038/ncb2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hooper AT, Butler JM, Nolan DJ, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell. 2009;4:263–74. doi: 10.1016/j.stem.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poulos MG, Guo P, Kofler NM, et al. Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 2013;4:1022–34. doi: 10.1016/j.celrep.2013.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding BS, Nolan DJ, Guo P, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. 2011;147:539–53. doi: 10.1016/j.cell.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–16. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu J, Lee K, Chuang PY, et al. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol. 2015;308:F287–97. doi: 10.1152/ajprenal.00533.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abonnenc M, Nabeebaccus AA, Mayr U, et al. Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c. Circ Res. 2013;113:1138–47. doi: 10.1161/CIRCRESAHA.113.302400. [DOI] [PubMed] [Google Scholar]
- 45.Ding BS, Cao Z, Lis R, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. doi: 10.1038/nature12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pottier N, Cauffiez C, Perrais M, et al. FibromiRs: translating molecular discoveries into new anti-fibrotic drugs. Trends Pharmacol Sci. 2014;35:119–26. doi: 10.1016/j.tips.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 47.Yu Y, Wang Y, Niu Y, et al. Leukemia inhibitory factor attenuates renal fibrosis through Stat3-miR-29c. Am J Physiol Renal Physiol. 2015;309:F595–603. doi: 10.1152/ajprenal.00634.2014. [DOI] [PubMed] [Google Scholar]
- 48.Bienaimé F, Muorah M, Yammine L, et al. Stat3 Controls Tubulointerstitial Communication during CKD. J Am Soc Nephrol. 2016;27:3690–3705. doi: 10.1681/ASN.2015091014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sis B, Tasanarong A, Khoshjou F, et al. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int. 2007;71:218–26. doi: 10.1038/sj.ki.5002039. [DOI] [PubMed] [Google Scholar]
- 50.Melk A, Schmidt BM, Braun H, et al. Effects of donor age and cell senescence on kidney allograft survival. Am J Transplant. 2009;9:114–23. doi: 10.1111/j.1600-6143.2008.02500.x. [DOI] [PubMed] [Google Scholar]
- 51.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parrinello S, Coppe JP, Krtolica A, et al. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005;118:485–96. doi: 10.1242/jcs.01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–72. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salama R, Sadaie M, Hoare M, et al. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi: 10.1101/gad.235184.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–90. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 57.O’Riordan E, Mendelev N, Patschan S, et al. NOS inhibition actuates endothelial-mesenchymal transformation. Am J Physiol Heart Circ Physiol. 2007;292:H285–94. doi: 10.1152/ajpheart.00560.2006. [DOI] [PubMed] [Google Scholar]
- 58.Zeisberg EM, Tarnavski O, Zeisberg M, et al. Nat Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Med. 2007;13:952–61. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 59.Li J, Qu X, Bertram JF. Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am J Pathol. 2009;175:1380–8. doi: 10.2353/ajpath.2009.090096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kato T, Mizuno S, Ito A. A Decrease in Glomerular Endothelial Cells and Endothelial-mesenchymal Transition during Glomerulosclerosis in the Tensin2-deficient Mice (ICGN strain) Acta Histochem Cytochem. 2014;47:265–71. doi: 10.1267/ahc.14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.LeBleu VS, Taduri G, O’Connell J, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–53. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maddaluno L, Rudini N, Cuttano R, et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature. 2013;498:492–6. doi: 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- 63.Bravi L, Rudini N, Cuttano R, et al. Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proc Natl Acad Sci USA. 2015;112:8421–6. doi: 10.1073/pnas.1501352112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu X, Friehs I, Zhong Hu T, et al. Endocardial fibroelastosis is caused by aberrant endothelial to mesenchymal transition. Circ Res. 2015;116:857–66. doi: 10.1161/CIRCRESAHA.116.305629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Medici D, Kalluri R. Semin Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Cancer Biol. 2012;22:379–84. doi: 10.1016/j.semcancer.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaengel K, Genové G, Armulik A, et al. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–8. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 67.Ubil E, Duan J, Pillai IC, et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. 2014;514:585–90. doi: 10.1038/nature13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin CH, Chen J, Ziman B, et al. Endostatin and kidney fibrosis in aging: a case for antagonistic pleiotropy? Am J Physiol Heart Circ Physiol. 2014;306:H1692–9. doi: 10.1152/ajpheart.00064.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin CH, Chen J, Zhang Z, et al. Endostatin and transglutaminase 2 are involved in fibrosis of the aging kidney. Kidney Int. 2016;89:1281–92. doi: 10.1016/j.kint.2016.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Welch-Reardon KM, Wu N, Hughes CC. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol. 2015;35:303–8. doi: 10.1161/ATVBAHA.114.303220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen PY, Qin L, Barnes C, et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012;2:1684–96. doi: 10.1016/j.celrep.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maarouf OH, Aravamudhan A, Rangarajan D, et al. Paracrine Wnt1 Drives Interstitial Fibrosis without Inflammation by Tubulointerstitial Cross-Talk. J Am Soc Nephrol. 2016;27:781–90. doi: 10.1681/ASN.2014121188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu J, Krautzberger AM, Sui SH, et al. Cell-specific translational profiling in acute kidney injury. J Clin Invest. 2014;124:1242–54. doi: 10.1172/JCI72126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kramann R, Tanaka M, Humphreys BD. Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. J Am Soc Nephrol. 2014;25:1924–31. doi: 10.1681/ASN.2013101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kang HR, Lee CG, Homer RJ, et al. Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–93. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Otani A, Kinder K, Ewalt K, et al. Bone marrow-derived stem cells target retinal astrocytes and can promote or inhibit retinal angiogenesis. Nat Med. 2002;8:1004–10. doi: 10.1038/nm744. [DOI] [PubMed] [Google Scholar]
- 77.Bechtel W, McGoohan S, Zeisberg EM, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–50. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tampe B, Tampe D, Müller CA, et al. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J Am Soc Nephrol. 2014;25:905–12. doi: 10.1681/ASN.2013070723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Margariti A, Winkler B, Karamariti E, et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue-engineered vessels. Proc Natl Acad Sci U S A. 2012;109:13793–8. doi: 10.1073/pnas.1205526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Montesano R, Pepper MS, Orci L. Paracrine induction of angiogenesis in vitro by Swiss 3T3 fibroblasts. J Cell Sci. 1993;105:1013–24. doi: 10.1242/jcs.105.4.1013. [DOI] [PubMed] [Google Scholar]
- 81.Villaschi S, Nicosia RF. Paracrine interactions between fibroblasts and endothelial cells in a serum-free coculture model. Modulation of angiogenesis and collagen gel contraction. Lab Invest. 1994;71:291–9. [PubMed] [Google Scholar]
- 82.Masur SK, Dewal HS, Dinh TT, et al. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc Natl Acad Sci U S A. 1996;93:4219–23. doi: 10.1073/pnas.93.9.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li P, Wang D, Lucas J, et al. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ Res. 2008;102:185–92. doi: 10.1161/CIRCRESAHA.107.157677. [DOI] [PubMed] [Google Scholar]