

Abstract
The design, synthesis, and evaluation of bifunctional linchpins, conformationally anchored on six-membered rings to achieve efficient [1,5]-Brook rearrangements involving vinyl silanes have been achieved. The restrained linchpins were subsequently exploited in type II anion relay chemistry (ARC) to permit both alkylations and Pd-mediated cross-coupling reactions (CCR) of sp2 stabilized carbanions. DFT calculations were then employed to understand the mechanism and reactivity trends of [1,4]- and [1,5]-vinyl Brook rearrangements to provide insight on the role of the required copper reagent and the substrate geometry.
Graphical abstract
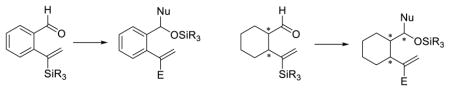
INTRODUCTION
In 2005, we defined a new platform for the construction of molecular complexity termed anion relay chemistry (ARC; Scheme 1).1 Critical to the success of this tactic is the ability to migrate charge in a controlled manner from an oxygen to a carbon atom, bearing an anion stabilizing group (ASG), during a reversible Brook rearrangement.2
Scheme 1.

Anion Relay Chemistry: Type I and Type II
The dithiane group, introduced by Corey and Seebach3 for umpolung reactions and subsequently exploited by the Schaumann4 and Tietze5 groups for what we now view as early versions of the ARC tactic, has proven to be an excellent ASG for both type I and type II ARC transformations involving [1,4]-Brook rearrangements.6 Indeed, by employing a series of diverse dithiane ARC linchpins, a number of architecturally complex targets have been achieved, as demonstrated in synthetic ventures toward spirastrellolide E,7 (−)-secu’amamine A,8 rhizopodin,9 enigmazole A,10 and most recently mandelalide A.11
Following the success of the ARC tactic exploiting the dithiane as an sp3 carbon ASG, several linchpins were subsequently designed, synthesized, and validated to exploit sp2-hybridized carbons in [1,4]-Brook rearrangements. Anions derived from furans,12 arenes, and olefins were employed.13 During this study, the stability of the sp2 carbanion derived from arenes and olefins during the reversible O to C(sp2) Brook rearrangement, not surprisingly, proved less favorable compared to the C(sp3) dithiane stabilized carbanions. To augment the sp2 based ARC tactic, we introduced a stabilization factor, via the formation of an organocuprate.13,14
The latter permitted the successful validation of vinyl silanes bearing β or γ electrophilic carbonyl sites for type II ARC transformations based on [1,4]-Brook rearrangements.13 During these earlier studies, we recognized however that the Brook rearrangement was quite sensitive to the specific location of the silyl group on the double bond. The conditions that proved effective for linchpin 10 (1.2 equiv of n-BuLi and CuI; Scheme 2) did not permit facile [1,4]-silyl migration with linchpin 12. In this case, 2 equiv of both n-BuLi and CuI were required to complete the 1,4-silyl C(sp2) to O migration over the course of a 2 h period at room temperature (Scheme 2).13 At the time, it was not clear why a second equivalent of n-BuLi and CuI significantly improved the [1,4]-vinyl Brook rearrangement of 12.
Scheme 2.

Type II ARC with [1,4]-Brook Rearrangement
Recognizing that [1,5]-Brook rearrangements with vinyl linchpins, and in particular linchpins with terminal olefins held considerable promise for the construction of molecular complexity, we began to investigate an ARC tactic based on [1,5]-silyl C(sp2) to O migrations (Figure 1).15 We reasoned that anchoring the requisite ARC functionality on a phenyl or cyclohexyl ring in a 1,2 fashion would limit bond rotation and thereby potentially enhance the [1,5]-Brook rearrangement.
Figure 1.

Previous and present work: union reaction exploiting [1,5]-Brook rearrangements.
RESULTS AND DISCUSSION
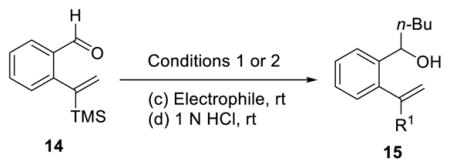
To initiate this study, we prepared phenyl linchpin 14 via Negishi cross coupling of (1-trimethylsilyl)-vinyl zinc with 2-bromo-benzaldehyde (see Supporting Information). Our initial conditions for the [1,5]-Brook rearrangement were derived from our previous work involving [1,4]-silyl C(sp2) to O migration,13 as well as from earlier observations of the Takeda16 and Song groups.17 Specifically, after addition of 2 equiv of n-BuLi to aldehyde 14, the resulting lithium alkoxide was added into a suspension of CuBr·DMS in HMPA and THF (1:1) via cannula to trigger the Brook rearrangement, presumably permitting the formation of a vinyl-cuprate. Importantly, we subsequently discovered that in place of adding 2 equiv of the required nucleophile, an additive (i.e., 1 equiv of a tert-butoxide salt) could be employed to achieve similar results, thus eliminating the need for 2 equiv of the potentially valuable nucleophile. The vinyl-cuprate was then subjected to alkylation with a variety of electrophiles to furnish three-component adducts in modest to good yields (Table 1).
Table 1.
Three-Component Coupling of Linchpin 14 via ARC/Alkylation
| ||
|---|---|---|
|
| ||
| electrophile | R1 | yield |
|
|
|
73%a (74%)b |
|
 15b
15b
|
68%a (73%)b |
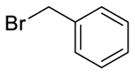
|
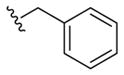 15c
15c
|
70%a (73%)b |
| Mel |
|
(77%)b |
|
|
|
61%a (56%)b |
Conditions 1: (a) 2.0 equiv of n-BuLi, Et2O, −78 °C; (b) 2.0 equiv of CuBr·DMS, HMPA/THF (1:1), rt; then (c) and (d).
Conditions 2: (a) 1.1 equiv of n-BuLi, THF, −78 °C; (b) 2.0 equiv of CuBr·DMS, 1.0 equiv of t-BuOK, HMPA, rt; then (c) and (d).
The organocuprate generated from the [1,5]-Brook rearrangement was next evaluated for cross coupling with various electrophilic partners, employing Pd(PPh3)4 as the catalyst (Table 2). Both electron-deficient and -rich aryl iodides proved viable substrates under the ARC/CCR protocol. Vinyl bromide could also be employed as the cross-coupling partner to provide diene 16e. In addition to n-BuLi, methyl, phenyl, 2-methyl vinyl, 2-furanyl, and 2-methyl-1,3-dithianyl lithiums were examined as the initiating nucleophile for the ARC process; all worked, leading to the desired three-component adducts 16f–16j.
Table 2.
Three-Component Coupling of Linchpin 14 via ARC/CCR

| ||||
|---|---|---|---|---|
|
| ||||
| nucleophile | electrophile | R2 | R3 | yield |
| n-BuLi |
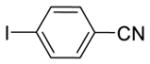
|
n-Bu |
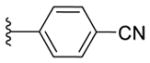 16a
16a
|
72%a (70%)b |
| n-BuLi |
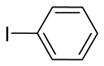
|
n-Bu |
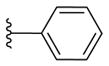 16b
16b
|
77%a (71%)b |
| n-BuLi |
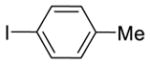
|
n-Bu |
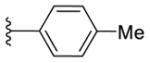 16c
16c
|
73%a (78%)b |
| n-BuLi |
|
n-Bu |
 16d
16d
|
72%a (76%)b |
| n-BuLi |
|
n-Bu |
 16e
16e
|
75%a (78%)b |
| MeLi |
|
Me |
16f
|
70%a (69%)b |
| PhLi |
|
Ph |
16g
|
63%a (57%)b |
|
|
|
16h
|
(69%)b |
|
|
|
16i
|
(57%)b,c |
|
|
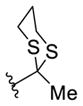
|
16j
|
(53%)b,d |
Conditions 1: (a) 2.0 equiv of nucleophile, Et2O, −78 °C; (b) 2.0 equiv of CuBr·DMS, HMPA/THF (1:1), rt; then (c) and (d).
Conditions 2: (a) 1.1 equiv of nucleophile, THF, −78 °C; (b) 2.0 equiv of CuBr·DMS, 1.0 equiv of t-BuOK, HMPA, rt; then (c) and (d).
For conditions 2, step b, 60 °C.
for conditions 2, step a, rt.
Encouraged by the viability of the ARC alkylation and cross coupling protocols exploiting a [1,5]-Brook rearrangement with phenyl linchpin 14, we turned to evaluate cyclohexyl-based linchpins, constructed via conjugate addition18,19 of the vinyl-cuprate derived from 1720 to aldehyde 18 (Scheme 3); a 1:1 mixture of cis- and trans-linchpins (19 and 20), separable via flash chromatography with the stereochemistry assigned by NMR, and subsequent chemical conversion (vide infra) resulted in a combined yield of 63%. Importantly, cis-linchpin 19 could be readily converted to the trans-congener (20) by treatment with DBU in MeOH.
Scheme 3.

Preparation of Cyclohexyl Incorporated Linchpins 19 and 20
With both cyclohexyl-linchpins (19 and 20) in hand, we first conducted an optimization study with cis-cyclohexyl linchpin 19 exploiting the conditions outlined in Table 3. Compared to the phenyl linchpin (14), an increase in reaction temperature was required to effectively trigger the [1,5]-Brook rearrangement. Again, t-BuOK could be employed to promote Brook rearrangement, thus saving 1 equiv of the nucleophile, although a longer reaction time was required to complete the Brook process. In the case of the TMS linchpin, the longer reaction time unfortunately resulted in several side reactions (i.e., loss of TMS group and/or decomposition of the vinyl-cuprate). To shorten the required reaction time and thus side product formation, we increased the solvent polarity to further promote the Brook rearrangement efficiency (entry 7).
Table 3.
Optimization of [1,5]-Brook Rearrangement with cis Linchpin 19
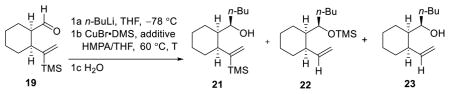
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | n-BuLi (equiv) | Cu(I) (equiv) | additive (equiv) | HMPA/THE | T (h) | ratioa (21/22/23) |
| 1b | 1.1 | 1:1 | 0.5 | 1:0: 0 | ||
| 2 | 1.1 | 1.2 | 1:1 | 6 | 1:0: 0 | |
| 3 | 1.1 | 2.0 | 1:1 | 6 | 1:0.2:0.1 | |
| 4 | 1.5 | 2.0 | 1:1 | 3 | 0.1:1: 0.7 | |
| 5 | 2.0 | 2.0 | 1:1 | 0.75 | 0:1: 0 | |
| 6 | 1.1 | 2.0 | 1.0c | 1:1 | 1.5 | 0.1:1: 0.7 |
| 7d | 1.1 | 2.0 | 1.0c | 1:0 | 0.75 | 0:1: 0 |
Ratio of major products (21/22/23) by NMR analysis of the reaction mixture: values are reported as 0 if less than 0.05.
Isolated yield for product 21: 86%.
t-BuOK.
Isolated yield for product 22: 73%.
Continuing with the envisioned vinyl-cuprate intermediate derived via the [1,5]-Brook rearrangement, we introduced an electrophile to complete the tricomponent ARC tactic. As with phenyl linchpin 14, the vinyl-cuprate was evaluated for both alkylation (Table 4) and cross coupling (Table 5); both reactions proceeded, leading to the three-component adducts in modest to good yield.
Table 4.
Three-Component Coupling of Linchpin 19 via ARC/Alkylation

| ||
|---|---|---|
|
| ||
| electrophile | R4 | yield |
|
|
|
63%a (63%)b |
|
|
24b
|
54%a (67%)b |
|
|
24c
|
66%a (71%)b |
| Mel |
|
(65%)b |
|
|
|
45%a (53%)b |
Conditions 1: (a) 2.0 equiv of n-BuLi, THF, −78 °C; (b) 2.0 equiv of CuBr·DMS, HMPA/THF (1:1), 60 °C; then (c) and (d).
Conditions 2: (a) 1.1 equiv of n-BuLi, THF, −78 °C; (b) 2.0 equiv of CuBr·DMS, 1.0 equiv of t-BuOK, HMPA, 60 °C; then (c) and (d).
Table 5.
Three-Component Coupling of Linchpin 19 via ARC/CCR

| ||||
|---|---|---|---|---|
|
| ||||
| nucleophile | electrophile | R5 | R6 | yield |
| n-BuLi |
|
n-Bu |
25a
|
49%a (53%)b |
| n-BuLi |
|
n-Bu |
25b
|
61%a (57%)b |
| n-BuLi |
|
n-Bu |
25c
|
54%a (59%)b |
| n-BuLi |
|
n-Bu |
25d
|
55%a (62%)b |
| n-BuLi |
|
n-Bu |
25e
|
(55%)b |
| n-BuLi |
|
n-Bu |
25f
|
(68%)b |
| MeLi |
|
Me |
25g
|
55%a (51%)b |
| PhLi |
|
Ph |
25h
|
63%a (52%)b |
Conditions 1: (a) 2.0 equiv of nucleophile, THF, −78 °C; (b) 2.0 equiv of CuBr·DMS, HMPA/THF (1:1), 60 °C; then (c) and (d).
Conditions 2: (a) 1.1 equiv of nucleophile, THF, −78 °C; (b) 2.0 equiv of CuBr·DMS, 1.0 equiv of t-BuOK, HMPA, 60 °C; then (c) and (d).
Notably, nucleophilic additions to cis-linchpin 19 proved highly diastereoselective (>20:1), yielding the Felkin–Anh product,21 which was confirmed in the case of three-component adduct 25h by X-ray analysis of the derived 4-nitrobenzoic ester (26) (Figure 2).
Figure 2.

Elucidation of relative configuration via X-ray analysis.
We next extended the [1,5] ARC tactic to trans-linchpin 20. High Felkin–Anh selectivity upon addition of the nucleophile to the aldehyde was achieved. The stereochemistry of the isolated product was again confirmed by X-ray analysis of the p-nitrobenzoic ester derivative 27 (Figure 2).
The [1,5]-Brook rearrangement of trans-linchpin 20 however proved more challenging than the cis-linchpin. In particular, difficulties were encountered in retaining the TMS silyl group during the [1,5]-Brook protocol. As a result, multiple alkylation byproducts were observed due to competing O-alkylation when an electrophile was introduced. To resolve this issue, we turned to the more robust TBS counterpart 31, which was prepared in similar fashion as the TMS linchpin (Scheme 4). As expected, loss of the silyl group was no longer observed, but a higher reaction temperature was required to trigger Brook rearrangement, presumably due to the increased steric hindrance of the TBS group. As mentioned earlier during the optimization of cis-linchpin, longer reaction times resulted in multiple side reactions. In this case, decomposition of a significant amount of the vinyl-cuprate occurred. To decrease the reaction time, we employed a 5 min pulse of microwave irradiation (Biotage: high setting) at 100 °C. The derived vinyl-cuprate was then employed for three-component unions, as demonstrated by alkylation with benzyl bromide and CCR with 4-iodobenzonitrile (Scheme 4).
Scheme 4.

Synthesis and Evaluation of trans Linchpin 31
To understand the mechanism and reactivity trends of the [1,4]- and [1,5]-vinyl Brook rearrangements, we conducted computational studies using DFT (M06 with solvation) calculations,22,23 in which MeLi was used as a computational model for the experimentally employed n-BuLi. First, three [1,4]-vinyl Brook rearrangements were compared to reveal the effect on the energetics of the position of the C═C double bond and the copper reagent employed. The results are summarized in Scheme 5, with the key structures illustrated in Figure 3.
Scheme 5.

Comparisons of [1,4]-Vinyl Brook Rearrangements Involving Different Substrates and Copper Reagents
Figure 3.

DFT-optimized structures of intermediates and transition states for [1,4]-vinyl Brook rearrangements; carbon gray, hydrogen white, oxygen red, silicone yellow, lithium blue, copper green, and iodine purple.
For the [1,4]-Brook rearrangement of substrate A having an internal C═C double bond (Scheme 5a), coordination of the CuI to both the C═C bond and oxygen atom of A generates an intermediate Ac; this step is exergonic by 28.5 kcal/mol in THF. Subsequently, the oxygen atom attacks the trimethylsilyl group via transition state TSA (Figure 3), in which the forming O–Si bond distance is 1.85 Å and the breaking C–Si bond is 2.37 Å, while the C–Cu distance is decreased to 1.96 Å. The free energy barrier for this process is 16.0 kcal/mol (from Ac to TSA). Importantly, the resultant [1,4]-Brook rearrangement product Ap is more stable than Ac by 6.3 kcal/mol.
As reported above, our experimental results demonstrate that the [1,4]-Brook rearrangement is sensitive to the position of C═C double bond (Scheme 2). The computed reaction pathway of the [1,4]-Brook rearrangement of B having a terminal C═C double bond with 1 equiv of CuI is illustrated in Scheme 5b. In this case, the formation of intermediate Bc via coordination of CuI to both the C═C bond and the oxygen atom of B is exergonic by 35.5 kcal/mol, which is 7 kcal/mol more favorable than that of Ac (−35.5 versus −28.5 kcal/mol). Subsequent [1,4]-Brook free energy of 29.8 kcal/mol. This barrier is 13.8 kcal/mol higher than that for Ac with an internal C═C double bond (29.8 versus 16.0 kcal/mol). As shown in Figure 3, the coordination of the internal C═C double bond forms a strained C–Cu–O–C four membered ring in Ac, with C–Cu and O–Cu distances of 2.10 and 2.22 Å. However, in the Brook rearrangement transition state TSA, the corresponding distances are significantly increased to 2.75 and 2.70 Å, respectively. This indicates that there is remarkable strain-release in transition state TSA, leading to a low barrier.24 However, when the position of the C═C double bond is changed from internal to terminal, the strained C–Cu–O–C four membered ring in Ac evolves to an unstrained C–Cu–O–C–C–C six membered ring in Bc. As a result, there is no strain-release in transition state TSB, making the barrier much higher, and in the product (Bp), which is even 0.2 kcal/mol less stable than Bc (−35.3 versus −35.5 kcal/mol). This result explains why the [1,4]-Brook rearrangement of B having a terminal C═C double bond does not occur using 1 equiv of CuI.
To achieve efficiency with the [1,4]-Brook rearrangement of B, experiments demonstrate that 2 equiv of a Cu(I) salt and a second equivalent of lithium reagent are required (Scheme 2). However, it was not clear what copper reagent is involved. Our calculations reveal that the most favorable reaction between 1 equiv of MeLi with 2 equiv of CuX (X = I or Br) is to form MeCu (eq 1),
| (1) |
which is exergonic by more than 50 kcal/mol in THF. This suggests that under these reaction conditions, MeCu instead of CuX will trigger Brook rearrangement.
In Scheme 5c, we outline the computed reaction pathway of [1,4]-Brook rearrangement of B with 1 equiv of MeCu. Here the MeCu is also coordinated by both the C═C bond and the oxygen atom of B to form intermediate Cc. However, this process is much less exergonic than that using CuI (−20.4 versus −35.5 kcal/mol). The subsequent [1,4]-Brook rearrangement via transition state TSC thus requires an activation free energy of 25.8 kcal/mol. This barrier is 4 kcal/mol lower than that using CuI (25.8 versus 29.8 kcal/mol). According to the frontier molecular orbital (FMO) analysis, the LUMO energies of CuI and MeCu are −15.3 and 1.3 kcal/mol, respectively. This indicates that CuI is a much better electron acceptor than MeCu, leading to a much stronger coordination or bonding interaction. As shown in Figure 3, the geometries of intermediates Bc and Cc, and transition states TSB and TSC are very similar. In the Brook rearrangement transition state TSB or TSC, one C–Cu bond (TSB, 1.96 Å; TSC, 1.97 Å) is forming, and one O···Cu and the other C···Cu coordinations (shown as green lines in the right structures of Figure 3) disappear. When MeCu is employed instead of CuI, two weaker O···Cu and C···Cu interactions are needed to be overcome in the transition state, although one weaker C–Cu bond is forming. Overall, this decreases the barrier for the Brook rearrangement. Therefore, the alkyl cuprate is more active than the Cu(I) salt to trigger Brook rearrangement, in agreement with the experimental results.
Four [1,5]-vinyl Brook rearrangements were next compared to reveal the role of the conformational constraints of substrates. The results are summarized in Scheme 6, with the key structures illustrated in Figure 4.
Scheme 6.

Comparisons of [1,5]-Vinyl Brook Rearrangements Involving Dierent Substrates
Figure 4.

DFT-optimized structures of intermediates and transition states for [1,5]-vinyl Brook rearrangements; carbon gray, hydrogen white, oxygen red, silicone yellow, lithium blue, and copper green.
For the MeCu mediated [1,5]-Brook rearrangement of substrate D having a terminal C═C double bond (Scheme 6a), the reaction free energy for the coordination step is −20.1 kcal/mol, which is very close to that of [1,4]-Brook rearrangement substrate C (−20.4 kcal/mol, Scheme 5c). However, the barrier for the [1,5]-Brook rearrangement is 3.7 kcal/mol higher than that for the [1,4] reaction (29.5 versus 25.8 kcal/mol). As shown in Figures 3 and 4, the forming and breaking bond distances in transition states TSC and TSD are almost identical; the main difference is the conformational change of the tether. In intermediate Dc, the dihedral angle along the (CH2)2 tether is −73.2°, but this angle becomes −57.9° in transition state TSD. This means a 15.3° conformational change, requiring additional energy as compared to [1,4]-Brook rearrangement from Cc to TSC, in which the tether is CH2 without the dihedral angle change. This explains why efficient [1,5]-Brook rearrangements are more challenging.
In Scheme 6b–d, we illustrate the computed reaction pathways of the three experimentally studied [1,5]-vinyl Brook rearrangements involving phenyl, cis-cyclohexyl, and trans-cyclohexyl linchpins. The activation free energies for these rearrangements are 23.3 (from Ec to TSE), 25.6 (from Fc to TSF), and 29.7 (from Gc to TSG) kcal/mol, respectively. This reactivity trend is in accordance with the increase in reaction temperature for Brook rearrangement observed experimentally. More importantly, the calculations demonstrate that there is a good correlation between the conformational change of the tether and the barrier for the [1,5]-Brook rearrangement. As illustrated in Figure 4, when the tether is switched from phenyl, to cis-cyclohexyl, and to trans-cyclohexyl, the corresponding conformational change is increased from 4.8° (from −1.4° to −6.2°), to 6.1° (from −41.4° to −47.5°), and to 13.5° (from 39.8° to 53.3°). Consequently, the barrier is increased from 23.3 to 25.6 and 29.7 kcal/mol (Scheme 6b–d). These results indicate that using the tether with conformational constraints can efficiently lower the [1,5]-Brook rearrangement barrier.
CONCLUSIONS
In summary, we have achieved extension of the ARC tactic exploiting more challenging [1,5]-Brook rearrangements with a series of conformationally anchored bifunctional vinyl linchpins. Phenyl, cis-cyclohexyl, and trans-cyclohexyl linchpins were systematically evaluated via both experimental and computational approaches. Of importance, we demonstrated that introduction of an additional amount of organolithium or t-BuOK as additive lowers the energy barrier to facilitate the requisite Brook rearrangement. As such, this ARC protocol now holds considerable synthetic promise for the rapid elaboration of advanced intermediates for complex molecule synthesis.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (National Cancer Institute) through Grant No. CA-19033 and the National Science Foundation (CHE-1361104). Y.L. thanks the National Thousand Young Talents Program and Jiangsu Specially-Appointed Professor Plan in China for financial support. We also thank China Scholarship Council for a scholarship for Q.L. We thank Drs. George Furst and Jun Gu and Dr. Rakesh Kohli at the University of Pennsylvania for help in obtaining the NMR and high-resolution mass spectral data, respectively, and Dr. Patrick J. Carroll for X- ray crystallographic data.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b04149.
Experimental procedures and computational details (PDF)
NMR spectra for obtained compounds (PDF)
X-ray data for compound 26 (CIF)
X-ray data for compound 27 (CIF)
References
- 1.Smith AB, Xian M. J Am Chem Soc. 2006;128:66. doi: 10.1021/ja057059w. [DOI] [PubMed] [Google Scholar]
- 2.Brook AG. Acc Chem Res. 1974;7:77. [Google Scholar]
- 3.Seebach D, Corey EJ. J Org Chem. 1975;40:231. [Google Scholar]
- 4.Fischer MR, Kirschning A, Michel T, Schaumann E. Angew Chem, Int Ed Engl. 1994;33:217. [Google Scholar]
- 5.Tietze LF, Geissler H, Gewert JA, Jakobi U. Synlett. 1994;1994:511. [Google Scholar]
- 6.For reviews on anion relay chemistry, see: Smith AB, III, Adams CM. Acc Chem Res. 2004;37:365. doi: 10.1021/ar030245r.Smith AB, III, Wuest WM. Chem Commun. 2008:5883. doi: 10.1039/b810394a.
- 7.Sokolsky A, Wang X, Smith AB. Tetrahedron Lett. 2015;56:3160. doi: 10.1016/j.tetlet.2014.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han H, Smith AB. Org Lett. 2015;17:4232. doi: 10.1021/acs.orglett.5b02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melillo B, Chen MZ, Forestieri R, Smith AB. Org Lett. 2015;17:6242. doi: 10.1021/acs.orglett.5b03235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ai Y, Kozytska MV, Zou Y, Khartulyari AS, Smith AB. J Am Chem Soc. 2015;137:15426. doi: 10.1021/jacs.5b11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen MH, Imanishi M, Kurogi T, Smith AB. J Am Chem Soc. 2016;138:3675. doi: 10.1021/jacs.6b01731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devarie-Baez NO, Kim WS, Smith AB, Xian M. Org Lett. 2009;11:1861. doi: 10.1021/ol900434k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith AB, Kim WS, Tong R. Org Lett. 2010;12:588. doi: 10.1021/ol902784q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taguchi H, Takami K, Tsubouchi A, Takeda T. Tetrahedron Lett. 2004;45:429. [Google Scholar]
- 15.For previous success of a coupling reaction exploiting [1,5]-Brook rearrangement, see: Smith AB, Xian M, Kim WS, Kim DS. J Am Chem Soc. 2006;128:12368. doi: 10.1021/ja065033e.Gao L, Lu J, Song Z, Lin X, Xu Y, Yin Z. Chem Commun. 2013;49:8961. doi: 10.1039/c3cc45002c.
- 16.Tsubouchi A, Itoh M, Onishi K, Takeda T. Synthesis. 2004;2004:1504. [Google Scholar]
- 17.Yan L, Sun X, Li H, Song Z, Liu Z. Org Lett. 2013;15:1104. doi: 10.1021/ol400145z. [DOI] [PubMed] [Google Scholar]
- 18.Corey EJ, Boaz NW. Tetrahedron Lett. 1985;26:6015. [Google Scholar]
- 19.Zou Y, Melvin JE, Gonzales SS, Spafford MJ, Smith AB. J Am Chem Soc. 2015;137:7095. doi: 10.1021/jacs.5b04728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boeckman RKJ, Blum DM, Ganem B, Halvey N. Org Synth. 1978;58:152. [Google Scholar]
- 21.Anh NT, Eisenstein O. Tetrahedron Lett. 1976;17:155. [Google Scholar]
- 22.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, revision D.01. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 23.Geometry optimizations and frequency calculations were performed at the M06/6-31G(d)[SDD, for Cu, Br, and I] level. Single-point energy calculations in THF using the CPCM model were performed at the M06/6-311+G(d,p)[SDD, for Cu, Br, and I] level. For details, see the SI.
- 24.(a) Hong X, Liang Y, Griffith AK, Lambert TH, Houk KN. Chem Sci. 2014;5:471. [Google Scholar]; (b) Zou L, Paton RS, Eschenmoser A, Newhouse TR, Baran PS, Houk KN. J Org Chem. 2013;78:4037. doi: 10.1021/jo400350v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.