

Abstract
Technical advances in the development of organoid systems enable cell lines, primary adult cells, or stem or progenitor cells to develop into diverse, multicellular entities, which can self-renew, self-organize, and differentiate. These 3D organoid cultures have proven to be of value in increasing our understanding of the biology of disease and offer the potential of regenerative and genetic therapies. The successful application of 3D organoids derived from adult tissue into urological cancer research can further our understanding of these diseases and could also provide preclinical cancer models to realize the precision medicine paradigm by therapeutic screening of individual patient samples ex vivo. Kidney organoids derived from induced pluripotent stem cells provide personalized biomarkers, which can be correlated with genetic and clinical information. Organoid models can also improve our comprehension of aspects of particular diseases; for example, in prostate cancer, 3D organoids can aid in the identification of tumour-initiating cells from an epithelial cell lineage. Furthermore, kidney organoid differentiation from human pluripotent stem cells enables gene editing to model disease in kidney tubular epithelial cells. State-of-the-art human organoid cultures have potential as tools in basic and clinical research in renal, bladder, and prostatic diseases.
Urological cancer encompasses cancers of the kidney, urothelium (including the bladder, ureters, and urethra), prostate, testicles, and penis. Urological cancers are major causes of morbidity: the American Cancer Society estimates that 318,980 new instances of cancer will be diagnosed and that 59,690 deaths will occur from urological cancers in 2017 in the USA. Prostate cancer is the most common malignancy and the third leading cause of mortality from cancer in men, and kidney cancer and urinary bladder cancer are among the ten most common malignancies in the USA1.
Developing in vitro and in vivo model systems that accurately reflect both the genetic diversity and the lineage specificity of cancer is crucial in order to understand the role of cancer-specific genetic alterations in tumorigenesis, tumour maintenance, and therapeutic sensitivity. Advances in next-generation sequencing technologies2 have enabled an improved understanding of the molecular classification of cancer, which could provide benefits for clinical practice in oncology in areas such as diagnosis, prognosis, and treatment decision making. The Cancer Genome Atlas (TCGA) included comprehensive genomic analyses such as whole-exome sequencing, mRNA and microRNA sequencing, DNA methylation analysis, and/or proteomic analysis of each of the four major urological malignancies (prostate, kidney, bladder, and testicular cancer), including separate studies of three distinct histological subtypes of renal cell carcinoma (RCC): clear cell (cc)RCC, papillary (p)RCC, and chromophobe (ch)RCC3–7. Data from these studies have highlighted the fact that each tumour type harbours a distinct mutational profile. For example, translocations in ETS family transcription factors ERG, FLI1, ETV1, and ETV4 are observed in half of all prostate cancers but not in other urological malignancies6. Among prostate cancers that lack ETS translocations, the most common genetic alterations are mutations in the E3-ligase SPOP and the pioneer transcription factor FOXA1, which are also two highly prostate-specific events. However, ccRCC is characterized by mutations involving von Hippel–Lindau (VHL) and chromatin modifiers including SETD2, BAP1, PBRM1, ARID1A, and SMARCA45. In addition to disease-defining lesions, even tumours within the same histological category are molecularly heterogeneous and each individual tumour harbours a unique set of potentially targetable alterations7,8. These observations highlight the importance of understanding genetic alterations within the context of correct lineage.
Cell lines have traditionally been the tools used for understanding the role of genetic and molecular alterations in cancer. For example, cell lines have been instrumental in the discovery of biomarkers of therapeutic response, highlighting the role of BRAF mutations in sensitivity to dual specificity mitogen-activated protein kinase kinase (MEK) inhibitors or BRCA1/2 mutations in sensitivity to poly [ADP-ribose] polymerase 1 (PARP) inhibitors9,10. Large-scale studies initiated at the Broad Institute, the National Cancer Institute, and the Sanger Institute aim to genetically characterize >1000 cell lines of diverse histology and test them for sensitivity against a large panel of drugs and also use genetic knockdown to understand both lineage and mutational determinants of therapeutic sensitivity11,12. However, cancer cell lines have many limitations, including genetic drift caused by being in long-term culture, a lack of annotated clinical data, and, most importantly, the fact that only a subset of tumours grow in 2D on plastic. For example, despite prostate cancer being the most common malignancy in men, only a few prostate cancer cell lines have been established because the vast majority of prostate cancer cells do not grow in traditional culture conditions.
Many researchers have used in vivo models — including genetically engineered mouse models and primary patient-derived xenografts (PDXs) — to overcome the limitations associated with cell lines. Genetically engineered mouse models are genetically well-defined and can be used to delineate the minimal set of genetic alterations that can cause tumorigenesis and affect therapeutic sensitivity. PDX models have an increased ‘take rate’ compared with cancer cell lines and better recapitulate the histology and therapeutic response of a patient’s tumours. However, these preclinical cancer models are expensive to create, and public cell-line repositories are limited in number. Moreover, these models have considerable limitations as models of human cancer, including species-specific differences and inaccurate recapitulation of in vivo human tumour biology13. PDX models are not very amenable to genetic manipulation, making mechanistic studies challenging.
Several technical breakthroughs have occurred in the past decade in in vitro culture technology, which have facilitated the culturing of both benign and malignant cells. Classic tissue culture conditions do not enable the growth of most nontransformed cells. When grown in classic serum-containing tissue culture media, nontransformed cells can grow for a finite number of cell passages and eventually senesce, a phenomenon termed the ‘Hayflick limit’ (REF. 14). However, the Hayflick limit is not universal to all cell types, and specific stem-cell populations, such as embryonic stem cells and neural stem cells, can proliferate indefinitely15,16. In 2009, Sato and colleagues17 observed that a single leucine-rich repeat-containing G-protein-coupled receptor 5 (Lgr5)-positive intestinal stem cell can generate a continuously expanding, self-organizing, physiological epithelial structure similar to that of normal gut tissue17, and they termed this novel culture system ‘organoid’ culture. Independently, building on work showing that treatment with Rho-associated protein kinase (ROCK) inhibitors can induce immortality in primary keratinocytes growing on fibroblast feeders, Liu and colleagues18,19 demonstrated that the combination of a ROCK inhibitor (Y-27632) and feeder fibroblast culture conditions enables the indefinite growth of multiple primary human epithelial cell types18,19. Cells treated in this manner are termed ‘conditionally reprogrammed cells’ and this method is an alternative culture strategy. Based on this finding, organoids from normal and tumour epithelial cells might be able to proliferate indefinitely in vitro, without requiring transduction of exogenous viral or cellular genes. Organoid cultures have now been adapted to cells of multiple different epithelial lineages; in this Review, we will focus on the potential uses of organoids in urological cancer research.
Development of organoids
2D culture techniques that were developed at the turn of the 20th century enabled biologists to observe and manipulate mammalian cells20. Subsequently, scientists began to understand mammalian organ development at a cellular and molecular level. However, in vitro 2D cultures do not accurately recapitulate real-world in vivo organ systems, which is especially important in cancer research. Cellular behaviour is strongly influenced by the microenvironment, specifically the extracellular matrix21. In order to mimic the structural relationships between epithelial cells and stroma in vitro, and to model the development and function of organs, various 3D in vitro model systems have been developed22. These 3D, organotypic systems are derived from cell lines, primary tissues, embryonic stem cells, induced pluripotent stem cells, and embryonic whole organs such as organ explants consisting of multiple tissue types22. The 3D organoids formed from primary tissue, embryonic stem cells, and induced pluripotent stem cells with extra cellular matrix are thought to closely recapitulate the in vivo environment, and are capable of self-renewal and self-organization. As a functional unit, an organoid is composed of multiple cell types and contains multicellular organ structures, mimicking the tissue of origin and functioning in a similar manner23. Different approaches for generating organoids are available, especially for urological organs (TABLE 1).
Table 1.
Advantages and disadvantages of different cellular inputs for organoid culture
| Organoids | Advantages | Disadvantages | Refs |
|---|---|---|---|
| Cell line | |||
| Prostate (RWPE-1) | Recapitulates acinar morphogenesis in vitro, forming structures representative of adult prostate glands, unlimited growth | Genotypic drift, limited number of cell lines available, loss of tumour heterogeneity, acquisition of mutations during immortalization | 29 |
| Kidney (MDCK) | A useful tool to study cyst formation and kidney structures | Genotypic drift, limited number of cell lines available, loss of tumour heterogeneity, acquisition of mutations during immortalization | 24–26 |
| Bladder (5637) | Expresses specific markers and structures observed in differentiated human urothelium | Genotypic drift, limited number of cell lines available, loss of tumour heterogeneity, acquisition of mutations during immortalization | 27 |
| Primary adult cells | |||
| Prostate (primary human prostate epithelial cells) | Reproduce the structural and functional differentiation of human prostatic acini that is observed in vivo | Lack of evidence for the possibility of serial passage | 30,31 |
| Prostate spheres | Serial passage possible, enable study of prostate basal progenitors | Lack luminal progenitor information | 32 |
| Prostate organoids | Long-term expansion possible, retain both basal and luminal layers and include all luminal differentiation, enable elucidation of both basal and luminal progenitors | Only established from metastatic prostate cancer | 51–53 |
| Bladder (mouse bladder epithelial cells) | Mimic the bladder injury model, enable study of bladder pluripotent progenitors | Lack information on the effects of long-term culture | 80 |
| Human pluripotent stem cells | |||
| Kidney | Contain eight different cell types, enable the study of the origin of progenitor cells of the vasculature of the kidney, enable study of human kidney development, enable modelling of renal diseases and screening of nephrotoxic drugs | Owing to immaturity, these organoids only mimic the first-trimester human kidney | 66,68 |
Cell lines
Several immortalized cell lines are able to form polarized 3D structures when grown in 3D culture conditions. For example, Madin–Darby canine kidney (MDCK) cells embedded into 3D Matrigel proliferate and assemble into cyst structures consisting of a polarized spherical monolayer surrounding a central lumen, enabling the molecular network of cellular polarization in cyst formation to be studied24–26. Smith and colleagues27 cultured human bladder 5637 cells under microgravity conditions within a rotating wall vessel bioreactor27, which enables cells to remain in suspension and form organoids that reflect the characteristics of in vivo tissue-specific differentiation (FIG. 1a). The researchers used this system to investigate how terminally differentiated human urothelial cells interact with uropathogenic Escherichia coli (UPEC)27. In this model, human bladder cells develop into 3D constructs that express specific markers and structures that are also observed in differentiated human urothelium. Importantly, this system mimics the interactions between well-differentiated urothelial cells and UPEC that are observed in vivo28. Tyson and colleagues29 showed that in vitro recapitulation of acinar morphogenesis is possible using 3D culture of the benign prostate epithelial cell lines RWPE-1, pRNS-1-1, PZ-HPV-7, PNT1A, BPH-1, and PrEC. Specifically, almost all RWPE-1 cells seeded in Matrigel undergo acinar morphogenesis, forming consistent structures that are representative of nonmalignant adult prostate glands.
Figure 1. Different organoid development processes.
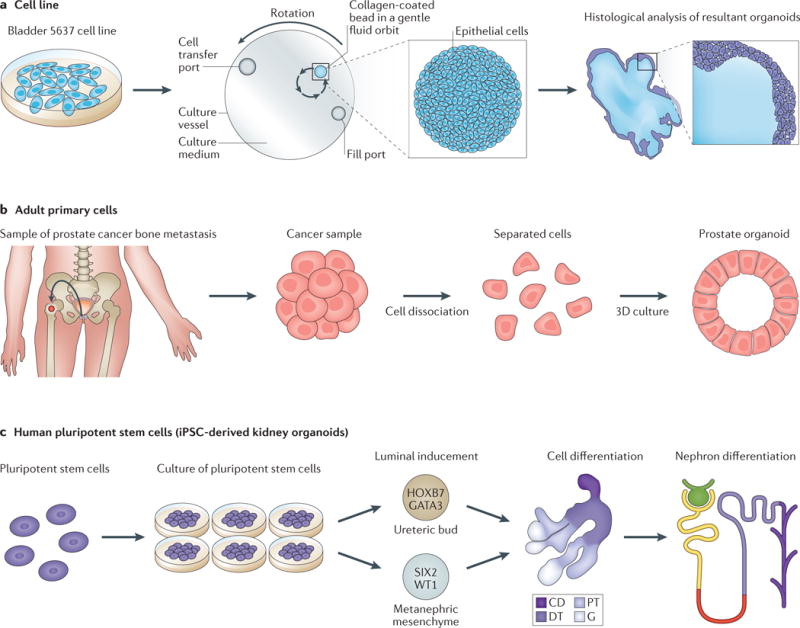
a| Organoids from cell lines. Bladder cancer organoids have been developed from 5637 cells, which were cultured in a rotating wall vessel bioreactor. Organoids were formed under microgravity conditions, and provide a robust model that mimics a bladder microenvironment. Histological analysis of these organoids showed they developed a tissue-like arrangement of closely packed layers of four to six cells that were heterogeneous in size and shape. The researchers used this system to investigate how terminally differentiated human urothelial cells interact with uropathogenic Escherichia coli (UPEC)27 b | Organoids from primary adult tissue. Tissue from a bone biopsy sample from a patient with metastatic prostate cancer can be dissociated into single cells and cultured. These cells can regenerate into gland-like organoids in 3D culture. These organoids can be used to study the cell of origin, cell lineage differentiation, and drug response in order to enable effective individualized treatment53. c | Organoids from induced pluripotent stem cells (iPSCs). The differentiation of human pluripotent stem cells to into kidney organoids is feasible owing to the ability of these cells to differentiate into any cell type. The kidney is derived from the intermediate mesoderm and forms through interactions between ureteric bud and metanephric mesenchyme. The formation of kidney organoids occurs after in-vitro-directed differentiation of luminal-induced pluripotent stem cells. Within these kidney organoids different structures can be detected including collecting ducts (CD), distal tubules (DT), proximal tubules (PT), and glomeruli (G), and nephron differentiation is also observed66.
Primary adult cells
Organoids can be established using primary adult stem cells; for example, reproduction of the structural and functional differentiation of human prostatic acini that occurs in vivo is possible in vitro using coculture of primary human prostatic epithelial and stromal cells, or by growing prostatic epithelial cells in androgen-treated, stromal-conditioned medium30,31. However, successful serial passage has not been demonstrated in these studies, which limits their usage as a functional tool. Xin and colleagues32 demonstrated that mouse prostate progenitor cells cultured in Matrigel had self-renewal capacity and could be serially passaged. They called these clonal spheroids grown in Matrigel ‘prostate spheres’. Rinkevich and colleagues33 established a culture system for growing renal epithelial organoids in suspension to investigate the in vitro fates of individual renal precursors. In this study, serial passaging of renal spheres over three sequential passages suggested the self-renewal ability of progenitors was preserved in organoid culture.
The discovery that intestinal stem cells are marked by Lgr5 expression and that R-spondin is the endogenous ligand of Lgr5 enabled Sato and colleagues17 to design a culture system that sustains the long-term expansion and multilineage differentiation of a single Lgr5-positive intestinal stem cell. In this method, stem cells are embedded into Matrigel with three growth factors: R-spondin 1, which activates protein WNT (WNT) signalling through binding with Lgr5; Noggin, which inhibits bone morphogenetic protein (BMP) signalling; and epidermal growth factor (EGF), an important growth factor, is required for crypt growth17. WNT signalling is a requirement for crypt proliferation: intestinal epithelial proliferation was greatly reduced with the loss of crypts by ectopically expressing WNT inhibitor Dickkopf1 (Dkk1) in a transgenic mouse model34. Systemic administration of human R-spondin 1 to mice potently increased proliferation of intestinal epithelial cells through activation of β-catenin35. Germline mutations in SMAD4 and BMPR1A have been discovered in some subsets of patients with juvenile polyposis, indicating a role for the BMP pathway in the pathogenesis of this disease36,37. Loss of function of these inhibitory signals in BMP pathway means that epithelial cells establish ectopic crypts, resulting in juvenile polyposis. Transgenic expression of Noggin in a mouse model also induced an expansion of crypt numbers38. EGF is a potent stimulator of cell proliferation in epithelial and non-epithelial cell types produced in salivary glands and Brunner glands in the duodenum39. EGF has a range of roles including modulating the expression of enzymes involved in the production of polyamines, increasing intestinal electrolyte and nutrient transport in the enterocyte, attenuating intestinal damage, and promoting intestinal healing40. Based on the important functions in the gastrointestinal tract, R-spondin-1, Noggin and EGF have been included in organoid medium17. These organoids recapitulate normal crypt villus structure, with Lgr5-positive cells occupying the crypts and undergoing asymmetric division, transiently amplifying into cells that divide and differentiate into mature villus cells. These organoids can perform physiological functions of the intestinal epithelium, such as Cl− secretion. Individual patient-derived intestinal organoids can be used to investigate Cl− secretion defects in diseases such as cystic fibrosis and screen drugs that could correct the defect41.
This organoid culture system can also be adapted to generate organoids by culturing stem or pro genitor cells from many other types of epithelial organs. Human adult epithelial-derived stem or progenitor cell organoid cultures have already been established using cells derived from lung42, small intestine43, colon43, pancreas44, liver45,46, and stomach47 tissue. For the urological organs, organoid systems of the kidney48,49 and prostate51,52 are most well developed. Prostate organoids can be derived from nonmalignant human and mouse prostate stem cells and mimic the morphology of a nonmalignant prostate. The use of patient-derived organoids to study cancers is expanding, with studies in bladder50, prostate53, kidney54,55, pancreatic44, and colorectal cancers56 already published (FIG. 1b). Within the field of urology, Matulay and collegaues50 collected bladder tumour specimens during routine cystoscopy and enzymatically digested them into single cells and cell clusters and then cultured them in organoid-promoting, embedded-cell culture conditions. Organoids successfully underwent serial passage and were subjected to freezing and thawing cycles. The researchers used these bladder organoids to analyse genetic mutations in the bladder tumours. Gao and co-workers53 successfully performed long-term culture of prostate cancer organoids from biopsy specimens and circulating tumour cells. These patient-derived organoids recapitulated the molecular diversity of prostate cancer subtypes, such as TMPRSS2–ERG fusion, SPOP mutation, SPINK1 overexpression, and CHD1 loss. The most common feature that the different organoid lines shared was loss of function in the TP53–PTEN–RB1 tumour-suppressor pathway. Batchelder and colleagues54 successfully established 20 tumour organoid lines from 25 RCC samples and 22 nonmaliganant lines from non-tumour samples using a 3D-scaffold system. The gene expression pattern of these cells was maintained for up to 21 days in 3D cultures, but was lost in cells in 2D culture. Moreover, Lobo and colleagues55 established patient-matched malignant and nonmalignant primary cell cultures from sample from patients with ccRCC with VHL mutations. Surprisingly, the unselected tumour cell lines derived from primary ccRCC samples failed to show the VHL mutations that matching their parental tumour samples, which means that these cell lines are not cancer cells. The investigators purified those cell lines using carbonic anhydrase 9 (CA9) (a biomarker that is highly expressed in VHL-mutant cells) and established six pure VHL-mutant and VHL-wild-type cell culture pairs grown in 10% FBS, but not in serum-free medium. Transcriptional profiles recapitulated the parental ccRCC tissues and demonstrated the key pathways involved in ccRCC cell biology.
The clinical practice of oncology is changing rapidly towards precision medicine. The biopsy sampling and molecular analysis of metastatic lesions has become increasingly common practice. Patient-derived organoids or cell cultures from individual patients could faithfully reproduce the molecular and histological phenotype of the parental tumour, which supplements the preclinical models for studying cancer biology and efficiently assessing patient-specific personalized therapies. However, the limitations of this technology are also obvious; for example prostate cancer and kidney cancer organoids are easily outgrown by nonmalignant cells, highlighting the importance of genotypic validation of newly established organoid lines by comparing them with the patient tissues from which they are derived. Another issue with these organoids is the absence of nerves, blood vessels, and immune cells in the system. For example, a big difference in transcriptional profiles between VHL-mutant cells and their matched primary tumour specimens was observed by Lobo and colleagues55. However, further analysis indicated that the differentially expressed genes in the tumour component were mainly involved in immune functions, meaning that these genes were lost in VHL-mutant cultures owing to the absence of the immune cells. Immune checkpoint therapy is now included in the treatment options for patients with cancer along with chemo therapy, surgery, radiation, and targeted therapy57. Results of a single-arm, multicentre, phase II trial evaluating the programmed death ligand 1 (PD-L1) inhibitor atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who had progressed on platinum-based chemotherapy demonstrated that approximately 15% of patients had at least a partial shrinkage58. The FDA approved atezolizumab (Tecentriq) for the treatment of select patients with urothelial carcinoma in 2016. The FDA also approved nivolumab (Opdivo) for patients with advanced RCC based on a randomized, open-label, phase III study of nivolumab in comparison with everolimus59. Patients treated with nivolumab lived an average of 25 months compared with 19.6 months for those treated with everolimus. Thus, future studies will focus on how immune response is regulated in the tumour microenvironment to improve the survival for more cancer patients. Coculture of immune cells with patient-derived kidney, bladder, and prostate cancer cells in 3D will provide another means of bringing personalized immunotherapy to fruition.
Pluripotent stem cells
Epithelial cells can differentiate into subspecialized epithelial compartments (such as basal or luminal prostatic cells, or crypt or villus intestinal cells), but pluripotent stem cells can form all cellular components of an organ, including epithelial, stromal, and endothelial cells. The organoid culture system can be adapted to generate organoids from embryonic cells or pluripotent stem cells. Human embryonic-based or induced pluripotent stem-cell-based organoids have already been established for the thyroid60, inner ear61, retina62, pituitary gland63, and kidney48,49. The cellular complexity of the kidney has, for decades, limited the ability of researchers to grow a patient’s kidney in a dish. This particular organ has >20 different types of epithelial cell, which complicates the regenerative medicine approaches because humans are incapable of generating new nephrons after birth64. However, the isolation of human pluripotent stem cells and, especially the success in inducing pluripotent stem cells, means that generating specific cell types is possible. Renewable, expandable, and patient-derived stem cells can then be used to derive the required cell types. Using the 3D organoid system, researchers can induce the pluripotent stem cells to form organoids of the developing eye, the cerebral cortex, liver, stomach, and intestine, making it possible to form a model of the organ from progenitor cells in 3D culture65.
In the field of urological research, Takasato et al.66 have successfully induced human pluripotent stem cells using the small molecule inhibitor CHIR99021 to alter WNT signalling. A complex multicellular kidney organoid formed from these cells, which had more than eight distinct cell types, including segmenting nephrons comprised of distal tubules, a loop of Henle, a proximal tubule, and Bowman capsules containing parietal epithelial cells and podocytes (FIG. 1c). These organoids also contain stromal cells that express key transcription factors specific for the cortical stroma of the developing kidney. Moreover, a vascular network with capillaries entering primitive glomeruli was also observed, which provided a robust tool for studying the origin of the progenitor cells of the vasculature of the kidney. The transcription profiles of these kidney organoids showed close similarity to the profiles of first-trimester human kidney tissue. Kidney tissue is derived from the definitive mesoderm of the embryo; the intermediate mesoderm differentiates into the ureteric bud that forms the collecting ducts and a metanephric mesenchyme, which generates all the cell types of the nephrons64,67. In order to establish a model to recapitulate the differentiation of human pluripotent stem cells into kidney tissue, Takasato and colleagues68 set up a protocol including four pro genitor populations: nephron progenitors, ureteric epithelial progenitors, renal interstitial progenitors, and endothelial progenitors, from human kidney material to generate kidney organoids within which segmented nephrons are connected with collecting ducts and are surrounded by renal interstitium and an endothelial network. The value of this work is that they not only included two key progenitors of ureteric epithelium and metanephric mesenchyme but also the progenitors of renal interstitium and endothelium, which have been suggested to be the progenitors all anticipated cell types69 in kidney tissue, and that these cells simultaneously develop in the organoids.
Considerable work has been conducted in the past 5 years to generate kidney organoids from human pluripotent stem cells to study the mechanism of kidney injury and repair and inherited diseases. Undoubtedly, further work to model kidney diseases using kidney organoids derived from human pluripotent stem cells will be conducted in the future. Progress made in approaches to induce differentiation of human pluripotent stem cells means that recapitulating the complexity of cell types in kidney regeneration is no longer a big issue. However, many challenges still need to be overcome (such as improving the efficiency nephron generation using differentiation protocols) before kidney organoids can realize their full potential and can be used to generate functional bioengineered kidney tissues.
Calderon-Gierszal and colleagues70 successfully induced differentiation of human embryonic stem cells into human prostate organoids in vitro. Brief exposure of cells to WNT10B and FGF10 to promote prostate fate, followed by a cocktail of prostate epithelial and stromal cell media containing exogenous factors including R-spondin 1, Noggin, and EGF resulted in the growth and maturation of prostate organoids. Similar to small intestinal organoid culture17, prostate organoids also require these three factors. R-spondin 1 is required because the endogenous WNT pathway is expressed in a spacio-temporal manner by the prostate during development and maturation; BMP signalling inhibition by Noggin could promote prostate growth and branching morphogenesis; EGF is included for regulation of budding and proliferation of mesenchymal and epithelial cells. Under these conditions, the prostate organoids generated the architecture of columnar epithelium with central lumen and expressed NKX3.1, AR, TMPRSS2 and PSA, recapitulating the typical morphology and functional properties observed in human prostate tissue. Low doses of the endocrine disruptor bisphenol-A disrupted human prostate morphogenesis and perturbed prostate stem cell homeostasis in the maturing prostate structures. This model mimics the key developmental event in human embryonic prostate tissue and provides robust evidence of the relevance of environmental endocrine disruptors in human prostate development.
Simon71 first described a urinary diversion using intestinal segments in 1852 and continent urinary diversion is currently the method of choice for a large number of patients who undergo radical cystectomy. However, the incorporated bowel segments do not have the mechanical properties and innervation to function as a urinary bladder, such as storage and emptying, which results in a high risk of complications including UTI and hydronephrosis72. Thus, a new approach to replace the bowel, namely urinary bladder regeneration is required and organoid technology seems to be promising in this context. Two protocols to induce human embryonic stem cells or human pluripotent stem cells into urothelium were reported in 2014 (REFS 73,74). Osborn et al.73 developed a matrix-free and cell-contact-free in vitro culture system to induce human embryonic stem cells or human pluripotent stem cells into definitive endoderm and then urothelium using directed differentiation in a urothelial-specific medium. Kang et al.74 developed a differentiation protocol to induce human pluripotent stem cells into definite endoderm and then bladder urothelial cells using a chemically defined culture system. These differentiation protocols will be useful for studying normal and pathological development of the human bladder urothelium in vitro, and could potentially be used to produce cells for bladder regeneration for urinary diversion. Presently, only limited bladder cell types can be induced74, but we believe the 3D organoid culture system that has been developed for kidney organoids66 could be adapted for bladder urothelium regeneration.
In general, organoids derived from human pluripotent stem cells have a key role in understanding the developmental biology of different organs including kidney, bladder, and prostate.
Application in urological diseases
Drug screening and nephrotoxicity
Large-scale screens of human cancer cell lines — including gene expression profiling, chromosomal copy number profiling, genomic sequencing data, and pharmacological sensitivity profiling — indicate that genetic information can predict a response to treatment and could guide future precision medicine regimens11. Studying the mechanisms of drug sensitivity and resistance in vitro requires cancer cell lines possessing the advantages of immortality, straightforward maintenance, amenability to high-throughput screening, and the ability to be xenografted9. Increased numbers of cell lines representing each tumour type are thought to be required in order to understand the role of specific genetic alterations in drug sensitivity. However, as an example, only seven prostate cancer cell lines are currently established, and many do not harbour the recurrent genetic lesions commonly observed in patients with this disease (such as TMPRSS2–ERG interstitial deletion, SPOP mutation, FOXA1 mutation, and CHD1 loss). Furthermore, gene expression profiles can differ substantially between low-passage and high-passage cells75. A 3D-organoid-based drug sensitivity screen to identify molecular signatures associated with altered drug responses in colorectal cancer has been developed that correlates drug sensitivity with genomic features56. Using this drug-screening platform, researchers confirmed the activity of cetuximab in KRAS-wild-type organoids and the effectiveness of nutlin-3a treatment in TP53-wild-type organoids: a similar strategy could be used for drug screening in prostate cancer, kidney cancer, and bladder cancer organoid lines in the future.
The successful establishment of seven prostate cancer organoid lines (six from biopsy samples from patients with metastatic prostate cancer and one from circulating tumour cells) has been achieved using an organoid culture system described for nonmalignant human basal and luminal prostate epithelial cells53. The culture medium used in this system enables both nonmalignant and malignant prostate cells to self-renew without artificial transformation. Characterization of these seven new organoid lines using whole-exome sequencing of germ line and organoid DNA, array comparative genomic hybridization, and paired-end RNA sequencing (FIG. 2a) showed that, after 3 months in in vitro culture, the patient-derived organoid lines had a mutational landscape identical to that of the tumour tissue. The organoids harboured recurrent disease-specific genomic alterations, such as ETS translocations, SPOP mutations, FOXA1 mutations, and CHD1 loss. Furthermore, tumour grafts derived from these organoid lines showed histological patterns that were similar to those of the primary tumours. In order to investigate the suitability of these lines for investigating therapeutic responses, all seven lines were treated with the antiandrogen enzalutamide and the PI3-kinase pathway inhibitors everolimus and BKM-120. One of the seven lines — MSK-PCa2, which harbours an AR amplification, PTEN loss, and PIK3R1 mutation — demonstrated sensitivity to enzalutamide with an IC50 of 50 nM and to the PI3-kinase inhibitors, which strengthens the conclusion that molecular subtyping is essential for targeting therapy76. Another of the organoid lines, MSK-Pca5, is derived from prostate cancer circulating tumour cells, suggesting that liquid biopsy, by taking a blood sample instead of a core biopsy of metastatic sites, is a distinct clinical possibility. The value of using circulating tumour cells as an efficacy-response surrogate for overall survival77 has also been demonstrated. CTC-derived organoids have advantages. Firstly, they can be used when culture from tissue biopsy fails or is not possible. For example, in instances in which access to bone marrow lesions of patients with metastatic prostate cancer is impaired, CTCs from their blood could be used to culture organoids to guide targeted therapy. Secondly, intratumour heterogeneity means that a single biopsy might not contain all tumour clonal lineages. Thus, CTCs could be derived from lesions that were not sampled and provide further genomic information. Data from another study also show that genetic diversity is preserved in 3D organoid culture. Matulay and colleagues50 successfully established urothelial cancer organoids from patient-derived tissue samples. Using Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Target (MSK–IMPACT) DNA sequencing analysis78,79, the researchers showed that the generated organoid lines have similar mutational profiles to those of tumour samples, and can provide a platform for personalized drug-response assays in urothelial cancers. For individual patients, 3D organoid systems are the first step toward achieving precision medicine that identifies therapeutic targets that will confer maximum benefit based on genetic changes that are specific to an individual’s cancer.
Figure 2. Applications of organoid culture in human prostate cancer.
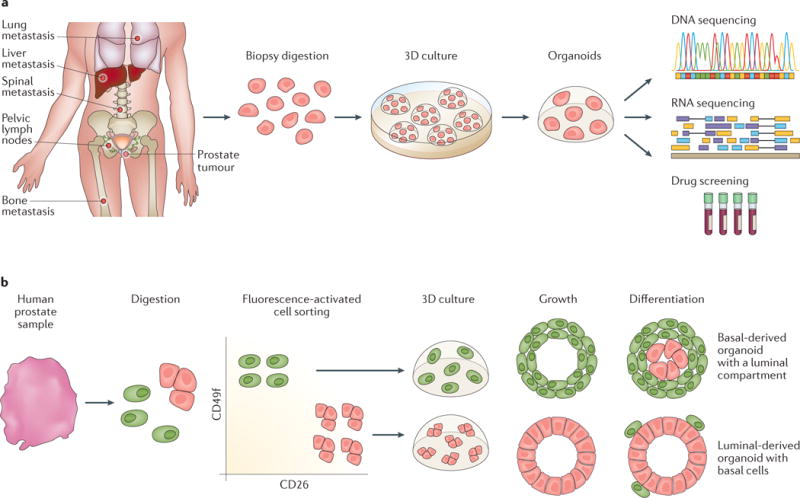
a| Seven established organoid lines derived from metastatic prostate cancer biopsy samples and circulating tumour cells were sent for genetic characterization and drug screening. The results provided important therapeutic information, which is a first step towards using these organoids in precision medicine53. b | Prostate progenitor basal and luminal cells were separated by fluorescence-activated cell sorting followed by 2–3 weeks of organoid culture. Basal-derived organoids developed a luminal compartment showing CK8 expression and luminal-derived organoids can generate CK5-positive, CK8-negative basal cells, indicating that either cell type could be a prostate progenitor51,90.
Traditionally, mouse models have been used as preclinical pharmaceutical tools for nephrotoxicity screening; however, these models cannot recapitulate the behaviour of human cells in a completely accurate way. Thus, human-derived, in vitro models are required for nephrotoxicity screening. Kidney organoids derived from induced pluripotent stem cells have been used to test the response of renal proximal tubules (which have a vital role in reabsorption) to nephrotoxic drugs such as cisplatin66 at a dose of 5 μM. Results showed that these drugs could induce specific acute apoptosis in mature proximal tubular cells but not in immature cells. Hence, kidney organoids could be useful preclinical models for nephrotoxicity screening.
Investigation of the cell of origin
Tumour stem cells have been proposed to promote resistance to chemotherapy. Thus, establishing models of the cancer stem cell of origin could help identify effective prognostic biomarkers and guide an individual’s treatment and management.
For bladder cancer, the identification of cancer stem cells has been inconclusive. One study describes Sonic hedgehog (Shh)-expressing and Cytokeratin-5 (Ck5)-expressing basal urothelial cells from ShhCreER/WT;R26mTmG/WT mice in organoid culture. These cells included multipotent stem cells that were capable of self-renewal and regeneration of all cell types within the urothelium in response to bacterial infection or chemical injury within the bladder80. Furthermore, single Shh-expressing cells formed cyst-like organoids after 5–7 weeks of 3D culture, expressing Ck5 in the outer layer and forming a luminal space in which Ck5 and Shh were not expressed. The single cells from these organoids were capable of self-renewing by generating new organoids in subsequent cultures, demonstrating that the Shh-expressing basal urothelial cell population includes multipotent stem cells that are capable of self-renewal and differentiation80. The origins and formation of bladder cancer stem cells remain unknown81, but 3D-based organoid culture could provide a model system for studying this topic82.
ccRCC and pRCC are thought to arise from cells in the proximal convoluted renal tubule83 and patient-derived kidney cancer cultures generated from defined serum-free medium consisted of nonmalignanant renal proximal tubule epithelial cells, which indicated that even rare residual nonmalignant cells could out compete tumour cells in this growth medium. This culture condition is similar to the conditions used to generate for prostate organoids culture52, highlighting that this organoid culture system could be suitable for the study of the cell of origin as malignant cells could arise under these conditions.
Several models have been proposed to study the cell of origin of prostate cancer84. Oncogenic transformation of basal cells can initiate prostate tumorigenesis, which suggests a basal cell origin for mouse and human prostate cancer. Lawson and colleagues85 showed that overexpression of Erg or activated RAC-alpha serine/threonine-protein kinase (Akt) in Lin−Sca-1+CD49hi basal cells resulted in prostatic intraepithelial neoplasia (PIN) lesions, and co-activation of Akt and AR signalling gave rise to adenocarcinoma. Stoyanova86 and colleagues showed that overexpression of myc proto-oncogene protein (MYC) and activated AKT in human basal cells initiated adenocarcinoma with squamous phenotype. However, an in vivo lineage-tracing study in genetically engineered mouse models demonstrated that both basal and luminal cells are of largely self-sustained cellular lineages, and that either cell type can initiate prostate cancer development87,88. Wang and colleagues87 showed that initiation of prostate cancer in basal or luminal epithelial cells in this model resulted in tumours with different molecular signatures that are predictive of human patient outcomes. Cross-species bioinformatic analysis demonstrated increased aggressiveness of tumours derived from luminal cells. Considering the different aggressiveness between basal-cell-derived and luminal-cell-derived tumours, identification of patients who need immediate treatment and those who could be benefit from active surveillance might be possible. Castration-resistant Nkx3-1-expressing cells (CARNs) can reside in the luminal compartment of an androgen-deprived prostate. These cells are bipotential stem cells and can give rise to both basal and luminal cells on prostate tissue regeneration, indicating that they can have stem and/or progenitor properties during prostate regeneration. Deletion of Pten in CARNs resulted in high-grade PIN and carcinoma after androgen-mediated regeneration, suggesting that CARNs in the regressed prostate can act as a cell of origin. These findings also give insight into the emergence of castration-resistant disease by identifying a castration-resistant stem cell population as a cell of origin, showing that the castration-resistant clones have been there since prostate tumorigenesis89.
Advances in 3D organoid culture such as extracellular-matrix culture and defined, serum-free medium provide new experimental platforms to investigate the cell of origin for prostate cancer. Organoids derived from human or mouse prostate luminal cells can generate basal cells and demonstrate bipotentiality51,90 (FIG. 2b). Results from a 2015 study using a Pten–Tp53-null mouse model of prostate cancer also demonstrate that luminal cells can be prostate cancer progenitors: culturing organoids derived from luminal cells initiated adenocarcinoma or tumours with multilineage histological phenotypes91. Liu and colleagues92 discovered that low expression of CD38 could be used as a biomarker to identify progenitor-like, inflammation-associated luminal cells that can initiate human prostate cancer. They demonstrated that CD38-low luminal cells expressing both basal (K5 and p63) and luminal (K8) markers were enriched by fourfold to fivefold in organoid formation compared with CD38-high luminal cells. The CD38-low luminal organoids could regenerate into prostatic glands in vivo, expressing both luminal and basal markers. Moreover, the CD26-positive and CD38-high luminal cell population showed low organoid-forming activity, 99% of cells in this group did not exhibit progenitor features. However, CD38-low luminal cells are enriched for progenitor activity in 2D and 3D progenitor assays with 5% efficiency, higher than the 1–2% reported for CD26-high, CD49f-low luminal cells51. Park and colleagues93 demonstrated that both primary human basal and luminal epithelial cells are cells of origin for prostate cancer using a prostate organoid culture system. These investigators overexpressed MYC and activated AKT1 in human prostate basal and luminal cells and then performed the prostate regeneration and transformation assay. They found MYC–myrAKT1-transduced luminal xenografts showed well-differentiated acinar adenocarcinoma, whereas the basal xenografts were histologically more aggressive. Their findings suggest that both basal and luminal cells could respond to the same oncogenic insults to initiate tumorigenesis, but the tumour phenotypes are different.
Organoid systems could be used to study organ development and also to discern the mechanisms of carcinogenesis and identify the cancer cell of origin. Organoids derived from primary benign cells could be used in combination with regeneration and transformation models to study development and malignant transformation. Furthermore, the organoid system enables direct phenotypic observation after introducing the oncogenic events into the system. For example, both the basal-cell-derived and luminal-cell-derived prostate organoids transduced with MYC–myrAKT1 did not display any vague glandular structures or lumen, but some MYC–myrAKT1-transduced luminal organoids showed adenosquamous differentiation93. The systems developed for patient-derived bladder cell culture system and kidney organoids55,80, could be used for studying prostate cancer oncogenic events, regeneration, and transformation to investigate the cell of origin.
Studying tumour microenvironments
Historically, oncologists have concentrated their research efforts on the tumour epithelial compartment as carcinoma develops from epithelial cells. However, gradually, the realization that cancer is a complex set of aberrant interactions between malignant cells within the stromal compartment has occurred, and this awareness has changed the approach to therapeutic design94. Cancer-associated stromal cells (which secrete growth factors, cytokines, and chemokines) have a prominent role in tumour growth and progression95. 3D organoid cultures of nonmalignant human urothelial cells and stroma have been important for investigating urothelial–stromal cell interactions during homeostasis and disease. These organoids were maintained at an air–liquid interface for up to 20 weeks and expressed markers of urothelial differentiation such as cytokeratin 20 from superficial cells96. The organoids were constructed by combining established human cell lines derived from bladder cancer with de-epithelialized stromal tissues and provide an important system for investigating urothelial–stroma inter actions in the context of paracrine signalling mechanisms. Organoids constructed using urothelial carcinoma cell lines recapitulated the invasive characteristics of the original tumour; however, the fidelity of these cells to their tumour of origin was not confirmed97.
In metastatic prostate cancer, disseminated tumour cells predominantly metastasize to bone, therefore, targeting cancer cell–bone interactions can be an effective strategy for treating patients with prostate cancer that has metastasized to bone98. Fong and colleagues99 engineered a bone-metastatic micro environment co encapsulated with tumour cells derived from patient-derived xenografts and osteoblastic cells using a 3D scaffold to provide a robust system for studying the mechanisms of tumour–stromal interactions and to enable drug evaluation. This 3D culture system uses hyaluronan hydrogel to form the scaffold, maintains the expression of key phenotypic tumour markers (such as fibroblast growth factor receptor 1 (FGFR1) and fibroblast growth factor 9 (FGF9), both of which are highly expressed by these cells in vivo), and mimics the FGFR-mediated tumour–stromal interactions of tumour cells grown in bone. Thus, osteoblasts proliferate in response to FGF9 produced by PDXs and FGFR1 is overexpressed in response to FGF9, aberrant FGF signalling could, therefore, promote prostate cancer progression and metastasis100. Moreover, the osteoblastic cells and the prostate cancer cells in the organoid system closely mimicked the architecture of prostate cancer metastasis in bone. Åkerfelt and colleagues101 used a real-time, live-cell platform consisting of tissue from prostate cancer cells and cancer-associated fibroblasts in extracellular matrix to quantitatively monitor the dynamics of cancer-associated fibroblasts in 3D cell cultures. Cancer-associated fibroblasts were observed to initiate tumour cell invasion and promote tumour organoid growth and invasiveness. Disruption of proteins that are associated with tumorigenesis using small-molecule inhibitors was found to perturb interactions between the tumours and cancer-associated cells. For example, focal adhesion kinase (FAK) inhibitors (Y11 and PF-573228) affected both the tumour and the stroma, constraining tumour growth and the dissemination of stromal cells. Y11 and PF-573228, which both target the same phosphorylation site of FAK, have therapeutic potential in vitro without notable cytotoxicity. Thus, this 3D organoid system enables mechanistic studies of tumour–stromal interactions in the prostate.
The microenvironment includes both the encapsulating matrix as well as the different cell types, which may be key determinants of organoid functionality. Organoids could be useful tools for studying the importance and influence of microenvironment interactions and their influence on cells in urological disorders.
Patient-derived organoids
Genetic manipulation
The ability to genetically manipulate organoids is one of the major strengths of this approach. The introduction of oncogenic lesions into organoids derived from non-malignant tissue can be used to model tumorigenesis and define the minimal requirement for neoplasia and metastasis. Nontransformed cell cultures have been extensively used to study oncogenesis102. For example, the focus formation assay, in which retroviral transduction of the oncogene of interest into cells is used to assess one aspect of that oncogene’s transforming potential, is widely used to assess tumorigenesis103,104. In prostate cancer, the RWPE-2 cell line has been used to study tumorigenesis using the focus formation assay105. However, truly nonmalignant cells do not propagate in traditional tissue culture media, therefore, these experiments use immortalized cells, generated either spontaneously through acquisition of mutations in TP53 or CDKN2A, or by use of viral oncogenes that inactivate tumour suppressors such as p53 and retinoblastoma-associated protein and are subject to the Hayflick limit. Thus, genetically engineered mouse models have become the gold standard for studying tumour initiation. Genetically engineered mouse models could be used to investigate cell types of origin by introducing oncogenic lesions in different epithelial lineages in vivo, followed by lineage-tracing to study their progeny and potential transformation. The lack of the microenvironmental factors in vitro (such as a functional immune system) is overcome, as all studies are performed in vivo84. However, the genetically engineered mouse model has several of the limitations. For example, the true phenotype could be affected by Cre-mediated apoptosis. Zhu and colleagues106 reported that Cre-induced apoptosis is Tp53 dependent, demonstrating that apoptosis is stimulated by Tp53 activation in response to DNA damage incurred during the process of Cre-mediated recombination106. Another limitation is the low number of Cre drivers, and generating new Cre drivers is difficult.
Organoid cultures self-renew and can be cultured from unmanipulated, nonmalignant cells enabling study of tumorigenesis that reflects how cancers naturally arise. Results from two studies involving colonic and intestinal organoids that have been manipulated by sequentially introducing genes that are commonly mutated in colorectal cancer using CRISPR–Cas9 technology have been reported107,108. Organoids generated from human normal colonic tissue were engineered to express mutations in the tumour suppressor genes APC, SMAD4, and TP53, and in the oncogenes KRAS and PIK3CA. These organoids grew independently of different factors in vitro, and they formed tumours after implantation under the kidney subcapsule in mice107,108. Organoids generated from nonmalignant human small intestinal stem cells were manipulated to express APC, TP53, KRAS, and/or SMAD4 and were subcutaneoulsy injected into mice. Mice injected with organoids containing KRAS, APC, and TP53 mutations developed adenoma nodules, whereas mice injected with organoids containing KRAS, APC, TP53, and SMAD4 mutations had more invasive carcinomas at 8 weeks after injection108. These results demonstrate that the introduction of oncogenic mutations enables nonmalignant human colonic or intestinal stem cell organoids to develop into tumours in vivo, this technique could be useful for organoids derived from urological tissue.
In prostate cancer, silencing of Pten expression with short hairpin (sh)RNA in prostate organoids derived from PBCre Rosa26LSL–ERG mice caused organoids to display hyperplastic phenotypes ex vivo and generate hyperplastic prostate glandular structures in a urogenital sinus mesenchyme recombination assay, which is consistent with PtenloxP/loxP–Rosa26LSL–ERG grafts51. Organoids are easily manipulated with genome-editing tools, and provide straightforward tool for studying the effect of genetic changes on growth, invasion, and drug sensitivity and are inexpensive and robust in comparison with xenografts. In kidney cancer, Freedman and colleagues109 applied the CRISPR–Cas9 technique to introduce biallelic, truncating mutations into PKD1 or PKD2 in human pluripotent stem cells cultured in organoid systems to model polycystic kidney disease. Large, translucent, cyst-like structures in tubular organoids were observed at 9 weeks after genetic manipulation. These data demonstrate that genetic manipulation using lentiviral or retroviral gene expression or CRISPR–Cas9-mediated gene knockout can be used to identify and study gene function in ex vivo cultured cancer cells. Currently the work in bladder cancer is in its early stages and conclusions with regards to bladder organoids in this context cannot yet be made.
The genetic manipulation of 3D organoids enables early observation of morphological phenotype, which has proven difficult in traditional cell culture. For example, prostate organoids derived from PBCre Rosa26LSL–ERG mice showed no signs of neoplasia in vitro51, consistent with in vivo data110, but Pten deletion and Erg overexpression induced a hyperplastic phenotype in these organoids with protrusion into the Matrigel matrix, suggesting aggressive behaviour.
Patient-derived cancer organoids, like organoids derived from nonmalignant tissue, are amenable to genetic manipulation, giving them an advantage over PDX models. This amenability to manipulation facilitates the study of the effects of alterations on tumour maintenance. For example, mutant genes could be knocked down or repaired, but this research has not yet been conducted in the field of urology. Furthermore, large libraries, including shRNA libraries, CRISPR–Cas9 guides, and cDNA libraries, could be introduced into organoids to enable genetic screens to be performed. Thus, they are powerful models for mechanistic studies and can help increase our understanding of the drivers and development of urological cancers.
Regeneration and gene therapy
Adult stem-cell therapy has been successfully applied in animal models of intestinal and hepatic damage. Long-term engraftment (>6 months) of transplanted organoids derived from a single Lgr5-positive colon stem cell from a donor mouse subjected to extensive in vitro expansion has been observed in superficially damaged mouse colonic epithelium111. In a mouse model of tyrosinaemia type I liver disease induced by a fumarylacetoacetate hydrolase (Fah−/−) mutation, organoids clonally expanded from single Lgr5-positive liver stem cells, which expressed Fah, were integrated into damaged livers by intrasplenical injection. After 2–3 months, immunohistochemical analysis of serial whole-liver sections showed Fah-positive nodules that consisted of cells with hepatocyte morphology, indicating that the cells had acquired a fully mature hepatocyte phenotype in vivo. Engraftment of organoids also improved the survival rate of recipient mice46. These techniques can be adapted for studying regeneration and gene therapy in urological diseases.
Toyohara and colleagues112 successfully ameliorated acute kidney injury (AKI) in mice by delivering renal progenitors derived from human induced pluripotent stem cells. The researchers developed a protocol in which renal progenitors with structures similar to 3D proximal renal tubules in vitro and in vivo were generated from human induced pluripotent stem cells. Renal subcapsular transplantation of these organoids alleviated ischaemia–reperfusion-induced AKI: areas of kidney injury containing tubular necrosis, urinary casts, tubular dilatation, loss of tubular borders, and interstitial fibrosis were smaller in treated mice. A second study also showed similar results113: investigators created a two-stage, reproducible protocol to generate renal progenitors from human induced pluripotent stem cells. Mice with cisplatin-induced kidney injury treated intravenously with the renal progenitors showed evidence of kidney localization of these organoids 24 h after injection, which was sustained after 4 days. Treated mice had improved renal function and histological characteristics compared with control mice. The results of these studies indicate the feasibility of using organoids in regenerative medicine for kidney diseases and for the bladder after cystectomy.
Patient-derived organoids also have the potential to respond to gene therapy. Correction of the most common cystic fibrosis transmembrane conductor receptor (CFTR) mutation, CTFR F508del, was achieved using CRISPR–Cas9 genome editing technology in organoids derived from intestinal stem cells from two patients with cystic fibrosis, using a homologous recombination approach41. The corrected allele is expressed in organoids and restores the functional transport defect of the CFTR. This study offers proof of concept for gene correction of adult stem cells using organoids cultured from patients with a single genetic defect. In the field of urology, no disease only has single genetic defect, therefore, single-gene editing by CRISPR–Cas9 seems to be limited in application. Common medical problems are likely associated with effects of multiple genes defects. However, CRISPR technology has now been adapted to enable multiplexing to target multiple loci114. This mu ltiple hit-method could be promising if combined with organoid technology to repair diseases with multiple genes defects.
The use of differentiated 3D organoid cultures derived from stem cells or progenitor cells for regenerative therapy indicate that organ function (such as for diseased or damaged kidneys, damaged urothelium, and bladder regeneration) can be restored by the integration of ex vivo genetically modified units of tissue rather than whole-organ transplantation.
Personalized medicine
The number of targeted agents for the treatment of urological diseases is currently increasing, therefore, finding predictive biomarkers of clinical benefit to aid precision therapy is of upmost importance. The lack of adequate ex vivo cancer models is a major limitation to identifying predictive biomarkers. Cancer cells cultured in 3D organoid systems have different phenotypes — including differences in, growth, proliferation, migration, and gene expression — from those in 2D culture systems115–117. Results of some studies show that cancer cells in 3D culture systems respond differently from those in 2D culture115–117, perhaps better recapitulating the in vivo response as the cellular microenvironment is more similar to that observed in vivo than for 2D culture.
The development of organoids derived from patients with castration-resistant prostate cancer enables analysis of primary biospecimen and potentially provides personalized information regarding response to therapy ex vivo53. The follow-up validation of predictive information from an organoid-based drug sensitivity assay will require the design of new clinical trials that test whether outcomes achieved using this assay-guided clinical decision are superior to those achieved using the standard-of-care approach.
Cheung and colleagues118 have developed an assay for 3D organoids derived from primary breast cancer samples to identify the most invasive cancer cells, showing that these cells require a conserved basal epithelial cell gene expression programme118. This assay provides an explanation for the crucial question of which mechanisms enable the small group of adherent epithelial cancer cells to acquire the property of collective invasion. Moreover, this 3D organoid culture system enables motile phenotypic observation analyses that could predict individualized prognosis outcomes (such as whether the disease is indolent or highly invasive) based on tissue samples from patients. In a colorectal cancer study, researchers defined poor-prognosis subtypes by evaluating expression profiles of the stromal cells and blocking a crucial signalling pathway (using transforming growth factor β inhibitors) in cancer-associated fibroblasts within the 3D organoid system, which halted disease progression by blocking crosstalk between cancer cells and the microenvironment119. These techniques can be adapted for use in urological systems. Patient-derived prostate cancer organoids have been successfully developed from both metastases and from CTCs53. MSK-PCa4 is a prostate cancer organoid line isolated from a pleural effusion and is capable of ADT-induced neuroendocrine differentiation. In 3D culture, organoids from this line display aggressive phenotypes including a high nuclear:cytoplasmic ratio and multiple mitotic figures indicative of rapid proliferation, which are similar to the original tumour biology. These results show that organoids based on 3D culture can, at least in part, reflect an individual patient’s tumour biology, and could be used to elicit prognostic information. However, further characterization of generated organoid lines is required. However, a major challenge to the risk stratification of patients or personalizing treatment is emerging: whole-genome, whole-exome, and deep-transcriptome sequencing studies in prostate cancer have identified distinct intrapatient molecular alterations in the different multifocal lesions within primary120,121 and different metastatic foci122. These studies have important clinical implications, which indicate that the origin of prostate cancer could be polyclonal. Thus, organoids derived from a single malignant lesion might not be enough to reflect the entire biological diversity of a patient’s cancer, and more tumour sites might need sampling. Modelling different regions in individual tumours to investigate tumour heterogeneity, and testing combined treatment modalities to overcome drug resistance are all promising applications of organoid technology that might aid patient risk stratification and assessment of an individual’s prognosis.
Limitations of organoids
The findings of studies on organoids demonstrate that this technology has a potentially important role in urological research, clinical decision making, and treatment of patients with urological cancers. However, the limitations and technical challenges for this system cannot be ignored. As pure epithelial cultures, organoids lack many cellular components that are contained within an in vivo system (such as stromal, vascular endothelial, or immune cells). The biology and behaviour of cancer can be influenced by microenvironmental factors including the cell types present, which can explain observations such as differences in drug sensitivity for the same cancer cells grown in 2D or 3D cultures or monoculture or coculture with fibroblasts101. Increasing emphasis is being placed on the role of tumour–stromal inter actions123 and several models based on the combination of 3D organoid systems and stromal cells have been developed99,101, which could aid in this research. Models that incorporate multiple cell types, such as fibroblasts, immune cells, and endothelial cells, into 3D culture systems need to be established to reflect the effects of extracellular matrix, epithelial–stromal communications, cell–matrix interactions, and cell–cell crosstalk. To facilitate model development, the many protocols that are currently in use for growing primary tissue in vitro need to be optimized and standardized in each laboratory that cultures organoids to enable comparison of results. Standardized characterization, performed using whole-exome sequencing, copy number analysis, RNA sequencing, and drug sensitivity assays, is necessary to achieve reproducibility.
A number of prostate cancer organoid cell lines have been successfully established; however, all of these organoids are derived from biopsy samples from patients with metastatic prostate cancer, and establishing renewable primary prostate cancer cell lines is technically challenging owing to the overgrowth of nonmalignant prostate epithelial cells that are present within each sample53. Also, generating organoid lines from metastatic biopsies has an efficiency rate of 15–20%, and failure is most frequently associated with the overgrowth of tumour-associated spindle cells. Ongoing efforts to overcome these technical problems include modification of the human organoid medium, purification of cancer samples, removal of the benign and stromal cells, and decreasing tissue processing time.
The identification of the tissue-specific stem cell marker Lgr5 in the formation and maintenance of gastrointestinal tract organs means that a single Lgr5-positive intestinal stem cell can be used to generate adult organoid culture systems. However, the exact nature of the stem or progenitor cells in the kidney23, bladder81, and prostate84 is still unclear and even if the stem or progenitor cells for these organs are discovered, these cells might not survive in culture. Despite this limitation, development of organoids could aid in the discovery of the tumour cells of origin for these organs.
Conclusions
The development of organoids for urological organs creates exciting possibilities for urological research. Organoid culture can facilitate personalized medicine as differentiation of epithelial cells is maintained in this culture system. For example, high-throughput drug screening for therapeutic and toxic effects is possible using renal epithelial cells in a 3D culture system from a kidney biopsy. The development of an organoid biobank means that patient-derived organoids are available for testing tumour sensitivity to drugs, either for personalized medicine or research purposes. Future studies using 3D organoid cultures will also aid the modelling of urological cancer initiation and the molecular characterization of tumour phenotypes in order to discover biomarkers of the tumour cell of origin for various urological organs and also tumour lineages. The use of organoid platforms in research creates exciting possibilities for the development of innovative new therapies and the identification of predictive or prognostic biomarkers, development screening systems, and the identification of patient-specific treatments.
Key points.
The development of in vitro and in vivo modelling systems that accurately reflect the diverse mutational profiles of urological diseases is crucial
Patient-derived organoids in 3D culture systems enable the culturing of stem or progenitor cells for urological organs and they also mimic the in vivo microenvironment and stromal interactions of cancer cells
Nephron progenitor cells derived from human pluripotent stem cells have been successfully used to form complex multicellular kidney organoids, which have potential in disease modelling and drug screening
Organoids can be used to model disease; use of gene editing tools enables the study of the mechanisms of tumorigenesis and restoration of the functional defects caused by genetic lesions
Acknowledgments
We thank Margaret McPartland for providing editorial input. Funding was providing by the National Institutes of Health (5K08CA140946 and NIH Cancer Center Core Grant P30 CA008748, and SPORE in Prostate Cancer (5P50CA092629-15)), US Department of Defense (W81XWH-10-1-0197), the Geoffrey Beene Cancer Center, the STARR Cancer Consortium (I8-A722), a Stand Up To Cancer–Prostate Cancer Foundation Prostate Dream Team Translational Research Grant (SU2CAACR-DT0712), and a Prostate Cancer Foundation Movember Challenge Grant.
Footnotes
Author contributions
All the authors researched data for the article, substantially contributed to discussion of the content, wrote and reviewed and/or edited the article before submission.
Competing interests statement
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–145. doi: 10.1056/NEJMoa1505917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis CF, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson D, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 11.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garnett MJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheon DJ, Orsulic S. Mouse models of cancer. Annu Rev Pathol. 2011;6:95–119. doi: 10.1146/annurev.pathol.3.121806.154244. [DOI] [PubMed] [Google Scholar]
- 14.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 15.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 16.McKay R. Stem cells in the central nervous system. Science. 1997;276:66–71. doi: 10.1126/science.276.5309.66. [DOI] [PubMed] [Google Scholar]
- 17.Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapman S, Liu X, Meyers C, Schlegel R, McBride AA. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J Clin Invest. 2010;120:2619–2626. doi: 10.1172/JCI42297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison RG, Greenman MJ, Mall FP, Jackson CM. Observations of the living developing nerve fiber. Anat Rec (Hoboken) 1907;1:116–128. [Google Scholar]
- 21.Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840:2506–2519. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shamir ER, Ewald AJ. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol. 2014;15:647–664. doi: 10.1038/nrm3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rookmaaker MB, Schutgens F, Verhaar MC, Clevers H. Development and application of human adult stem or progenitor cell organoids. Nat Rev Nephrol. 2015;11:546–554. doi: 10.1038/nrneph.2015.118. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien LE, et al. Rac1 orientates epithelial apical polarity through effects on basolateral laminin assembly. Nat Cell Biol. 2001;3:831–838. doi: 10.1038/ncb0901-831. [DOI] [PubMed] [Google Scholar]
- 25.Bryant DM, et al. A molecular network for de novo generation of the apical surface and lumen. Nat Cell Biol. 2010;12:1035–1045. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yagi S, Matsuda M, Kiyokawa E. Suppression of Rac1 activity at the apical membrane of MDCK cells is essential for cyst structure maintenance. EMBO Rep. 2012;13:237–243. doi: 10.1038/embor.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith YC, Grande KK, Rasmussen SB, O’Brien AD. Novel three-dimensional organoid model for evaluation of the interaction of uropathogenic Escherichia coli with terminally differentiated human urothelial cells. Infect Immun. 2006;74:750–757. doi: 10.1128/IAI.74.1.750-757.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Unsworth BR, Lelkes PI. Growing tissues in microgravity. Nat Med. 1998;4:901–907. doi: 10.1038/nm0898-901. [DOI] [PubMed] [Google Scholar]
- 29.Tyson DR, Inokuchi J, Tsunoda T, Lau A, Ornstein DK. Culture requirements of prostatic epithelial cell lines for acinar morphogenesis and lumen formation in vitro: role of extracellular calcium. Prostate. 2007;67:1601–1613. doi: 10.1002/pros.20628. [DOI] [PubMed] [Google Scholar]
- 30.Lang SH, et al. Experimental prostate epithelial morphogenesis in response to stroma and three-dimensional matrigel culture. Cell Growth Differ. 2001;12:631–640. [PubMed] [Google Scholar]
- 31.Garraway LA, et al. Intermediate basal cells of the prostate: in vitro and in vivo characterization. Prostate. 2003;55:206–218. doi: 10.1002/pros.10244. [DOI] [PubMed] [Google Scholar]
- 32.Xin L, Lukacs RU, Lawson DA, Cheng D, Witte ON. Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells. 2007;25:2760–2769. doi: 10.1634/stemcells.2007-0355. [DOI] [PubMed] [Google Scholar]
- 33.Rinkevich Y, et al. In vivo clonal analysis reveals lineage-restricted progenitor characteristics in mammalian kidney development, maintenance, and regeneration. Cell Rep. 2014;7:1270–1283. doi: 10.1016/j.celrep.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17:1709–1713. doi: 10.1101/gad.267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim KA, et al. Mitogenic influence of human R-spondin1 on the intestinal epithelium. Science. 2005;309:1256–1259. doi: 10.1126/science.1112521. [DOI] [PubMed] [Google Scholar]
- 36.Howe JR, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- 37.Howe JR, et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat Genet. 2001;28:184–187. doi: 10.1038/88919. [DOI] [PubMed] [Google Scholar]
- 38.Haramis AP, et al. De novo crypt formation and juvenile polyposis on BMP inhibition in mouse intestine. Science. 2004;303:1684–1686. doi: 10.1126/science.1093587. [DOI] [PubMed] [Google Scholar]
- 39.Konturek JW, Bielanski W, Konturek SJ, Bogdal J, Oleksy J. Distribution and release of epidermal growth factor in man. Gut. 1989;30:1194–1200. doi: 10.1136/gut.30.9.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dignass AU, Sturm A. Peptide growth factors in the intestine. Eur J Gastroenterol Hepatol. 2001;13:763–770. doi: 10.1097/00042737-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Schwank G, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 42.Rock JR, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci USA. 2009;106:12771–12775. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 44.Boj SF, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huch M, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160:299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huch M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–250. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barker N, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 48.Xia Y, et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat Cell Biol. 2013;15:1507–1515. doi: 10.1038/ncb2872. [DOI] [PubMed] [Google Scholar]
- 49.Takasato M, et al. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat Cell Biol. 2014;16:118–126. doi: 10.1038/ncb2894. [DOI] [PubMed] [Google Scholar]
- 50.Matulay JT, et al. Genetic mutations in patient-derived bladder tumor organoids mimic parental tumor samples [abstract PD38-07] J Urol. 2016;195:e926. [Google Scholar]
- 51.Karthaus WR, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014;159:163–175. doi: 10.1016/j.cell.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Drost J, et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc. 2016;11:347–358. doi: 10.1038/nprot.2016.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gao D, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Batchelder CA, Martinez ML, Duru N, Meyers FJ, Tarantal AF. Three dimensional culture of human renal cell carcinoma organoids. PLoS ONE. 2015;10:e0136758. doi: 10.1371/journal.pone.0136758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lobo NC, et al. Efficient generation of patient-matched malignant and normal primary cell cultures from clear cell renal cell carcinoma patients: clinically relevant models for research and personalized medicine. BMC Cancer. 2016;16:485. doi: 10.1186/s12885-016-2539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van de Wetering M, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 58.Rosenberg JE, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Motzer RJ, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Antonica F, et al. Generation of functional thyroid from embryonic stem cells. Nature. 2012;491:66–71. doi: 10.1038/nature11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koehler KR, Mikosz AM, Molosh AI, Patel D, Hashino E. Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature. 2013;500:217–221. doi: 10.1038/nature12298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eiraku M, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472:51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 63.Suga H, et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature. 2011;480:57–62. doi: 10.1038/nature10637. [DOI] [PubMed] [Google Scholar]
- 64.Little MH, McMahon AP. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol. 2012;4:a008300. doi: 10.1101/cshperspect.a008300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ader M, Tanaka EM. Modeling human development in 3D culture. Curr Opin Cell Biol. 2014;31:23–28. doi: 10.1016/j.ceb.2014.06.013. [DOI] [PubMed] [Google Scholar]
- 66.Takasato M, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526:564–568. doi: 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- 67.Takasato M, Little MH. The origin of the mammalian kidney: implications for recreating the kidney in vitro. Development. 2015;142:1937–1947. doi: 10.1242/dev.104802. [DOI] [PubMed] [Google Scholar]
- 68.Takasato M, Er PX, Chiu HS, Little MH. Generation of kidney organoids from human pluripotent stem cells. Nat Protoc. 2016;11:1681–1692. doi: 10.1038/nprot.2016.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mugford JW, Sipila P, McMahon JA, McMahon AP. Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev Biol. 2008;324:88–98. doi: 10.1016/j.ydbio.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calderon-Gierszal EL, Prins GS. Directed differentiation of human embryonic stem cells into prostate organoids in vitro and its perturbation by low-dose bisphenol A exposure. PLoS ONE. 2015;10:e0133238. doi: 10.1371/journal.pone.0133238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simon J. Ectropia vesicæ; (absence, of the anterior walls of the bladder and pubic abdominal parietes); operation for directing the orifices of the ureters into the rectum; temporary success; subsequent death; autopsy. Lancet. 1852;60:568–570. [Google Scholar]
- 72.van Hemelrijck M, Thorstenson A, Smith P, Adolfsson J, Akre O. Risk of in-hospital complications after radical cystectomy for urinary bladder carcinoma: population-based follow-up study of 7608 patients. BJU Int. 2013;112:1113–1120. doi: 10.1111/bju.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Osborn SL, et al. Induction of human embryonic and induced pluripotent stem cells into urothelium. Stem Cells Transl Med. 2014;3:610–619. doi: 10.5966/sctm.2013-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kang M, Kim HH, Han YM. Generation of bladder urothelium from human pluripotent stem cells under chemically defined serum- and feeder-free system. Int J Mol Sci. 2014;15:7139–7157. doi: 10.3390/ijms15057139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Driscoll L, et al. Phenotypic and global gene expression profile changes between low passage and high passage MIN-6 cells. J Endocrinol. 2006;191:665–676. doi: 10.1677/joe.1.06894. [DOI] [PubMed] [Google Scholar]
- 76.Beltran H, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1:487–495. doi: 10.1158/2159-8290.CD-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scher HI, et al. Circulating tumor cell biomarker panel as an individual-level surrogate for survival in metastatic castration-resistant prostate cancer. J Clin Oncol. 2015;33:1348–1355. doi: 10.1200/JCO.2014.55.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gao J, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shin K, et al. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature. 2011;472:110–114. doi: 10.1038/nature09851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ohishi T, Koga F, Migita T. Bladder cancer stem-like cells: their origin and therapeutic perspectives. Int J Mol Sci. 2016;17:43. doi: 10.3390/ijms17010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li Z, et al. 3D culture supports long-term expansion of mouse and human nephrogenic progenitors. Cell Stem Cell. 2016;19:516–529. doi: 10.1016/j.stem.2016.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pavlovich CP, Schmidt LS. Searching for the hereditary causes of renal-cell carcinoma. Nat Rev Cancer. 2004;4:381–393. doi: 10.1038/nrc1364. [DOI] [PubMed] [Google Scholar]
- 84.Lee SH, Shen MM. Cell types of origin for prostate cancer. Curr Opin Cell Biol. 2015;37:35–41. doi: 10.1016/j.ceb.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 85.Lawson DA, et al. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci USA. 2010;107:2610–2615. doi: 10.1073/pnas.0913873107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stoyanova T, et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proc Natl Acad Sci USA. 2013;110:20111–20116. doi: 10.1073/pnas.1320565110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang ZA, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. 2013;15:274–283. doi: 10.1038/ncb2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Choi N, Zhang B, Zhang L, Ittmann M, Xin L. Adult murine prostate basal and luminal cells are self-sustained lineages that can both serve as targets for prostate cancer initiation. Cancer Cell. 2012;21:253–265. doi: 10.1016/j.ccr.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461:495–500. doi: 10.1038/nature08361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chua CW, et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nat Cell Biol. 2014;16:951–961. doi: 10.1038/ncb3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agarwal S, et al. Identification of different classes of luminal progenitor cells within prostate tumors. Cell Rep. 2015;13:2147–2158. doi: 10.1016/j.celrep.2015.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu X, et al. Low CD38 identifies progenitor-like inflammation-associated luminal cells that can initiate human prostate cancer and predict poor outcome. Cell Rep. 2016;17:2596–2606. doi: 10.1016/j.celrep.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Park JW, et al. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc Natl Acad Sci USA. 2016;113:4482–4487. doi: 10.1073/pnas.1603645113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sluka P, Davis ID. Cell mates: paracrine and stromal targets for prostate cancer therapy. Nat Rev Urol. 2013;10:441–451. doi: 10.1038/nrurol.2013.146. [DOI] [PubMed] [Google Scholar]
- 95.Gaggioli C, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 96.Scriven SD, Booth C, Thomas DF, Trejdosiewicz LK, Southgate J. Reconstitution of human urothelium from monolayer cultures. J Urol. 1997;158:1147–1152. doi: 10.1097/00005392-199709000-00115. [DOI] [PubMed] [Google Scholar]
- 97.Varley CL, Southgate J. Organotypic and 3D reconstructed cultures of the human bladder and urinary tract. Methods Mol Biol. 2011;695:197–211. doi: 10.1007/978-1-60761-984-0_13. [DOI] [PubMed] [Google Scholar]
- 98.Wan X, et al. Prostate cancer cell-stromal cell crosstalk via FGFR1 mediates antitumor activity of dovitinib in bone metastases. Sci Transl Med. 2014;6:252ra122. doi: 10.1126/scitranslmed.3009332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fong EL, et al. A 3D in vitro model of patient-derived prostate cancer xenograft for controlled interrogation of in vivo tumor-stromal interactions. Biomaterials. 2016;77:164–172. doi: 10.1016/j.biomaterials.2015.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Valta MP, et al. FGF-8 is involved in bone metastasis of prostate cancer. Int J Cancer. 2008;123:22–31. doi: 10.1002/ijc.23422. [DOI] [PubMed] [Google Scholar]
- 101.Åkerfelt M, et al. Automated tracking of tumor-stroma morphology in microtissues identifies functional targets within the tumor microenvironment for therapeutic intervention. Oncotarget. 2015;6:30035–30056. doi: 10.18632/oncotarget.5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hahn WC, et al. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 103.Johnson PJ, Coussens PM, Danko AV, Shalloway D. Overexpressed pp60c-src can induce focus formation without complete transformation of NIH 3T3 cells. Mol Cell Biol. 1985;5:1073–1083. doi: 10.1128/mcb.5.5.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bonner TI, et al. Structure and biological activity of human homologs of the raf/mil oncogene. Mol Cell Biol. 1985;5:1400–1407. doi: 10.1128/mcb.5.6.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis. 1997;18:1215–1223. doi: 10.1093/carcin/18.6.1215. [DOI] [PubMed] [Google Scholar]
- 106.Zhu J, Nguyen MT, Nakamura E, Yang J, Mackem S. Cre-mediated recombination can induce apoptosis in vivo by activating the p53 DNA damage-induced pathway. Genesis. 2012;50:102–111. doi: 10.1002/dvg.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Matano M, et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–262. doi: 10.1038/nm.3802. [DOI] [PubMed] [Google Scholar]
- 108.Drost J, et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 2015;521:43–47. doi: 10.1038/nature14415. [DOI] [PubMed] [Google Scholar]
- 109.Freedman BS, et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun. 2015;6:8715. doi: 10.1038/ncomms9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carver BS, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41:619–624. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yui S, et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5+ stem cell. Nat Med. 2012;18:618–623. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 112.Toyohara T, et al. Cell therapy using human induced pluripotent stem cell-derived renal progenitors ameliorates acute kidney injury in mice. Stem Cells Transl Med. 2015;4:980–992. doi: 10.5966/sctm.2014-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Imberti B, et al. Renal progenitors derived from human iPSCs engraft and restore function in a mouse model of acute kidney injury. Sci Rep. 2015;5:8826. doi: 10.1038/srep08826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42:e147. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Deiss F, et al. Platform for high-throughput testing of the effect of soluble compounds on 3D cell cultures. Anal Chem. 2013;85:8085–8094. doi: 10.1021/ac400161j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hakanson M, Textor M, Charnley M. Engineered 3D environments to elucidate the effect of environmental parameters on drug response in cancer. Integr Biol (Camb) 2011;3:31–38. doi: 10.1039/c0ib00074d. [DOI] [PubMed] [Google Scholar]
- 117.Weigelt B, Lo AT, Park CC, Gray JW, Bissell MJ. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res Treat. 2010;122:35–43. doi: 10.1007/s10549-009-0502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155:1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Calon A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47:320–329. doi: 10.1038/ng.3225. [DOI] [PubMed] [Google Scholar]
- 120.Wyatt AW, et al. Heterogeneity in the inter-tumor transcriptome of high risk prostate cancer. Genome Biol. 2014;15:426. doi: 10.1186/s13059-014-0426-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Boutros PC, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;47:736–745. doi: 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- 122.Kumar A, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22:369–378. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dayyani F, Gallick GE, Logothetis CJ, Corn PG. Novel therapies for metastatic castrate-resistant prostate cancer. J Natl Cancer Inst. 2011;103:1665–1675. doi: 10.1093/jnci/djr362. [DOI] [PMC free article] [PubMed] [Google Scholar]