

Abstract
Conventional TopN data-dependent acquisition (DDA) LC–MS/MS analysis identifies only a limited fraction of all detectable precursors because the ion-sampling rate of contemporary mass spectrometers is insufficient to target each precursor in a complex sample. TopN DDA preferentially targets high-abundance precursors with limited sampling of low-abundance precursors and repeated analyses only marginally improve sample coverage due to redundant precursor sampling. In this work, advanced precursor ion selection algorithms were developed and applied in the bottom-up analysis of HeLa cell lysate to overcome the above deficiencies. Precursors fragmented in previous runs were efficiently excluded using an automatically aligned exclusion list, which reduced overlap of identified peptides to ∼10% between replicates. Exclusion of previously fragmented high-abundance peptides allowed deeper probing of the HeLa proteome over replicate LC–MS runs, resulting in the identification of 29% more peptides beyond the saturation level achievable using conventional TopN DDA. The gain in peptide identifications using the developed approach translated to the identification of several hundred low-abundance protein groups, which were not detected by conventional TopN DDA. Exclusion of only identified peptides compared with the exclusion of all previously fragmented precursors resulted in an increase of 1000 (∼10%) additional peptide identifications over four runs, suggesting the potential for further improvement in the depth of proteomic profiling using advanced precursor ion selection algorithms.
Keywords: data-dependent acquisition, DDA, proteomics, indexed exclusion, dynamic exclusion, LC–MS retention time alignment
Graphical abstract
Introduction
In a typical precursor-driven “TopN” LC–MS data-dependent acquisition (DDA) bottom-up experiment, precursor ions are automatically selected for fragmentation, without a priori knowledge, using simple rules. The precursor selection logic includes highest intensity priority, intensity thresholds, exclusion of specified m/z ranges (e.g., 445.12 Th, polysiloxane), dynamic exclusion,1 and charge-state selection.2 The most important goals of precursor selection logic are noise filtering based on charge and intensity of ion species (e.g., z > + 1 for tryptic peptides and intensity above a set threshold) and deeper sample probing using dynamic exclusion, which prevents redundant selection of a precursor for a set period of time after a fragment (MS2) scan has been acquired. Although commonly used, the conventional TopN data acquisition strategy is limited by stochastic and biased sampling,3,4 fragmentation of precursors before and after the chromatographic peak maximum,5 cofragmentation of near isobaric coeluting precursors resulting in difficult to interpret chimeric spectra,6 and redundant selection of the same precursors between replicate analyses or within individual LC–MS experiments. In bottom-up proteomic analysis of complex samples, these deficiencies lead to the identification of only a minority of detectable peptides within individual experiments (typically <30%).7 Additional peptides can be identified by repeated analysis, but due to sampling bias toward high-intensity features, high-abundance peptides are redundantly identified while low-abundance peptides are neglected. Thus, repeated analysis eventually leads to saturation where few new peptides are identified by additional technical replicates.8
To circumvent the deficiencies of conventional DDA, multiplexed and data-independent acquisition (DIA) strategies have been implemented in the bottom-up proteomic analysis.9 In multiplexed data acquisition, several precursors are simultaneously selected and fragmented to increase the rate of precursor sampling. In DIA experiments, fragment information is acquired for all eluting precursors also by parallel fragmentation of multiple precursors. DIA does not require precursor selection but rather uses a systematic scheme for scanning the entire m/z range of interest (e.g., MSE,10 SWATH,11,12 MSX-DIA,13 and pSMART14). A given peptide is more likely to be identified from a spectrum obtained by isolation in a corresponding narrow m/z window (e.g., 2 Th) such as in conventional DDA than from a multiplexed DDA or DIA spectrum (e.g., 20–25 Th isolation windows) due to challenges in interpreting multiplexed spectra and due to improved selectivity and sensitivity (for the individual precursor) in narrow isolation windows. Furthermore, DIA is incompatible with isobaric tag quantitation due to intermixing of reporter ion signals when multiple precursors are cofragmented.15,16 However, DIA can serve as a means for obtaining fragmentation spectra for peptides that would otherwise be neglected by conventional TopN analysis, which results in complementary coverage between the two strategies.17
Aside from improving physical separation of analytes by multidimensional and high-resolution liquid chromatography,18 the depth of DDA-based proteomic profiling can be enhanced with strategies for improved precursor selection. The most straightforward solution for sampling peptides missed by conventional DDA is targeted analysis (e.g., parallel reaction monitoring) to subject peptide-like precursors unidentified in the initial LC–MS run(s) to scheduled fragmentation in subsequent replicate analyses. Several versions of this solution have been demonstrated to be advantageous over the conventional approach.19 One notable example, Post Analysis Data Acquisition (PAnDA), performs an automated database search after conventional DDA analysis to generate a list of precursor targets that were not identified in the conventional analysis, followed by targeted MS analysis to fragment these features.20 The authors reported a 30.9% gain (3849 vs 2941) and 20.5% gain in peptide and protein identifications, respectively, in the bottom-up analysis of C. elegans proteome after six iterations.20 A related strategy implemented on an Orbitrap Elite mass spectrometer combined targeted and untargeted analysis by fragmenting preset peptides when they were detected and performing conventional Top 15 DDA simultaneously to identify other peptides in the sample in an untargeted manner.21 Although effective, the targeted strategy is dependent on the reproducible appearance of the precursors at the same retention time between runs. This is, unfortunately, not always the case for low-intensity peptides in complex mixtures, where ion suppression from high-abundance species and chromatographic variability can mask the precursor in a somewhat stochastic manner.22 If a peptide is not detected in the initial survey, it will not be targeted, and if the elution time of the peptide changes during targeted analysis, it will likely not be detected.
An alternative to targeted analysis for increasing protein coverage in shotgun proteomics is scheduled exclusion of m/z ranges corresponding to previously identified peptides for set retention time intervals in previous gradient runs. Accurate Mass Exclusion-based DDA (AMEx), has been reported to increase the number of identified peptides by 26% (4490 vs 3564) over conventional DDA by iteratively excluding peptides, which were identified in previous LC–MS/MS runs (six iterations).23 However, the inability to adjust for retention time shifts and the lack of on-the-fly deconvolution algorithms for both the charge and isotopic states of precursor ions have hampered the benefits of conventional interexperiment exclusion (enabled through Xcalibur) in our hands. It is possible to extend the control of LTQ Orbitrap-based mass spectrometers (Thermo Fisher Scientific) beyond the standard functionality allotted by Xcalibur through LTQ COM Object, which communicates with the instrument's low-level data acquisition software. A previous version of COM Object, instrument OCX, has been implemented by MaxQuant Real-Time, which has allowed for the identification of peptides and SILAC pairs on-the-fly.24 In the current paper, the implementation of COM Object, Smart MS2 (Spectroglyph, Kennewick, WA), creates a user interface software that customizes the control of LTQ Orbitrap-based mass spectrometers and expands on the iterative precursor exclusion strategy implemented in AMEx.23 Several additional algorithms were developed and implemented through Smart MS2 to improve the efficacy of precursor exclusion: noise filtering based on persistent precursor detection, deconvolution of peptide precursors in multiple charge and isotopic states, an indexed dynamically aligned exclusion list, dynamic exclusion extended for the entire precursor elution profile, a percent chimeric intensity (PCI) filter,6 and an integrated postacquisition database search that generates an exclusion list of only identified peptides. The application of the developed algorithms results in deeper probing of the HeLa proteome than possible with the conventional DDA strategy. The algorithms were tested on an LTQ Orbitrap XL as a proof-of-concept, and similar strategies can benefit the performance of more advanced and faster duty cycle mass spectrometry platforms.
Materials and Methods
Materials
HPLC-grade water, acetonitrile, formic acid, and Pierce HeLa tryptic digest standard were acquired from Thermo Fisher Scientific (San Jose, CA). The column was prepared in-house by polymerizing a frit from a 3:1 mixture of Kasil 1 (29.1% potassium silicate solution) from PQ Corporation (Valley Forge, PA) and formamide from Sigma-Aldrich (St. Louis, MO) inside a 75 μm internal diameter, 360 μm outer diameter polyimide-coated fused silica capillary from Polymicro/Molex (Phoenix, AZ). The frit was cut to ∼0.2 mm, and the end was polished. The fritted capillary was packed with Magic C18AQ, 3 μm diameter, 200 Å pore size beads from Michrom Bioresources (Auburn, CA) to 30 cm of stationary phase. Liquid chromatography (LC) separation was performed on an Ultimate 3500 system from Thermo Fisher Scientific, and mass spectrometry data were acquired on an LTQ Orbitrap XL ETD (Tune Plus version 2.5.5) from Thermo Fisher Scientific. The sample was electrosprayed using a distal-coated 20 μm internal diameter, 360 μm outer diameter tip with a 10 μm opening from New Objective (Woburn, MA) connected directly to the column head using a Teflon sleeve.
Liquid Chromatography
Reversed-phase liquid chromatography was carried out using mobile phase A: 0.1% formic acid in water and mobile phase B: 0.1% formic acid in acetonitrile. Twenty μg of HeLa digest was dissolved in 100 μL of 2% acetonitrile, 0.1% formic acid in water. One μL (200 ng HeLa lysate) was injected directly onto the analytical column using 2% B at 250 nL/min. The sample was loaded and desalted over 10 min in 2% B and separated at 250 nL/min flow rate using the following linear gradient: 2– 37% B over 120 min, 37–95% B over 10 min, 95% B hold for 9 min, 95% to 2% B in 1 min, 2% B hold for 10 min.
Mass Spectrometry
Data were acquired on an LTQ Orbitrap XL mass spectrometer using matching parameters for conventional analysis enabled through Xcalibur (v. 2.0.7) and intelligent, user-defined DDA analysis enabled through Smart MS2 (Spectroglyph, Kennewick, WA). Full precursor scans were acquired over a 400–1700 Th range in the Orbitrap at 60 000 resolution (at 400 Th), with the AGC set to 1 000 000 and 500 ms maximum ion accumulation time. The 10 highest intensity eligible precursors (Top 10) were serially selected in 2 Th wide isolation windows centered on the monoisotopic peak and activated for a maximum of 30 ms by CID at 35% normalized energy. In fragment ion accumulation, the AGC target was set to 40 000, the maximum injection time was 100 ms, and the ion trap was scanned at 0.5 Da resolution. Noise filtering and exclusion of previously (during the same LC–MS run) fragmented precursors were accomplished differently between the two approaches. In the conventional analysis, ions with a +1 charge state and lower than 500 intensity units were considered chemical noise, and precursors were excluded for 60 s after fragmentation to prevent redundant sampling. In Smart MS2 analysis, precursors with a charge of +2 or higher and at least two isotopic peaks detected in at least three consecutive precursor scans were considered fragmentation candidates (not noise). In Smart MS2 analysis, previously fragmented precursors in all isotope and charge states were excluded for a minimum of 30 s with the exclusion period extended until the end of the precursor's elution profile.
Exclusion of Identified Peptides
Completion of data acquisition for each LC–MS run triggered execution of an in-house developed Python script based on the Pyteomics library25 to perform an X!Tandem26 (version 2013.09.01.1 Sledgehammer) search against the UniProt human database described below, appended with an equal number of reverse decoy sequences. Carbamidomethylation of cysteine was set as a static modification, and up to one tryptic missed cleavage was allowed. The error tolerances were set at 10 ppm for the precursor mass (with the monoisotopic peak mismatch enabled) and 0.4 Da fragment mass error tolerance. The scored spectral matches were filtered to 1% FDR, and the accepted precursors remained in the Smart MS2 exclusion database. The precursors that were not identified were removed from the exclusion list, which made them eligible for refragmentation in subsequent runs. Smart MS2 data acquisition was initiated 8 min after injection and terminated at 140 min (before column washing and equilibrating) to allow sufficient time to complete the database search and exclusion list generation before the subsequent replicate.
Data Analysis
The acquired data was analyzed in Proteome Discoverer 1.4 (Thermo Fisher Scientific) with Sequest HT27 peptide spectral match scoring and Percolator28 validation and filtering (q < 0.01), and with MaxQuant (v. 1.5.2.8) with 1% FDR filtering.29 Both searches were conducted against a human UniProt database containing canonical proteins and known variants from March 2014 appended with 47 common contaminants (88 894 total entries). The maximum precursor mass error was set to 10 ppm, and the maximum fragment mass error was set to 0.6 Da. Carbamidomethylation of cysteine was set as a static modification, and oxidation of methionine and deamidation of glutamine and asparagine were set as dynamic modifications. Up to two missed tryptic cleavages were allowed. Evaluation of overlapping identifications and cumulative identifications were carried out in Excel.
Results and Discussion
Precursor Selection Algorithms Applied through Smart MS2
The Smart MS2 flow-control process is diagrammed in Figure 1. Elution profiles of all fragmentation candidates were monitored in real-time by Smart MS2 software. Once fragmented, precursors in all charge and isotopic states were excluded for a user-defined minimum exclusion period (30 s in the current experiment). If the software determined that an elution profile of a previously fragmented precursor ion had extended past the minimum exclusion period, the exclusion time of that precursor ion was then automatically extended until elution of the precursor was complete. Nonredundant fragmentation of precursors between replicate experiments was accomplished by an indexed exclusion list where retention times of precursor ions were dynamically aligned during data acquisition based on the retention times of automatically selected anchor peptides using a 2 min retention time adjustment window. Anchor peptides were automatically identified by Smart MS2 from the initial experiment in each series by selecting the highest intensity ion species in each 5 min long interval and identifying their chromatographic peak apex positions. The percent chimeric intensity filter (PCI)6 was set to 500%, meaning that if a second precursor appeared within the isolation window of an excluded precursor, this second precursor would be considered an eligible candidate for MS2 only if its intensity is at least 5 times greater than the intensity of the excluded precursor. This threshold was selected based on the definition of chimeric spectra used in Houle et al., which stated that the spectrum match score is significantly reduced at a PCI ≥ 20%. Because we are interested in the “contaminant” precursor, the inverse value is used.6
Figure 1.
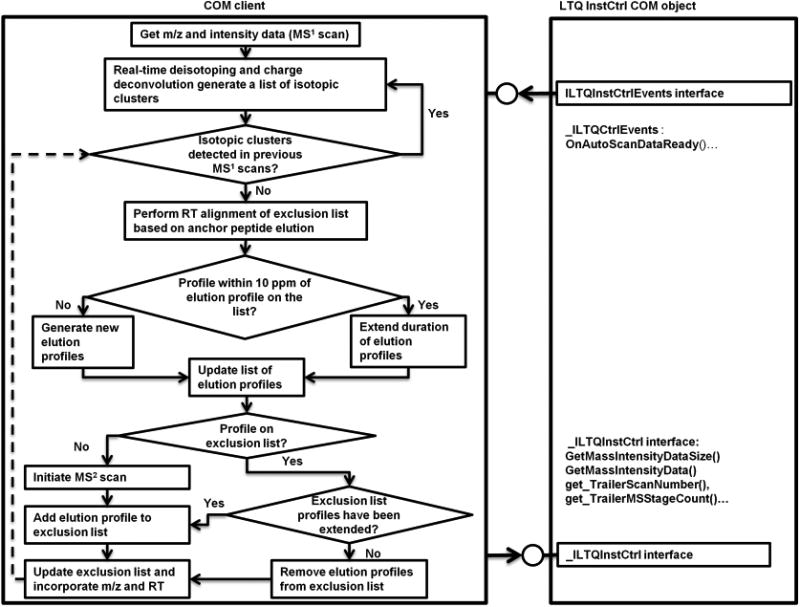
Smart MS2 process workflow. The flow control diagram of the Smart MS2 algorithm (left side) and programmatic interfacing to Thermo's COM Object (right side). The latter incorporates interface “ILTQInstCtrl”, which controls the instrument and dispatch interface “_ILTQInstCtrlEvents” used to receive instrument status updates. Upon receiving an event from “_ILTQInstCtrlEvents”, Smart MS2 adds a list of m/z and intensity data from the latest scan to the previously generated dynamic list, performs real-time isotope deconvolution, and then updates the dynamic list of elution profiles, which also incorporates information on the retention time and charge states of the deconvolved features. A feature represented by, at least, C12 and C13 isotopes is considered to be an elution profile (non-noise precursor) if observed in, at least, two consecutive MS1 scans at a mass accuracy of 10 ppm. The algorithm performs real-time retention domain alignment using higher intensity anchor peptides and then determines whether an elution profile is off the exclusion list, and the profile intensity matches user-defined criteria (e.g., top 10, bottom 10, middle 10–20, etc.) for collisional activation. If the above conditions are met, the profile of interest is added to the MS2 attention list to be passed over the instrument through the “ILTQInstCtrl” interface. Redundant elution profiles of the same species represented by lower intensity charge states are excluded. Upon completion of the MS2 event, the profile is added to the dynamic exclusion list (which can be expanded with profiles from previous experiments) and then monitored real-time during the experiment. Once eluted off the column, the profile is dynamically removed from the exclusion list. If an elution profile, which is not on the exclusion list, overlaps with nearly isobaric excluded profile in the retention time domain and conforms to percent chimeric intensity (PCI) filter and user-defined criteria for precursor ion fragmentation (e.g., top 10, etc.), the elution profile would be chosen for MS2 fragmentations. In summary, Smart MS2 algorithm monitors all elution profiles concurrently, dynamically puts them on or removes from the exclusion list, performs real-time alignment of retention time domains, and conducts MS2 experiments based on user-defined activation criteria.
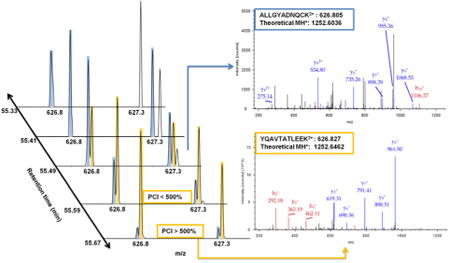
Figure 2A presents an example of a precursor being recovered despite coelution with a near-isobaric (i.e., within the same 2 Th isolation window) previously fragmented precursor using the PCI filter. Conventional dynamic exclusion could mask the presence of near-isobaric coeluting species by acquiring one spectrum when one species may be underrepresented and then excluding both precursors until both have eluted. Compounding dynamic exclusion with the exclusion of precursors from previous experiments can quickly deplete the allowed sampling space (a situation where most m/z windows are ineligible for precursor ion selection for most of the LC- gradient to prevent redundant fragmentation of previously selected precursors). The implemented PCI filter recovered sampling of 300–900 precursors in each run. While this translates to <10% of the total acquired MS2 scans, without the PCI filter the number of acquired MS2 spectra is significantly decreased at the third iteration (data not shown). This decrease is delayed until the fourth iteration by application of the PCI filter.
Figure 2.
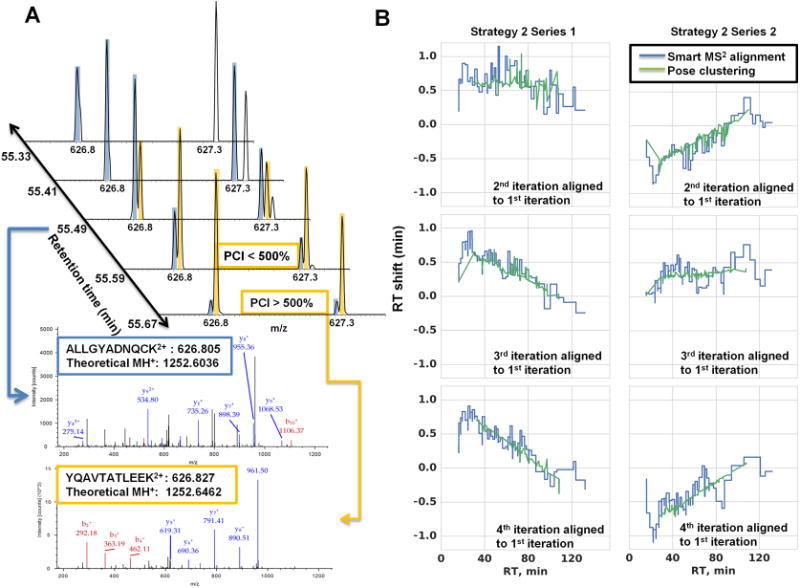
Application of PCI candidate recovery and on-the-fly exclusion list adjustment. (A) Example of a candidate (YQAVTATLEEK, orange) which would not be eligible for fragmentation due to coelution with a candidate that has been previously fragmented (ALIGYADNQCK, blue) within the same 2 Th isolation window. However, the precursor is fragmented when its intensity surpasses the previously fragmented candidate by the 500% PCI threshold. (B) Agreement between Smart MS2 on-the-fly retention time adjustment of the indexed exclusion list and pose-clustering alignment relative to the first run in the exclusion series. Pose-clustering did not align the earliest and latest regions of the gradient due to high variability in elution of the most hydrophilic and hydrophobic peptides.
The Smart MS2 noise filtering strategy permits the selection of very low-abundance features, while the conventional intensity threshold approach does not consider features below the intensity threshold. The filter implemented in Smart MS2 relies on the persistent appearance of the isotopic envelope of the precursor in at least three sequential precursor scans independent of intensity. After detection in two consecutive scans, the precursor is considered a candidate and may be selected for fragmentation in the third scan if it is present. At least two isotopic peaks (i.e., 12C and 13C peaks) were considered an isotopic envelope because for many low-abundance features the M+2 peak (i.e., Two 13C) was not consistently detectable. This approach allowed the mass spectrometer to sample precursors that would be considered noise by a conventional threshold filter and, in some cases, acquire informative MS2 spectra.
Smart MS2 real-time precursor ion charge deconvolution treats all m/z peaks derived from the same peptide species as one entity, so that higher intensity ion species, for example, [M +2H]2+, would be fragmented, while lower intensity species (e.g., [M+3H]3+ and [M+4H]4+) would be excluded. In addition, extended dynamic exclusion prevents redundant fragmentation of a precursor for a minimum exclusion duration that is reset if the precursor is persistently detected after the minimum set duration. This is in contrast with conventional DDA, where a set exclusion duration time, which is the same for every precursor, is the only means to control redundant fragmentation of precursor ions.
The most important feature of Smart MS2 is the automatic alignment of the indexed exclusion list during data acquisition using a set of high-intensity anchor peptides automatically selected from the first run. The database of precursors to be excluded is dynamically adjusted by the difference between the reference and observed retention times of the anchor peptide chromatographic apexes. Figure 2B demonstrates the agreement in the on-the-fly alignment of the exclusion list performed by Smart MS2 with the pose-cluster alignment30 implemented in OpenMS31 of the reference LC–MS run (the first iteration of exclusion series) and each subsequent LC–MS iteration in the series. While the two alignment strategies are inherently different, the general agreement indicates that the adjustments made by Smart MS2 adequately address the retention time shifts between replicate experiments.
The postacquisition database analysis, triggered by Smart MS2 at the end of each LC–MS experiment, executed an X!Tandem database search. The peptides identified at FDR < 1% remained in the exclusion list, while the unidentified precursors were removed from the exclusion list and were made eligible for fragmentation in subsequent runs. A relatively short time interval during column washing and equilibration was allocated for the database search so X!Tandem was used because it is a fast and readily available open-access search algorithm. To further increase the benefits of the postacquisition database search and exclusion of only identified peptides, more comprehensive search strategies can be implemented in the future.
Performance Metrics of Smart MS2 and Conventional DDA
Three DDA strategies were compared: (1) conventional Xcalibur driven Top 10 approach, to be referred as Strategy 1, (2) Smart MS2-driven analysis in which all previously fragmented precursors were iteratively excluded in subsequent replicate LC–MS/MS analyses, to be referred as Strategy 2, and (3) Smart MS2-driven analysis, in which only identified peptides (FDR < 1%) were iteratively excluded in subsequent replicates, to be referred as Strategy 3. The differences in precursor selection algorithms implemented in the three data acquisition strategies are summarized in Table 1. Strategy 1 was evaluated using eight LC–MS/MS runs to approximate the saturation of analysis (the maximum number of peptide species identified using the analytical platform) in duplicate using two batches of HeLa lysate (Replicate 1 and Replicate 2). Three series of four iterations were used to evaluate the performance of Strategy 2. In a Strategy 2 series, the precursors fragmented during the first iteration (i.e., the initial analysis of the sample) were excluded from selection in the second iteration (i.e., second replicate LC–MS/MS analysis of the same sample), the precursors fragmented in the first and second iteration were excluded in the third iteration, and all previously fragmented precursors were excluded in the fourth iteration. Three Strategy 2 series were benchmarked against the extrapolated Strategy 1 saturation. Finally, Strategies 2 and 3 were compared using the same four-iteration series experimental design, in triplicate.
Table 1. Precursor Selection Logic Implemented in Each Data Acquisition Strategy.
| precursor selection logic | Strategy 1: conventional DDA | Strategy 2: smart MS2 iterative exclusion of all precursors | Strategy 3: smart MS2 iterative exclusion of identified peptides |
|---|---|---|---|
| intensity-based precursor selection | 10 highest intensity eligible precursors from each MS1 scan are selected for MS2 (top 10) | ||
| noise filtration | charge and intensity threshold | charge and defined elution profile (isotopic envelope and consecutive scan appearance) | |
| percent chimeric intensity (PCI) filter | N/A | precursor becomes an eligible MS2 candidate if PCI filter threshold is exceeded | |
| intraexperiment (dynamic) exclusion | 60 s exclusion after MS2 | 30 s minimum m/z exclusion extended until end of elution | |
| interexperiment exclusion | N/A | all precursors fragmented in previous experiments | only peptides identified in previous experiments |
| postacquisition database search | N/A | N/A | X!Tandem (1% FDR) |
Table 2 presents the performance metrics of the tested data acquisition strategies. It is possible to schedule exclusion of specified m/z ranges in Xcalibur, but our attempt to implement exclusion of all identified peptides between two replicate analyses of complex proteomic samples found no benefit in this strategy compared with simply performing two replicate analyses (Supplementary Figure 1). To be effective, exclusion of previously fragmented or identified precursors requires exclusion of all charge and isotopic states and highly reproducible chromatography (or sufficiently wide retention time exclusion windows) as with AMEx23 or on-the-fly exclusion list alignment as in our approach. Thus, exclusion was not implemented in the two sets of experiments evaluating the saturation of Strategy 1 (conventional DDA). The identification results of Strategy 1 improved in the second replicate, as indicated by an increase in MS2 scans, peptide spectral matches, and peptide identifications, which can possibly be attributed to differences in sample batches or experimental variables. However, even with the improved performance, the cumulative peptide identification increased by <4% over eight runs. Smart MS2 performance in the first exclusion iteration (no interexperiment precursor exclusion is applied) acquired fewer precursor (MS1) and MS2 scans than the conventional analysis due to (i) the inability to perform parallel MS1 and MS2 experiments with Thermo's COM Object and (ii) latency in data transfer between the external and internal PCs controlling the mass spectrometer. Evaluation of MaxQuant Real-Time has also observed these latency issues.24 However, the ratio of acquired MS2 scans to precursor scans was higher in the Smart MS2 experiments: 6.3, 7.0, and 7.2 (Strategy 2 replicates 1 and 3 and Strategy 3, respectively) compared with 4.7 and 3.4 in Strategy 1 (see Table 2). This suggests that the Smart MS2 data acquisition approach was more efficient, but somewhat slower in acquiring MS2 scans.
Table 2. Performance Metrics for Each Data Acquisition Strategya.
| DDA strategy | HeLa lysate batch | iteration | full scans | MS2 scans | PSMs | peptides | protein groups |
|---|---|---|---|---|---|---|---|
| Strategy 1: Conventional DDA | Replicate 1 | average of 8 | 2942 ± 39 | 13 720 ± 483 | 9217 ± 294 | 6758 ± 247 | 1611 ± 45 |
| cumulative 4 runs | 9673 | 2005 | |||||
| cumulative 8 runs | 11 003 | 2448 | |||||
| Replicate 2 | average of 8 | 4660 ± 74 | 15 850 ± 81 | 10 130 ± 112 | 6998 ± 66 | 1764 ± 19 | |
| cumulative 4 runs | 9810 | 2157 | |||||
| cumulative 8 runs | 11 443 | 2597 | |||||
| Strategy 2: Smart MS2 iterative exclusion of all precursors | Replicate 1 | 1st iteration | 2525 ± 72 | 15871 ± 288 | 7209 ± 159 | 6283 ± 170 | 1754 ± 30 |
| 2nd iteration | 3019 ± 83 | 13 887 ± 461 | 2957 ± 282 | 2719 ± 275 | 1627 ± 118 | ||
| 3rd iteration | 3690 ± 359 | 11 563 ± 1368 | 2021 ± 884 | 1839 ± 817 | 1240 ± 348 | ||
| 4th iteration | 4326 ± 851 | 8178 ± 1541 | 1556 ± 878 | 1398 ± 795 | 965 ± 381 | ||
| cumulative | 10 549 ± 285 | 2817 ± 17 | |||||
| Replicate 3 | 1st iteration | 2110 ± 114 | 14 854 ± 393 | 6948 ± 93 | 6004 ± 79 | 1739 ± 19 | |
| 2nd iteration | 3079 ± 30 | 11 416 ± 249 | 2854 ± 309 | 2558 ± 265 | 1569 ± 98 | ||
| 3rd iteration | 4528 ± 395 | 7255 ± 1160 | 1365 ± 332 | 1237 ± 312 | 933 ± 181 | ||
| 4th iteration | 5174 ± 835 | 5389 ± 2323 | 1402 ± 1158 | 1257 ± 1016 | 838 ± 470 | ||
| cumulative | 9757 ± 244 | 2661 ± 35 | |||||
| Strategy 3: Smart MS2 iterative exclusion of identified peptides | Replicate 3 | 1st iteration | 2072 ± 50 | 14 844 ± 136 | 7170 ± 277 | 6160 ± 272 | 1781 ± 24 |
| 2nd iteration | 2069 ± 43 | 14 547 ± 335 | 4106 ± 335 | 3579 ± 262 | 1661 ± 69 | ||
| 3rd iteration | 2017 ± 72 | 14 445 ± 487 | 3305 ± 332 | 2847 ± 260 | 1467 ± 55 | ||
| 4th iteration | 2071 ± 47 | 14 793 ± 107 | 3036 ± 371 | 2626 ± 338 | 1416 ± 109 | ||
| cumulative | 10 768 ± 483 | 2563 ± 12 |
Performance of Strategy 1 was evaluated using two batches of HeLa lysate (Replicate 1 and 2), and the results are presented as an average of eight runs and cumulative results from four and eight runs. Strategy 2 was evaluated using the Replicate 1 HeLa lysate batch, and the results are presented as an average of three series. Strategies 2 and 3 were compared using the Replicate 3 batch of HeLa lysate, and the results are again presented as an average of three series.
Effects of Iterative Exclusion (Strategy 2) on Precursor Selection
Iterative exclusion implemented in Smart MS2 effectively prevented redundant fragmentation of precursors between technical replicates. Figure 3A demonstrates the overlap in peptides identified by Sequest HT with Percolator rescoring and filtering (q < 0.01, equivalent to FDR < 1%) between three runs acquired using Strategy 1 (left) and Strategy 2 (right). The typical Strategy 1 overlap in peptide identifications between runs of roughly 70% is decreased to 10–20% when iterative exclusion is applied (Table 3). Iterative exclusion also affects the intensity distribution of features sampled. Figure 3B was generated through label-free quantitative analysis (MaxQuant), which (1) identified all peptide-like LC–MS features (at least three peaks in the isotopic envelope, and charge +2 to +5), (2) determined which features were fragmented, (3) identified peptides using the Andromeda search engine, and (4) filtered to FDR < 1%. The peptide-like features were sorted by intensity into 20 5-percentile bins, and the percentage of sampled and identified peptide-like features in each bin was determined. The orange line represents the combined feature identifications from all eight Strategy 1 runs with the expected high-intensity bias observed for conventional DDA in other studies.7 The Strategy 2 iterations, presented as an average of three experiments, show a reduction in sampling bias with each iteration of precursor exclusion. The first iteration resembles the Strategy 1 distribution, the second iteration shows a shift toward sampling of medium intensity features, and the third and fourth iterations show evenly distributed sampling across the intensity range. As expected, the ratio of identified to sampled features correlated with precursor ion intensity in both Strategies 1 and 2 (not shown) because low-intensity precursor ion signals are less likely to produce an identifiable fragmentation spectrum. Another caveat of iterative exclusion is the eventual depletion of the eligible MS2 candidate pool, as manifested in a decrease in the number of acquired MS2 scans and identified unique peptides in the third and fourth iterations (Table 2).
Figure 3.
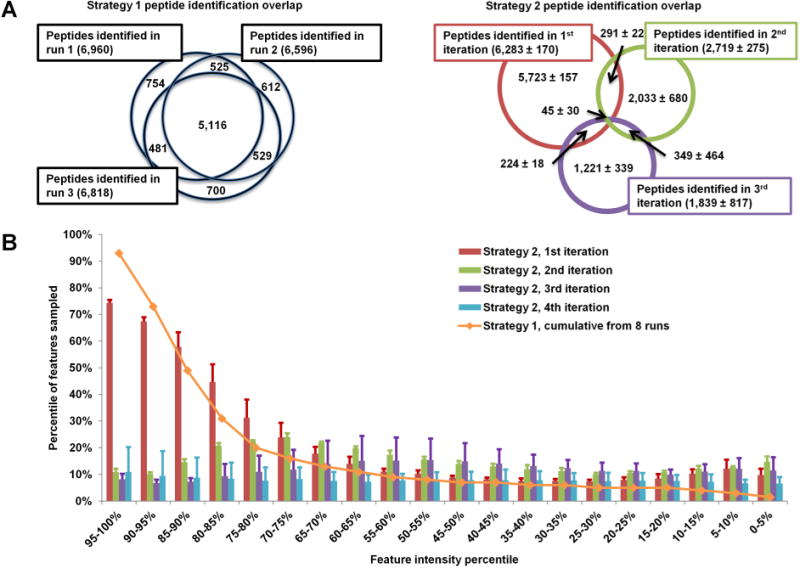
Effect of iterative exclusion. In conventional analysis (Strategy 1), over 70% of identified peptides are redundant between replicates (A, left). In iterative exclusion (Strategy 2), this overlap is reduced to 10–30% (A, right). This decrease in redundant fragmentation allows the MS instrument to target medium and low-abundance peptides, as shown in panel B, where the blue line presents the sampling coverage by Strategy 1 from eight replicate runs and the bar graphs represent the iterations of Strategy 2 (average of three runs).
Table 3. Overlap in Peptide Identifications between Replicate LC–MS Experimentsa.
| experimental condition | second iteration, peptide IDs | third iteration, Peptide IDs | fourth iteration, peptide IDs | |||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| overlap | cumulative | overlap | cumulative | overlap | cumulative | |
| Strategy 1 (Rep. 1) | 79.4% | 8322 | 88.4% | 9113 | 91.9% | 9673 |
| Strategy 1 (Rep. 2) | 79.2% | 8395 | 88.2% | 9221 | 91.5% | 9810 |
| Strategy 2 (Rep. 1) | 12.5% ± 1.4% | 8666 ± 290 | 30.7% ± 10.0% | 9886 ± 51 | 46.3% ± 18.5% | 10549 ± 285 |
| Strategy 2 (Rep. 3) | 11.0% ± 1.4% | 8281 ± 186 | 25.2% ± 4.5% | 9198 ± 27 | 47.1% ± 15.6% | 9757 ± 244 |
| Strategy 3 (Rep. 3) | 33.6% ± 1.4% | 8539 ± 484 | 55.8% ± 4.6% | 9789 ± 459 | 62.4% ± 3.5% | 10768 ± 483 |
Strategy 1 Replicates 1 and 2 demonstrate typical overlap determined from the first four replicates in each set. The Strategy 2 and 3 results are averaged across 3 series. The low overlap in the second iteration of Strategy 2 indicates that almost 90% of the peptides identified in the second iteration were not detected in the first iteration. In Strategies 2 and 3 the overlap increases in subsequent iterations because the population of previously observed peptides grows while the number of identified peptides decreases due to increased sampling of low-abundance species.
Differences in Identifications between Strategies 1 and 2
Figure 4A presents the cumulative peptide identifications using Strategy 1 (Replicates 1 (dark blue) and 2 (purple)) and Strategy 2. Extrapolation of experimental data (dotted lines) shows that Strategy 1 would reach the saturation level of approximately 12 500 peptides (dotted light-blue line) after 12+ replicates. After four iterations of Strategy 2, 84.4 ± 2.2% of all peptides identified at the approximate saturation level are identified. When peptide identifications from two Strategy 2 series are combined, the saturation level is exceeded by 13%. When results from three Strategy 2 series are combined, the saturation is exceeded by 29% (16 156 total peptide identifications). There is a noticeable decrease in the gain of new identifications in the fourth iteration of Strategy 2 in comparison with preceding iterations. While the fourth iteration provides a substantial number of novel peptide identifications, if the identifications from only the first three iterations in each Strategy 2 series are combined, then a total of 15 168 peptides (21% above saturation) are identified over nine runs (not shown), providing a quicker alternative for in-depth analysis. Figure 4B presents the overlap between the cumulative protein group identifications using Sequest HT and Percolator (q < 0.01, equivalent to FDR < 1%) in each of the Strategy 2 series (top left Venn diagram) and the overlap in protein identifications between Strategy 1 and Strategy 2 (bottom left Venn diagram). The same Venn diagrams are presented in the right panel when a minimum of two peptides per protein group filter are applied in addition to q < 0.01 for more stringent protein identifications. Without the two-peptide filter, over 1000 more protein groups are identified with Strategy 2. However, this advantage diminishes to several hundred when the filter is included. Many single-peptide protein groups were identified with high confidence in multiple Strategy 2 series, as reflected by the drop in overlapped protein group identifications when the two-peptide filter is applied, which improves confidence in the identifications despite reliance on a single peptide. The same data were also processed using MaxQuant (FDR < 1%). MaxQuant identified a total of 2963 protein groups with 304 protein groups unique to Strategy 2 and 23 protein groups unique to Strategy 1. The majority of proteins unique to either strategy are within the lowest 20 intensity percentile (data not shown). Depending on the stringency of data analysis (minimum of either 2 or 1 peptide per protein group, q < 0.01), Strategy 2 identified between 300 and 1000 additional low-abundance protein groups undetected by Strategy 1, indicating that the dynamic range of analysis is increased with more efficient data acquisition. The low-abundance protein identifications vary between the Strategy 2 series, suggesting that the ability to detect these low-abundance species is stochastic even with efficient precursor sampling, so the flexibility of an untargeted approach (i.e., Strategies 2 and 3) may be better for discovering these species than the targeted strategies described in the Introduction (e.g., PAnDA).
Figure 4.
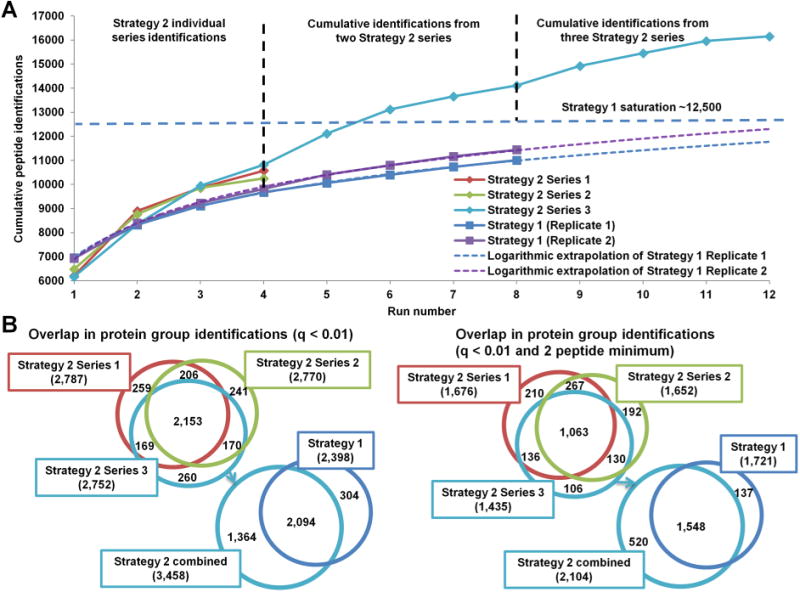
Peptide and protein identification gains with iterative exclusion. (A) Cumulative peptide identifications using either Strategy 1 or Strategy 2. Each 4-iteration Strategy 2 series identified more peptides than four replicates of Strategy 1. The logarithmic extrapolation of Strategy 1 from eight runs estimates the saturation at approximately 12 500 unique peptides (light-blue line). Combined results from two Strategy 2 series exceed Strategy 1 saturation by ∼1500 unique peptides. Combined results from all three Strategy 2 series show that peptide identification does not plateau after over 16 000 peptides identification due to sampling and identification of low-intensity peptides. (B) Overlap in protein group identifications between Strategy 2 series and the combined identifications between Strategies 1 and 2 with single peptide identifications accepted on the left and a 2 peptide minimum filter applied on the right.
Iterative Exclusion of Identified Precursors, Strategy 3 versus All Fragmented Precursors, Strategy 2
One of the major deficiencies of Strategy 2, presented in Table 2, is a drop in MS2 scan acquisition in the third and fourth iterations caused by the depletion of the candidate pool. Many ion precursors already fragmented in the initial LC–MS iteration(s) that were therefore excluded from fragmentation in subsequent iterations did not result in informative MS2 spectra and hence successful peptide identifications. In Strategy 3, to address this issue, an automated database search is executed at the completion of each LC–MS run using an in-house developed Python script to exclude only precursors identified by X!Tandem (FDR < 1%). Strategy 3 shows uniform numbers of MS2 scans acquired across all four LC–MS iterations and allows a second opportunity to acquire better quality spectra for previously fragmented but not identified precursors. Presented in Figure 5A is a significant gain (p < 0.05) of 1000 peptides (∼10%) over four exclusion iterations in Strategy 3 compared with Strategy 2. The performance of Strategy 3 can be further improved with a more comprehensive automated search. As presented in Table 3, the overlap in identified peptides between the first and second iterations is over 30% in Strategy 3 (compared with ∼10% with Strategy 2). This is due to redundant fragmentation of peptides that were not identified by the postacquisition X!Tandem search but are identified by Sequest HT and Percolator (Figure 5B). While a more thorough postacquisition database search would improve performance by reducing the overlap between iterations to the ∼10% level observed with Strategy 2, we were limited by the system resources of the instrument-controlling PC and the allotted search time (duration of column cleaning and equilibration). Thus, X!Tandem was used without the benefits of postsearch validation and rescoring with Percolator. However, even under suboptimal conditions Strategy 3 is superior to Strategy 2 and demonstrates even greater potential after addressing the above-mentioned limitations.
Figure 5.
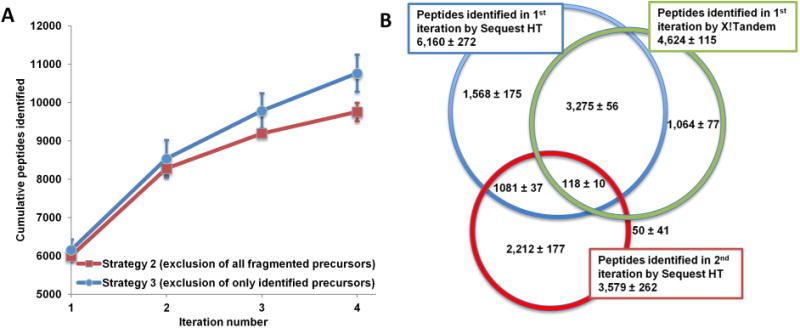
Exclusion of all precursors versus exclusion of identified precursors. Exclusion of identified precursors (Strategy 3) identified on average (n = 3) ∼1000 (or 10%) more peptides over four iterations than the exclusion of all fragmented candidates (Strategy 2) (A). The identification gains were hampered by limitations of the postacquisition search resulting in higher redundant peptide identifications between replicates. The majority of peptides redundantly identified between the first and second iteration of Strategy 3 were not identified in the postacquisition (X!Tandem) search (B).
Conclusions
This work has demonstrated that improved precursor ion selection increases the number of identified peptides in DDA analysis, which results in the identification of low-abundance proteins, previously undetectable with the conventional approach (TopN DDA, Strategy 1). The conventional strategy reaches saturation due to an inefficient sampling of medium and low-abundance species in proteome-complexity samples, while the application of efficient data acquisition provides deeper proteomic profiling. Specifically, to increase the efficiency of iterative DDA analyses, we have developed combined real-time tracking and intelligent handling of all elution profiles of isotopically and charge-state deconvolved precursor ions signals. The proposed strategy includes a dynamic (signal-to-noise and persistent precursor detection based) exclusion list of elution profiles of all precursor ion candidates, alignment of retention time domains of different LC runs, nonredundant triggering of MS2 events using the most abundant charge states of the peptides of interest, use of a percent chimeric intensity (PCI) filter, and automated postdata acquisition database searching performed immediately after completion of the run (integrated into the Smart MS2 workflow to exclude identified precursors from the following analyses). Without the postacquisition database search, 29% (16 156 vs ∼12 500, FDR < 1%) more peptides were identified above the saturation level of conventional data acquisition after 12 runs. The postacquisition search improved the performance of iterative exclusion by an average gain of 1,000 peptides (∼10%) over four exclusion iterations. While a direct comparison is beyond the scope of this publication, these improvements in identifications are comparable to those reported for AMEx and PAnDA (26 and 31% peptide identification gains, respectively, over six iterations).
Recent generations of mass spectrometers are certainly capable of faster precursor sampling rates than an LTQ Orbitrap XL. However, these sampling rates are still insufficient for fragmentation of every eluting precursor in complex samples. Furthermore, newer mass spectrometers also tend to offer higher sensitivity, which translates to more detectable candidates that require even faster sampling rates for comprehensive sampling. As such, even the most advanced mass spectrometers would benefit from improved precursor ion selection strategies such as those presented in this proof-of-concept study. Alternatively, DIA allows comprehensive sampling, but identification of low-abundance precursors from multiplexed spectra can be problematic. While numerous software packages for interpreting DIA data are now available,32 the fundamental obstacle of accumulating sufficient signal for fragments from low-intensity precursors cofragmented with high-intensity precursors is still a limitation. Comparisons of DDA and DIA analysis demonstrated that the two approaches identify complementary peptide populations.14,33 Furthermore, DIA data analysis by targeted data extraction (peptide-centric analysis) requires a comprehensive spectral library that is typically obtained through exhaustive DDA analysis of the sample of interest. The described approach can obtain spectra for peptides beyond the saturation point of conventional DDA in LC–MS proteomic profiling with replicate injections, allowing the opportunity for detection of these low-abundance species in DIA analysis. The depth of the proteomic coverage is commonly increased by higher efficiency separation of analytes using multidimensional and high-resolution liquid chromatography. The efficient exclusion of previously fragmented precursors allows an additional dimension for pseudoseparation by manipulating the precursor ion sampling in MS data acquisition. These developed precursor ion selection and exclusion algorithms will be instrumental in any experiments where liquid-phase separation (e.g., liquid chromatography, capillary electrophoresis, capillary isoelectrofocusing, etc.) is coupled to mass spectrometry to enable deep molecular profiling (i.e., proteomic, lipidomic, metabolomics, etc.) of complex biological samples and especially limited samples, where all steps toward increasing the coverage of profiling are important.34
The performance of the developed algorithms was noticeably decreased by latency in data transfer between the data acquisition PC and the on-board computer. Additionally, the postacquisition database search was limited by the PC system resources. We expect to see even greater advantages in sample analysis by the developed precursor ion selection algorithms with improvements in computational hardware. In summary, better precursor selection can provide a gain in depth of proteomic profiling by reducing the inefficiencies of conventional precursor sampling. Similar gains can be expected in the analysis of post-translational modifications when modified peptides are adequately enriched. It should be noted that quantitation by spectral counting is not possible when high- abundance precursor bias is reduced by iterative exclusion. However, we anticipate that quantitation by precursor peak integration will be further improved by identification of additional peptides per protein. The DDA algorithms reported here were implemented in the Smart MS2 platform, which was developed in collaboration with and can be licensed from Spectroglyph LLC. The required LTQ COM Object instrument control library can be licensed from Thermo Fisher Scientific. The Python script enabling postacquisition database search is available upon request from the Barnett Institute.
Supplementary Material
Acknowledgments
This work was supported by the Barnett Institute of the Chemical and Biological Analysis at Northeastern University, ASMS 2015 Research Award (ARI), NIH 1R01GM120272-01, and the Dana-Farber Cancer Institute/Northeastern University Joint Seed Funding Program in Cancer Drug Development (ARI). We thank Thermo Fisher Scientific for providing access and licensing the LTQ COM Object instrument control library. This is contribution number 1058 from the Barnett Institute.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteo-me.6b00312.
The supplement describes an experiment in which the efficacy of precursor exclusion implemented through the conventional instrument control (Xcalibur) was tested. In this experiment, the overlap in peptide identifications is compared between an initial run, a second run, and a run where all peptides identified in the initial replicate (Sequest HT with Percolator q < 0.01) are excluded. The experiment found that the exclusion implemented through the standard control did not efficiently exclude identified peptides, and there was no benefit in cumulative identifications, which supported our decision not to use the exclusion feature when evaluating conventional DDA (Strategy 1). (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Wang N, Li L. Exploring the precursor ion exclusion feature of liquid chromatography-electrospray ionization quadrupole time-of-flight mass spectrometry for improving protein identification in shotgun proteome analysis. Anal Chem. 2008;80(12):4696–710. doi: 10.1021/ac800260w. [DOI] [PubMed] [Google Scholar]
- 2.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 3.Berg M, Parbel A, Pettersen H, Fenyo D, Bjorkesten L. Reproducibility of LC–MS-based protein identification. J Exp Bot. 2006;57(7):1509–14. doi: 10.1093/jxb/erj139. [DOI] [PubMed] [Google Scholar]
- 4.Stalder D, Haeberli A, Heller M. Evaluation of reproducibility of protein identification results after multidimensional human serum protein separation. Proteomics. 2008;8(3):414–24. doi: 10.1002/pmic.200700527. [DOI] [PubMed] [Google Scholar]
- 5.Bern M, Finney G, Hoopmann MR, Merrihew G, Toth MJ, MacCoss MJ. Deconvolution of mixture spectra from ion-trap data-independent-acquisition tandem mass spectrometry. Anal Chem. 2010;82(3):833–41. doi: 10.1021/ac901801b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Houel S, Abernathy R, Renganathan K, Meyer-Arendt K, Ahn NG, Old WM. Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J Proteome Res. 2010;9(8):4152–60. doi: 10.1021/pr1003856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michalski A, Cox J, Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC–MS/MS. J Proteome Res. 2011;10(4):1785–93. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 9.Chapman JD, Goodlett DR, Masselon CD. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom Rev. 2014;33(6):452–470. doi: 10.1002/mas.21400. [DOI] [PubMed] [Google Scholar]
- 10.Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ, McKenna T, Nold MJ, Richardson K, Young P, Geromanos S. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77(7):2187–200. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 11.Rardin MJ, Schilling B, Cheng LY, MacLean BX, Sorensen DJ, Sahu AK, MacCoss MJ, Vitek O, Gibson BW. MS1 Peptide Ion Intensity Chromatograms in MS2 (SWATH) Data Independent Acquisitions. Improving Post Acquisition Analysis of Proteomic Experiments. Mol Cell Proteomics. 2015;14:2405. doi: 10.1074/mcp.O115.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11(6):O111.016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egertson JD, Kuehn A, Merrihew GE, Bateman NW, MacLean BX, Ting YS, Canterbury JD, Marsh DM, Kellmann M, Zabrouskov V, Wu CC, MacCoss MJ. Multiplexed MS/MS for improved data-independent acquisition. Nat Methods. 2013;10(8):744–6. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prakash A, Peterman S, Ahmad S, Sarracino D, Frewen B, Vogelsang M, Byram G, Krastins B, Vadali G, Lopez M. Hybrid data acquisition and processing strategies with increased throughput and selectivity: pSMART analysis for global qualitative and quantitative analysis. J Proteome Res. 2014;13(12):5415–30. doi: 10.1021/pr5003017. [DOI] [PubMed] [Google Scholar]
- 15.Ow SY, Salim M, Noirel J, Evans C, Rehman I, Wright PC. iTRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly”. J Proteome Res. 2009;8(11):5347–5355. doi: 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]
- 16.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS (vol 15, pg 1895, 2003) Anal Chem. 2003;75(18):4942–4942. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 17.Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, Nesvizhskii AI. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12(3):258–64. 7. doi: 10.1038/nmeth.3255. following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stasyk T, Huber LA. Zooming in: fractionation strategies in proteomics. Proteomics. 2004;4(12):3704–16. doi: 10.1002/pmic.200401048. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt A, Gehlenborg N, Bodenmiller B, Mueller LN, Campbell D, Mueller M, Aebersold R, Domon B. An integrated, directed mass spectrometric approach for in-depth characterization of complex peptide mixtures. Mol Cell Proteomics. 2008;7(11):2138–50. doi: 10.1074/mcp.M700498-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoopmann MR, Merrihew GE, von Haller PD, MacCoss MJ. Post analysis data acquisition for the iterative MS/MS sampling of proteomics mixtures. J Proteome Res. 2009;8(4):1870–5. doi: 10.1021/pr800828p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailey DJ, McDevitt MT, Westphall MS, Pagliarini DJ, Coon JJ. Intelligent data acquisition blends targeted and discovery methods. J Proteome Res. 2014;13(4):2152–61. doi: 10.1021/pr401278j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabb DL, Vega-Montoto L, Rudnick PA, Variyath AM, Ham AJ, Bunk DM, Kilpatrick LE, Billheimer DD, Blackman RK, Cardasis HL, Carr SA, Clauser KR, Jaffe JD, Kowalski KA, Neubert TA, Regnier FE, Schilling B, Tegeler TJ, Wang M, Wang P, Whiteaker JR, Zimmerman LJ, Fisher SJ, Gibson BW, Kinsinger CR, Mesri M, Rodriguez H, Stein SE, Tempst P, Paulovich AG, Liebler DC, Spiegelman C. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J Proteome Res. 2010;9(2):761–76. doi: 10.1021/pr9006365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudomin EL, Carr SA, Jaffe JD. Directed sample interrogation utilizing an accurate mass exclusion-based data-dependent acquisition strategy (AMEx) J Proteome Res. 2009;8(6):3154–60. doi: 10.1021/pr801017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graumann J, Scheltema RA, Zhang Y, Cox J, Mann M. A framework for intelligent data acquisition and real-time database searching for shotgun proteomics. Mol Cell Proteomics. 2012;11(3):M111.013185. doi: 10.1074/mcp.M111.013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goloborodko AA, Levitsky LI, Ivanov MV, Gorshkov MV. Pyteomics–a Python framework for exploratory data analysis and rapid software prototyping in proteomics. J Am Soc Mass Spectrom. 2013;24(2):301–4. doi: 10.1007/s13361-012-0516-6. [DOI] [PubMed] [Google Scholar]
- 26.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 27.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5(11):976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 28.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4(11):923–5. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 29.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 30.Lange E, Gropl C, Schulz-Trieglaff O, Leinenbach A, Huber C, Reinert K. A geometric approach for the alignment of liquid chromatography-mass spectrometry data. Bioinformatics. 2007;23(13):i273–81. doi: 10.1093/bioinformatics/btm209. [DOI] [PubMed] [Google Scholar]
- 31.Sturm M, Bertsch A, Gropl C, Hildebrandt A, Hussong R, Lange E, Pfeifer N, Schulz-Trieglaff O, Zerck A, Reinert K, Kohlbacher O. OpenMS - an open-source software framework for mass spectrometry. BMC Bioinf. 2008;9:163. doi: 10.1186/1471-2105-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bilbao A, Varesio E, Luban J, Strambio-De-Castillia C, Hopfgartner G, Muller M, Lisacek F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics. 2015;15(5–6):964–80. doi: 10.1002/pmic.201400323. [DOI] [PubMed] [Google Scholar]
- 33.Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, Nesvizhskii AI. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12(3):258. doi: 10.1038/nmeth.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li S, Plouffe BD, Belov AM, Ray S, Wang X, Murthy SK, Karger BL, Ivanov AR. An Integrated Platform for Isolation, Processing and Mass Spectrometry-based Proteomic Profiling of Rare Cells in Whole Blood. Mol Cell Proteomics. 2015;14:1672. doi: 10.1074/mcp.M114.045724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.