

Abstract
Anti-mitotic cancer drugs include classic microtubule-targeting drugs, such as taxanes and vinca alkaloids, and the newer spindle-targeting drugs, such as inhibitors of the motor protein, Kinesin-5 (aka KSP, Eg5, KIF11), and Aurora-A, Aurora-B and Polo-like kinases. Microtubule-targeting drugs are among the first line of chemotherapies for a wide spectrum of cancers, but patient responses vary greatly. We still lack understanding of how these drugs achieve a favorable therapeutic index, and why individual patient responses vary. Spindle-targeting drugs have so far shown disappointing results in the clinic, but it is possible that certain patients could benefit if we understand their mechanism of action better. Pre-clinical data from both cell culture and mouse tumor models showed that the cell death response is the most variable point of the drug action. Hence, in this review we focus on current mechanistic understanding of the cell death response to anti-mitotics. We first draw on extensive results from cell culture studies, and then cross-examine them with the more limited data from animal tumor models and the clinic. We end by discussing how cell-type variation in cell death response might be harnessed to improve anti-mitotic chemotherapy by better patient stratification, new drug combinations and identification of novel targets for drug development.
Despite the development of new targeted anticancer drugs and immunotherapeutics, classic chemotherapeutics, including DNA damaging drugs, anti-metabolites and anti-mitotic drugs, remain the primary treatment modality for cancer patients. Anti-mitotic drugs currently used in the clinic are mostly taxanes (paclitaxel and its derivatives) and vinca alkaloids (vinblastine, vincristine and their derivatives) [1–3]. These classic anti-mitotics are called “microtubule poisons”, as they bind to microtubules and block their polymerization dynamics. In the absence of microtubule dynamics, mitotic spindle cannot be properly formed, resulting in prolonged mitotic arrest and subsequent growth arrest and cell death [4]. Taxanes and vincas are widely used, mostly in combination therapies with other classes of chemotherapeutics; and taxanes in particular have shown a significant positive effect on solid tumors. However, these drugs suffer significant limitations: they are ineffective for many types of cancer; sensitive cancers tend to acquire resistance; and they cause serious toxicity, particularly to dividing cells in the bone marrow and gut, and also to neurons [1, 5–8].
In a large effort to improve anti-mitotic chemotherapy, inhibitors of more mitotic spindle-specific proteins, such as motor proteins (e.g., Kinesin-5) and several kinases (e.g., Aurora and Polo-like kinases), were developed [9, 10]. We will refer to these as spindle-targeting drugs. Unlike microtubules, which play diverse roles not only in cell division but also cell dynamics in interphase, the spindle proteins targeted by these drugs only, or mainly, function in mitosis. Hence, targeted inhibition of these proteins is believed to mainly affect, and selectively kill, dividing cells, but not the majority of non-proliferating tissues in our body. At least conceptually, this should render a better efficacy-to-toxicity ratio (“therapeutic index”) compared to classic microtubule inhibitors. For instance, the significant neurotoxicity of taxanes and vincas, presumably due to their inhibition of microtubule dynamics in non-dividing neurons, should be eliminated with spindle-targeting anti-mitotic drugs. However, clinical trials of the spindle-targeting inhibitors have so far shown disappointing results, fairing no better, and often time less effective than the classic microtubule-targeting drugs [11–15].
This failure divided the anti-mitotic field into two camps: some now argue mitosis is not a useful target for cancer chemotherapy, due to slow proliferation of tumor cell compared to normal proliferating compartments; and by extension, that microtubule poisons must promote tumor regression by killing interphase cancer cells [12]. Others argue that mitosis is still a useful target, but the details matter, for example, it may be more effective to promote chromosome mis-segregation without mitotic arrest than to strongly block mitotic progression [16, 17]. We see merit in both arguments, but will not rehash them here. Rather, we will focus on how cells that try to divide in the presence of anti-mitotic drugs make decisions, and in particular on differences between cell types in these decisions and how those might affect therapy.
The therapeutic index of a cytotoxic cancer drug depends on variation between cell types in cell death responses. For the drug to succeed, cancer cells of some particular genotype and phenotype must die at a level of drug exposure (combining dose and duration) that does not kill normal cells. In this review, we will explore this kind of variability specifically for cell passing through an abnormal division event in drug. We will start by summarizing recent results on cellular mechanism by which anti-mitotic drugs induce cell death, and then examining the points of variation in mechanism of action between classic anti-microtubule drugs and the spindle-targeting inhibitors. By further comparing the cell culture results with those from mouse tumor models, we would discuss potential therapeutic and toxic mechanisms of anti-mitotic drugs seen in the clinic.
In our discussion of cell type variability, we will focus on decisions made at saturating drug concentrations, and not on variation in the minimal drug concentration needed to trigger mitotic arrest. Both types of variability are likely to be clinically important, but most reviews of drug sensitivity focus only on variation in IC50 values, i.e., on variation in the concentration-dependence of response, and assume similar behavior once drug concentration is saturated, which is clearly not that case, as we would discuss below. A discussion of factors that alter IC50 values, which includes drug efflux pump activity, lies outside the scope of this review.
Overall response dynamics to anti-mitotic drugs in cultured cancer cells
Single-cell live imaging revolutionized the analysis of anti-mitotic drug responses by avoiding the need for synchronization protocols, and revealed considerable cell-to-cell variation even in clonal populations [18–22]. Microtubule-targeting drugs, like paclitaxel, and spindle-targeting inhibitors of Kinesin-5 and Polo-like kinase 1 (Plk1) induced a similar sequence of cellular alterations, as summarized in Figure 1 [18, 23]. Despite difference in the primary targets of these anti-mitotic drugs, they all disrupt formation of the mitotic spindle, which subsequently leads to activation of the Spindle Assembly Checkpoint (SAC) that arrests the cells in mitosis for prolonged duration (~10–20 hours). Depending on the cell type, cells may die during the prolonged mitotic arrest or slip out of the arrest (i.e., mitotic slippage) into an abnormal G1 state without division. From the 4N G1 state, cells may die, arrest or progress again through cell cycle. Different from the other spindle-targeting inhibitors, Aurora A kinase inhibitors only induced a transient mitotic arrest [24]; and inhibitors of Aurora B kinase inhibit the SAC itself so drug-treated cells do not arrest in mitosis, despite spindle defects [25]. Because Aurora B kinase activity is required for cytokinesis, these cells subsequently become polyploidy and undergo apoptosis for unclear mechanism [26].
Figure 1.
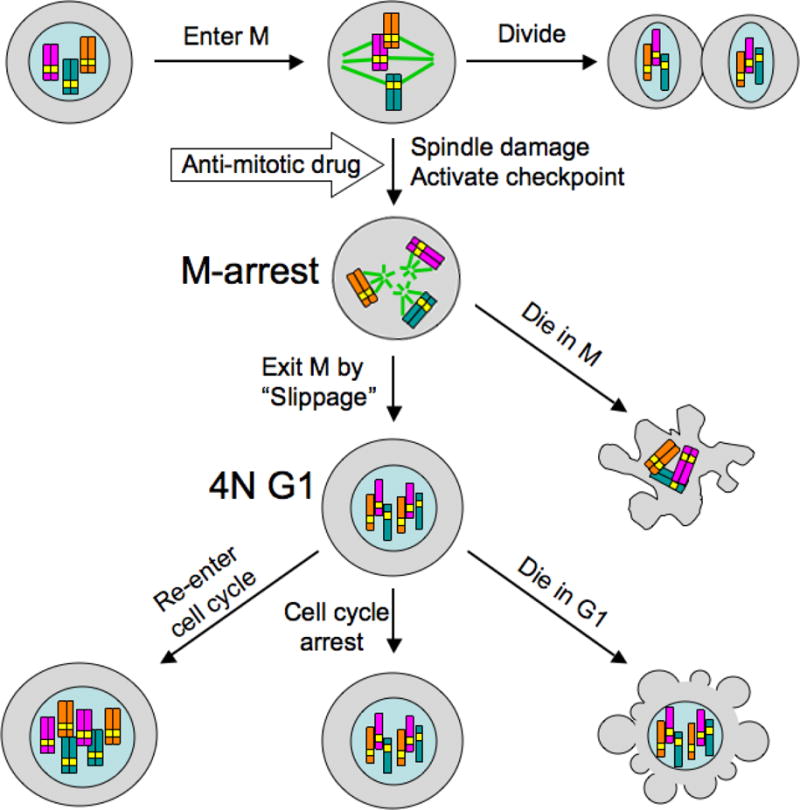
This diagram illustrates anti-mitotic drug responses at the phenotypic level observed by time-lapse microscopy [18–22]. Similar responses are seen with microtubules-targeting drugs, Kinesin-5 inhibitors and Plk1 inhibitors. The timing of events, and the fraction of cells that undergo alternative outcomes, vary between cell lines and drug types. Duration of mitotic arrest varies from 10–20 hours, and fraction of cells that die from <5% to 100%. The 4N G1-arrest cells that slip out of mitotic arrest progress to distinct cellular state, likely depending on p53 activity (see text).
Figure 1 illustrates how cells treated with anti-mitotic drugs choose between different final outcomes, which are determined by alternative cell behaviors at two major decision points. The first decision point occurs during mitotic arrest, between cell death during the arrest and slippage out of the arrest to an abnormal G1 state without division. The second decision point occurs during the abnormal G1 state after mitotic slippage, between again cell death, cell-cycle arrest and progression through cell cycle.
The question of how cells make choices between alternative cell fates has been much studied across biology, most recently using single-cell analysis. Some decisions, such as a gene turning on or off, depend on two states of the same biochemical circuit. For choosing different fates during abnormal cell division, a different model was proposed, in which alternative decisions are due to competing actions of alternative pathways from different, non-interacting molecular circuits. The fastest process to execute wins, and the biology is such that when one pathway executes, the competing alternative is immediately blocked. For example, cells that die cannot exit mitosis, and cells that exit mitosis may turn off some timer that will eventually trigger apoptosis in mitosis. In this type of model, alternative decisions arises from variable rates of the competing processes. The choice between cell death and mitotic exit during prolonged mitotic arrest depends on two simultaneously active pathways that function independently, one promoting cell death via intrinsic apoptosis and the other promoting gradual proteolysis of cyclin B1 that leads to mitotic slippage to G1, overcoming a still active Spindle Assembly Checkpoint (SAC) [27, 28]. Cell types with a faster rate of cell death induction than cyclin B1 degradation tend to reach the threshold of Mitochondrial Outer Membrane Permeabilization (MOMP) before they slip out of mitosis, and thus exhibiting mostly death in mitotic arrest in response to saturating dosage of anti-mitotic drugs. Similarly, cells with a slow rate of cell death activation, or fast cyclin B1 degradation, mostly slip out of mitotic arrest to the 4N G1 state. Large cell-to-cell variation in these rates causes variable decision making, so individual cells may make different choices even in clonal populations. The second major point of cell fate decision occurs in the abnormal 4N G1 state after mitotic slippage, and is most likely determined by the status of DNA damage response, which we would discuss below in more details.
Cell death response to anti-mitotic drugs in cultured cancer cells
While anti-mitotic drugs at sufficiently high concentration can induce mitotic arrest in all proliferating cells, kinetics and the extent of cell death induction during or after the arrest are highly variable across different cancer cell types, as observed by studies of both syngeneic mouse tumors [29] and cultured human cell lines [18]. Therefore, the cell death pathways shown in Figure 1 are arguably the most important with respect to understanding tumor response to anti-mitotic drugs, and drug toxicity. Many studies have shown that paclitaxel and other microtubule-targeting drugs promote apoptosis through the mitochondrial, or intrinsic pathway [18, 30–32]. The hallmark of this pathway is Mitochondrial Outer Membrane Permeabilization (MOMP), which leads to release of Cytochrome C and other pro-apoptotic proteins into the cytoplasm, subsequently activating caspases that execute cell death (Fig. 2). The spindle-targeting Kinesin-5 inhibitor and Plk1 inhibitor also activates this pathway similarly [18, 33, 34]. Cell death induced by anti-mitotics can occur both during mitotic arrest, and after mitotic slippage, depending on the cell type and drug. Although some studies concluded that anti-mitotics promote cell death by non-apoptotic pathways [35, 36], in our hands, cell death occurring at both cell cycle states are mediated by mitochondrial apoptosis, though with very different upstream regulation and triggers of MOMP, as detailed below [18, 21, 22].
Figure 2.
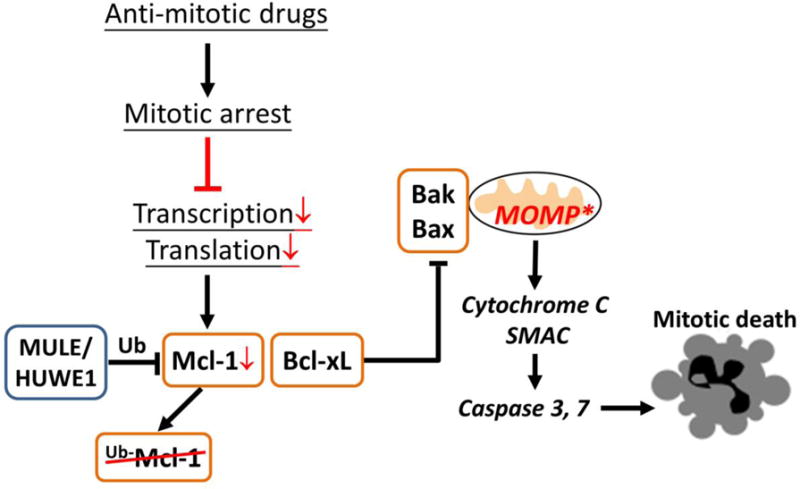
Cell death induced by anti-mitotic drugs during mitotic arrest is mediated by mitochondrial, intrinsic apoptosis triggered by depletion of an anti-apoptotic protein, Mcl-1. The other major negative regulator of mitotic death is Bcl-xL, whose expression level largely determines the sensitivity of different cancer cell lines to anti-mitotics-induced cell death in mitotic arrest.
Apoptotic response in mitotic arrest
Time-lapse microscopy data revealed long and variable delays from cells entering anti-mitotics-induced mitotic arrest to induction of MOMP and subsequent cell death in the arrest, typically 10–20 hours. A key pro-apoptotic signal that slowly accumulates during mitotic arrest and eventually triggers MOMP was identified to be the depletion of an anti-apoptotic protein, Mcl-1 [21, 37–39] (Fig. 2). Transcription is silenced during mitosis due to chromosome condensation, and translation is attenuated due to phosphorylation of elongation factors (eEF1 & eEF2) [40–42]. Therefore, proteins with short half-life are significantly depleted during anti-mitotics-induced prolonged mitotic arrest. Uniquely among the Bcl-2 family proteins, Mcl-1 turns over rapidly through ubiquitination by E3 ligases, such as MULE/HUWE1 [43], with a typical half-life of < 1 hour [44, 45]. Rescuing Mcl-1 by knockdown of MULE/HUWE1 significantly attenuates cell death in mitotic arrest, confirming loss of Mcl-1 is a key pro-apoptotic signal during mitotic arrest [21].
However, loss of Mcl-1 alone cannot explain the variable cell death response during mitotic arrest between different cancer cell lines. Both cell lines that are sensitive and resistant to mitotic death showed similar kinetics and extent of Mcl-1 depletion [21]. Further experiments showed that another anti-apoptotic protein, Bcl-xL, acts with Mcl-1, as the major negative regulators of apoptosis during mitotic arrest [21, 46]. For cell lines that express low basal level of Bcl-xL, such as HeLa, loss of Mcl-1 alone due to mitotic arrest is sufficient to release adequate amount of Bax/Bak to form pores on the mitochondria membrane and trigger MOMP. In cancer cell lines with high levels of Bcl-xL, e.g., U-2 OS, OVCAR-5 and A549, loss of Mcl-1 alone is insufficient to release Bax/Bak to a level that can cross the MOMP threshold, due to significant Bax/Bak sequestration by Bcl-xL. These results suggest inhibiting Bcl-xL is potentially an effective combinatorial strategy with anti-mitotic drugs to enhance and accelerate cell death even for cancer cells that are resistant to anti-mitotics alone. This indeed was confirmed in both cell culture and mouse tumor model that combined anti-mitotics, such as paclitaxel and Kinesin-5 inhibitor, with a Bcl-xL inhibitor, Navitoclax [21, 47, 48].
Although the mechanism of apoptosis regulation during anti-mitotics-induced mitotic arrest was partly elucidated by studies as discussed above, it was not solved. Combined inhibition of Mcl-1 and Bcl-xL induced significant cell death only in mitosis, but not in interphase [21], indicating there is additional, mitosis-specific pro-apoptotic signal that contributes to MOMP induction in prolonged mitotic arrest. Given that mitotic arrest is a hyper-phosphorylated state resulting from, e.g., prolonged activation of Cyclin-dependent kinase-1 (Cdk1) [49, 50], we suspect the additional pro-apoptotic signal could be triggered by activation of kinase signaling pathways. Phospho-proteome analysis of mitotic arrest cells may help to shed light on additional proteins/pathways involved in mitotic death control.
As the pro-apoptotic signals that trigger death during mitotic arrest are mitosis specific, the process of slippage, which moves cells out of mitotic arrest, protects cells against initiation of death in mitosis. This led to the proposal that inhibiting mitotic slippage is a better strategy to induce cell death than anti-mitotic drugs, as it renders a nearly permanent mitotic arrest that allows the mitosis-specific pro-apoptotic signals to accumulate indefinitely. Compared to anti-mitotic drugs that typically activate mitotic arrest of 10–20 hours before cells exit the arrest, inhibiting the mitotic slippage pathway, e.g., by Cdc20 knockdown, induced significantly longer arrest, about 30–40 hours [51]. This long arrest efficiently activates death in even the slippage-prone cancer cell lines that exhibit little mitotic death in response to anti-mitotic drugs, e.g., MCF7 and A549 [51]. Similar effect of Cdc20 knockdown in attenuating cancer growth was also observed in mouse tumor model [52]. The type of cell death activated in this ultra-long arrest is again apoptosis mediated by the mitochondrial, intrinsic apoptotic pathway. Considering the measured half-life of Bcl-xL is about 10 hours [53], mitotic arrest of 30–40 hours should have enabled Bcl-xL to deplete to a significantly low level, thus triggering apoptosis in cells with even high level of Bcl-xL. Therefore, we think similar cell death mechanism is likely to operate in mitotic arrest, irrespective whether it arises from abnormal spindle assembly due to anti-mitotic drug treatment or inhibition of mitotic slippage.
Apoptotic response after mitotic slippage
Anti-mitotic drugs, in particularly microtubule inhibitors, also trigger interphase cell death after slippage out of mitotic arrest into an abnormal, 4N G1-like state. For some cancer cell lines, e.g., U-2 OS, apoptosis after mitotic slippage is the major cell death response to anti-mitotic drug treatment and highly effective (~90% death after slippage) [18]. The key pro-apoptotic signal that activates cell death after slippage was identified to be DNA damage [22, 54–59] (Fig. 3). Several studies have shown that caspase activities, which were partially activated during prolonged mitotic arrest, engender DNA fragmentation in the post-slippage cells and thus induce p53-mediated DNA damage response [56–59]. Our own results confirmed the involvement of DNA damage in post-slippage cell death and further illustrated that the degree of DNA damage induced by microtubule-targeting drugs, e.g., paclitaxel, is substantially higher than the spindle-targeting Kinesin-5 inhibitor [22]. This variation in DNA damage level well explains the much higher cytotoxicity of paclitaxel than Kinesin-5 inhibitor, in particular to the post-slippage population of cells. The critical role of DNA damage in triggering post-slippage apoptosis was also confirmed by experiments that showed attenuation of DNA damage or knockdown of p53 significantly reduced cell death after mitotic slippage in response to paclitaxel [22].
Figure 3.
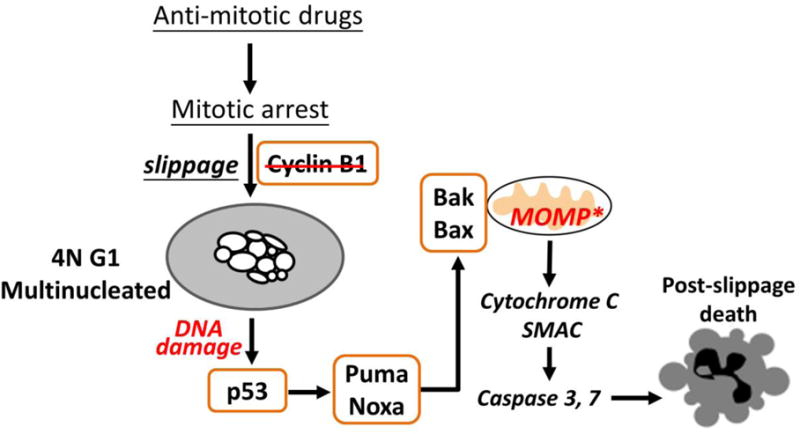
Post-slippage cell death induced by anti-mitotic drugs is mediated by mitochondrial, intrinsic apoptosis triggered by DNA damage in the 4N G1-arrest cells. The degree of multinucleation in the post-slippage 4N cells determines the level of DNA damage and subsequent p53-mediated cell death activation. Taxanes engender significantly more mutlinucleation than the spindle-targeting Kinesin-5 inhibitor. Therefore, while different anti-mitotic drugs induce similar degree of cell death during mitotic arrest, the extent of post-slippage death that they trigger vary significantly.
Although the involvement of DNA damage in post-slippage apoptosis is clear, ambiguity remains regarding the triggers of this DNA damage. Results from our lab showed combined treatment of paclitaxel and a pan-caspase inhibitor, zVAD-FMK, only reduced the level of DNA damage in post-slippage U-2 OS cells by 40%. And MOMP was still triggered in nearly 70% of these post-slippage cells in the absence of functioning caspases. The data suggest induction of DNA damage after mitotic slippage cannot be attributed to caspase activity alone. One distinct feature of the post-slippage cells rendered by paclitaxel treatment is they consist of multiple small nuclei (Fig. 4). Work by Crasta et al showed defective and asynchronous replication of small micronuclei results in DNA fragmentation [60]. To test whether this mechanism contributes to DNA damage seen in the paclitaxel-induced multinucleated cells after mitotic slippage, we inhibited DNA replication in the post-slippage cells using thymidine or mimosine. However, we did not observe significant reduction in DNA damage with either treatment. In addition, bromodeoxyuridine (BrdU) staining indicated no chromosome replication in the post-slippage cells, indicating cells arresting in G1. Hence, defective replication of the multiple small nuclei is unlikely a major contributor to DNA damage after mitotic slippage. Further investigation is clearly needed to establish the potential links between multinucleation and DNA damage, which appears to determine apoptotic response to anti-mitotic drugs after mitotic slippage, at least in cell culture.
Figure 4.
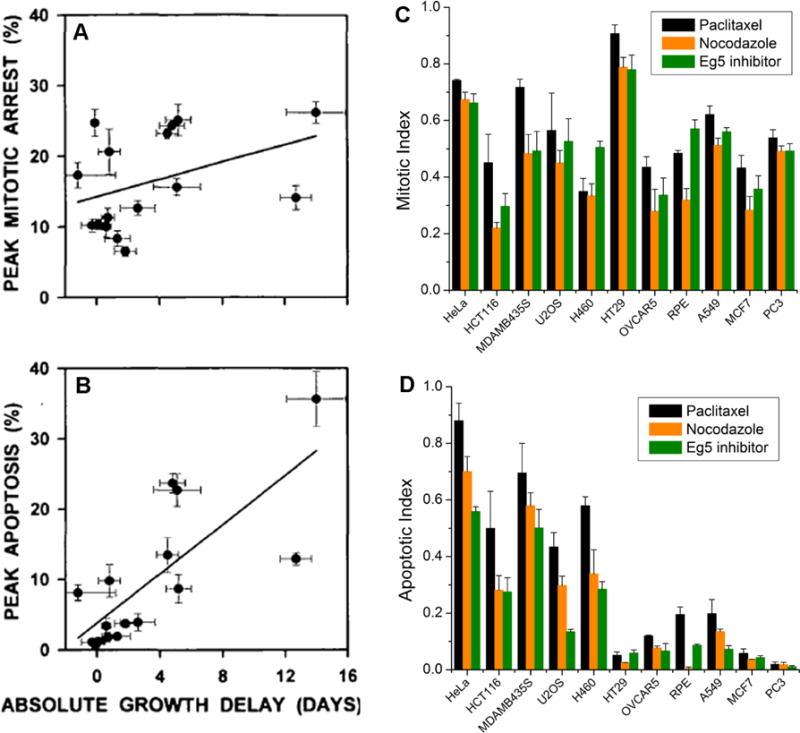
Variation of apoptotic response to anti-mitotic drugs is much more significant than that of mitotic response between both different mouse tumors and culture human cancer cell lines. (A, B) Fifteen different mouse cancers (propagated by serial passage in mice) were treated with a single dose of paclitaxel. Tumor response was measured as growth delay (x-axis). Mitotic arrest and apoptosis were scored by quantitative histology at different times after drug injection, and peak responses were scored as % of cells in the tumor. (A) plots the average peak mitotic arrest response, and (B) the average peak apoptotic response, against average growth delay for different tumors. Note that most tumors showed a strong mitotic arrest response, but only tumors that respond well to drug show a strong apoptotic response. These figures are taken from Fig. 1 by Milross et al (1996) [29]. (C, D) Comparison of mitotic response (C) and apoptotic response (D) to three different anti-mitotic drugs, including paclitaxel (150nM, shown in black), nocodazole (500nM, shown in orange), and Kinesin-5 (Eg5) inhibitor (500nM, shown in green), for a panel of human cancer cell lines. The mitotic response corresponds to the percentage of mitotic cells after 24 hours of drug treatment, probed by phospho-Histone 3 antibody, and the apoptotic response corresponds to the percentage of apoptotic cells after 48 hours of drug treatment, probed by Parp1 cleavage. Cell lines are arranged in descending order of sensitivity to anti-mitotics-induced cell death. These figures are taken from Fig. 1 by Shi et al (2008) [18].
p53 monitors the duration of mitosis independent of DNA damage
p53 is the most commonly mutated gene in cancer, and is thought to act as a “guardian of the genome” by triggering apoptosis or senescence in cells where the genome is compromised. Recently, an exciting new circuit was discovered, in which p53 monitors the duration of mitosis independent of DNA damage and triggers senescence in cells that take too long to progress through mitosis. In normal RPE cells, any lengthening of mitosis beyond ~90 minutes was shown to cause G1 arrest in the following cell cycle [61]. In 2016 three independent groups used CRISPR-based loss-of-function genetic screens to uncover the underlying circuitry [62–64]. These screens revealed four key genes required to halt cells in G1 after an abnormally long mitosis, including TP53 (p53), CDKN1A (p21), TP53BP1 (53BP1) and USP28. p21 is a known transcriptional target of p53 that halts cell cycle progression in G1 by inhibiting CDKs. 53BP1 and USP28 somehow measure mitotic duration and activate p53 upon mitotic exit. The mechanism of the timing measurement is not yet known, but it likely involves transient localization of 53BP1 to kinetochores, which is linked to stabilization of p53 by USP28 removing ubiquitin. This pathway is lost in p53-null cancer cells, where it likely contributes to development of aneuploidy. Whether this pathway helps to activate mitosis in p53-positive cancer cells, and more generally its role in response to anti-mitotic drugs, are currently unknown.
Cell death responses in tumors: mechanistic data from animal model and the clinic
Microtubule-targeting anti-mitotic drugs have been extensively studied in mouse tumor models, although most early investigations only acquired mechanistically uninformative data of tumor mass vs. dose/time. The Milas group performed a more informative series of studies, where they treated mice carrying a diverse panel of syngeneic mouse tumors with a single doses of taxane (either paclitaxel or docetaxel), sacrificed animals at multiple time points for quantitative histology, and related the histological data to overall tumor responses across a panel of tumors [29, 65, 66]. The kinetics of drug response that they observed were consistent with the sequence of events summarized in Figure 1, with peaks of mitotic arrest typically occurring 3–12 hours after drug injection in most tumors, and peaks of cell death at 6–36 hours in drug-sensitive tumors [29]. Comparing between tumor lines, peak mitotic arrest levels following a single dose of paclitaxel varied to a limited extent between tumors, while peak apoptosis levels varied greatly. Only the apoptosis response correlated with the “clinical response”, measured as delay in growth of tumor volume (Fig. 4A & B). These data showed that efficient triggering of apoptosis is required for effective treatment of the tumor, and that different tumors are highly variable in this response. Remarkably, the tendency to die from mitotic arrest as well as the bulk growth delay correlated well with the rate of spontaneous apoptosis in untreated animals across the tumor panel, suggesting that the more treatable tumors were generally more apoptosis prone. Both conclusions are consistent with our cell line study [18] (Fig. 4C & D), pointing to variation in apoptosis sensitivity, rather than mitotic arrest, as the determining factor in anti-mitotic drug response. In a broader sense, the conclusions from the Milas mouse tumor studies match the concept advanced by Letai and colleagues that the response of tumors to chemotherapy depends on the extent to which their mitochondria are “primed” to undergo MOMP and initiate apoptosis [67]. Higher priming is expected to promote spontaneous apoptosis as well as drug-induced apoptosis. In support of this model, Letai and colleagues showed that the degree of priming predicts survival across a broad range of human cancers, including multiple myeloma, acute myelogenous and lymphoblastic leukemia, and ovarian cancer [68].
Milas et al went on to compare the effects of two taxanes, paclitaxel and docetaxel, using a dose of docetaxel that promoted stronger average tumor responses than the respective dose of paclitaxel [66]. At these doses, docetaxel caused mostly death in mitosis, while paclitaxel caused mostly death after slippage (Fig. 5). This analysis is one of the few detailed head-to-head comparisons of these widely used taxanes in a tumor model, and their effects, as scored by histopathology, were surprisingly different. Interestingly, death in mitosis released more cellular debris, including condensed chromosomes, and was histologically classified as necrosis rather than apoptosis. This was different from results from cell culture studies that suggest both death in mitosis and after slippage are caused by mitochondrial apoptosis. Considering Mcl-1 and Bcl-xL were also found to be the major predictors of tumor response to docetaxel in mouse xenograft [48], similar to our data from cell culture, we think tumor death in mitosis in vivo is also mediated by apoptosis. We suspect the distinct histological classification of the two types of cell death by Milas et al does not correspond to different molecular mechanisms of death. Rather, we interpret these data as suggesting that the downstream aspects of the mitochondrial apoptotic program, for example, the tendency of apoptotic cells to elicit their own phagocytosis, differs if the death program is executed in mitosis compared to interphase (i.e., after mitotic slippage). Milas et al observed massive recruitment of mononuclear leukocytes in response to docetaxel, but not paclitaxel, in some of their treated tumors, which they interpreted as a response to release of cell debris following tumor cell lysis in mitosis. They went on to argue that this leukocyte recruitment amplified the therapeutic effect of the drug, because the leukocytes killed non-dividing tumor cells in the treated tumor [69]. Moreover, the extent of leukocyte recruitment also correlates with the extent of tumor shrinkage following taxane treatment in human tumors [70], though it is not clear how much this is a cause vs. a consequence of cell death. The idea that when a dividing tumor cell dies, it recruits leukocytes to help kill its non-dividing neighbors is an example of bystander effect. Bystander effects have long been recognized in ionizing radiation responses [71]. If they occur with anti-mitotic drugs, they might amplify drug responses in tumors with a low mitotic index, e.g., due to slow proliferation. Paclitaxel-treated cells have been shown to damage untreated neighbors in culture by emitting reactive oxygen species (ROS) [72], a second bystander mechanism that might be important in tumors. Intra-vital imaging of tumor response in mouse xenograft had shown paclitaxel treatment did not induce mitotic arrest in most tumor cells, nonetheless the tumor still significantly regressed [73]. This suggests killing by bystander effect could be an important mechanism by which taxanes exert cytotoxicity in vivo. Further investigation of bystander effects induced by anti-mitotic drugs is clearly warranted.
Figure 5.
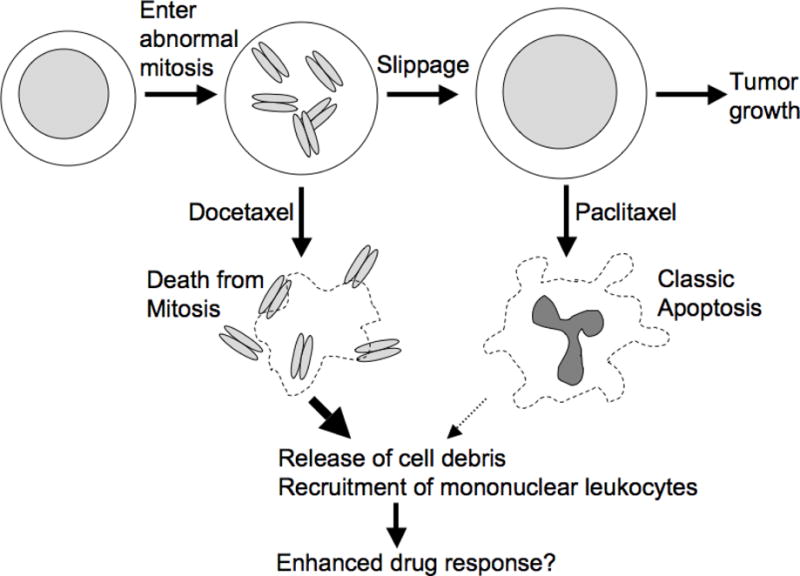
This cartoon summarizes the work of the Milas group on the effect of two taxanes on mouse model cancers [29, 65, 66]. Docetaxel was overall more effective than paclitaxel at the does used, though sensitivity of different tumor lines to the two drugs strongly correlated. In cancers that responded well, docetaxel promoted mainly death in mitosis, and paclitaxel mainly apoptosis after slippage. Death in mitosis following docetaxel treatment was correlated with extensive release of cell debris, including mitotic chromosomes, and massive recruitment of mononuclear leukocytes to the shrinking tumor. Subsequent work suggested that leukocyte recruitment enhances the therapeutic response [69]. Therefore, timing of cell death may be important in tumors for anti-mitotic response.
What then do we know about the sequence of events that following tumor cell mitosis in the presence of an anti-mitotic drug in the clinic? Sampling human tumors or tissues at multiple time points after drug treatment is difficult. Symmans et al were able to take fine needle biopsies at 24-hour intervals from a panel of breast cancer patients treated with paclitaxel prior to surgery [74]. Their data were consistent with the response kinetics shown in Figure 1, and also with conclusion from the Milas et al that clinical response depends on apoptosis, not just mitotic arrest. They found large variation between patients in mitotic index as well as in apoptotic index. As with the Milas et al mouse tumor data, apoptotic index correlated with overall response much better than mitotic index, again reflecting the importance of apoptosis-sensitivity in overall tumor response. Peak mitotic index values in drug reported by Symmans et al were quite low (<6%), presumably reflecting slow proliferation in human solid tumors. An unanswered question is what fractions of the cells that went on to die in response to drug did so after experiencing drug-induced mitotic arrest or chromosome mis-segregation, versus dying independent of mitosis via poorly characterized pathways that convert microtubule stabilization to cell death in interphase.
A different approach to obtaining histological data from human tumors responding to drug is to explant human tumor samples directly into nude mice (i.e, Patient-derived Xenograft, PDX), and treat them with anti-mitotic drug in that context. This may preserve proliferation rates and drug responses of primary tumors more reliably than cell culture. Perez-Soler et al used this approach to investigate paclitaxel responses in non-small cell lung cancers [75]. They found no correlation between drug response and proliferation rate, or with the increase in mitotic arrest after drug treatment, again suggesting that the critical factor is efficient triggering of cell death. A striking aspect of this study is the slow proliferation rate of the directly explanted human tumors. Efficient drug-induced shrinkage in tumors with low proliferation rate, and therefore low mitotic index following drug treatment, provide credence to the concept of bystander killing discussed above and/or other unknown mechanism of killing independent of mitotic arrest, which were reviewed previously [76].
Clinical responses to microtubule-targeting vs. spindle-targeting anti-mitotic drugs
Clinical trials of the spindle-targeting drugs provide an important test of whether perturbation of mitosis alone can be an effective therapeutic strategy to treat cancer. Results from phase I, and limited phase II, trials so far showed rather disappointing results. At the neutropenia-determined exposure limit, the spindle-targeting drugs showed little anticancer effect in shrinking tumor mass, though the drugs may stabilize tumor growth in some patients [10–15]. It is of course problematic to evaluate the anti-cancer efficacy of these new drugs on the basis of tumor response data from heavily pre-treated patients. Nonetheless, we probably can conclude from a lack of tumor regression reports that none of the spindle-targeting drugs tested so far could be much more effective than taxanes for treating solid tumors. Toxicity data are more reliable, and an interesting surprise has emerged in that end. At doses that are effective for treating cancer, taxanes and vincas damage all proliferating tissues, including bone marrow, gut and hair follicles, as well as non-dividing neurons. The dose-limiting toxicity is spread between these effects, and varies for particular compounds, with most taxanes and vincas causing some gut toxicity and hair loss. The spindle-targeting drugs tested so far are, in contrast, dose limited by only one toxicity, neutropenia. At this dose limit, patients suffer no hair loss, and little, if any, gut toxicity [10]. Remarkably, the spindle-targeting drugs are similar to each other in this respect and thus different from all the microtubule-targeting drugs, despite their different targets and pharmacokinetics. Lack of neurotoxicty of the spindle-targeting drugs was expected, but their apparent selectivity for bone marrow damage, with little evidence of gut damage or hair loss at the dose limit, is a surprise that was not predicted by studies in cell culture or mice.
Neutrophils are among the shortest-lived cells in our body, and their level depends on continuous proliferation of promyelocytes in the bone marrow. Neutropenia is diagnostic of cytotoxic effects that are specific for dividing cells, and this dose-limiting toxicity strongly suggests that all the candidate spindle-targeting drugs are doing exactly what they were designed to do, that is, to kill dividing cells without neurotoxicity. The lack of gut toxicity and hair loss at the neutropenia dose limit implies an altered selectivity profile among dividing cells for the spindle-targeting agents compared to taxanes and vincas, which is currently difficult to explain. This altered selectivity profile seems to point in the wrong direction for treatment of epithelial cancers, but might be an advantage for hematological cancers. Why might mitosis-specific inhibitors show a different selectivity profile compared to microtubule-targeting drugs? One possibility is that effects on non-dividing cells in tumors are important; these occur only with the microtubule-targeting drugs. For example, vincas and taxanes have potent effects on the cytoskeleton of interphase endothelial cells that may contribute to their effects on solid tumors [77, 78]. Alternatively, the anti-mitotic drug classes may differ in the responses that they elicit from dividing cells. Consistent with the latter possibility, we observed that while a Kinesin-5 inhibitor and paclitaxel are equal in their ability to kill cells in mitotic arrest, taxanes are significantly more able to trigger apoptosis after mitotic slippage to the G1 state, due to induction of DNA damage [18, 22]. Why paclitaxel triggers more DNA damage after slippage than a kinesin-5 inhibitor is not known. An interesting hypothesis based on recent data is that micronucleation is important. When cells slip out of mitosis in paclitaxel, their chromosomes are spread far apart, and as a result, many micronuclei from. When they slip in a Kinesin-5 inhibitor, the chromosomes tend to be more clumped, and often a single, large nucleus forms [22]. DNA in micronuclei tends to undergo extensive fragmentation for reasons that are not fully understood [60]. The tendency to drive cells into a highly multinucleated state after slippage might account for part of the extra efficacy of taxanes compared to spindle-targeting drugs.
Implications for improving anti-mitotic therapy
It is not clear whether major improvements in anti-mitotic therapy are possible; if they are, progress is likely to come from either new methods for identifying sub-populations of patients that respond well to current drugs, developing improved combination therapies, or developing new anti-mitotic drugs with novel targets that give rise to more cancer-selective actions. In order to answer the basic question of whether improvement is possible, and to rationally pursue these avenues towards improvement, we definitely need to further improve our understanding of how anti-mitotic drugs work in the complex environment of tumor in vivo.
For current therapies in the clinic involving taxanes and vincas, a pressing need is to identify patients most likely to respond well or poorly by molecular diagnosis, so as to avoid unnecessary treatment with the highly toxic drugs. If we could predict highly responsive patients, and avoid treating non-responders, these drugs would become “precision medicines”, with a much higher therapeutic index averaged across the treated population. The identification of Mcl-1 and Bcl-xL as the major negative regulators of apoptotic response to anti-mitotic drugs, at least in cell culture and mouse tumor models, suggests that basal expression of Mcl-1 and Bcl-xL, in particular Bcl-xL, may be employed as diagnostic predictors of tumor response. However, we already know additional mechanisms are likely involved in mediating the anticancer effect of anti-mitotics in tumor, e.g., bystander effects and interphase toxicity, so we still need more mechanistic data to curate a comprehensive set of molecular biomarkers. We advocate grounding the search of predictive biomarkers on phenotypic and mechanistic data, rather than the more popular approaches using only genomic data (profiles of mutation profiles, mRNA and miRNA). This popular approach allows for easy measurement, but suffers from the large amount of uninformative data that is inevitable with broad profiling measurements, which may swamp the useful data in noise. We believe it would be very helpful if we first have more mechanistic data on what makes tumors respond well or poorly, and then use that understanding to filter the genomic data, or even replace it with a smaller number of mechanism-relevant measurements at the protein level, akin to measuring receptor status for hormonal drugs. We argue that finding such predictive measurements will require not only elucidating the pathways that connect spindle damage and alteration of tumor microenvironment to induction of mitochondrial apoptosis and tumor regression, but also understanding how these pathways vary and are differentially activated in distinct cancer types so as to exploit these differential points as diagnostic markers and also potential new targets for drug combination.
One encouraging example of a phenotypic measurement that predicts tumor responses is Letai’s mitochondrial priming assay [68]. Measurement of priming in untreated cells predicts general chemo-sensitivity, although not which specific drugs a given patient will best respond to. Recently, this approach was extended to measurement of changes in mitochondrial priming in response to ex-vivo drug treatment [79]. This can predict specific drug responses, but with the usual caveat that the drug exposure occurs under standard tissue culture conditions, which poorly mimic the complex tumor environment.
Since taxanes are only partially effective on a variety of solid tumors, it requires additional strategies, such as new drug combinations, to increase its efficacy and combat acquired resistance. One potentially viable combination is taxanes plus a Bcl-xL inhibitor, e.g., Navitoclax (previously known as ABT-263), as both cell culture and mouse tumor data illustrated that this combination can significantly enhance and accelerate cell death, even for cancer types that are resistant to taxanes alone. A phase I trial of Navitoclax plus paclitaxel showed both hematological and non-hematological toxicity, including neutropenia, thrombocytopenia, small intestinal obstruction and pulmonary embolism [80]. It remains to be seen whether this combination indeed can improve patient response. A major challenge to clinical development of Navitoclax is that Bcl-xL inhibition tends to kill platelets, whose lifespan is naturally limited by apoptosis [81]. This is a classic case of on-target toxicity in an off-target cell type. Given the huge potential of Bcl-xL inhibition to sensitize tumors to chemotherapy, we wonder if it might be possible to generate drugs that specifically protect platelets as part of a two-drug strategy. Combining taxanes with other reagents that directly enhance apoptosis sensitivity of tumor cells, such as TRAIL, may also synergize with anti-mitotic drugs. In addition, a new modality of anticancer therapy is immuno-oncology drugs, such as checkpoint inhibitors (e.g., PD-1/PD-L1 and CTLA-4 neutralizing antibodies), and cancer vaccines [82–85]. Combination of taxane with either checkpoint inhibitor or cancer vaccine has shown notable improvement in progression-free survival in certain solid tumors than taxanes treatment alone [86–88]. Given tumor response to anti-mitotic drugs clearly involves leukocytes and changes in the tumor microenvironments, and cell death induced by anti-mitotics is strongly inflammatory, stimulating anti-tumor immunity is certainly a promising combinatorial strategy to improve anti-mitotic therapy. However, it is still early stage of this line of research; we need more mechanistic and clinical data to determine whether an improved efficacy-to-toxicity ratio can indeed be achieved by such combination; and if so, what patient population would benefit and what would be the optimal dose, schedule and sequence [89].
Compared to better patient stratification and new drug combinations, developing new anti-mitotic drugs of novel targets seem to be less promising in terms of therapeutic development. The similar clinical profiles, so far, of Kinesin-5, Aurora and Plk-1 inhibitors suggest that targeting another highly conserved mitotic spindle protein is unlikely to yield a more selective drug. However, this does not mean that we should ignore the mitotic spindle and cleavage apparatus in the search for better targets. It is possible that certain oncogenic pathways generate specific, druggable vulnerabilities in mitosis, although none has yet been found. Recent work suggested MELK kinase activity is required for successful mitosis in certain breast cancer lines, but not others [90, 91]. Whether MELK is a useful target remains to be seen, but this work is encouraging in the context that it is at least theoretically possible to perturb mitosis selectively in some cancer genotypes. A protein required for spindle assembly in epithelial lineages, but not hematopoietic lineages, might make an excellent target for a novel anti-mitotic that lacks bone marrow toxicity. Although no such protein is yet known, the confluence of RNAi data and informatics might reveal one. The systems that position the spindle may be more complex in epithelial cells compared to hematopoietic cells, arguing such proteins may exist. We also believe that further research into the pathways that connect spindle damage to induction of apoptosis will reveal targets, whose inhibition could more efficiently trigger apoptosis downstream of spindle damage. Given the complexity and variation in apoptosis regulating pathway as well as its secondary effects on immune response, it is reasonable to hope that aspects of this pathway may differ between normal cell types, and change in certain cancers. So it may be possible to develop small molecules that exploit this variation to selectively block apoptosis downstream of spindle damage in bone marrow cells to reduce toxicity, or selectively trigger it in particular cancers to increase efficacy.
Acknowledgments
We thank members of our research groups, whose work and efforts have helped shed light on our thoughts on anti-mitotic drug response. Our work on anti-mitotic drugs was supported by the Hong Kong Research Grant Council (#261310) to J Shi and the National Cancer Institute (CA139980 & CA078048-09) to TJ Mitchison.
Footnotes
The authors declare no conflict of interest.
References
- 1.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 2.Perez EA. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol Cancer Ther. 2009;8(8):2086–95. doi: 10.1158/1535-7163.MCT-09-0366. [DOI] [PubMed] [Google Scholar]
- 3.Mukhtar E, Adhami VM, Mukhtar H. Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther. 2014;13(2):275–84. doi: 10.1158/1535-7163.MCT-13-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Med Res Rev. 2011;31(3):443–81. doi: 10.1002/med.20242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crown J, O’Leary M, Ooi WS. Docetaxel and paclitaxel in the treatment of breast cancer: a review of clinical experience. Oncologist. 2004;9(Suppl 2):24–32. doi: 10.1634/theoncologist.9-suppl_2-24. [DOI] [PubMed] [Google Scholar]
- 6.Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85(9):1219–26. doi: 10.1016/j.bcp.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 7.Rowinsky EK. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu Rev Med. 1997;48:353–74. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- 8.Canta A, Chiorazzi A, Cavaletti G. Tubulin: a target for antineoplastic drugs into the cancer cells but also in the peripheral nervous system. Curr Med Chem. 2009;16(11):1315–24. doi: 10.2174/092986709787846488. [DOI] [PubMed] [Google Scholar]
- 9.Marzo I, Naval J. Antimitotic drugs in cancer chemotherapy: promises and pitfalls. Biochem Pharmacol. 2013;86(6):703–10. doi: 10.1016/j.bcp.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007;7(2):107–17. doi: 10.1038/nrc2049. [DOI] [PubMed] [Google Scholar]
- 11.Purcell JW, Davis J, Reddy M, Martin S, Samayoa K, Vo H, Thomsen K, Bean P, Kuo WL, Ziyad S, Billig J, Feiler HS, Gray JW, Wood KW, Cases S. Activity of the kinesin spindle protein inhibitor ispinesib (SB-715992) in models of breast cancer. Clin Cancer Res. 2010;16(2):566–76. doi: 10.1158/1078-0432.CCR-09-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol. 2011;8(4):244–50. doi: 10.1038/nrclinonc.2010.228. [DOI] [PubMed] [Google Scholar]
- 13.Katayama H, Sen S. Aurora kinase inhibitors as anticancer molecules. Biochim Biophys Acta. 2010;1799(10–12):829–39. doi: 10.1016/j.bbagrm.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bavetsias V, Linardopoulos S. Aurora Kinase Inhibitors: Current Status and Outlook. Front Oncol. 2015;5:278. doi: 10.3389/fonc.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McInnes C, Wyatt MD. PLK1 as an oncology target: current status and future potential. Drug Discov Today. 2011;16(13–14):619–25. doi: 10.1016/j.drudis.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 16.Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6(229):229ra43. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–81. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68:3269–76. doi: 10.1158/0008-5472.CAN-07-6699. [DOI] [PubMed] [Google Scholar]
- 19.Orth JD, Tang Y, Shi J, Loy CT, Amendt C, Wilm C, Zenke FT, Mitchison TJ. Quantitative live imaging of cancer and normal cells treated with Kinesin-5 inhibitors indicates significant differences in phenotypic responses and cell fate. Mol Cancer Ther. 2008;7:3480–9. doi: 10.1158/1535-7163.MCT-08-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–22. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Shi J, Zhou Y, Huang HC, Mitchison TJ. Navitoclax (ABT-263) Accelerates Apoptosis during Drug-Induced Mitotic Arrest by Antagonizing Bcl-xL. Cancer Res. 2011;71:4518–26. doi: 10.1158/0008-5472.CAN-10-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y, Zhou Y, Shi J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle. 2014;13(11):1756–64. doi: 10.4161/cc.28672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17(4):304–15. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 24.Kaestner P, Stolz A, Bastians H. Determinants for the efficiency of anticancer drugs targeting either aurora-A or aurora-B kinases in human colon carcinoma cells. Mol Cancer Ther. 2009;8(7):2046–56. doi: 10.1158/1535-7163.MCT-09-0323. [DOI] [PubMed] [Google Scholar]
- 25.Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters JM. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161(2):281–94. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, Crafter C, Foster JR, Brady MC, Bigley A, Brown E, Byth KF, Barrass NC, Mundt KE, Foote KM, Heron NM, Jung FH, Mortlock AA, Boyle FT, Green S. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res. 2007;13(12):3682–8. doi: 10.1158/1078-0432.CCR-06-2979. [DOI] [PubMed] [Google Scholar]
- 27.Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16(12):1194–200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang HC, Mitchison TJ, Shi J. Stochastic competition between mechanistically independent slippage and death pathways determines cell fate during mitotic arrest. PLoS One. 2010;5(12):e15724. doi: 10.1371/journal.pone.0015724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milross CG, Mason KA, Hunter NR, Chung WK, Peters LJ, Milas L. Relationship of mitotic arrest and apoptosis to antitumor effect of paclitaxel. J Natl Cancer Inst. 1996;88:1308–14. doi: 10.1093/jnci/88.18.1308. [DOI] [PubMed] [Google Scholar]
- 30.Woods CM, Zhu J, McQueney PA, Bollag D, Lazarides E. Taxol-induced mitotic block triggers rapid onset of a p53-independent apoptotic pathway. Mol Med. 1995;1(5):506–26. [PMC free article] [PubMed] [Google Scholar]
- 31.Wang LG, Liu XM, Kreis W, Budman DR. The effect of antimicrotubule agents on signal transduction pathways of apoptosis: a review. Cancer Chemother Pharmacol. 1999;44(5):355–361. doi: 10.1007/s002800050989. [DOI] [PubMed] [Google Scholar]
- 32.Park SJ, Wu CH, Gordon JD, Zhong X, Emami A, Safa AR. Taxol induces caspase-10-dependent apoptosis. J Biol Chem. 2004;279(49):51057–67. doi: 10.1074/jbc.M406543200. [DOI] [PubMed] [Google Scholar]
- 33.Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L, Lobell RB. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005;8(1):49–59. doi: 10.1016/j.ccr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Choi M, Kim W, Cheon MG, Lee CW, Kim JE. Polo-like kinase 1 inhibitor BI2536 causes mitotic catastrophe following activation of the spindle assembly checkpoint in non-small cell lung cancer cells. Cancer Lett. 2015;357(2):591–601. doi: 10.1016/j.canlet.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 35.Broker LE, Huisman C, Span SW, Rodriguez JA, Kruyt FA, Giaccone G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004;64(1):27–30. doi: 10.1158/0008-5472.can-03-3060. [DOI] [PubMed] [Google Scholar]
- 36.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res. 2005;11(9):3155–62. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 37.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29(14):2407–20. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tunquist BJ, Woessner RD, Walker DH. Mcl-1 stability determines mitotic cell fate of human multiple myeloma tumor cells treated with the kinesin spindle protein inhibitor ARRY-520. Mol Cancer Ther. 2010;9(7):2046–56. doi: 10.1158/1535-7163.MCT-10-0033. [DOI] [PubMed] [Google Scholar]
- 39.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC, Dixit VM. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–4. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 40.Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends Biochem Sci. 1997;22(6):197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- 41.Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle. 2007;6:70–4. doi: 10.4161/cc.6.1.3682. [DOI] [PubMed] [Google Scholar]
- 42.Sivan G, Elroy-Stein O. Regulation of mRNA Translation during cellular division. Cell Cycle. 2008;7:741–4. doi: 10.4161/cc.7.6.5596. [DOI] [PubMed] [Google Scholar]
- 43.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–95. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 44.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17(12):1475–86. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams KW, Cooper GM. Rapid turnover of Mcl-1 couples translation to cell survival and apoptosis. J Biol Chem. 2007;282(9):6192–200. doi: 10.1074/jbc.M610643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, Ridgway RA, Huels D, Littler S, Schandl C, Sun Y, Bechi B, Procter DJ, Sansom OJ, Cleveland DW, Taylor SS. MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell. 2015;28(1):129–40. doi: 10.1016/j.ccell.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bennett A, Sloss O, Topham C, Nelson L, Tighe A, Taylor SS. Inhibition of Bcl-xL sensitizes cells to mitotic blockers, but not mitotic drivers. Open Biol. 2016;6(8):pii, 160134. doi: 10.1098/rsob.160134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan N, Malek M, Zha J, Kassees R, Yue P, Berry L, Fairbrother W, Sampath D, Belmont LD. Navitoclax Enhances the Efficacy of Taxanes in Non-Small Cell Lung Cancer Models. Clin Cancer Res. 2011;17(6):1394–404. doi: 10.1158/1078-0432.CCR-10-2353. [DOI] [PubMed] [Google Scholar]
- 49.Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9(12):1287–93. doi: 10.1038/sj.cdd.4401130. [DOI] [PubMed] [Google Scholar]
- 50.Enserink JM, Kolodner RD. An overview of Cdk1-controlled targets and processes. Cell Div. 2010;5:11. doi: 10.1186/1747-1028-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16(4):347–58. doi: 10.1016/j.ccr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manchado E, Guillamot M, de Cárcer G, Eguren M, Trickey M, García-Higuera I, Moreno S, Yamano H, Cañamero M, Malumbres M. Targeting mitotic exit leads to tumor regression in vivo: Modulation by Cdk1, Mastl, and the PP2A/B55α,δ phosphatase. Cancer Cell. 2010;18(6):641–54. doi: 10.1016/j.ccr.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 53.Kutuk O, Temel SG, Tolunay S, Basaga H. Aven blocks DNA damage-induced apoptosis by stabilising Bcl-xL. Eur J Cancer. 2010;46(13):2494–505. doi: 10.1016/j.ejca.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Hayashi MT, Karlseder J. DNA damage associated with mitosis and cytokinesis failure. Oncogene. 2013;32(39):4593–601. doi: 10.1038/onc.2012.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lanni Jennifer S, Jacks Tyler. Characterization of the p53-Dependent Postmitotic Checkpoint following Spindle Disruption. Mol Cell Biol. 1998;18(2):1055–64. doi: 10.1128/mcb.18.2.1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quignon F, Rozier L, Lachages AM, Bieth A, Simili M, Debatisse M. Sustained mitotic block elicits DNA breaks: one-step alteration of ploidy and chromosome integrity in mammalian cells. Oncogene. 2007;26(2):165–72. doi: 10.1038/sj.onc.1209787. [DOI] [PubMed] [Google Scholar]
- 57.Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell. 2012;23(4):567–76. doi: 10.1091/mbc.E11-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lai SK, Wong CH, Lee YP, Li HY. Caspase-3-mediated degradation of condensin Cap-H regulates mitotic cell death. Cell Death Differ. 2011;18:996–1004. doi: 10.1038/cdd.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hain KO, Colin DJ, Rastogi S, Allan LA, Clarke PR. Prolonged mitotic arrest induces a caspase-dependent DNA damage response at telomeres that determines cell survival. Sci Rep. 2016;6:26766. doi: 10.1038/srep26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–8. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uetake Y, Sluder G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol. 2010;20(18):1666–71. doi: 10.1016/j.cub.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fong CS, Mazo G, Das T, Goodman J, Kim M, O’Rourke BP, Izquierdo D, Tsou MF. 53BP1 and USP28 mediate p53-dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife. 2016;5:pii, e16270. doi: 10.7554/eLife.16270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ. A USP28-53BP1-p53-p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol. 2016;214(2):143–53. doi: 10.1083/jcb.201604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK, Oegema K. 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol. 2016;214(2):155–66. doi: 10.1083/jcb.201604081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mason KA, Hunter NR, Milas M, Abbruzzese JL, Milas L. Docetaxel enhances tumor radioresponse in vivo. Clin Cancer Res. 1997;3:2431–8. [PubMed] [Google Scholar]
- 66.Schimming R, Mason KA, Hunter N, Weil M, Kishi K, Milas L. Lack of correlation between mitotic arrest or apoptosis and antitumor effect of docetaxel. Cancer Chemother Pharmacol. 1999;43(2):165–72. doi: 10.1007/s002800050879. [DOI] [PubMed] [Google Scholar]
- 67.Del Gaizo Moore V, Letai A. BH3 profiling–measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. 2013;332(2):202–5. doi: 10.1016/j.canlet.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P, Tai YT, Mitsiades CS, Matulonis UA, Drapkin R, Stone R, Deangelo DJ, McConkey DJ, Sallan SE, Silverman L, Hirsch MS, Carrasco DR, Letai A. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334(6059):1129–33. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mason K, Staab A, Hunter N, McBride W, Petersen S, Terry N, Milas L. Enhancement of tumor radioresponse by docetaxel: Involvement of immune system. Int J Oncol. 2001;18:599–606. doi: 10.3892/ijo.18.3.599. [DOI] [PubMed] [Google Scholar]
- 70.Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, Muggia F, Symmans WF. Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin Cancer Res. 2001;7(10):3025–30. [PubMed] [Google Scholar]
- 71.Little JB. Cellular radiation effects and the bystander response. Mutat Res. 2006;597(1–2):113–8. doi: 10.1016/j.mrfmmm.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 72.Alexandre J, Hu Y, Lu W, Pelicano H, Huang P. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res. 2007;67(8):3512–7. doi: 10.1158/0008-5472.CAN-06-3914. [DOI] [PubMed] [Google Scholar]
- 73.Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res. 2011;71(13):4608–16. doi: 10.1158/0008-5472.CAN-11-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Symmans WF, Volm MD, Shapiro RL, Perkins AB, Kim AY, Demaria S, Yee HT, McMullen H, Oratz R, Klein P, Formenti SC, Muggia F. Paclitaxel-induced apoptosis and mitotic arrest assessed by serial fine-needle aspiration: implications for early prediction of breast cancer response to neoadjuvant treatment. Clin Cancer Res. 2000;6(12):4610–7. [PubMed] [Google Scholar]
- 75.Perez-Soler R, Kemp B, Wu QP, Mao L, Gomez J, Zeleniuch-Jacquotte A, Yee H, Lee JS, Jagirdar J, Ling YH. Response and determinants of sensitivity to paclitaxel in human non-small cell lung cancer tumors heterotransplanted in nude mice. Clin Cancer Res. 2000;6(12):4932–8. [PubMed] [Google Scholar]
- 76.Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell. 2012;23(1):1–6. doi: 10.1091/mbc.E10-04-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nihei Y, Suzuki M, Okano A, Tsuji T, Akiyama Y, Tsuruo T, Saito S, Hori K, Sato Y. Evaluation of antivascular and antimitotic effects of tubulin binding agents in solid tumor therapy. Jpn J Cancer Res. 1999;90(12):1387–95. doi: 10.1111/j.1349-7006.1999.tb00724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hotchkiss KA, Ashton AW, Mahmood R, Russell RG, Sparano JA, Schwartz EL. Inhibition of endothelial cell function in vitro and angiogenesis in vivo by docetaxel (Taxotere): association with impaired repositioning of the microtubule organizing center. Mol Cancer Ther. 2002;1(13):1191–200. [PubMed] [Google Scholar]
- 79.Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D, Piao H, Horowitz NS, Berkowitz RS, Matulonis U, Jänne PA, Amrein PC, Cichowski K, Drapkin R, Letai A. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015;160(5):977–89. doi: 10.1016/j.cell.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vlahovic G, Karantza V, Wang D, Cosgrove D, Rudersdorf N, Yang J, Xiong H, Busman T, Mabry M. A phase I safety and pharmacokinetic study of ABT-263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors. Invest New Drugs. 2014;32(5):976–84. doi: 10.1007/s10637-014-0116-3. [DOI] [PubMed] [Google Scholar]
- 81.Zhang H, Nimmer PM, Tahir SK, Chen J, Fryer RM, Hahn KR, Iciek LA, Morgan SJ, Nasarre MC, Nelson R, Preusser LC, Reinhart GA, Smith ML, Rosenberg SH, Elmore SW, Tse C. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007;14(5):943–51. doi: 10.1038/sj.cdd.4402081. [DOI] [PubMed] [Google Scholar]
- 82.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13(5):273–90. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol. 2015;33:23–35. doi: 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 84.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11(9):509–24. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 85.Hodge JW, Ardiani A, Farsaci B, Kwilas AR, Gameiro SR. The tipping point for combination therapy: cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin Oncol. 2012;39(3):323–39. doi: 10.1053/j.seminoncol.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Neal J, Lu H, Cuillerot JM, Reck M. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–54. doi: 10.1200/JCO.2011.38.4032. [DOI] [PubMed] [Google Scholar]
- 87.Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Lu H, Cuillerot JM, Lynch TJ. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24(1):75–83. doi: 10.1093/annonc/mds213. [DOI] [PubMed] [Google Scholar]
- 88.Slovin S. Chemotherapy and immunotherapy combination in advanced prostate cancer. Clin Adv Hematol Oncol. 2012;10(2):90–100. [PubMed] [Google Scholar]
- 89.Antonia SJ, Larkin J, Ascierto PA. Immuno-oncology combinations: a review of clinical experience and future prospects. Clin Cancer Res. 2014;20(24):6258–68. doi: 10.1158/1078-0432.CCR-14-1457. [DOI] [PubMed] [Google Scholar]
- 90.Wang Y, Lee YM, Baitsch L, Huang A, Xiang Y, Tong H, Lako A, Von T, Choi C, Lim E, Min J, Li L, Stegmeier F, Schlegel R, Eck MJ, Gray NS, Mitchison TJ, Zhao JJ. MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. Elife. 2014;3:e01763. doi: 10.7554/eLife.01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Y, Begley M, Li Q, Huang HT, Lako A, Eck MJ, Gray NS, Mitchison TJ, Cantley LC, Zhao JJ. Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proc Natl Acad Sci U S A. 2016;113(35):9810–5. doi: 10.1073/pnas.1606862113. [DOI] [PMC free article] [PubMed] [Google Scholar]