

Abstract
AIM
To investigate interleukin (IL)-26 expression in the inflamed mucosa of patients with inflammatory bowel disease (IBD) and the function of IL-26.
METHODS
Human colonic subepithelial myofibroblasts (SEMFs) were isolated from colon tissue surgically resected. The expression of IL-26 protein and its receptor complex was analyzed by immunohistochemistry. The gene expression induced by IL-26 was evaluated by real-time polymerase chain reaction. Intracellular signaling pathways were evaluated by immunoblotting and specific small interfering (si) RNA transfection.
RESULTS
The mRNA and protein expression of IL-26 were significantly enhanced in the inflamed mucosa of patients with IBD. IL-26 receptor complex was expressed in colonic SEMFs in vivo and in vitro. IL-26 stimulated the mRNA expression of IL-6 and IL-8 in colonic SEMFs. The inhibitors of mitogen-activated protein kinases and phosphoinositide 3-kinase, and siRNAs for signal transducers and activator of transcription 1/3, nuclear factor-kappa B and activator protein-1 significantly reduced the mRNA expression of IL-6 and IL-8 induced by IL-26.
CONCLUSION
These results suggest that IL-26 plays a role in the pathophysiology of IBD through induction of inflammatory mediators.
Keywords: Inflammatory bowel disease, Interleukin-26, Myofibroblasts
Core tip: We investigated interleukin (IL)-26 expression in the inflamed mucosa of patients with inflammatory bowel disease (IBD) and characterized the biological function of IL-26 using human colonic subepithelial myofibroblasts. To our knowledge, this is the first report to state that IL-26 activates STAT1/3 and leads to the induction of IL-6 and IL-8 expression in non-transformed cells derived from human colon. We suggest that IL-26 plays a role in the pathophysiology of IBD through the induction of inflammatory mediators.
INTRODUCTION
Inflammatory bowel diseases (IBD) comprise two major phenotypes, Crohn’s disease (CD) and ulcerative colitis (UC). Recent studies suggest that the chronic inflammation in IBD is mediated by uncontrolled immune responses against a subset of luminal bacteria and dietary factors[1-3]. This hypothesis is supported by the recent finding that the genes encoding innate immune responses are responsible for the susceptibility to IBD[4-6]. However, the precise etiology of IBD still remains unclear.
Interleukin (IL)-26 is a member of the IL-10 cytokine family. IL-26 was first discovered in herpesvirus saimiri-transformed T-cell lines by subtractive hybridization[7]. IL-26 has been reported to be co-expressed with IL-17 and IL-22 by Th17 cells[8-10], and recent studies have reported that natural killer cells[11,12], macrophages[13], and fibroblast-like cells[14,15] are sources of IL-26. A murine IL-26 homologue has not been identified[16,17], limiting the experimental opportunities to study the phenotypic consequences of IL-26 gene knockout and IL-26-mediated functions in murine models in vivo.
Although IL-19, IL-20, and IL-24, members of IL-10 cytokine family, are located in proximity to the IL-10 gene on human chromosome 1q32, IL-26 is located on chromosome 12q15 between the genes encoding IFN-γ and IL-22[18]. For its signaling, IL-26 requires the heterodimeric receptors composed of IL-20R1 and IL-10R2[19]. The transmembrane protein IL-20R1 has been shown to possess the specific ligand-binding site for IL-26, whereas the IL-10R2 subunit acts as an essential second chain that completes the assembly of its active receptor complex and signaling[20]. IL-22, IL-26, IL-28A/B and IL-29 also use IL-10R2 for their signaling. IL-10R2 is ubiquitously expressed in various tissues, but the expression of IL-20R1 is absent in hematopoietic cells and is restricted within non-hematopoietic cells[19,21].
Previous reports have suggested that intracellular signaling of IL-26 is mediated by the Janus kinase-signal transducer and activator of transcription (STAT) pathway[16,20]. Additionally, IL-26 has been reported to activate extracellular signal-related kinase (ERK)-1/2, stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), p38 mitogen-activated protein kinase and phosphoinositide 3-kinase (PI3K) in various kinds of cells[7,16,20,22].
Human colonic subepithelial myofibroblasts (SEMFs) are located immediately subjacent to the basement membrane in the normal intestinal mucosa, juxtaposed against the bottom of the epithelial cells[23-25]. These cells are considered to play a role in the regulation of a number of epithelial cell functions, such as epithelial proliferation and differentiation. Isolated SEMFs retain their representative and differentiated phenotypes[23].
Recent studies have demonstrated that IL-26 is involved in the pathophysiology of chronic inflammatory disorders, such as rheumatoid arthritis[15] and chronic hepatitis C infection[26]. Concerning IBD, the pathological role of IL-26 has been reported in CD, but remains unclear in UC[16]. In addition, functional analysis of IL-26 has been studied using a transformed cell line, but there are no reports using primary culture cells. In the present study, to explore the potential role of IL-26 in IBD, we investigated the expression of IL-26 in the inflamed mucosa of UC and CD patients. Furthermore, the biological functions and the intra-cellular signal pathways activated by IL-26 were investigated in human colonic SEMFs.
MATERIALS AND METHODS
Reagents
All reagents and antibiotics used in this study were commercially purchased as shown in Supplementary Table 1.
Table 1.
Oligonucleotides used in this study
| Gene name | Accession number | Primers | |
| IL-26 | NM_018402.1 | Sense | 5’-GGCAGAAATTGAGCCACTGT-3’ |
| Antisense | 5’-TCCAGTTCACTGATGGCTTTG-3’ | ||
| IL-10R2 | NM_000628.4 | Sense | 5’-GGCTGAATTTGCAGATGAGCA-3’ |
| Antisense | 5’-GAAGACCGAGGCCATGAGG-3’ | ||
| IL-20R1 | NM_014432.3 | Sense | 5’-TACACCCCTCAGCTCCAAGACT-3’ |
| Antisense | 5’-GAAGGAATTACACAGCCTGCCAG-3’ | ||
| IL-6 | NM_000600.4 | Sense | 5’-GGTACATCCTCGACGGCATCT-3’ |
| Antisense | 5’-GTGCCTCTTTGCTGCTTTCAC-3’ | ||
| IL-8 | NM_000584.3 | Sense | 5’-ATGACTTCCAAGCTGGCCGTGGCT-3’ |
| Antisense | 5’-TCTCAGCCCTCTTCAAAAACTTCTC-3’ | ||
| β-actin | NM_001101.3 | Sense | 5’-TGACCCAGATCATGTTTGAGACCT-3’ |
| Antisense | 5’-CCACGTCACACTTCATGATGGAG-3’ | ||
| STAT1 | NM_139266 | Sense | 5’-GGAAGGGGCCATCACATTCA-3’ |
| Antisense | 5’-GTAGGGTTCAACCGCATGGA-3’ | ||
| STAT3 | NM_003150.3 | Sense | 5’-GGAGGAGTTGCAGCAAAAAG-3’ |
| Antisense | 5’-GGAGGAGTTGCAGCAAAAAG-3’ | ||
| c-jun | NM_002228.3 | Sense | 5’-CAGGTGGCACAGCTTAAACA-3’ |
| Antisense | 5’-GTTTGCAACTGCTGCGTTAG-3’ | ||
| NF-κBp65 | NM_003998.3 | Sense | 5’-CGCATCCAGACCAACAACAA-3’ |
| Antisense | 5’-GCATTCAGGTCGTAGTCCCC-3’ |
Tissue samples
The diagnosis of IBD was based on conventional clinical and endoscopic criteria. Surgically-resected specimens and biopsy specimens from 49 patients with UC and 19 patients with CD were used after obtaining written informed consent. All experiments were approved by the local ethics committee of the Shiga University of Medical Science (Permit number: 27-27).
The clinical activity of IBD was determined according to the colitis activity index for UC[27] and CD activity index[28]. Normal colonic tissues were obtained from the distal part of the surgically resected sample of colon cancer (n = 3) and using colonoscopy (n = 17).
Immunohistochemistry
Immunohistochemical analyses were performed according to the method described in our previous report[29]. Briefly, goat anti-IL-26, goat anti-IL-20R1 and rabbit anti-IL-10R2 were used as the primary antibodies. After incubation with the primary antibodies, the sections were treated with HRP-labeled anti-goat or anti-rabbit antibodies. Diaminobenzidine was used as a substrate for color development.
For double-staining procedures, anti-IL-20R1 or anti-IL-10R2 antibodies were applied and incubated overnight in a humidified chamber. Subsequently, anti-α-smooth muscle actin (SMA) antibodies were applied and incubated overnight. Dylight488-labeled anti-goat IgG, Dylight549-labeled anti-mouse IgG, or Dylight549-labeled anti-mouse IgG were used as secondary antibodies. Images were obtained with a digital confocal laser scanning microscope LSM510 version 3.0 (Carl Zeiss Microscopy, Tokyo, Japan).
Culture of human colonic SEMFs
Primary cultures of colonic SEMFs were prepared according to the method reported by Mahida et al[30]. The cellular characteristics and culture conditions have also been described in our previous report[31]. The studies were performed on passages 3-6 of SEMFs.
Reverse transcription-polymerase chain reaction and Real-time polymerase chain reaction
The expression of mRNA in the samples was assessed by reverse transcription polymerase chain reaction (RT-PCR) and real-time PCR analysis. RT-PCR was performed according to the methods described in our previous report[32]. Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, United States) and was reverse transcribed using SuperScript II (Invitrogen). Subsequently, cDNAs were generated using SYBR Premix Ex Taq (TAKARA, Shiga, Japan), and real-time PCR was performed using a LightCycler480 System II (Roche Diagnostics, Basel, Switzerland) with specific primers for target genes. The PCR primers used in this study are shown in Table 1.
Enzyme-linked immunosorbent assay
Concentrations of IL-6 and IL-8 in cell culture supernatants were determined using ELISA kits (R&D systems, Minneapolis, MN, United States).
Silencing gene expression in human colonic SEMFs
Human colonic SEMFs were transfected with siRNA specific for STAT1, STAT3, nuclear factor (NF)-κBp65, and c-Jun according to the instructions for Lipofectamine RNAiMAX (Invitrogen). Briefly, human colonic SEMFs were cultured in complete medium without antibiotics in the presence of a mixture of an RNAi duplex and Lipofectamine RNAiMAX for 24 h, and were then stimulated with or without IL-26 for 3 h.
Nuclear and cytoplasmic protein extraction and immunoblot analysis
Nuclear proteins were extracted using the CelLytic NuCLEAR Extraction Kit (Sigma-Aldrich, St. Louis, MO, United States). Extracted nuclear proteins were subjected to immunoblotting with rabbit anti-NF-κBp65 (C-20) antibody or mouse anti-phospho (P)-c-Jun (KM-1) antibody, followed by incubation with HRP-labeled anti-rabbit antibody or HRP-labeled anti-mouse antibody. Immunoblots were performed according to a method previously described[33,34]. Signal detection was performed using the enhanced chemiluminescence Western blot system (GE Healthcare, Little Chalfont, United Kingdom).
Cytoplasmic protein was extracted using a lysis buffer [50 mmol/L Tris pH 8.0, 0.5% Nonidet P-40, 1 mmol/L EDTA, 150 mmol/L NaCl, 2 mmol/L Na3VO4, 1 mmol/L NaF, 20 mmol/L Na4P2O7, 1 mmol/L PMSF, 10% glycerol and complete Mini Protease Inhibitor Cocktail (Roche Diagnostics)]. Extracted protein was subjected to immunoblotting with antibodies against phospho-p44/42 MAPK (ERK1/2), p38 MAPK, or SAPK/JNK, Akt, STAT1, or STAT3 followed by incubation with HRP-labeled anti-rabbit antibody or HRP-labeled anti-mouse antibody. After detection as described above, the membrane was stripped using Restore Western Blot Stripping Buffer (Thermo Scientific Inc., Waltham, MA) and was then incubated with antibodies against total-p44/42 MAPK (ERK1/2), p38 MAPK, SAPK/JNK, Akt, STAT1, or STAT3.
Statistical analysis
Single comparisons were analyzed using the nonparametric Mann-Whitney U test. Differences resulting in P values of less than 0.05 were considered to be statistically significant. The statistical methods of this study were reviewed by a biomedical statistician from Shiga University of Medical Science.
RESULTS
IL-26 expression in IBD mucosa
The mRNA expression of IL-26 in the inflamed mucosa of IBD patients was evaluated using real-time PCR. As shown in Figure 1, IL-26 mRNA expression was faintly detected in normal mucosa. The mucosal mRNA expression of IL-26 was significantly higher in active UC patients than in the inactive UC mucosa and normal mucosa. Similar findings were also observed in the inflamed mucosa of CD patients. The average level of IL-26 mRNA expression was significantly higher in active CD mucosa than in active UC mucosa.
Figure 1.
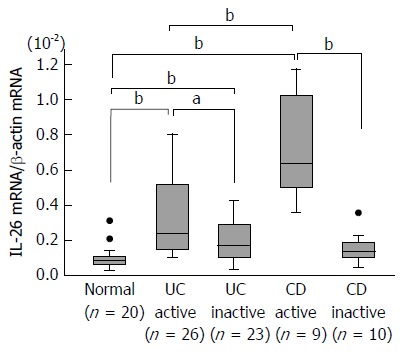
Expression of interleukin-26 mRNA in the inflamed mucosa of patients with inflammatory bowel disease. Total RNA was extracted from biopsied samples, and the mRNA expression of IL-26 was evaluated using real-time PCR. IL-26 mRNA expression was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for normal mucosa. Data are expressed as mean ± SE. Normal mucosa, n = 20; active UC, n = 26; inactive UC, n = 23; active CD, n = 9; inactive CD, n = 10. aP < 0.05 UC active vs UC inactive, bP < 0.01 Normal vs UC active, Normal vs UC inactive, Normal vs CD active, UC active vs CD active, CD active vs CD inactive. IL: Interleukin; CD: Crohn’s disease; UC: Ulcerative colitis.
IL-26 protein expression was evaluated by immunohistochemical analysis. IL-26 positive cells were not detected in normal mucosa, but the number of IL-26 positive cells markedly increased in the inflamed mucosa of UC and CD patients (Figure 2A). Moreover, the number of IL-26 positive cells more increased in the inflamed mucosa of CD patients as compared to in the inflamed mucosa of UC patients (Figure 2A). This result was confirmed by the results from Figure 1. Furthermore, to identify the cellular source of IL-26, double staining was performed. As shown in Figure 2B, double staining studies indicated that IL-26-positive cells were positive for CD4 (T cell), CD56 (NCAM; NK cell), or CD68 (macrophage). These findings indicated that CD4+ T cells, NK cells, and macrophages were the cellular sources of IL-26 in the inflamed mucosa of UC and CD patients.
Figure 2.
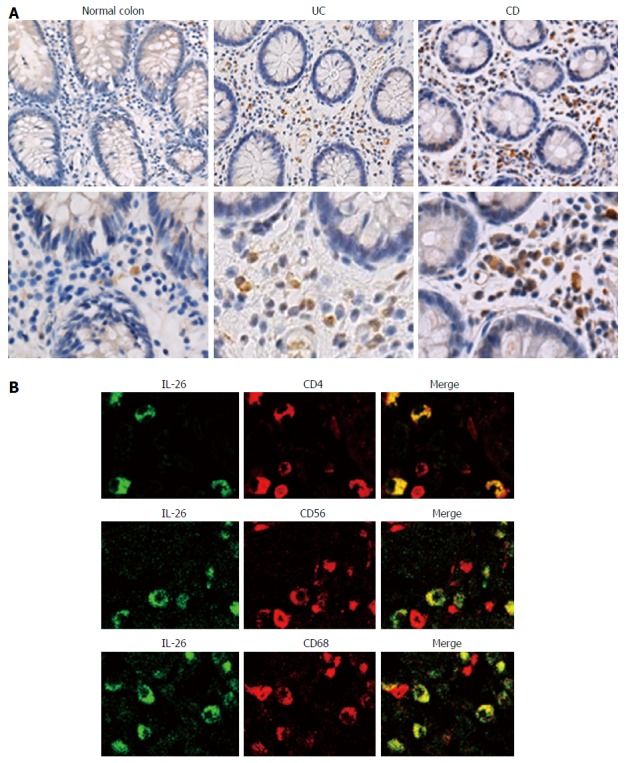
Immunohistochemical analyses of interleukin-26 in the inflamed mucosa of inflammatory bowel disease patients. A: Immunostaining for IL-26 in normal mucosa and inflamed mucosa of UC and CD. Pictures are shown from one of four independent samples with similar results. Magnification; upper panel × 200, lower panel × 400; B: Dual-colored immunofluorescence was used to determine the expression of IL-26 (green fluorescence) and CD4, CD56 and CD68 (red fluorescence) in inflamed mucosa of UC patients. Double-positive cells were detected by yellow fluorescence. Pictures are shown from one of four independent samples with similar results. IL: Interleukin; CD: Crohn’s disease; UC: Ulcerative colitis.
IL-26 receptor expression in SEMFs
We looked for the presence of either IL-20R1 or IL-10R2 in the inflamed mucosa of IBD patients. It has been reported that the expression of IL-20R1 is restricted in non-hematopoietic cells, while IL-10R2 is ubiquitously expressed[19,35]. The tissue samples from the normal, the active IBD and inactive IBD were double stained with anti-α-SMA, a marker for myofibroblasts, and IL-20R1 or IL-10R2. As shown in Figure 3A, IL-20R1 was expressed in epithelial cells and in some of the other cells in the submucosa, and the cells in the subepithelial region also stained with α-SMA. IL-10R2 expression was detected in various cells including epithelial cells and leukocytes, and the cells at the subepithelial region also stained with α-SMA. The expression levels of IL-20R1 and IL-10R2 were not different between in the mucosa from the normal and in the active or inactive mucosa from IBD patients. These results suggested that human colonic SEMFs are expressing the IL-26 receptor.
Figure 3.
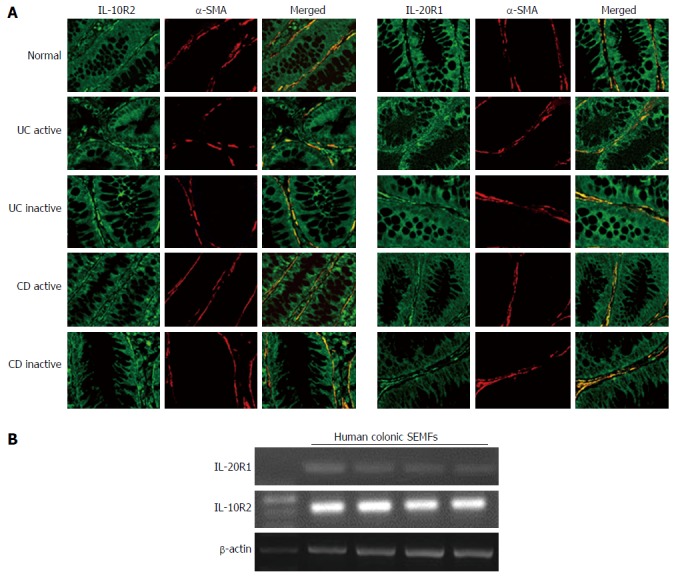
Immunohistochemical analyses of interleukin-26 receptor expression in normal and active/inactive inflammatory bowel disease mucosa. A: Dual-colored immunofluorescence was used to localize α-SMA (red fluorescence) and IL-26 receptor, IL-10R2 and IL-20R1 (green fluorescence). Dual positive immunostaining can be seen as yellow fluorescence in the merged images. Pictures are shown from one of four independent samples with similar results. Magnification × 200; B: The expression of IL-26 receptor in human colonic SEMFs isolated from four different surgical samples. The expression of IL-26 receptor was evaluated by RT-PCR. SMA: Smooth muscle actin; IL: Interleukin; SEMFs: Subepithelial myofibroblasts.
We also confirmed the IL-20R1 and IL-10R2 expression in isolated human colonic SEMFs. As shown in Figure 3B, isolated human colonic SEMFs expressed IL-20R1 and IL-10R2 mRNAs.
Effects of IL-26 in SEMFs
Based on the expression of IL-26 receptor in colonic SEMFs, we examined the biological effect of IL-26 on human colonic SEMFs in vitro. The cells were incubated with IL-26 (100 ng/mL) for 12 h, and then IL-6 and IL-8 levels in supernatants were evaluated using ELISA. As shown Figure 4A, IL-26 induced a significant increase in the secretion of IL-6 and IL-8. These responses were also confirmed at the mRNA levels. IL-26 dose- and time-dependently induced the mRNA expression of IL-6 and IL-8 (Figure 4B and C).
Figure 4.
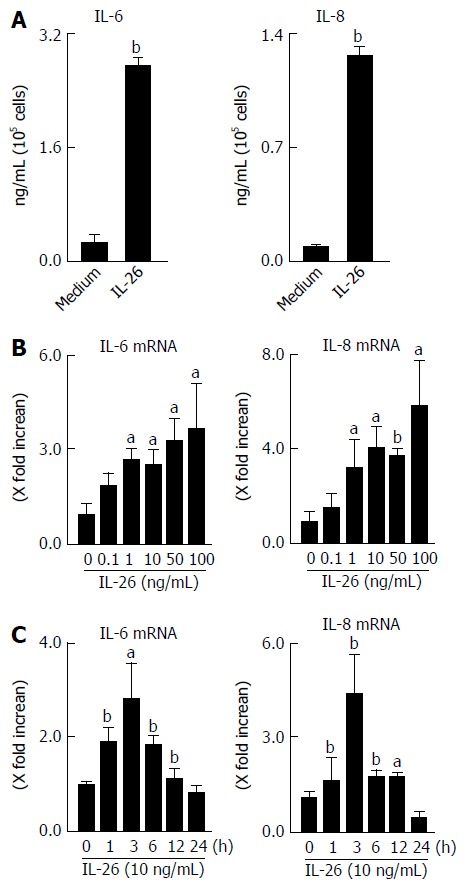
Secretion of interleukin-6 and interleukin-8 in response to interleukin-26 stimulation in human colonic subepithelial myofibroblasts. A: The cells were stimulated for 12 h with 100 ng/mL of IL-26, and the secretion of IL-6 and IL-8 in supernatants were determined using ELISA; B: Dose-dependent effects of IL-26 on IL-6 and IL-8 mRNA expression. Cells were stimulated for 3 h with increasing concentrations of IL-26, and the mRNA expression of IL-6 and IL-8 was then determined using real-time PCR. The mRNA expression of IL-6 and IL-8 was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for medium alone; C: The kinetics of IL-6 and IL-8 induction by IL-26. Human colonic SEMFs were stimulated with 100 ng/mL of IL-26 for the indicated predetermined times, and the mRNA expression of IL-6 and IL-8 was then determined using real-time PCR. The mRNA expression of IL-6 and IL-8 was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for medium alone (no stimulation). Data are expressed as mean ± SE of four independent experiments. aP < 0.05, bP < 0.01 vs control. IL: Interleukin; SEMFs: Subepithelial myofibroblasts.
Activation of STAT1 and STAT3 by IL-26 in human colonic SEMFs
It has been previously reported that the activation of STAT1 and STAT3 is induced by IL-26[16,22]. Therefore, we examined whether IL-26 induced the phosphorylation of STAT1 and STAT3 in human colonic SEMFs. IL-26 induced the phosphorylation of STAT1 and STAT3 as early as 5 min after stimulation with IL-26 (Figure 5A).
Figure 5.
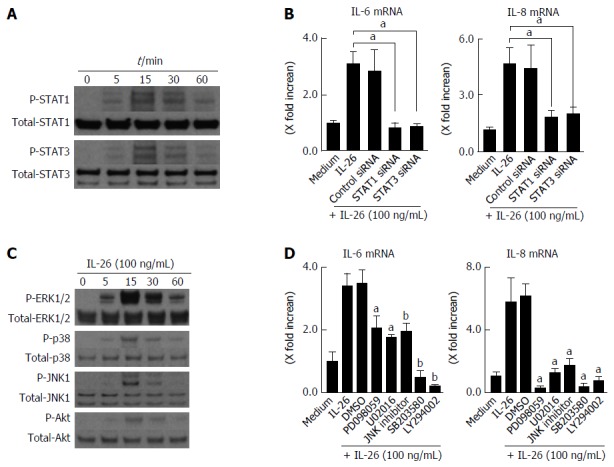
Involvement of STAT1, STAT3, MAPKs and PI3K/Akt activation in interleukin-26-stimulated interleukin-6 and interleukin-8 induction. A: STAT1 and STAT3 activation in response to IL-26. Human colonic SEMFs were stimulated with 100 ng/mL of IL-26 for the indicated pre-determined times, and the phosphorylation (P-) of STAT1 and STAT3 was evaluated by immunoblot analyses. The data are representative of two independent experiments; B: The effects of siRNAs for STAT1 and STAT3 on IL-26-induced mRNA expression of IL-6 and IL-8. Cells were transfected with siRNA for STAT1 and STAT3 or control siRNA, and were incubated for 3 h with or without 100 ng/mL of IL-26. The mRNA expression of IL-6 and IL-8 was then evaluated using real-time PCR. The mRNA expression of IL-6 and IL-8 was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for medium alone (no stimulation); C: MAPKs and PI3K/Akt activation in response to IL-26. Human colonic SEMFs were stimulated with 100 ng/mL of IL-26 for the indicated pre-determined times, and the phosphorylation (P-) of MAPKs and PI3K was evaluated by immunoblot analyses. The data are representative of two independent experiments; D: Effects of inhibitors of MAPKs and PI3K/Akt on IL-6 and IL-8 induction by IL-26 in human colonic SEMFs. The cells were pretreated with 10 μmol/L of a p38 MAPK inhibitor (SB203580) or an MEK1/2 inhibitor (U0216 or PD098059), or with 3 μmol/L of a JNK inhibitor (JNK inhibitor I) or 25 μmol/L of a PI3K inhibitor (LY294002) for 30 min, and were then incubated with or without 100 ng/mL of IL-26 for 3 h. The mRNA expression of IL-6 and IL-8 was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for medium alone. Data are expressed as mean ± SE of four independent experiments. aP < 0.05, bP < 0.01 vs IL-26 stimulation. IL: Interleukin; SEMFs: Subepithelial myofibroblasts.
Involvement of STAT1 and STAT3 activation in IL-26-induced IL-6 and IL-8 was tested using siRNA specific for STAT1 and STAT3. As shown in Figure 5B, the siRNA specific for STAT1 and STAT3 significantly suppressed IL-26-induced mRNA expression of IL-6 and IL-8 effectiveness of siRNA for STAT1 and STAT3 is presented in Figure 6A. These findings indicated that the activation of STAT1 and STAT3 is involved in the induction of IL-6 and IL-8 by the stimulation of IL-26 in human colonic SEMFs.
Figure 6.
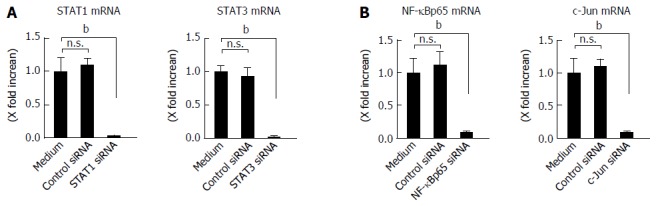
Effect of siRNA. A: Effect of siRNAs specific for STAT1 and STAT3 in human colonic subepithelial myofibroblasts. The cells were transfected with siRNAs specific for STAT1 and STAT3. A control siRNA was also used. The effect of siRNAs was evaluated using real-time PCR. The expression of STAT1 and STAT3 was significantly suppressed by specific siRNAs as compared to a control siRNA. STAT1 and STAT3 mRNA expression was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for medium alone (no stimulation); B: Effect of siRNAs specific for NF-κBp65 and c-Jun (AP-1) in human colonic subepithelial myofibroblasts. The cells were transfected with siRNAs specific for NF-κBp65 and c-Jun (AP-1). A control siRNA was also used. The effect of siRNAs was evaluated using real-time PCR. The expression of NF-κBp65 and c-Jun (AP-1) was significantly suppressed by specific siRNAs as compared to a control siRNA. The mRNA expression of NF-κBp65 and c-Jun (AP-1) mRNA was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for medium alone (no stimulation). Data are expressed as mean ± SE of four independent experiments. bP < 0.01 vs medium. n.s.: Not significant.
Activation of ERK1/2, SAPK/JNK1/2 and Akt by IL-26 in colonic SEMFs
The MAPKs and Akt are involved in the cytokine signaling in various kinds of cells. We examined whether IL-26 activates MAPKs and Akt using immunoblot analysis. As shown in Figure 5C, IL-26 induced a phosphorylation of MAPKs, including p42/44MAPK, SAPK/JNK, and p38MAPK, and Akt as early as 5 min after stimulation with IL-26. Moreover, MEK1/2 inhibitors (PD098059 and U0216), a p38 MAPK inhibitor (SB203580), a JNK inhibitor (JNK inhibitor 1) and a PI3K inhibitor (LY294002) significantly suppressed IL-26-induced IL-6 and IL-8 mRNA expression (Figure 5D). These findings indicate that the activation of MAPKs and the PI3K/Akt pathway is involved in the IL-26-induced IL-6 and IL-8 expression in human colonic SEMFs.
Activation of NF-κB and AP-1 by IL-26
The expression of a number of inflammatory genes is regulated by the activation of transcription factors such as NF-κB and AP-1[36]. In the nuclear proteins, the expression of NF-κB and phosphorylated c-Jun was clearly detected after IL-26 stimulation (Figure 7A). The siRNAs specific for NF-κBp65 and c-Jun (AP-1) significantly suppressed the mRNA expression of IL-6 and IL-8 effectiveness of siRNA for NF-κBp65 and c-Jun (AP-1) is presented in Figure 6B, (Figure 7B). These findings indicate an involvement of NF-κB and AP-1 activation in IL-26-induced IL-6 and IL-8 expression. As shown in Figure 7C, IL-26-induced NF-κBp65 phosphorylation was suppressed by PI3K inhibitor (LY294002), but was not affected by MAPKs. On the other hand, IL-26-induced AP-1 (c-Jun) phosphorylation was suppressed by both MAPKs and PI3K inhibitor. These findings indicate that PI3K, but not MAPKs, plays a role in IL-26-induced NF-κB activation, but that both MAPKs and PI3K activation are involved in IL-26-induced AP-1 activation.
Figure 7.
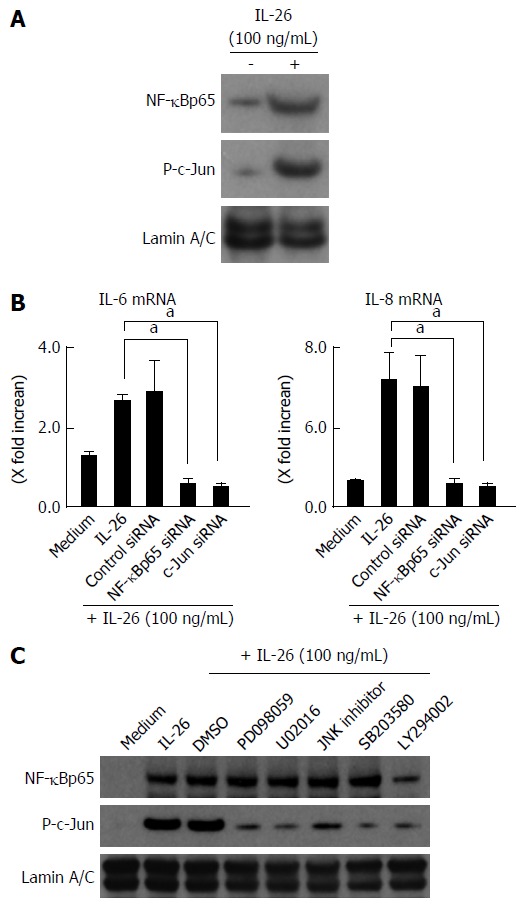
Involvement of nuclear factor-κB and AP-1 activation in the induction of inflammatory mediators induced by interleukin-26. A: Activation of NF-B and AP-1 (c-Jun) in response to IL-26 in human colonic SEMFs. The cells were stimulated with 100 ng/mL of IL-26 for 15 min, following which nuclear proteins were extracted and analyzed by immunoblotting for NF-κB (p65) and phosphorylated (P-) c-Jun. Lamin A/C was used as a loading control. The data are representative of two individual experiments; B: The effects of siRNAs specific for NF-κBp65 and c-Jun (AP-1) on IL-26-induced expression of IL-6 and IL-8 in human colonic SEMFs. The cells were transfected with siRNA specific for NF-κBp65, c-Jun (AP-1) or a control siRNA, and were incubated for 24 h with or without 100 ng/mL of IL-26. The mRNA expression of IL-6 and IL-8 was evaluated using real-time PCR. The mRNA expression of IL-6 and IL-8 mRNA was converted to a value relative to β-actin mRNA expression and presented as fold-increase relative to the results for medium alone (no stimulation). Data are expressed as mean ± SE of four independent experiments. aP < 0.05 vs IL-26 stimulation; C: The effect of MAPKs and PI3K/Akt on the activation of NF-κBp65 and c-Jun (AP-1). The cells were pretreated with 10 μmol/L of a p38 MAPK inhibitor (SB203580) or an MEK1/2 inhibitor (U0216 or PD098059), or with 3 μmol/L of a JNK inhibitor (JNK inhibitor I) or 25 μmol/L of a PI3K inhibitor (LY294002) for 30 min, and were then incubated with or without 100 ng/mL of IL-26 for 15 min. Nuclear proteins were then extracted and analyzed by immunoblotting for NF-κBp65 and phosphorylated (P-) c-Jun. Lamin A/C was used as a loading control. The data are representative of two individual experiments. IL: Interleukin.
DISCUSSION
In the present study, we demonstrated that: (1) the expression of IL-26 is enhanced in the inflamed mucosa of UC and CD patients; (2) human colonic SEMFs express IL-26 receptor complex IL-10R2/IL-20R1 in vivo and in vitro; (3) human colonic SEMFs secrete inflammatory mediators in response to IL-26 stimulation; and (4) IL-26 induced inflammatory mediators via the activation of STAT1/3 and MAPKs/PI3K followed by the activation of NF-κB/AP-1. These observations suggest that IL-26 plays a role in the pathophysiology of IBD.
In this study, we found that IL-26 mRNA expression was enhanced in the inflamed mucosa of UC and CD patients. Its expression was higher in the active mucosa of CD patients than in the active mucosa of UC patients. These findings are similar to our previous observation that mucosal mRNA expression of Th17 cytokines, such as IL-17 and IL-22, was enhanced in the inflamed mucosa of UC and CD patients[29,37]. The mRNA expression of either IL-17 or IL-22 was higher in CD patients than UC patients[29,37]. Th17 cells are now recognized as one of the cellular sources of IL-26[15]. These findings suggest that the IL-26-expressing CD4+ T cells in this study are probably Th17 cells and that Th17 cells are more closely associated with the pathophysiology of CD patients than UC patients.
In the intestinal mucosa, CD56+NCR+ type 3 innate lymphoid cells (ILC3s), a subclass of CD56+ NKp44+ NK cells, have been reported to concomitantly express IL-22 and IL-26, especially following stimulation with IL-23[11]. This suggests that the IL-26-producing CD56+ cells in our study may be NCR+ ILC3s. Our observation of IL-26 expression by CD68+ macrophages is supported by the recent report that CD68+ macrophages are the main IL-26-producing cells in joints with rheumatoid arthritis[15]. Thus, our observations in this study indicate that various types of immune cells are producing IL-26 in the inflamed mucosa of IBD. Further investigation using more precise cellular markers should be performed to more clearly identify the cellular source of IL-26 in the inflamed mucosa of IBD.
There are some reports concerning the in vivo expression of IL-26 under normal and pathological conditions. Corvaisier et al[15] demonstrated a pathogenic role of IL-26 in rheumatoid arthritis on the basis of its capacity to induce pro-inflammatory cytokines. IL-23-induced IL-26 plays a role in the pathophysiology of psoriasis[8]. Moreover, Dambacher et al[16] have reported that IL-26 modulated proliferation and pro-inflammatory gene expression in colon cancer cells and that IL-26 expression was upregulated in active CD, suggesting a role of IL-26 in the innate host cell response during intestinal inflammation. In addition, a genome-wide association study identified IL-26 as one of the susceptibility genes associated with UC[38], suggesting a pathophysiological role of IL-26 in patients with UC. However, previous studies of the clinical role of IL-26 mainly focused on CD rather than UC[16]. In this study, we found that IL-26 was enhanced in the inflamed mucosa of patients with UC, as well as patients with CD. In addition, human colonic SEMFs were expressing functionally-active IL-10R2 and IL-20R1 and secreted inflammatory cytokines in response to IL-26. This phenomenon is supported by previous reports that the expression of IL-20R1 is restricted within non-hematopoietic cells[19,21]. These observations suggest an interaction between IL-26 and colonic SEMFs in inflammatory responses in the colonic mucosa. IL-26 may stimulate the induction of inflammatory mediators from colonic SEMFs and possibly contributes to the inflammatory responses in IBD mucosa. However, recent studies have revealed that Th17-derived cytokines, such as IL-17 and IL-22, possess conflicting (pro-inflammatory and protective) roles in the mucosa[39]. So, further investigations to determine whether IL-26 production is ultimately tissue protective or a significant source of tissue damage in IBD mucosa are required in the future.
There are a few reports concerning the signaling pathway of IL-26 in the colonic tissue. A previous report using transformed epithelial cell lines suggested that IL-26 induces activation of STAT1/3, ERK1/2, SAPK/JNK1/2 and Akt[16]. To our knowledge, this is the first report to state that IL-26 activates STAT1/3 and leads to the induction of IL-6 and IL-8 expression in non-transformed cells derived from human colon. We have also revealed that IL-26 induced the activation of MAPKs, including ERK1/2, p38MAPK and SAPK/JNKL1/2, and PI3K/Akt. Furthermore, we found that PI3K, but not MAPKs, plays a role in IL-26-induced NF-κB activation, but that both MAPKs and PI3K activation were required in IL-26-induced AP-1 activation. These results indicate that IL-26-induced inflammatory responses in the colonic mucosa are mediated by various signaling pathways including STAT1/3, MAPKs and PI3K/Akt, followed by the activation of NF-κB and AP-1.
In conclusion, we demonstrated that the expression of IL-26 is increased in the inflamed mucosa of IBD patients. In human SEMFs, IL-26 induced an activation of STAT1/3 and MAPKs/PI3K, leading to an activation of NF-κB and AP-1. Since the IL-26 gene has not been identified in rodents, the experiments using human colonic SEMFs will contribute to the investigation of the true role of IL-26 in gut inflammation.
COMMENTS
Background
Recent studies have reported that interleukin (IL)-26 is involved in the pathophysiology of chronic inflammatory disorders. Concerning inflammatory bowel disease (IBD), the pathological role of IL-26 has been reported in Crohn’s disease (CD), but remains unclear in ulcerative colitis. Moreover, functional analysis of IL-26 has been studies using a transformed cell line, but there are no reports using primary culture cells.
Research frontiers
The authors found that the expression of IL-26 was enhanced in the inflamed mucosa of IBD as compared to in the normal mucosa. The cellular source of IL-26 in the inflamed mucosa are CD4+ T cells, NK cells, and macrophages. Human colonic subepithelial myofibroblasts (SEMFs) are target cells of IL-26 in the mucosa of IBD.
Innovations and breakthroughs
This study revealed that IL-26 enhanced the induction of inflammatory mediators, IL-6 and IL-8, in the human colonic SEMFs. The inhibitors of IL-26 signaling pathway significantly suppressed the induction of IL-6 and IL-8. The inhibition of IL-26 signaling may lead to the suppression of intestinal inflammation.
Applications
The results of this study indicated that IL-26 may an important role in the pathogenesis of IBD. The authors suggested that IL-26 can a therapeutic candidate for IBD.
Terminology
IL-26 is a member of the IL-10 cytokine family. IL-26 is located on chromosome 12q5. IL-26 requires the heterodimeric receptors composed of IL-20R1 and IL-10R2.
Peer-review
The authors demonstrated that an increased expression of IL-26 in the inflamed mucosa of IBD patients and explored its possible pathway in IBD pathology. The study is well designed and the demonstration seems sufficient.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Japan
Peer-review report classification
Grade A (Excellent): A, A
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study was reviewed and approved by the local ethics committee of the Shiga University of Medical Science (Permit number: 27-27).
Conflict-of-interest statement: The authors declare that they have no competing interests.
Data sharing statement: No additional data are available.
Peer-review started: April 10, 2017
First decision: June 8, 2017
Article in press: July 12, 2017
P- Reviewer: Marzaban R, Pastorelli R, Reyes VE, Wu ZQ, Zimmer V S- Editor: Ma YJ L- Editor: A E- Editor: Zhang FF
References
- 1.Goldsmith JR, Sartor RB. The role of diet on intestinal microbiota metabolism: downstream impacts on host immune function and health, and therapeutic implications. J Gastroenterol. 2014;49:785–798. doi: 10.1007/s00535-014-0953-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sheehan D, Moran C, Shanahan F. The microbiota in inflammatory bowel disease. J Gastroenterol. 2015;50:495–507. doi: 10.1007/s00535-015-1064-1. [DOI] [PubMed] [Google Scholar]
- 3.Sun M, Wu W, Liu Z, Cong Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J Gastroenterol. 2017;52:1–8. doi: 10.1007/s00535-016-1242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franke A, Balschun T, Karlsen TH, Sventoraityte J, Nikolaus S, Mayr G, Domingues FS, Albrecht M, Nothnagel M, Ellinghaus D, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–1323. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 5.Fuyuno Y, Yamazaki K, Takahashi A, Esaki M, Kawaguchi T, Takazoe M, Matsumoto T, Matsui T, Tanaka H, Motoya S, et al. Genetic characteristics of inflammatory bowel disease in a Japanese population. J Gastroenterol. 2016;51:672–681. doi: 10.1007/s00535-015-1135-3. [DOI] [PubMed] [Google Scholar]
- 6.Mizoguchi A, Takeuchi T, Himuro H, Okada T, Mizoguchi E. Genetically engineered mouse models for studying inflammatory bowel disease. J Pathol. 2016;238:205–219. doi: 10.1002/path.4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knappe A, Hör S, Wittmann S, Fickenscher H. Induction of a novel cellular homolog of interleukin-10, AK155, by transformation of T lymphocytes with herpesvirus saimiri. J Virol. 2000;74:3881–3887. doi: 10.1128/jvi.74.8.3881-3887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 9.Pène J, Chevalier S, Preisser L, Vénéreau E, Guilleux MH, Ghannam S, Molès JP, Danger Y, Ravon E, Lesaux S, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 lymphocytes. J Immunol. 2008;180:7423–7430. doi: 10.4049/jimmunol.180.11.7423. [DOI] [PubMed] [Google Scholar]
- 10.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes T, Becknell B, McClory S, Briercheck E, Freud AG, Zhang X, Mao H, Nuovo G, Yu J, Caligiuri MA. Stage 3 immature human natural killer cells found in secondary lymphoid tissue constitutively and selectively express the TH 17 cytokine interleukin-22. Blood. 2009;113:4008–4010. doi: 10.1182/blood-2008-12-192443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Che KF, Tengvall S, Levänen B, Silverpil E, Smith ME, Awad M, Vikström M, Palmberg L, Qvarfordt I, Sköld M, et al. Interleukin-26 in antibacterial host defense of human lungs. Effects on neutrophil mobilization. Am J Respir Crit Care Med. 2014;190:1022–1031. doi: 10.1164/rccm.201404-0689OC. [DOI] [PubMed] [Google Scholar]
- 14.Sziksz E, Pap D, Lippai R, Béres NJ, Fekete A, Szabó AJ, Vannay Á. Fibrosis Related Inflammatory Mediators: Role of the IL-10 Cytokine Family. Mediators Inflamm. 2015;2015:764641. doi: 10.1155/2015/764641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E, Hoppe E, Barré B, Audran M, Bouvard B, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. 2012;10:e1001395. doi: 10.1371/journal.pbio.1001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dambacher J, Beigel F, Zitzmann K, De Toni EN, Göke B, Diepolder HM, Auernhammer CJ, Brand S. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2009;58:1207–1217. doi: 10.1136/gut.2007.130112. [DOI] [PubMed] [Google Scholar]
- 17.Wang T, Díaz-Rosales P, Martin SA, Secombes CJ. Cloning of a novel interleukin (IL)-20-like gene in rainbow trout Oncorhynchus mykiss gives an insight into the evolution of the IL-10 family. Dev Comp Immunol. 2010;34:158–167. doi: 10.1016/j.dci.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Jones EA, Flavell RA. Distal enhancer elements transcribe intergenic RNA in the IL-10 family gene cluster. J Immunol. 2005;175:7437–7446. doi: 10.4049/jimmunol.175.11.7437. [DOI] [PubMed] [Google Scholar]
- 19.Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 20.Sheikh F, Baurin VV, Lewis-Antes A, Shah NK, Smirnov SV, Anantha S, Dickensheets H, Dumoutier L, Renauld JC, Zdanov A, et al. Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J Immunol. 2004;172:2006–2010. doi: 10.4049/jimmunol.172.4.2006. [DOI] [PubMed] [Google Scholar]
- 21.Blumberg H, Conklin D, Xu WF, Grossmann A, Brender T, Carollo S, Eagan M, Foster D, Haldeman BA, Hammond A, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell. 2001;104:9–19. doi: 10.1016/s0092-8674(01)00187-8. [DOI] [PubMed] [Google Scholar]
- 22.Hör S, Pirzer H, Dumoutier L, Bauer F, Wittmann S, Sticht H, Renauld JC, de Waal Malefyt R, Fickenscher H. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J Biol Chem. 2004;279:33343–33351. doi: 10.1074/jbc.M405000200. [DOI] [PubMed] [Google Scholar]
- 23.Andoh A, Bamba S, Brittan M, Fujiyama Y, Wright NA. Role of intestinal subepithelial myofibroblasts in inflammation and regenerative response in the gut. Pharmacol Ther. 2007;114:94–106. doi: 10.1016/j.pharmthera.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–C9. doi: 10.1152/ajpcell.1999.277.1.C1. [DOI] [PubMed] [Google Scholar]
- 25.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. II. Intestinal subepithelial myofibroblasts. Am J Physiol. 1999;277:C183–C201. doi: 10.1152/ajpcell.1999.277.2.C183. [DOI] [PubMed] [Google Scholar]
- 26.Miot C, Beaumont E, Duluc D, Le Guillou-Guillemette H, Preisser L, Garo E, Blanchard S, Hubert Fouchard I, Créminon C, Lamourette P, et al. IL-26 is overexpressed in chronically HCV-infected patients and enhances TRAIL-mediated cytotoxicity and interferon production by human NK cells. Gut. 2015;64:1466–1475. doi: 10.1136/gutjnl-2013-306604. [DOI] [PubMed] [Google Scholar]
- 27.Rachmilewitz D. Coated mesalazine (5-aminosalicylic acid) versus sulphasalazine in the treatment of active ulcerative colitis: a randomised trial. BMJ. 1989;298:82–86. doi: 10.1136/bmj.298.6666.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Best WR, Becktel JM, Singleton JW. Rederived values of the eight coefficients of the Crohn’s Disease Activity Index (CDAI) Gastroenterology. 1979;77:843–846. [PubMed] [Google Scholar]
- 29.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahida YR, Beltinger J, Makh S, Göke M, Gray T, Podolsky DK, Hawkey CJ. Adult human colonic subepithelial myofibroblasts express extracellular matrix proteins and cyclooxygenase-1 and -2. Am J Physiol. 1997;273:G1341–G1348. doi: 10.1152/ajpgi.1997.273.6.G1341. [DOI] [PubMed] [Google Scholar]
- 31.Andoh A, Fujino S, Bamba S, Araki Y, Okuno T, Bamba T, Fujiyama Y. IL-17 selectively down-regulates TNF-alpha-induced RANTES gene expression in human colonic subepithelial myofibroblasts. J Immunol. 2002;169:1683–1687. doi: 10.4049/jimmunol.169.4.1683. [DOI] [PubMed] [Google Scholar]
- 32.Nishida A, Andoh A, Imaeda H, Inatomi O, Shiomi H, Fujiyama Y. Expression of interleukin 1-like cytokine interleukin 33 and its receptor complex (ST2L and IL1RAcP) in human pancreatic myofibroblasts. Gut. 2010;59:531–541. doi: 10.1136/gut.2009.193599. [DOI] [PubMed] [Google Scholar]
- 33.Nishida A, Nagahama K, Imaeda H, Ogawa A, Lau CW, Kobayashi T, Hisamatsu T, Preffer FI, Mizoguchi E, Ikeuchi H, et al. Inducible colitis-associated glycome capable of stimulating the proliferation of memory CD4+ T cells. J Exp Med. 2012;209:2383–2394. doi: 10.1084/jem.20112631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shimada M, Andoh A, Hata K, Tasaki K, Araki Y, Fujiyama Y, Bamba T. IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J Immunol. 2002;168:861–868. doi: 10.4049/jimmunol.168.2.861. [DOI] [PubMed] [Google Scholar]
- 35.Andoh A, Shioya M, Nishida A, Bamba S, Tsujikawa T, Kim-Mitsuyama S, Fujiyama Y. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J Immunol. 2009;183:687–695. doi: 10.4049/jimmunol.0804169. [DOI] [PubMed] [Google Scholar]
- 36.Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol. 2014;10:44–56. doi: 10.1038/nrrheum.2013.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, Kitoh K, Kim-Mitsuyama S, Takayanagi A, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology. 2005;129:969–984. doi: 10.1053/j.gastro.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 38.Silverberg MS, Cho JH, Rioux JD, McGovern DP, Wu J, Annese V, Achkar JP, Goyette P, Scott R, Xu W, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41:216–220. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dige A, Støy S, Rasmussen TK, Kelsen J, Hvas CL, Sandahl TD, Dahlerup JF, Deleuran B, Agnholt J. Increased levels of circulating Th17 cells in quiescent versus active Crohn’s disease. J Crohns Colitis. 2013;7:248–255. doi: 10.1016/j.crohns.2012.06.015. [DOI] [PubMed] [Google Scholar]