

Abstract
Alzheimer’s disease (AD) has long been recognized as a heterogeneous illness, with a common clinical presentation of progressive amnesia and less common “atypical” clinical presentations, including syndromes dominated by visual, aphasic, “frontal”, or apraxic symptoms. Our knowledge of atypical clinical phenotypes of AD comes from clinicopathologic studies, but with the growing use of in vivo molecular biomarkers of amyloid and tau pathology, we are beginning to recognize that these syndromes may not be as rare as once thought. When a clinician is evaluating a patient whose clinical phenotype is dominated by progressive aphasia, complex visual impairment, or other neuropsychiatric symptoms with relative sparing of memory, the differential diagnosis may be broader and a confident diagnosis of an atypical form of AD may require the use of molecular biomarkers. Despite the evolving sophistication in our diagnostic tools, and the acknowledgement of atypical AD syndromes in the 2011 revised diagnostic criteria for AD, the assessment of such patients still poses substantial challenges. We use a case-based approach to review the clinical and imaging phenotypes of a series of patients with typical and atypical AD, discussing our current approach to their evaluation. One day, we hope that regardless of whether a patient exhibits typical or atypical symptoms of AD pathology, we will be able to identify the condition at a prodromal or preclinical phase and institute a combination of symptomatic and disease-modifying therapies to support cognitive processes, function and behavior, and slow or halt progression to dementia.
Keywords: Alzheimer’s disease, biomarkers, corticobasal syndrome, mild cognitive impairment, posterior cortical atrophy, primary progressive aphasia
As our ability to measure biomarkers specific to certain neurodegenerative diseases has advanced, it has become increasingly clear that we need to separate neuropathological disease entities (the “disease pathology”) from clinical syndromes of neuropsychiatric dysfunction (the “illness” or “clinical syndrome”). The neuropathological disease known as Alzheimer’s disease (AD), with hallmark amyloid-β neuritic plaques, tau neurofibrillary tangles and neuronal loss, is well-known to manifest clinically as a variety of diverse syndromes. The most common clinical syndrome associated with AD pathology is the “typical” amnesia-predominant multi-domain dementia syndrome that likely begins in most cases as amnesic mild cognitive impairment (MCI). In fact, this form of the illness is so common that for many years the diagnostic criteria required impairment of memory plus impairment of one or more other domains of cognitive function1. Clinicopathologic reports have called attention to the heterogeneity of AD2–4, including “atypical” variants of AD5–7, such as posterior cortical atrophy (PCA), sometimes known as the “visual variant” of AD, aphasic variants of AD, a behavioral-comportmental (“frontal”) variant of AD, a dysexecutive variant, and even motor variants including cases that meet clinical criteria for corticobasal syndrome (CBS)8. As such, AD might be considered the “great imitator” of our time, at least when it comes to other neurodegenerative diseases.
Clinicopathologic studies provide the foundation for knowledge of atypical clinical phenotypes of AD, but with the growing use of specific in vivo molecular biomarkers of amyloid and tau pathology, we are beginning to recognize that these syndromes may not be as rare as once thought. Approximately one third of patients with AD and onset of symptoms before age 65 present atypically with primary cognitive dysfunction in a domain other than episodic memory9,10, a phenomenon less common but still encountered in late-onset AD as well11. Clinical diagnosis is frequently delayed in cases with atypical presentations, and many questions remain about the pathogenesis, risk factors, natural history and response to treatments in comparison with typical AD8. In a patient older than 65 with insidiously progressive amnesia, executive dysfunction, and complex visual impairment who has lost independence in daily function to a degree consistent with dementia (the “typical” AD clinical phenotype), many clinicians would likely be highly confident in their diagnosis of probable AD dementia without using molecular biomarkers. In contrast, when a clinician is evaluating a patient whose clinical phenotype is predominanted by progressive aphasia, complex visual impairment, or other neuropsychiatric symptoms with relative sparing of memory, the differential diagnosis may be broader and a confident diagnosis of an atypical form of AD may require the use of molecular biomarkers12. Despite the evolving sophistication in our diagnostic tools, and the acknowledgement of atypical AD syndromes in the revised diagnosis criteria for AD in 201113, the determination that a patient with one of these syndromes likely has an atypical form of AD still poses substantial challenges in clinical and research settings. Here we use a case-based approach to review the clinical and imaging phenotypes of a series of patients with typical and atypical AD. First, however, we briefly discuss our current approach to the evaluation of such patients.
Goals of evaluation and nomenclature of diagnostic summary
When we evaluate a patient, our first goal is to determine whether the overall characteristics and severity of cognitive-behavioral symptoms are consistent with dementia; mild cognitive impairment; encephalopathy (e.g. chronic encephalopathies due to immune-mediated or infectious conditions, hormonal or vitamin deficiencies, substance abuse); a learning or attentional disorder; a mood, psychiatric or sleep disorder; subjective cognitive impairment; or normal cognition. We make this clinical judgment based on all the information gathered during the assessment (e.g. assessment of premorbid level/quality and changes in cognitive abilities, activities of daily living, socio-emotional behavior, comportment, sleep, mood, and other neuropsychiatric and medical context), and attempt to grade it, at a minimum, using a severity scale (e.g., Clinical Dementia Rating Scale) and a basic cognitive assessment instrument (e.g., Montreal Cognitive Assessment score). We will often refer patients at this stage for detailed neuropsychological assessment. This is critical for providing tailored psycho-education and recommendations regarding adaptive planning, safety, and care coordination. Next, we describe the clinical phenotype, including major cognitive, behavioral, and sensorimotor symptoms, and attempt to match it to contemporary syndromic diagnostic criteria. We then consider all of the aforementioned information in order to gauge primary suspected etiology. Finally, we integrate all of the aforementioned information with other indicated diagnostic studies to exclude potentially mimicking conditions (e.g., when indicated serum testing for vitamin B12 deficiency, thyroid hormone disorder, or Hashimoto’s or autoimmune encephalopathy, MRI for mass lesion or vascular cerebral damage/infarct; CSF for voltage-gated channelopathy or paraneoplastic encephlalopathy; EEG for subclinical seizures; sleep study for obstructive sleep apnea; urine toxicity or heavy metal screen) and with any available neurodegenerative or other biomarker data and reconsider primary suspected etiology. This approach to the formulation of neurocognitive cases can be summarized as hierarchically determining 1) the patient’s overall functional status along the MCI-dementia spectrum, followed by 2) a description of the major syndrome, followed by 3) a prediction of the likely neuropathology. The case formulation then guides treatment recommendations. When possible, we go through the exercise of stating our confidence in clinical syndrome and suspected etiology before and after diagnostic biomarker testing for the purposes of evaluating the current clinical diagnostic criteria and assessing the utility of current and future diagnostic tests.
A detailed discussion of biomarkers for AD and related neurodegenerative diseases is beyond the scope of this article14, but we will briefly summarize our current practice. Brain MRI is routinely obtained for most of these patients, or CT with three-dimensional reformatting in patients with contraindications to MRI. Regional brain atrophy can provide supportive evidence for the localization of atrophy consistent with neurodegenerative pathology (e.g., medial temporal and posterolateral temporoparietal atrophy vs. frontal and anterior temporal atrophy). FDG-PET can provide supportive evidence for the localization of hypometabolism consistent with neurodegenerative pathology similar to MRI, but in some cases is more obviously visually apparent. Cerebrospinal fluid (CSF) can be analyzed for a profile consistent with AD when Aβ is abnormally low and both total tau and hyperphosphorylated tau are abnormally high, or not consistent with AD when these measures are in the normal range15. In some patients, results are indeterminate. The tests providing these biomarker measures are often reimbursed by Medicare and other payors, although FDG-PET may not be reimbursed by private insurance providers (in patients younger than Medicare eligibility). Multiple amyloid PET tracers are now available and approved for clinical use but not yet reimbursed except in the context of some studies. Appropriate use criteria for amyloid imaging have been published16,17; in all cases summarized here, the clinicians felt that the patients fit with appropriate use criteria. We next provide a series of case studies to illustrate this approach.
Typical clinical syndromes associated with AD
Case 1 is a right-handed man who presented at age 62 with a two-year duration of symptoms. Symptoms included gradually progressive impairment in episodic memory (forgetting important information from recent experiences, including conversations at work and at home, with repetitive asking of questions), in spatial orientation (getting lost in familiar areas), and in judgment and problem solving (no longer able to reason about financial or other decision-making at work or at home), with no reported language, motor, or behavioral-psychiatric symptoms. His impairments resulted in the loss of his job and the need for assistance at home. Medical and family history were unremarkable except for mild hypertension. On exam, the patient demonstrated impaired episodic memory acquisition, retention and retrieval, impaired complex attention and executive function, and impaired visual construction. Neurological exam was normal. Montreal Cognitive Assessment (MoCA) score18 was 22; Clinical Dementia Rating (CDR) score19 was 1 with Sum of Boxes (CDR-sb) of 4.5. Brain MRI scan showed symmetrical atrophy in bilateral rostral hippocampal and medial temporal cortex, medial and lateral parietal cortex, and posterior lateral temporal cortex. At this point, a diagnosis was made of mild dementia, amnesia-predominant syndrome with executive and visuospatial dysfunction, likely typical AD dementia; the clinician rated his confidence in the clinical syndrome as 100% and the underlying etiology as 95%. An FDG-PET was obtained which showed bilateral inferior parietal, posterior cingulate, and superior temporal hypometabolism. As part of a research study, an amyloid PET scan was visually read as positive. CSF profile of Aβ and tau proteins was highly consistent with underlying AD pathology. These biomarkers brought diagnostic confidence in suspected etiology to 99%. The final clinical diagnosis was dementia, amnesia-predominant multi-domain syndrome, highly likely due to AD pathology.
This patient exhibited gradually progressive symptoms and signs of multi-domain cognitive impairment including memory, spatial function, and executive function, which had substantially impacted independent function. This clinical phenotype is the prototypical form of AD dementia20, also known as Major Neurocognitive Disorder due to AD. In our practice, structural brain imaging is the standard of care in a patient such as this, and in this case the findings clearly supported the suspected diagnosis. In many patients with dementia in whom a confident (>85–90%) diagnosis of AD can be made, we often do not pursue additional biomarkers in clinical practice. However, we are conducting research to better understand the utility of these biomarkers in a clinical setting, and discuss such studies with most patients. In this case, biomarkers increased certainty in diagnosis, which may be valuable, for example, for consideration of enrollment into a clinical trial of an amyloid-modifying agent.
Case 2 is a right-handed man who presented at age 64 with two years of gradually progressive memory loss. He was no longer able to remember details of conversations with colleagues and now needed to take copious notes. He was having trouble finding his way to places he had been before but to which he traveled infrequently, and needed to rely on prompts from a newly purchased navigation system. There were no reported difficulties with judgment and problem solving, language, visual, motor, or behavioral-psychiatric function. He was still working as a professor but the symptoms had resulted in the need for new support systems and greater reliance on an administrative assistant than previously. Medical and family history were unremarkable. On exam, there was normal cognitive test performance except subtle impairment with episodic memory retention and retrieval. Neurological exam was normal. MoCA was 27 (memory); CDR was 0.5, with CDR-sb of 1.5 (memory, spatial orientation, community affairs). Brain MRI scan showed mild left-greater-than-right atrophy in rostral hippocampal and medial temporal cortex with otherwise preserved brain structure (Figure 1). Neuropsychological testing demonstrated impaired verbal and visual memory storage and retrieval (< 1 percentile) with below average acquisition (10 percentile), while other cognitive domains were above average. At this point, a diagnosis was made of MCI, single-domain amnesic subtype. The suspected etiology was AD. The clinician rated his confidence in this syndrome as 100%, and confidence in the etiology as 60%. Additional clinical workup included an FDG-PET which showed left-greater-than-right inferior parietal and superior temporal hypometabolism without obvious posterior cingulate hypometabolism. The clinician then rated his confidence in suspected AD etiology as 80%. Because he and the patient and spouse desired greater confidence, CSF was obtained which demonstrated a profile of Aβ and tau proteins highly consistent with underlying AD pathology. These biomarkers brought diagnostic confidence to 99%. The final clinical diagnosis was MCI, amnesic syndrome, highly likely due to AD pathology.
Figure 1.
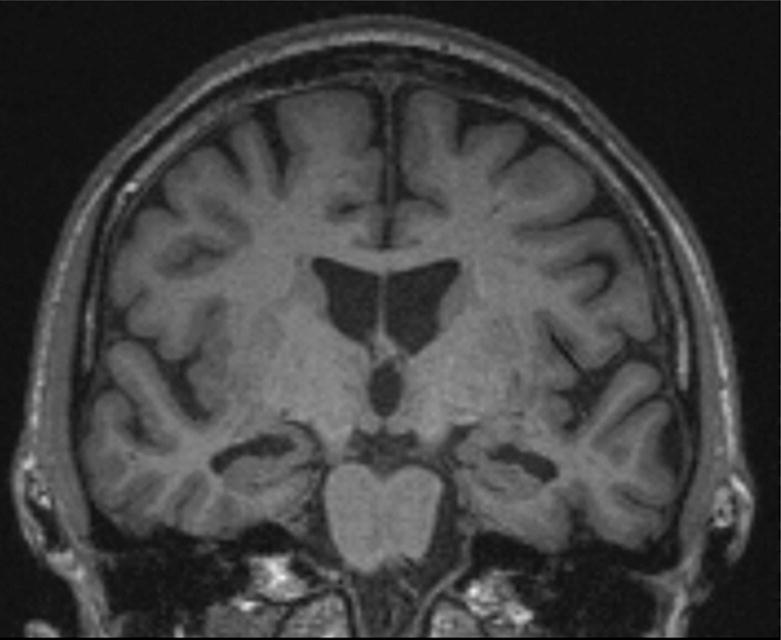
Coronal MRI of Case 2 showing moderate medial temporal cortical and hippocampal atrophy with relative sparing of other cortical regions, consistent with an amnesic neurodegenerative syndrome.
This patient exhibits the prototypical prodromal stage of AD, in which the amnesia typical of AD is present. Yet the patient has developed compensatory strategies and is managing to function independently at work and in usual daily activities; thus, he would not be considered to have dementia13. This is the clinical construct of MCI, originally described in 1999 and subsequently revised to specify cognitive subtypes—amnesic vs. non-amnesic21,22. When a patient experiences gradually progressive amnesia with characteristics suggestive of a “memory storage” problem (as opposed to acquisition or retrieval), there is a strong possibility that the underlying etiology is AD23, although other neurodegenerative diseases or cerebrovascular disease may also present this way24. While recent diagnostic criteria incorporating biomarkers into the formulation of likely etiology in patients with MCI specify that these are meant to be research criteria25,26, we and others are increasingly using them in specialty clinical practice. Using contemporary diagnostic criteria, the patient described here would be classified as having likely Prodromal AD25 or MCI due to AD with high likelihood26 or Mild Neurocognitive Disorder likely due to AD.
Atypical clinical syndromes associated with AD
Case 3 is a right-handed woman who presented at age 67 with a four-year history of progressive visuospatial impairment. She and her spouse reported difficulty with spatial orientation, including positioning the car correctly in parking spots or in the garage; difficulty with depth perception, including making mistakes on stairs, escalators, revolving doors, and curbs; trouble seeing objects that were “right in front of her,” especially when in the full refrigerator or on a crowded countertop. Her memory was intact. There were no reported symptoms involving judgment and problem solving, language, motor, or behavioral-psychiatric symptoms, except that she was mildly anxious. She was still working as an in-home visiting nurse with some difficulty due to the visual symptoms, but was otherwise functioning independently, doing a variety of community and home activities as usual. Medical and family history were unremarkable. On exam, the only abnormality was visuospatial deficits in figure-copying, clock-drawing, and complex visual perception (difficulty perceiving line drawings of overlapping objects). Neurological exam was unremarkable except for mild oculomotor apraxia, simultanagnosia but no optic ataxia, and very mild left limb apraxia; there was no extrapyramidal dysfunction. MoCA was 26 (visuospatial); CDR was 0.5, with CDR-sb of 1 (spatial orientation, community affairs). The patient had seen an ophthalmologist and was told that there was an inconsistent left partial hemianopia but otherwise normal basic vision. Neuropsychological testing confirmed that, despite normal acuity, complex visual function was significantly impaired (<1 percentile), while other cognitive domains were within normal limits. Brain MRI demonstrated right > left lateral parietal lobe atrophy with preserved medial and lateral temporal lobe structure (Figure 2). FDG-PET showed right > left posterior temporal, parietal and occipital hypometabolism. At this point, a diagnosis was made of MCI, non-amnesic single domain visual impairment, consistent with Posterior Cortical Atrophy (PCA). Although the clinician was >90% confident in the clinical syndromic diagnosis, confidence in the likely underlying pathology being AD was 65%. Therefore, CSF was obtained which showed a profile of Aβ and tau proteins highly consistent with underlying AD pathology. As part of a research protocol, an amyloid PET scan was obtained and visually read as positive. These biomarkers brought diagnostic confidence to 99% confidence that the underlying disease was likely AD. The final clinical diagnosis was MCI, PCA syndrome, highly likely due to AD pathology.
Figure 2.
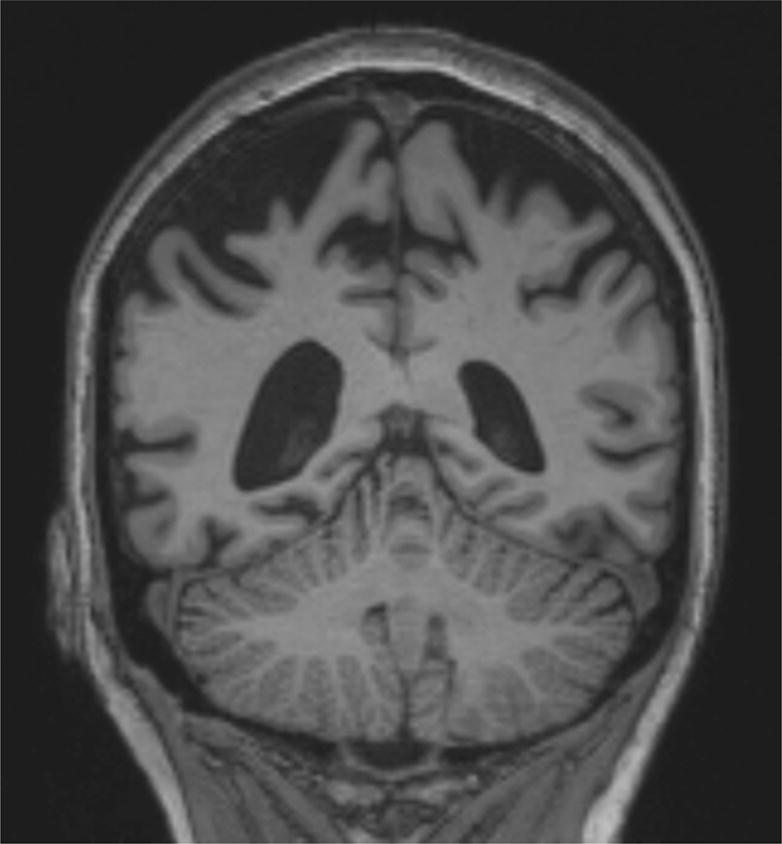
Coronal MRI of Case 3 showing severe bilateral parietal cortical atrophy, consistent with the pattern of atrophy typically seen in the PCA syndrome.
The original description of the posterior cortical atrophy syndrome is usually attributed to D.F. Benson27, but multiple earlier case reports describe patients with AD pathology who had prominent early visual disturbances28, such as Balint’s syndrome with atypical occipitoparietal pathology29. In 1993, a detailed clinicopathologic report describing “the visual variant of AD” called attention to the severe early visual and spatial impairment with occipito-temporoparietal plaque and tangle neuropathology30. Contemporary clinical diagnostic criteria emphasize the presence of progressive visual impairment with relative sparing of memory, language, behavior, and insight31,32; an international work group is currently refining clinical diagnostic criteria33. Although contemporary literature largely equates PCA with the visual variant of AD, there are hardly any clinicopathologic studies of PCA with more than 5 cases, with studies suggesting that AD neuropathology may account for 65%34 – 77%31 – 100%11. PCA may also be caused by corticobasal degeneration, Lewy body disease, or rarely other neurodegenerative diseases28.
Case 4 is a right-handed woman who presented at age 65 with a two-year history of progressive language difficulties. She and her daughter described gradually progressive difficulty finding words in conversation, increasing mispronunciation of words, and new difficulty spelling. Her memory was intact. There were no reported symptoms involving spatial or temporal orientation, judgment and problem solving, motor, or behavioral-psychiatric symptoms, except that she reported feeling mildly depressed. She had retired at age 60 but was actively volunteering for 20 hours each week at her local library with little difficulty, and was otherwise functioning independently, living by herself. Medical and family history were unremarkable. On exam, her speech was articulate and fluent at times but with word retrieval difficulties that would reduce fluency along with phonemic paraphasias; she was able to repeat short but not long phrases. Grammar and single word comprehension were normal. The remainder of the office-based cognitive exam was normal except for impairments in spelling, calculation, and verbal list encoding but retrieval and recognition were normal. Neurological exam was unremarkable except for mild right limb apraxia without rigidity or other extrapyramidal dysfunction. MoCA was 27 (naming, repetition); CDR was 0, with CDR supplemental language box of 0.5. Speech and language pathology assessment demonstrated variable fluency with impairments likely arising during word-retrieval difficulty, anomia, phrase length-dependent repetition impairment, normal verbal grammatical production and comprehension, normal single word comprehension, mildly impaired auditory comprehension for long phrases, normal reading, spelling errors on writing samples but normal grammar, normal motor speech. Progressive Aphasia Severity Scale (PASS)35,36 scores were 0.5 in fluency, 1 in word retrieval, 0.5 in repetition, 0.5 in auditory comprehension, 0.5 in writing; PASS sum of boxes was 3. Neuropsychological testing demonstrated mild verbal encoding impairment (5 percentile) but normal retention and retrieval with normal visual memory performance, and mildly impaired verbal fluency (5 percentile) but normal performance on executive function tasks and tests of other cognitive domains. Brain MRI demonstrated widening of the left Sylvian fissure due to posterior lateral temporal atrophy with preserved medial temporal lobe structure (Figure 3); FDG-PET showed left > right posterior superior temporal and inferior parietal hypometabolism with mild posterior cingulate hypometabolism. A diagnosis was made of MCI, non-amnesic single domain language impairment, consistent with Primary Progressive Aphasia (PPA), logopenic variant (lvPPA). Although the clinician was >90% confident in the clinical syndromic diagnosis, confidence in the likely underlying pathology being AD was 70%. Therefore, CSF was obtained which showed a profile of Aβ and tau proteins highly consistent with underlying AD pathology. As part of a research protocol, an amyloid PET scan was obtained and visually read as positive. These biomarkers brought diagnostic confidence to 99% confidence that the underlying disease was likely AD. The final clinical diagnosis was MCI, lvPPA syndrome, highly likely due to AD pathology.
Figure 3.
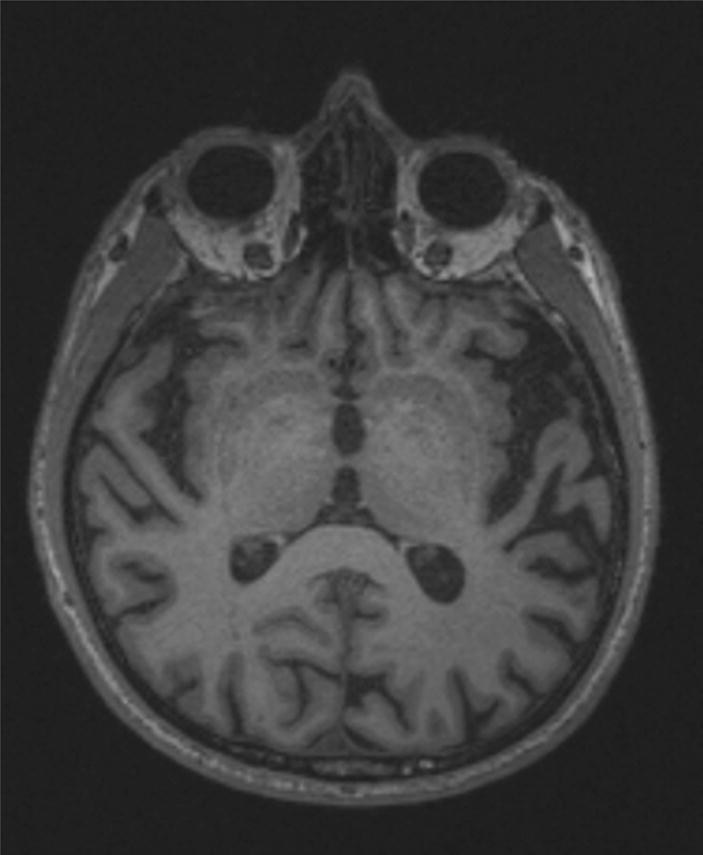
Axial MRI of Case 4 showing moderate left-lateralized temporal cortical atrophy, consistent with the pattern of atrophy typically seen in the logopenic PPA syndrome. Image right is patient left.
Early descriptions of patients presenting with progressive aphasia emphasized the observation that the aphasia sometimes remained isolated for years prior to the development of multi-domain impairment and functional loss consistent with dementia37. Many of these cases did not show AD pathology, but some did, leading to the idea that this could be an atypical form of AD38–41. Current clinical diagnostic criteria emphasize the presence of progressive language impairment with relative sparing of memory, visual abilities, and behavior42. Although many contemporary summaries suggest that the logopenic variant of PPA is essentially equivalent to a language variant of AD, the clinicopathologic investigations of PPA to date indicate that AD neuropathology may only account for about two-thirds of the cases43. Moreover, other clinical phenotypes of PPA may be associated with AD pathology44.
Case 5 is a right-handed woman who presented at age 62 with a one-year history of progressive cognitive and behavioral symptoms. She reported difficulty with concentration and memory, attributing difficulty at her job as a lab technician to recently diagnosed hypothyroidism. She denied other symptoms. In contrast, her sister reported that her memory was “pretty good,” but that the more notable problems included disorganization and poor judgment and decision-making. She had abruptly left a family gathering for no clear reason, and had recently made several purchases that were beyond her financial capacity (impulsivity). She had developed a new habit of repeatedly checking to make sure her house and car were locked and that she had the keys, and seemed to be collecting pairs of sunglasses (compulsivity). Her sister noticed that she had gained weight and always carried a bag of candy in her purse, a behavior she had never done before (hyperorality). Her sister was concerned that she did not seem to be aware of these unusual behaviors. There were no reported symptoms involving orientation in space or time, language, visual skills, or motor function. She was still working as a lab technician in a hospital but was on probation due to several errors. She was otherwise functioning largely independently, living at home and going on trips with friends, but recently had made two errors paying bills which were out of character and had come to her sister’s attention. Medical and family history were unremarkable except for recently-diagnosed hypothyroidism which was adequately treated. On exam, she had difficulty with performing alternating sequencing and verbal fluency tasks, as well as free recall of words but was able to correctly retrieve them with cues. Neurological exam was unremarkable except for impersistence when asked to maintain her gaze on an object or hold her arms in the air; there was no extrapyramidal dysfunction. MoCA was 25 (Trails, clock hands, continuous performance task, serial 7s, verbal recall); CDR was 0.5, with CDR-sb of 1.5 (memory, judgment and problem-solving, community affairs); supplemental behavior box score was 1. Social Impairment Rating Scale (SIRS)45 scores were 0.5 for lack of attention/response to social cues, 0.5 for difficulty with social norms; SIRS sum of boxes was 1. Neuropsychological testing demonstrated borderline performance on tasks of working memory, executive function, verbal and visual recall (5 percentile) but normal encoding and cued recall and recognition. She was impaired on verbal fluency (<1 percentile) but had normal performance in other language domains as well as visual perception and construction tasks. Brain MRI demonstrated right > left lateral, medial, and orbital frontal lobe atrophy with preserved medial and lateral temporal lobe structure (Figure 4). FDG-PET showed right > left frontal and lateral temporal with a lesser degree of posterior cingulate hypometabolism. At this point, a diagnosis was made of MCI, amnesic multi-domain cognitive impairment with behavioral symptoms, consistent with very mild behavioral variant Frontotemporal Dementia (bvFTD). Since she was still functioning largely independently, although substantial concerns had been raised, she was not yet considered to have dementia. The clinician was >85% confident in the clinical syndromic diagnosis. Although the patient reported a memory concern and neuropsychological testing showed memory retrieval difficulty, the patient’s sister denied that this was a prominent symptom and the clinician attributed the performance difficulty to frontal systems dysfunction. The clinician was 85% confident that Frontotemporal Lobar Degeneration (FTLD) was the likely underlying pathology. Nevertheless, CSF was obtained with the goal of “ruling out” AD as a possibility and it showed a profile of Aβ and tau proteins highly consistent with underlying AD pathology. As part of a research protocol, an amyloid PET scan was obtained and visually read as positive. These biomarkers changed the clinician’s thinking to 99% confidence that the underlying disease was likely AD. The final clinical diagnosis was MCI, bvFTD syndrome, highly likely due to AD pathology.
Figure 4.
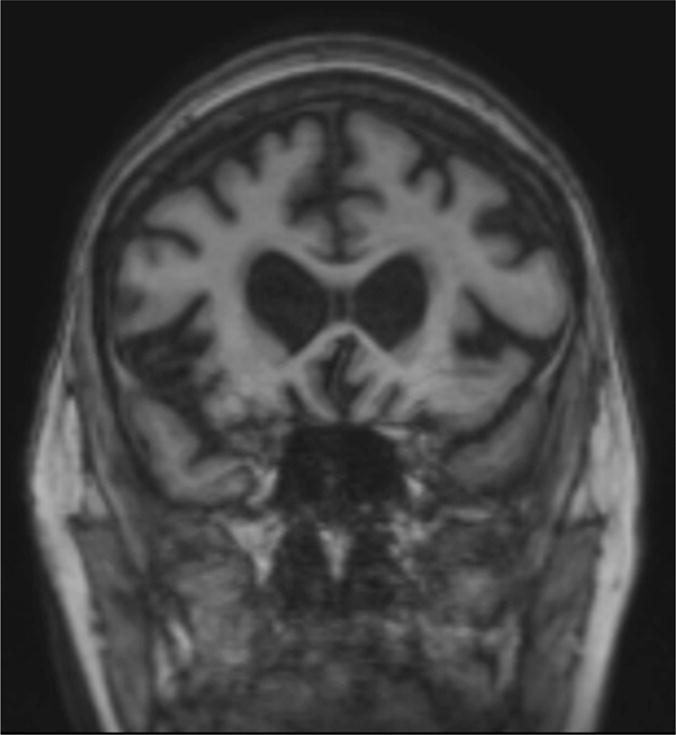
Coronal MRI of Case 5 showing moderate dorsolateral, ventrolateral, and ventromedial prefrontal and insular cortical atrophy, suggestive of the pattern of atrophy seen in Frontotemporal Dementia.
The original description of the “frontal variant” of AD is usually attributed to Johnson et al.46, but earlier case reports describe patients with AD pathology who had prominent early behavioral symptoms47. In the past 15 years, multiple clinicopathologic studies have described patients who had been diagnosed with FTD by expert clinicians but who at autopsy were shown to have solely AD pathology10,11,48–52. It appears much more common for patients with AD to present with a prominent dysexecutive syndrome53 with or without apathy than a full-blown socioaffective behavioral syndrome consistent with bvFTD54. Clinical diagnostic criteria have not yet been developed for the behavioral variant of AD (bvAD), but investigators are beginning to study whether clinical features may help distinguish bvAD from bvFTD52. In the end, we believe that clinical features in conjunction with MRI or FDG-PET may improve the probabilistic prediction of AD vs. FTLD pathology in a patient with a prominent behavioral syndrome, but molecular biomarkers will likely be necessary to make this discrimination confidently.
Case 6 is a right-handed man who presented at age 61 with an 18-month history of gradually progressive movement symptoms followed by cognitive and mood symptoms. He first started having difficulty using his left hand followed by the left leg despite no weakness; sometimes the arm would move “as if it had a mind of its own.” He then developed myoclonic jerks of the left foot and occasionally left arm. He required assistance shaving and getting dressed due to these motor symptoms, and struggled to write and to use utensils and the remote control. Concentration and memory then declined, such that he had to ask people to repeat themselves in conversation and needed reminders for his schedule. He began having difficulty with multi-tasking. Mild anxiety and depression had developed. He was still working as a manufacturing plant manager with some assistance required and was performing complex activities of daily living largely independently, with the exception of tasks that required the motor functions described above. Medical and family history were unremarkable except for hypercholesterolemia. On examination, difficulties were present with memory encoding, alternating sequences, verbal fluency, and serial sevens. His neurologic exam was notable for mild left-sided extrapyramidal dysfunction with rigidity, right hand dystonia, bilateral ideomotor apraxia, and bilateral agraphesthesia and astereognosia. MoCA was 28 (Trails, serial 7s); CDR was 0.5, with CDR-sb of 1.5 (memory, judgment and problem-solving, community affairs). Neuropsychological testing demonstrated borderline performance on tasks of working memory, executive function, verbal and visual encoding (5 percentile) but normal retention and retrieval, low average performance on verbal fluency (10 percentile) but normal performance in other language domains. Motor speed and dexterity were impaired, more prominently in the left hand (<1 percentile). He also had low average performance (10 percentile) on visual construction tasks. MRI showed right-greater-than-left precentral and postcentral gyrus atrophy (Figure 5). FDG-PET confirmed right-greater-than-left peri-Rolandic hypometabolism. A diagnosis was made of MCI, non-amnesic multi-domain cognitive impairment with motor impairment consistent with Corticobasal Syndrome (CBS). The clinician was >85% confident in the clinical syndromic diagnosis, and was less than 50% confident that the likely underlying pathology Corticobasal Degeneration (CBD). Therefore, CSF was obtained which showed a profile of Aβ and tau proteins highly consistent with underlying AD pathology. As part of a research protocol, an amyloid PET scan was obtained and visually read as positive. These biomarkers brought diagnostic confidence to 99% confidence that the underlying disease was likely AD. The final clinical diagnosis was MCI, non-amnesic multidomain syndrome with predominant motor-cognitive features consistent with CBS, highly likely due to AD pathology.
Figure 5.
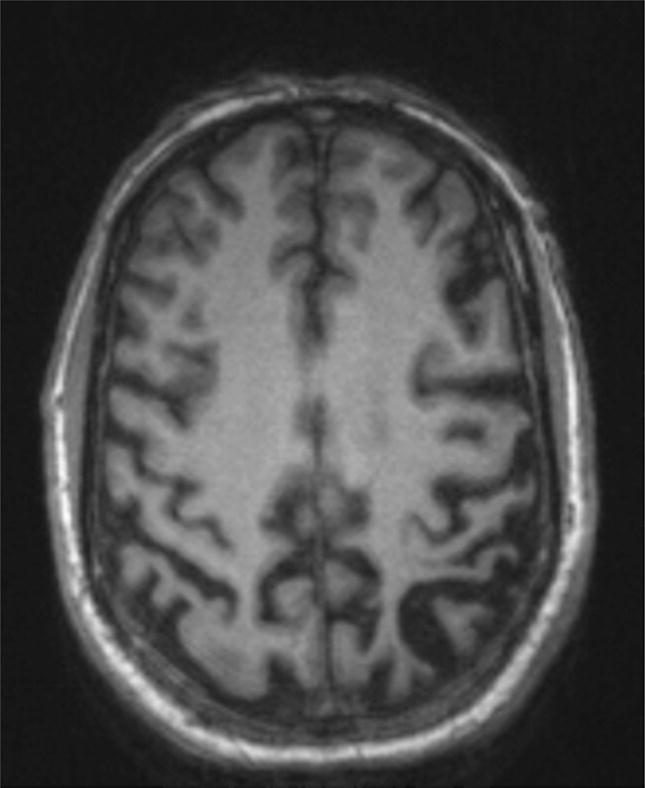
Axial MRI of Case 6 showing severe left-lateralized peri-Rolandic and parietal cortical atrophy, consistent with the pattern of atrophy typically seen in the CBS syndrome.
Although CBS was originally conceptualized as a distinct clinicopathological entity, numerous studies over the past 15 years have highlighted the fact that a substantial minority of cases with classical CBS syndromes arise as a result of AD pathology55,56. Recent studies have shown that a substantial proportion (35%) of patients presenting clinically with CBS ultimately are shown to have AD pathology57. Efforts are ongoing to revise clinical diagnostic criteria to better predict pathology58, but biomarkers of molecular pathology will almost certainly be an important element of future diagnostic criteria for CBS.
Discussion and conclusions
The clinical evaluation of AD and other dementias has evolved substantially in the past decade with the advent of a variety of biomarkers of the localization and molecular nature of neurodegenerative diseases. As this has occurred, it has become increasingly clear that we should separate our consideration of the clinical syndrome exhibited by the patient from the suspected underlying pathology, assessing each at least partially independently. Although a progressive amnesic and dysexecutive dementia may be relatively easy to accurately diagnose as likely due to AD pathology, non-amnesic syndromes are less common and present a broader pathological differential diagnosis, and thus are often more difficult to diagnose. In parallel, large pathology investigations have demonstrated that as many as 25% of cases of AD do not conform to the stereotypical progression of neurofibrillary tangle pathology described in the Braak pathology staging scheme59. Thus, one of the core principles of behavioral neurology is reinforced: it is not the molecular nature of the lesion that determines the clinical deficit, but rather its localization60.
Although fibrillar amyloid plaques are necessary for a pathological diagnosis of AD61, the density and distribution of plaques is weakly associated with clinical features in patients with symptoms of the illness62. In contrast, detailed neuropathological studies performed more than two decades ago showed that the topographical distribution and density of neurofibrillary tangles is closely linked to the clinical phenotype and severity of symptoms. The relationship between tau pathology and regional neurodegeneration and clinical symptoms has also been reported in atypical forms of AD, including posterior cortical atrophy29, behavioral (“frontal”) variant AD46,63, and primary progressive aphasia64. The observation that neither the localization of amyloid pathology nor its severity relate to clinical symptoms or markers of neurodegeneration in typical or atypical forms of AD has now been confirmed in vivo using amyloid PET imaging65–68. Amyloid PET studies to date have not demonstrated an ability to distinguish between typical and atypical AD67,69 despite results suggesting higher cortical amyloid burden in apolipoprotein E ε4 non-carriers vs. carriers70 and higher amyloid burden in the parietal cortex in early-onset AD vs. late-onset AD71. Furthermore, although amyloid PET has revolutionized our approach to the evaluation of patients with suspected AD, like most medical diagnostic tests it may produce false positive or false negative results.
The lack of a PET ligand specific for neurofibrillary tau pathology has rendered it impossible to test the hypotheses regarding relationships of tau to clinical features of AD in vivo. Although CSF tau measures are a validated biomarker of neurofibrillary tau pathology15, this biomarker does not enable the localization of tau. As of 2013, this is now changing with the development of new PET ligands to measure tau pathology in vivo72,73. The first case report of a patient with atypical AD (PCA) imaged using tau PET demonstrates the co-localization of tau pathology measured in vivo with regional hypometabolism, and the lack of correspondence with regional amyloid68. This and similar reports discussed at recent meetings74 demonstrate the we are now able to measure the localization and magnitude of both major pathological hallmarks of AD in living patients, a revolution that will almost certainly lead to improved diagnostics and therapeutics.
The question of what factors influence whether a patient may develop typical or atypical AD is largely unanswered. Data from both clinical and pathological studies indicate that younger age is associated with a greater likelihood of an atypical phenotype, as is the absence of an apolipoprotein E ε4 allele53,59,75–77. If AD originates and progresses through connections of distributed neural networks in the brain78–81, the organization of brain networks will shed light on the topographical differences between typical and atypical AD pathology8. However, it is still unclear why and how a critical node of one brain network rather than another becomes selectively vulnerable to AD pathology in the first place82. Further investigation is necessary to identify other genetic and environmental drivers of phenotypic diversity in AD, and the mechanisms by which age influences the biology and clinical expression of AD. It is also important to acknowledge that increasing age also makes mixed pathologies more common, such as AD with cerebrovascular disease or AD with cortical Lewy body disease; mixed cases present an additional layer of diagnostic challenge.
The treatment of symptoms of AD may in part be targeted toward specific circuits and the symptoms that arise when they fail, but future therapies will hopefully be able to modify the underlying disease proteinopathies. If this is the case, then determining that a patient’s clinical dementia syndrome is likely due to underlying AD will be a critical factor in guiding the therapeutic approach. One day, we hope that regardless of whether a patient exhibits typical or atypical symptoms of AD pathology, we will be able to identify the condition at a prodromal or preclinical phase and institute disease-modifying therapy, in combination with symptomatic treatments, to slow or halt progression to dementia.
Acknowledgments
The authors thank the patients and their families for allowing their stories to be told, and our colleagues and staff for assistance in evaluating these patients. We also appreciate our funding sources, including R21 NS077059, R21 NS084156, P50 AG005134, Cure Foundation New Vision Award and Gerstner Family Career Development Award.
Disclosure information:
Bradford Dickerson has the following disclosures:
National Institutes of Health, Grant recipient, Research grant P50 AG005134
National Institutes of Health, Grant recipient, Research grant R21 NS077059
National Institutes of Health, Grant recipient, Research grant R21 NS084156
Footnotes
This paper is based in part on presentations from a platform session at the 26th American Neuropsychiatric Association meeting, March 26, 2015, Orlando, Florida.
Scott McGinnis, Chenjie Xia, Bruce Price, Alireza Atri, Melissa Murray, Mario Mendez, and David Wolk do not have anything to disclose
Bibliography
- 1.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 2.Kanne SM, Balota DA, Storandt M, McKeel DW, Jr, Morris JC. Relating anatomy to function in Alzheimer’s disease: neuropsychological profiles predict regional neuropathology 5 years later. Neurology. 1998;50(4):979–985. doi: 10.1212/wnl.50.4.979. [DOI] [PubMed] [Google Scholar]
- 3.Becker JT, Huff FJ, Nebes RD, Holland A, Boller F. Neuropsychological function in Alzheimer’s disease. Pattern of impairment and rates of progression. Arch Neurol. 1988;45(3):263–268. doi: 10.1001/archneur.1988.00520270037018. [DOI] [PubMed] [Google Scholar]
- 4.Martin A, Brouwers P, Lalonde F, et al. Towards a behavioral typology of Alzheimer’s patients. J Clin Exp Neuropsychol. 1986;8(5):594–610. doi: 10.1080/01688638608405178. [DOI] [PubMed] [Google Scholar]
- 5.Galton CJ, Patterson K, Xuereb JH, Hodges JR. Atypical and typical presentations of Alzheimer’s disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain. 2000;123(Pt 3):484–498. doi: 10.1093/brain/123.3.484. [DOI] [PubMed] [Google Scholar]
- 6.Neary D, Snowden JS, Bowen DM, et al. Neuropsychological syndromes in presenile dementia due to cerebral atrophy. J Neurol Neurosurg Psychiatry. 1986;49(2):163–174. doi: 10.1136/jnnp.49.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price BH, Gurvit H, Weintraub S, Geula C, Leimkuhler E, Mesulam M. Neuropsychological patterns and language deficits in 20 consecutive cases of autopsy-confirmed Alzheimer’s disease. Arch Neurol. 1993;50(9):931–937. doi: 10.1001/archneur.1993.00540090038008. [DOI] [PubMed] [Google Scholar]
- 8.Warren JD, Fletcher PD, Golden HL. The paradox of syndromic diversity in Alzheimer disease. Nat Rev Neurol. 2012;8(8):451–464. doi: 10.1038/nrneurol.2012.135. [DOI] [PubMed] [Google Scholar]
- 9.Koedam EL, Lauffer V, van der Vlies AE, van der Flier WM, Scheltens P, Pijnenburg YA. Early-versus late-onset Alzheimer’s disease: more than age alone. Journal of Alzheimer’s disease: JAD. 2010;19(4):1401–1408. doi: 10.3233/JAD-2010-1337. [DOI] [PubMed] [Google Scholar]
- 10.Balasa M, Gelpi E, Antonell A, et al. Clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology. 2011;76(20):1720–1725. doi: 10.1212/WNL.0b013e31821a44dd. [DOI] [PubMed] [Google Scholar]
- 11.Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer’s disease. Brain. 2007;130(Pt 10):2636–2645. doi: 10.1093/brain/awm213. [DOI] [PubMed] [Google Scholar]
- 12.Wolk DA. Amyloid imaging in atypical presentations of Alzheimer’s disease. Curr Neurol Neurosci Rep. 2013;13(12):412. doi: 10.1007/s11910-013-0412-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging and the Alzheimer’s Association workgroup. Alzheimers Dement. 2011 doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dickerson BC. Neuroimaging, Cerebrospinal Fluid Markers, and Genetic Testing in Dementia. In: Dickerson BC, Atri A, editors. Dementia: Comprehensive Principles and Practice. New York: Oxford University Press; 2014. pp. 530–564. [Google Scholar]
- 15.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement. 2013;9(1):e-1–16. doi: 10.1016/j.jalz.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson KA, Minoshima S, Bohnen NI, et al. Update on appropriate use criteria for amyloid PET imaging: dementia experts, mild cognitive impairment, and education. Amyloid Imaging Task Force of the Alzheimer’s Association and Society for Nuclear Medicine and Molecular Imaging. Alzheimers Dement. 2013;9(4):e106–109. doi: 10.1016/j.jalz.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Nasreddine ZS, Phillips NA, Bedirian V, Charbonneau S, Whitehead V, Collin I. The montreal cognitive assessment, moca: A brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695–699. doi: 10.1111/j.1532-5415.2005.53221.x. [DOI] [PubMed] [Google Scholar]
- 19.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- 20.Atri A. Alzheimer’s disease and Alzheimer’s dementia. In: Dickerson BC, Atri A, editors. Dementia: Comprehensive Principles and Practice. New York: Oxford University Press; 2014. pp. 362–433. [Google Scholar]
- 21.Wicklund M, Petersen RC. Mild cognitive impairment. In: Dickerson BC, Atri A, editors. Dementia: Comprehensive Principles and Practice. New York: Oxford University Press; 2014. pp. 434–449. [Google Scholar]
- 22.Petersen RC. Clinical practice. Mild cognitive impairment. N Engl J Med. 2011;364(23):2227–2234. doi: 10.1056/NEJMcp0910237. [DOI] [PubMed] [Google Scholar]
- 23.Wolk DA, Budson AE. Memory systems. Continuum (Minneap Minn) 2010;16(4):15–28. doi: 10.1212/01.CON.0000368257.30791.3a. Behavioral Neurology. [DOI] [PubMed] [Google Scholar]
- 24.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63(5):665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 25.Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9(11):1118–1127. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- 26.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benson DF, Davis RJ, Snyder BD. Posterior cortical atrophy. Arch Neurol. 1988;45(7):789–793. doi: 10.1001/archneur.1988.00520310107024. [DOI] [PubMed] [Google Scholar]
- 28.Tang-Wai DF, Lake A, Graff-Radford N. Posterior cortical atrophy. In: Dickerson BC, Atri A, editors. Dementia: Comprehensive Principles and Practice. New York: Oxford University Press; 2014. pp. 210–221. [Google Scholar]
- 29.Hof PR, Bouras C, Constantinidis J, Morrison JH. Balint’s syndrome in Alzheimer’s disease: specific disruption of the occipito-parietal visual pathway. Brain Res. 1989;493(2):368–375. doi: 10.1016/0006-8993(89)91173-6. [DOI] [PubMed] [Google Scholar]
- 30.Levine DN, Lee JM, Fisher CM. The visual variant of Alzheimer’s disease: a clinicopathologic case study. Neurology. 1993;43(2):305–313. doi: 10.1212/wnl.43.2.305. [DOI] [PubMed] [Google Scholar]
- 31.Tang-Wai DF, Graff-Radford NR, Boeve BF, et al. Clinical, genetic, and neuropathologic characteristics of posterior cortical atrophy. Neurology. 2004;63(7):1168–1174. doi: 10.1212/01.wnl.0000140289.18472.15. [DOI] [PubMed] [Google Scholar]
- 32.Mendez MF, Ghajarania M, Perryman KM. Posterior cortical atrophy: clinical characteristics and differences compared to Alzheimer’s disease. Dement Geriatr Cogn Disord. 2002;14(1):33–40. doi: 10.1159/000058331. [DOI] [PubMed] [Google Scholar]
- 33.Crutch SJ, Schott JM, Rabinovici GD, et al. Shining a light on posterior cortical atrophy. Alzheimers Dement. 2013;9(4):463–465. doi: 10.1016/j.jalz.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 34.Renner JA, Burns JM, Hou CE, McKeel DW, Jr, Storandt M, Morris JC. Progressive posterior cortical dysfunction: a clinicopathologic series. Neurology. 2004;63(7):1175–1180. doi: 10.1212/01.wnl.0000140290.80962.bf. [DOI] [PubMed] [Google Scholar]
- 35.Sapolsky D, Bakkour A, Negreira A, et al. Cortical neuroanatomic correlates of symptom severity in primary progressive aphasia. Neurology. 2010;75(4):358–366. doi: 10.1212/WNL.0b013e3181ea15e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sapolsky D, Domoto-Reilly K, Dickerson BC. Use of the Progressive Aphasia Severity Scale (PASS) in monitoring speech and language status in PPA. Aphasiology. 2014;28(8–9):993–1003. doi: 10.1080/02687038.2014.931563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol. 1982;11(6):592–598. doi: 10.1002/ana.410110607. [DOI] [PubMed] [Google Scholar]
- 38.Karbe H, Kertesz A, Polk M. Profiles of language impairment in primary progressive aphasia. Arch Neurol. 1993;50(2):193–201. doi: 10.1001/archneur.1993.00540020069020. [DOI] [PubMed] [Google Scholar]
- 39.Pogacar S, Rubio A. Morphological features of Pick’s and atypical Alzheimer’s disease in Down’s syndrome. Acta Neuropathol. 1982;58(4):249–254. doi: 10.1007/BF00688605. [DOI] [PubMed] [Google Scholar]
- 40.Green J, Morris JC, Sandson J, McKeel DW, Jr, Miller JW. Progressive aphasia: a precursor of global dementia? Neurology. 1990;40(3 Pt 1):423–429. doi: 10.1212/wnl.40.3_part_1.423. [DOI] [PubMed] [Google Scholar]
- 41.Kempler D, Metter EJ, Riege WH, Jackson CA, Benson DF, Hanson WR. Slowly progressive aphasia: three cases with language, memory, CT and PET data. J Neurol Neurosurg Psychiatry. 1990;53(11):987–993. doi: 10.1136/jnnp.53.11.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer’s and frontotemporal pathology in primary progressive aphasia. Brain. 2014;137(Pt 4):1176–1192. doi: 10.1093/brain/awu024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grossman M. Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol. 2010;6(2):88–97. doi: 10.1038/nrneurol.2009.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bickart KC, Brickhouse M, Negreira A, Sapolsky D, Barrett LF, Dickerson BC. Atrophy in distinct corticolimbic networks in frontotemporal dementia relates to social impairments measured using the Social Impairment Rating Scale. J Neurol Neurosurg Psychiatry. 2014;85(4):438–448. doi: 10.1136/jnnp-2012-304656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol. 1999;56(10):1233–1239. doi: 10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- 47.von Gunten A, Bouras C, Kovari E, Giannakopoulos P, Hof PR. Neural substrates of cognitive and behavioral deficits in atypical Alzheimer’s disease. Brain Res Rev. 2006;51(2):176–211. doi: 10.1016/j.brainresrev.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 48.Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. 2006;59(6):952–962. doi: 10.1002/ana.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- 50.Knopman DS, Boeve BF, Parisi JE, et al. Antemortem diagnosis of frontotemporal lobar degeneration. Ann Neurol. 2005;57(4):480–488. doi: 10.1002/ana.20425. [DOI] [PubMed] [Google Scholar]
- 51.Snowden JS, Thompson JC, Stopford CL, et al. The clinical diagnosis of early-onset dementias: diagnostic accuracy and clinicopathological relationships. Brain. 2011;134(Pt 9):2478–2492. doi: 10.1093/brain/awr189. [DOI] [PubMed] [Google Scholar]
- 52.Ossenkoppele R, Pijnenburg YA, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain. 2015;138(Pt 9):2732–2749. doi: 10.1093/brain/awv191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dickerson BC, Wolk DA, Alzheimer’s Disease Neuroimaging I Dysexecutive versus amnesic phenotypes of very mild Alzheimer’s disease are associated with distinct clinical, genetic and cortical thinning characteristics. J Neurol Neurosurg Psychiatry. 2011;82(1):45–51. doi: 10.1136/jnnp.2009.199505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477. doi: 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999;53(4):795–800. doi: 10.1212/wnl.53.4.795. [DOI] [PubMed] [Google Scholar]
- 56.Litvan I, Agid Y, Goetz C, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997;48(1):119–125. doi: 10.1212/wnl.48.1.119. [DOI] [PubMed] [Google Scholar]
- 57.Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011;70(2):327–340. doi: 10.1002/ana.22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. doi: 10.1212/WNL.0b013e31827f0fd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10(9):785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weintraub S, Mesulam M. With or without FUS, it is the anatomy that dictates the dementia phenotype. Brain. 2009;132(Pt 11):2906–2908. doi: 10.1093/brain/awp286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathologica. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 63.Blennerhassett R, Lillo P, Halliday GM, Hodges JR, Kril JJ. Distribution of pathology in frontal variant Alzheimer’s disease. J Alzheimers Dis. 2014;39(1):63–70. doi: 10.3233/JAD-131241. [DOI] [PubMed] [Google Scholar]
- 64.Gefen T, Gasho K, Rademaker A, et al. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain. 2012;135(Pt 5):1554–1565. doi: 10.1093/brain/aws076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wolk DA, Price JC, Saxton JA, et al. Amyloid imaging in mild cognitive impairment subtypes. Ann Neurol. 2009;65(5):557–568. doi: 10.1002/ana.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rosenbloom MH, Alkalay A, Agarwal N, et al. Distinct clinical and metabolic deficits in PCA and AD are not related to amyloid distribution. Neurology. 2011;76(21):1789–1796. doi: 10.1212/WNL.0b013e31821cccad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lehmann M, Ghosh PM, Madison C, et al. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain. 2013;136(Pt 3):844–858. doi: 10.1093/brain/aws327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ossenkoppele R, Schonhaut DR, Baker SL, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol. 2015;77(2):338–342. doi: 10.1002/ana.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Formaglio M, Costes N, Seguin J, et al. In vivo demonstration of amyloid burden in posterior cortical atrophy: a case series with PET and CSF findings. Journal of neurology. 2011;258(10):1841–1851. doi: 10.1007/s00415-011-6030-0. [DOI] [PubMed] [Google Scholar]
- 70.Lehmann M, Ghosh PM, Madison C, et al. Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4-positive AD patients. J Neurol Neurosurg Psychiatry. 2014;85(3):266–273. doi: 10.1136/jnnp-2013-305858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ossenkoppele R, Zwan MD, Tolboom N, et al. Amyloid burden and metabolic function in early-onset Alzheimer’s disease: parietal lobe involvement. Brain: a journal of neurology. 2012;135(Pt 7):2115–2125. doi: 10.1093/brain/aws113. [DOI] [PubMed] [Google Scholar]
- 72.Johnson KA, Schultz A, Betensky RA, et al. Tau PET imaging in aging and early Alzheimer’s disease. Ann Neurol. 2015 doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol. 2015;14(1):114–124. doi: 10.1016/S1474-4422(14)70252-2. [DOI] [PubMed] [Google Scholar]
- 74.Dickerson BC, Makaretz S, Xia C, McGinnis S, Caso C, Johnson KA. An update on imaging typical and atypical AD and FTLD spectrum tauopathies with [18F] AV1451 PET and PiB PET; 10th Human Amyloid Imaging Conference; 2016; Miami, FL. [Google Scholar]
- 75.Wolk DA, Dickerson BC, Alzheimer’s Disease Neuroimaging I Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107(22):10256–10261. doi: 10.1073/pnas.1001412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barnes J, Dickerson BC, Frost C, Jiskoot LC, Wolk D, van der Flier WM. Alzheimer’s disease first symptoms are age dependent: Evidence from the NACC dataset. Alzheimers Dement. 2015 doi: 10.1016/j.jalz.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE varepsilon4 allele. Lancet Neurol. 2011;10(3):280–288. doi: 10.1016/S1474-4422(10)70306-9. [DOI] [PubMed] [Google Scholar]
- 78.Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82(6):1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62(1):42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 2012;73(6):1216–1227. doi: 10.1016/j.neuron.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mesulam MM. Neuroplasticity failure in Alzheimer’s disease: bridging the gap between plaques and tangles. Neuron. 1999;24(3):521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 82.Hof PR, Morrison JH. Hippocampal and neocortical involvement in normal brain aging and dementia: morphological and neurochemical profile of the vulnerable circuits. J Am Geriatr Soc. 1996;44(7):857–864. doi: 10.1111/j.1532-5415.1996.tb03748.x. [DOI] [PubMed] [Google Scholar]