

Abstract
Chemical inhibition of epigenetic regulatory proteins BrdT and Brd4 is emerging as a promising therapeutic strategy in contraception, cancer, and heart disease. We report an easily synthesized dihydropyridopyrimidine pan-BET inhibitor scaffold, which was uncovered via a virtual screen followed by testing in a fluorescence anisotropy assay. Dihydropyridopyimidine 3 was subjected to further characterization and is highly selective for the BET family of bromodomains. Structure-activity relationship data and ligand deconstruction highlight the importance of the substitution of the uracil moiety for potency and selectivity. Compound 3 was also cocrystallized with Brd4 for determining the ligand binding pose and rationalizing subsequent structure-activity data. An additional series of dihydropyridopyrimidines was synthesized to exploit the proximity of a channel near the ZA loop of Brd4, leading to compounds with submicromolar affinity and cellular target engagement. Given these findings, novel and easily synthesized inhibitors are being introduced to the growing field of bromodomain inhibitor development.
TOC image
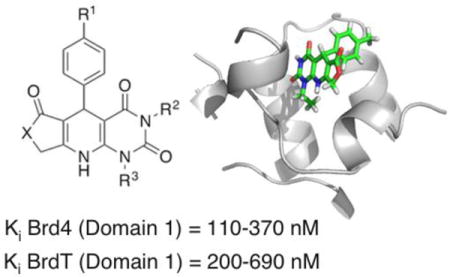
Introduction
Bromodomain modules are essential protein recognition domains for transcriptional regulation. Bromodomains function through selective binding to N-ε-acetylated lysine side-chains, most notably on histones and transcription factors. Additional enzymatic and scaffolding function can then be recruited to DNA via other domains within the bromodomain-containing protein for modulating gene transcription. The BET (bromodomain and extra-terminal) proteins are a subfamily of bromodomain-containing proteins consisting of Brd2, -3, -4, and -T, each of which contains tandem bromodomains, an extra-terminal motif, and in the case of Brd4, a C-terminal protein-protein interaction domain. BET proteins have attracted growing clinical interest due to their significant role in human health and disease. BrdT, which is testis-specific, has emerged as a promising target for male contraceptive agents due to its role in regulating spermatogenesis.1 The other BET bromodomains play a critical role in a number of pathologies.2 In particular, Brd4 regulates the transcription of the oncogene c-Myc.3 In the clinic, several pan-BET inhibitors are under study against various cancers including multiple myeloma, NUT-midline carcinoma and glioblastoma multiforme.3,4 Outside of cancer, Brd4 also plays a role in affecting viral activation and cardiac disease.3,5–7
Small molecules I-BET762 (1)8 and (+)-JQ1 (2)8,9, both based on a triazoloazepine scaffold, were the first described pan-BET inhibitors in the primary literature. Recently, compound 2 related memory deficits in mice10 and drug resistance against both triazoloazepines11 have been described. These issues motivate the development of additional BET inhibitors. New chemotypes are therefore desirable for the optimization of the pharmaceutical properties of BET inhibitors as alternatives for clinical development. The first reported synthesis of 2 was completed in four steps, including three transformations carried out in one pot in the final step.9 Subsequently, a scalable synthesis of 2 that utilized less toxic reagents and the same number of steps was described.12 Several other scaffolds have been reported as pan-BET inhibitors.2,13–17 The most accessible inhibitor reported to date was synthesized in two steps, the first of which was a multicomponent reaction, highlighting the demand for rapid, facile syntheses.18
In this report, we describe using a high-throughput virtual screen optimized for the first bromodomain of BrdT (hereafter referred to as BrdT(1)) to discover new binding scaffolds, followed by validation of these lead compounds (Figure 1). Structure-activity relationships (SAR), ligand deconstruction, and binding pose were subsequently explored for the most potent compound found from the virtual screen, compound 3, which is especially accessible due to its facile synthesis. Structure-guided attempts to access a new binding site on the protein surface of Brd4(1) based on the co-crystal structure with 3 led to additional gains in potency for analogs of 3. To facilitate future studies using 3 and subsequent analogs as chemical probes, a bromodomain selectivity screen was also carried out, demonstrating high selectivity for the BET class of bromodomains. Herein, we describe a facile synthesis of pan-BET inhibitors with submicromolar potency based on a novel dihydropyridopyrimidine scaffold.
Figure 1.
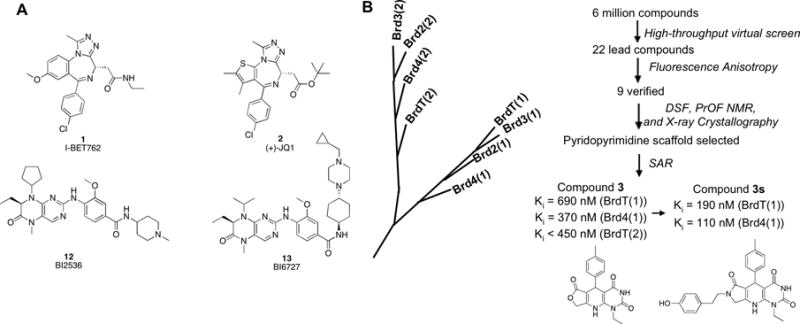
(A) BET bromodomain inhibitors 1, 2, BI2536 (12), and BI6727 (13). (B) Members of the BET branch of the bromodomain phylogenetic tree. Hits were identified by virtual screening, and binding potential of hits was assessed by fluorescence anisotropy (FA), differential scanning fluorimetry (DSF), and protein-observed 19F NMR (PrOF NMR). Structure-activity relationship (SAR) studies were subsequently conducted to probe the indicated dihydropyridopyrimidine hit compound. Optimization of compound 3 based on an x-ray co-crystal structure led to the development of compound 3s, with improved potency for both Brd4(1) and BrdT(1). The Ki values shown were determined by fluorescence anisotropy.
Results
We chose to pursue inhibition of BrdT because of its role in spermatogenesis, making BrdT a promising target for male contraception. The high-throughput virtual screen was performed with a library of six million compounds from the ZINC database that were docked against BrdT(1) using the PDB structure 4FLP in a low-precision mode. The molecules scoring in the top 0.4% were selected, refining the list to 24,000 hits. Increasing precision and applying computational filters for removal of potential pan-assay interference compounds (PAINS) and non-druglike compounds ultimately refined the number of hits to 200.19 A total of 22 commercially available compounds (Figure 2 and Figure S1) were selected from the 200 compounds for further testing. The choice of compounds was made qualitatively by visual inspection and applying multiple parameters, which included Lipinski and Weber rules, identification of toxicophores, scaffold novelty, and commercial availability at an affordable price. In addition, optimal shape complementarity in the binding pocket and the presence of multiple hits of the same scaffold were major factors in the selection of compounds for testing.
Figure 2.
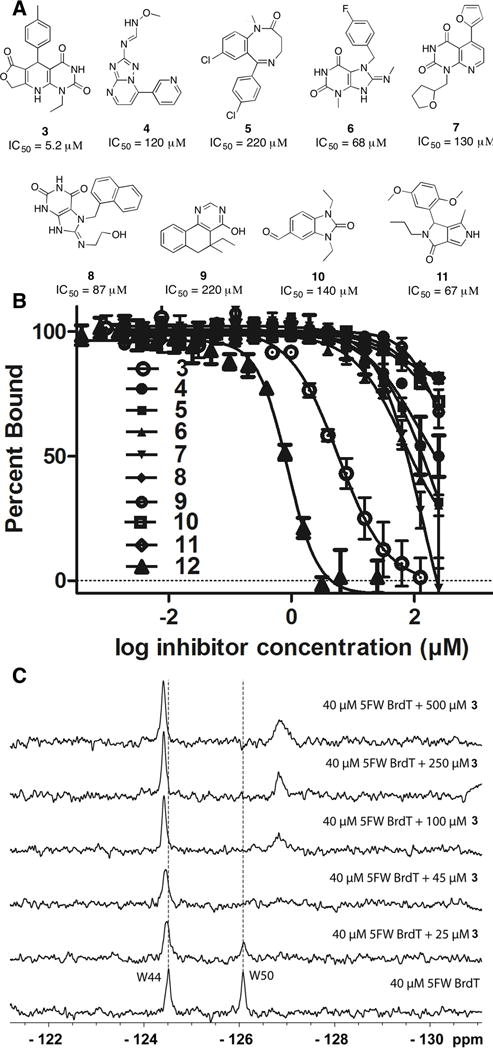
Compounds from high-throughput virtual screen tested for binding to BrdT(1) in a fluorescence anisotropy assay. (A) Of the 22 compounds tested, the nine shown here bound to BrdT(1). (B) Fluorescence anisotropy binding of compounds 3–12 to BrdT(1). Compound 3 (open circles), the tightest binder, was selected for further studies. (C) PrOF NMR titration of compound 3 into 5FW-BrdT(1). The dashed lines indicate the location of the unperturbed resonance.
The selected compounds were initially tested by fluorescence anisotropy because of the assay’s modest requirements of material and ability to provide quantitative binding information. We recently reported a fluorescence anisotropy assay for the first bromodomain of Brd4 using BI-BODIPY, a fluorescently-labeled version of pan-BET inhibitor 13.20–22 In that study, we limited ourselves to compounds soluble in ethylene glycol because of concerns regarding the known binding interactions of DMSO with bromodomains as a potential source of assay interference.23 Although fluorescence anisotropy assays on BET bromodomains have been described by others,13,24–26 we sought to find optimal screening conditions for first bromodomains of both Brd4 and BrdT. To this end, we tested experimental conditions with respect to chemical additives, incubation time, and protein stability. A detailed description of the optimization can be found in the Supporting Information. Optimization led us to a standard set of conditions: 4 mM CHAPS as detergent, 0.5% (v/v) DMSO, and a thirty min binding equilibration time.
Of the 22 compounds selected from the virtual screen for further testing by fluorescence anisotropy, nine showed unambiguous binding to BrdT(1) (Figure 2A and B). Six of the nine hits contained a pyrimidine moiety; the remaining three hits were a diazocinone (compound 5), a benzimidiazolone (compound 10), and a pyrrolopyrazolone (compound 11). Dihydropyridopyrimidine 3 was selected for further optimization due to its high binding potential for BrdT(1) (IC50 = 5.2 μM, Ki = 0.69 μM). For comparison, the known BET inhibitor 12 had an IC50 of 1.4 μM under these conditions. The S enantiomer of 3 was identified to bind in the virtual screen; however, in this study only racemates were tested based on commercial availability and synthetic accessibility. Although our starting point was a virtual screen against BrdT(1), we observed more potent binding to Brd4(1) with a Ki value of 0.37 μM.
Compound 3 binding was further verified by differential scanning fluorimetry (DSF), which yields an increase in protein melting temperature upon ligand binding.9,27 Compound 3 caused a 4.8 °C increase of the melting temperature of BrdT(1), consistent with the binding of 3 to BrdT(1) being significantly stabilizing. Finally, a competitive alpha-screen assay carried out against the compound 2-BET bromodomain interaction yielded IC50 values of 1.0 ± 0.2 μM and 2.3 ± 0.5 μM Brd4(1) and BrdT(1), respectively (Figure S2). Compound 3 also bound competitively with the native acetylated histone substrate in a similar alpha-screen assay (Figure S3), giving a comparable IC50 of 0.90 μM with Brd4 (1).
The structural effects of compound 3 on binding to BET bromodomains was verified by protein-observed 19F (PrOF) NMR, which is emerging as a structure-based technique for detecting binding events for small molecule discovery.21,28–31 PrOF NMR utilizes 19F NMR spectra of fluorine-labeled proteins, in this case 5-fluorotryptophan (5FW) labeled Brd4(1) and BrdT(1). Because of the high environmental sensitivity of the 19F protein resonances, perturbations to the 19F NMR spectrum of the protein, such as resonance broadening or shifting in the presence of ligand, correlate to ligand binding. Binding is minimally perturbed by the presence of the fluorinated residues, demonstrated by the similar affinities of 5FW-labeled Brd4(1) and unlabeled Brd4(1) for BI-BODIPY (0.055 μM and 0.11 μM, respectively).32 Similarly, the affinities of 5FW-BrdT(1) and BrdT(1) for BI-BODIPY (0.69 μM and 0.32 μM, respectively) are within approximately two-fold of each other (Figure S4), supporting the minimally perturbing nature of the fluorinated protein for quantifying bromodomain-ligand interactions.
PrOF NMR corroborated the results from the fluorescence anisotropy assay, with compound 3 exhibiting behavior consistent with slow to intermediate exchange (Figure 2C and 4A). Chemical exchange phenomenon in the slow to intermediate exchange regime, which is based on the residence time of the ligand bound to the protein and the relative frequency difference between the resonances for the bound and unbound states, is consistent with a low micromolar to submicromolar binding ligand.33,34 For both 5FW-BrdT(1) and 5FW-Brd4(1), the resonance corresponding to the tryptophan in the acetylated lysine binding pocket (W50 and W81, respectively) broadens into baseline at or below 50 μM of compound 3, with a new resonance growing in upfield of the original peak at higher ligand concentrations. The presence of serum did not interfere with the binding of compound 3 to 5FW-Brd4(1) (Figure S5) and for cell-based studies that are discussed below.
Figure 4.

(A) PrOF NMR confirms binding of compound 3 to 5FW-Brd4(1). W81 in Brd4 and W50 in BrdT are broadened at 25 μM 3, and a new resonance begins to grow in at higher concentrations. Dashed lines reference the position of the resonance in the absence of ligand. The protein concentration was 45 μM in all experiments. (B) Compound 3 does not bind to non-BET bromodomain BPTF, shown by the similar spectra in the presence and absence of ligand. Protein concentration was 50 μM in all experiments. (C) Selectivity of 3 against a panel of bromodomains. BET bromodomains are in bromodomain family II.
Neither compound 3 nor close structural analogs (as low as 70% similarity as defined by ChEMBL) have been reported as bioactive compounds for bromodomains. However, other dihydropyridopyrimidine scaffolds have been described as active against a variety of targets.35–38 While dihydropyridine moieties are prone to oxidation, they are commonly utilized in calcium channel blockers39 and the dihydropyridine moiety in 3 remains stable over several freeze-thaw cycles when stored as a DMSO solution at −20 °C as measured by ESI-MS (data not shown). After oxidation of compound 3 to a pyridine (3n) under forcing conditions with chloranil (Scheme 1), no binding was observed to BrdT(1) or Brd4(1) by fluorescence anisotropy.
Scheme 1. Synthesis of dihydropyridopyrimidines 3–3m and pyridine 3na.
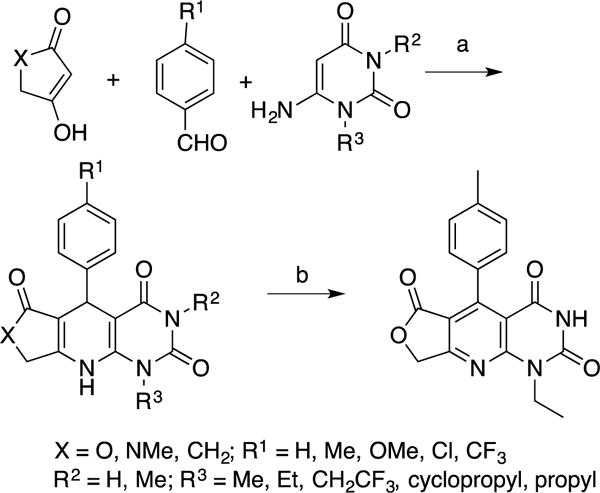
aReagents and reaction conditions: (a) Acetic acid, 110 °C, 6 h, N2, 11–81%; (b) Compound 3, chloranil, DMSO, rt, 2 h, 87%.
Compound 3 is an especially attractive lead due to its synthesis in a single step from commercially available starting materials (Scheme 1). The synthesis was performed using the Hantzsch dihydropyridine three component cyclization based on the methods described by Tanifum and colleagues.35,40 Pure compounds (> 95% purity for 11 of the 12 synthesized compounds) were obtained by simply washing the product; no chromatography was necessary. Analogs were generated by varying the substituents on the 1,3-dicarbonyl, benzaldehyde, and uracil starting materials.
To optimize the potency of 3, the substitution on the para-position of the phenyl ring and N-1 (R3) and N-3 (R2) of the pyrimidine ring were varied (Table 1). Additionally, the lactone from tetronic acid was exchanged for a lactam or a ketone. The lactone is the most potent of these variants, with a ketone leading to loss of detectable potency. The substituent on N-1 of the pyrimidine ring was critical for potency, with an ethyl group proving optimal. A propyl group was also tolerated, albeit with a substantial decrease in potency compared to an ethyl group. No binding was observed with a cyclopropyl group at this position. We observed a mild loss in potency against BrdT(1) upon exchanging the ethyl on N-1 for a 2,2,2-trifluoroethyl group. This compound, 3j, did not cause a positive change in melting temperature by DSF for BrdT(1) (Table 1 and Figure S7). Although tighter binding to Brd4(1) was measured in our fluorescence anisotropy assay (Ki = 0.16 μM for 3j vs 0.37 μM for 3), alpha-screen competition data did not reproduce this higher affinity and only showed higher micromolar inhibition (4.6 μM for 3j vs. 1.0 μM for 3) with a steeper Hill slope (−1.6 for 3j vs. −1 for 3), suggesting potential non-specific binding (Figures S2 and S3). PrOF NMR of 3j with 5FW-Brd4(1) also shows slow to intermediate exchange kinetics (Figure S8).
Table 1.
Effects of Varying the Substitution of Compound 3 on Potency Against Brd4 and BrdT, N/D = not determined

| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| compound number | R1 | R2 | R3 | X | Ki (μM) (BrdT(1)) |
Ki (μM) (Brd4(1)) |
ΔTm (°C) (150 μM) (BrdT(1)) |
| 3 | Me | H | Et | O | 0.69 ± 0.11 | 0.37 ± 0.13 | 4.8 |
| 3a | OMe | H | Me | O | > 40 μM | N/D | −0.35 |
| 3b | H | H | Me | O | > 40 μM | N/D | −1.1 |
| 3c | H | H | Et | O | 1.4 ± 0.7 | 0.35 ± 0.07 | 0.99 |
| 3d | H | H | Pr | O | 2.4 ± 0.9 | 1.2 ± 0.1 | 0.14 |
| 3e | Me | Me | Me | O | > 40 μM | N/D | −0.46 |
| 3f | OMe | Me | Me | O | > 40 μM | N/D | −0.48 |
| 3g | Cl | H | Et | O | 1.1 ± 0.07 | 0.30 ± 0.07 | 1.4 |
| 3h | CF3 | H | Et | O | 4.7 ± 3.8 | 1.2 ± 0.4 | −0.44 |
| 3i | Me | H |
|
O | > 40 μM | > 40 μM | 0.06 |
| 3j | Me | H | CH2CF3 | O | 0.85 ± 0.07 | 0.16 ± 0.07 | −0.54 |
| 3k | Me | H | Et | CH2 | 23 ± 12 | 10 ± 9 | 0.06 |
| 3l | Me | H | Et | NMe | 5.6 ± 1.8 | 2.5 ± 2.6 | −0.04 |
| 3m | Me | Me | Et | O | > 40 μM | > 40 μM | N/D |
| 3n (pyridine) | Me | H | Et | O | > 40 μM | > 40 μM | N/D |
Substitution of a methyl group at N-1 led to levels of binding too low (Ki > 40 μM) to be detected by fluorescence anisotropy. By comparison, varying the para-substituent on the phenyl ring had a smaller impact on potency, with chloro, methyl, trifluoromethyl, and no substituent all tolerated. Adding a methyl group at N-3 of the pyrimidine ring led to loss of detectable potency.
To determine the active pharmacophore of 3, this compound was deconstructed into its parent pyrimidine due to the high frequency of pyrimidine compounds binding to BrdT(1) in the virtual screen. A fragment corresponding to the pyrimidine moiety of 3, 6-amino-1-ethyluracil, was tested for binding to 5FW-BrdT(1) and 5FW-Brd4(1) by PrOF NMR to determine the dissociation constants. Titration of 6-amino-1-ethyluracil with 5FW-Brd4(1) yielded a Kd of 660 ± 60 μM and a ligand efficiency of 0.39 kcal/mol/non-hydrogen atom (Figure 3). W81, which is part of WPF shelf in the binding pocket (Figure 3), is perturbed significantly; the other 5FW resonances remain nearly unchanged, consistent with the binding mode of other known Brd4(1) ligands. 5FW-BrdT(1) binding was also detected with 6-amino-1-ethyluracil, in this case perturbing the two 5FW resonances with similar affinity. Non-linear regression analysis of the chemical shift perturbation for W50, which is the residue located in the WPF shelf of BrdT(1), yielded a Kd of 1.5 ± 0.2 mM. Similarly, analysis of the chemical shift perturbation for the resonance corresponding to W44, the residue located beneath W50, also yielded a Kd of 1.5 ± 0.5 mM, resulting in a slightly lower ligand efficiency of 0.33 kcal/mol/non-hydrogen atom. No binding was observed to 5FW-Brd4(1) when tetronic acid, a fragment corresponding to the lactone moiety of 3, was tested by PrOF NMR.
Figure 3.
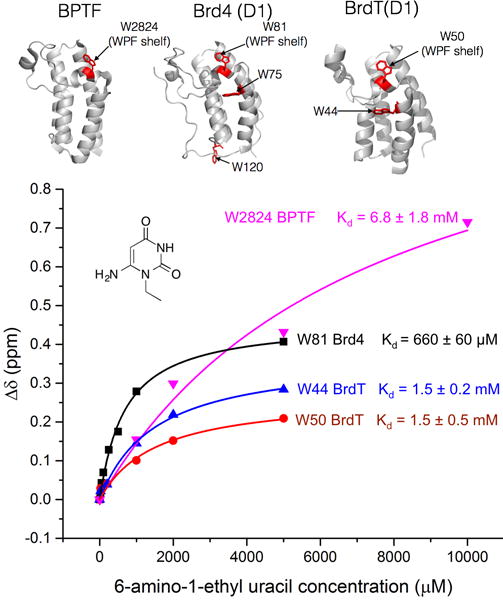
Binding isotherm showing chemical shift perturbation from a PrOF NMR titration of 6-amino-1-ethyluracil into 50 μM 5FW-Brd4(1), -BrdT(1), and -BPTF. Perturbed resonances for each protein and their respective Kd values are shown. The ribbon structure for each protein is also shown with tryptophan residues labeled in red. Residue assignment for 5FW-BrdT(1) is shown in Figure S9.
Other pyrimidine-containing compounds have been reported to bind bromodomains. ATAD2, a non-BET bromodomain-containing protein, binds to the pyrimidine thymine with 10 mM affinity.41 Several higher affinity pyrimidine-containing dual kinase-bromodomain inhibitors have also been described.27,42 Based on these results, we postulated that uracil derivatives are broad-spectrum bromodomain binders. In support of this hypothesis, we found that 6-amino-1-ethyluracil bound non-BET bromodomain 5FW-BPTF, albeit weakly (Kd = 6.8 mM ± 1.8 mM) (Figure 3).
Chemical probe molecules must have both high functional activity and high selectivity to provide useful biological information. Based on the binding of 6-amino-1-ethyluracil to BPTF, we tested binding of compound 3 to non-BET bromodomain BPTF by PrOF NMR as a representative off target (Figure 4B). In this case no binding was detected, suggesting that while 6-amino-1-ethyluracil may bind to multiple bromodomains, elaboration of the uracil into the substituted dihydropyridopyrimidine scaffold generates selectivity towards BET bromodomains. PrOF NMR experiments on both 5FW-BrdT(1) (Figure 2C) and 5FW-Brd4(1) (Figure 4A) are consistent with potent binding of 3. To test the selectivity of compound 3 against additional bromodomains, compound 3 was subjected to a selectivity screen against a panel of bromodomains in a bromodomain qPCR-based displacement assay (BromoScan, DiscoverX). Because Ki values are an indirect measure of dissociation constant, and may differ based on the protein’s interaction with the fluorescently labeled ligand and experimental conditions, a direct titration was first carried out. BromoScan titration of 3 against Brd4(1) and BrdT(1) provided attenuated but similar Kd values compared to Ki values determined by fluorescence anisotropy, within two-fold for BrdT(1) (Kd = 1.6 μM) and within three-fold for Brd4(1) (Kd = 0.84 μM) (Figure S10). Additionally, single point measurements at 20 μM showed that 3 is highly selective for BET bromodomains. Low levels of activity were observed against non-BET bromodomains, with the most significant binding observed to the WDR9(2) (72% inhibition), SMARCA2 (71% inhibition), CBP (68% inhibition), and p300 (66% inhibition) bromodomains (Figure 4C). We have yet to express the majority of these bromodomains for validating inhibition in complementary biophysical assays as described above for BPTF. Notably, the most significant inhibition apparent was for the second bromodomain of BrdT (BrdT(2)). To verify this inhibition, we further tested our fluorescence anisotropy conditions with BrdT(2). For these experiments, the lowest Ki value that can be determined is equal to the Kd of the fluorescent tracer.43 This limit of resolvable detection was reached in the interaction of 3 and 3j, yielding a Ki of 0.45 μM or less for 3 and 3j. Together these results support selective pan-BET inhibition with 3 and its analogs.
The high selectivity for BET bromodomains guided our attempts to obtain a co-crystal structure of Brd4(1) with 3 at atomic resolution. Brd4(1) was co-crystallized with compound 3 and the structure of the complex was determined at 1.5 Å resolution (Figure 5 and Table S2). Although the racemate of 3 was used for crystallization, only electron density corresponding to the S-stereoisomer was observed, consistent with our docking studies. The N-3 nitrogen and C-2 carbonyl oxygen directly interact with the conserved Asn140 side chain via H-bonds. Consequently, methylation at N-3 abolishes binding potential and renders compounds 3e, 3f, and 3m inactive. Additionally, water-mediated H-bonds are observed between the C-2 carbonyl oxygen and the side chain of Tyr97 and between N-9 and the carbonyl oxygen of Pro82. Oxidation of 3 to 3n may lead to the loss of the latter interaction, resulting in loss of potency in 3n. Several hydrophobic van der Waals interactions exist between the inhibitor and acetylated lysine binding site residues. The pyrimidine N-1 ethyl side-chain interacts with the aromatic ring of Phe83 which serves as a backstop for binding further into the histone binding pocket of Brd4(1). Such an interaction would be missed with shorter methyl analogs. Finally, the tolyl group of 3 in the S-configuration is 3.7 Å away from the indole of W81 at the WPF shelf, consistent with an offset pi-pi interaction. More extended aromatic ring systems may be able to capitalize on this interaction in future designs. This interaction may be responsible for the upfield shift of the W81 resonance in PrOF NMR spectra in the presence of 3, similar to that observed for 12.29 Together, the noncovalent interactions from the co-crystal structure support the N-ethyl pyrimidine ring of 3 being the dominant pharmacophore and help rationalize the SAR trends observed in our related analogs.
Figure 5. Co-crystal structure of Brd4(1) with compound 3.
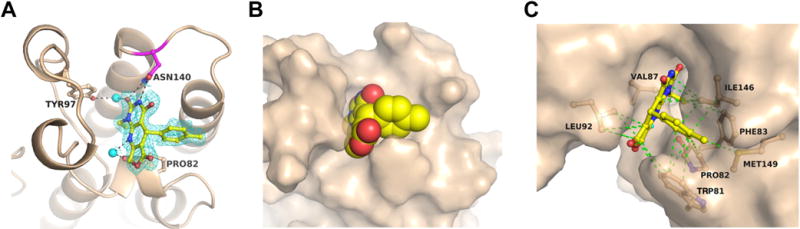
(A) Direct hydrogen bonding interactions (black dotted lines) of compound 3 with Asn140 and water-mediated interactions with Tyr97 and Pro82. The inhibitor is shown in yellow and the 2Fo−Fc electron density map (contoured at 1σ) is indicated as blue mesh. Water molecules are shown as cyan spheres. (B) Space filling model of the inhibitor bound to the acetylated lysine binding site. (C) Van der Waals interactions (green dotted lines) with mostly hydrophobic residues of the acetylated lysine binding site.
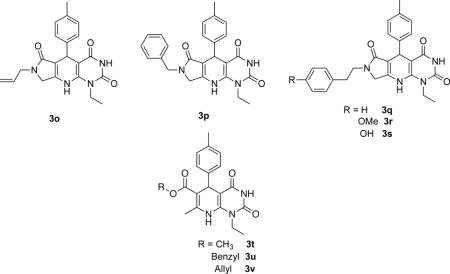
The x-ray co-crystal structure led us to take initial steps towards optimization of 3, leading to a series of more potent inhibitors (Scheme S1, Table 2). To take advantage of the proximity of a channel near Trp81 in the ZA loop observed in the co-crystal structure of 3 and Brd4(1), the N-methyl group of lactam 3l was extended to an allyl (3o), benzyl (3p), phenethyl (3q), phenethyl methoxy (3r), or phenethyl 4-hydroxyl (3s). These extensions capitalize on accessing potential interactions that extend into the channel beyond those of 2 (Figure S11). We also explored a series of ring-opened lactones 3t, 3u, and 3v, but these compounds either did not show gains in potency or only showed partial inhibition so were not investigated further (Figure S12). Compounds 3p, 3q, and 3r, and 3s, each of which contain a phenyl ring, were substantially more potent than compound 3 in binding to both Brd4(1) and BrdT(1). Competitive alpha-screen data using 2 as a displaceable probe revealed similar trends in binding behavior (Table 2). PrOF NMR experiments corroborated the potent binding observed by fluorescence anisotropy, showing slow exchange upon titration of 3s into 5FW-Brd4(1) (Figure S13) and affected both W81 near the histone binding site and W75 outside the pocket, indicating a subtle conformational change. Slow exchange binding behavior is also consistent with a long resonance time of 3s bound to Brd4(1).
Table 2.
Effects of Varying the N-Substitution of Lactam on Potency Against Brd4 and BrdT N/D = not determined
| |||||
|---|---|---|---|---|---|
|
| |||||
| compound number | Ki (μM) (BrdT(1)) |
Ki (μM) (Brd4(1)) |
IC50 (μM) (BrdT(1)) |
IC50 (μM) (Brd4(1)) |
IC50 (μM) MM1.S cells |
| 3o | N/D | 14± 9 | 2.2 | 0.63 | N/D |
| 3p | 0.23 ± 0.13 | 0.19 ± 0.04 | 0.55 | 0.24 | 2.1 ± 1.7 |
| 3q | 0.22 ± 0.12 | 0.14 ± 0.04 | 1.0 | 1.0 | 1.2 ± 0.1 |
| 3r | 0.20 ± 0.17 | 0.10 ± 0.01 | 1.0 | 0.48 | 2.1 ± 1.6 |
| 3s | 0.20 ± 0.18 | 0.11 ± 0.01 | 0.76 | 0.39 | 0.46 ± 0.04 |
We tested for cellular activity using the multiple myeloma cell line MM1.S, which is highly sensitive to Brd4 inhibition.3,44 Compound 2 strongly inhibited the growth of MM1.S cells with an IC50 value of 0.02 μM (Figure S14A), which was accompanied by a dose-dependent reduction of c-Myc levels and upregulation of p21Cip1 levels (Figure 6). Both lactone-containing inhibitors 3 and 3j lacked growth inhibition activity and were ineffective in altering c-Myc or p21Cip1 levels, indicating poor cellular uptake or stability. However, the lactam series of compounds 3p, 3q, 3r, and 3s, all of which are both more potent and more lipophilic than 3 and 3j, showed cellular activity with low to submicromolar IC50 values (Figure S15, Table 2). This data is supported mechanistically with this series of compounds showing dose-dependent target engagement via decreasing c-Myc expression levels and concomitant increase in p21Cip1 via western blot analysis (Figure 6).
Figure 6.

Western blot analysis of the effect of compounds 3p, 3q, 3r, and 3s on c-Myc and p21Cip1 levels. Compound 2 served as a positive control, and vinculin served as a loading control.
Discussion
The pan-BET inhibitor described here is based on a dihydropyridopyrimidine scaffold that is readily accessible through a one-step synthesis. Beyond BET bromodomains, our NMR studies show that uracil derivatives, including a deconstructed fragment of compound 3, can bind with bromodomains of wide sequence diversity. The uracil scaffold may therefore be a useful starting point for bromodomain inhibitor design. As we have shown in this work, elaboration upon this scaffold leads to the development of selectivity and potency. The dihydropyridopyrimidine compounds described herein maintain a molecular weight below 500 g/mol (339 g/mol for 3 and 458 g/mol for 3s) and favorable cLogP values (2.1–3.6). We have shown that the substituent on the para position of the aromatic ring on the dihydropyridine is solvent exposed and binding is not substantially perturbed for a variety of substituents. Therefore, either solubilizing groups, affinity tags for future proteomic studies, bivalent inhibitor development,45 or protein degradation motifs46 can readily be appended starting from commercially available benzaldehyde derivatives, adding to the utility of this scaffold.
McKeown and colleagues described an imidazopyrazine pan-BET inhibitor with a facile synthesis consisting of two steps: a multicomponent reaction followed by a Suzuki coupling.18 The potency of their most potent compound via isothermal titration calorimetry for Brd4 is 0.55 μM at 15 °C. Compound 3 compares favorably to the most potent compound from McKeown and colleagues in overall yield (70% vs. 45%) and number of synthetic steps (1 vs. 2), and compound 3s shows improvements in potency over compound 3. Although our compounds are selective for BET bromodomains, the imidazopyrazine also opens the door for targeting TAF1 if selectivity were optimized. Together, these two classes of compounds offer an opportunity for rapid structure-activity data to be generated against BET bromodomains using non-diazepine scaffolds.
Conclusions
Through performing a high-throughput virtual screen against bromodomain BrdT(1), we have discovered several new inhibitors, based on a dihydropyridopyrimidine scaffold from 3, that are highly selective for BET bromodomains with submicromolar affinity for the first and second bromodomains of BrdT. Although multiple BET inhibitors are known, compound 3 is already commercially available, and many analogs of 3 can be generated in a single step in a metal-free process. Diversity can be readily accomplished by varying the composition of the aldehyde, uracil, and 1,3-dicarbonyl compound starting materials. Compound 3 and related analogs such as molecules 3p to 3s can thus serve as starting points for improving potency, solubility, and cell permeability for future probe development.
The development of inhibitors selective for a particular member of the BET family represents a significant challenge due to the high sequence similarity between BET family members. Two domain 1 and one domain 2 BET bromodomain selective molecules have been reported.16,47,48 No potent, isoform selective inhibitor has yet been described, although a small molecule containing a uracil moiety with micromolar affinity for Brd4 has recently been reported.49 An optimized fluorescence anisotropy assay based on competition with a fluorescently labeled pan-BET inhibitor can be used to profile binding to multiple bromodomains, offering a path towards discovery and development of isoform-selective inhibitors.
EXPERIMENTAL SECTION
General Methods
Solvents and reagents were purchased from commercial suppliers and used without additional purification. 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz, respectively, in DMSO-d6. Chemical shifts, δ, are reported in ppm and coupling constants, J, are expressed in Hertz (Hz). Abbreviations for peaks are s = singlet, d = doublet, t = triplet, and m = multiplet. High-resolution mass spectrometry was performed using positive mode electrospray ionization methods (ESMS) with a Bruker BioTOF II spectrometer. The purity of compounds was determined by analytical reverse-phase high performance liquid chromatography (HPLC) analysis using Waters 2695 HPLC (column: Phenomenex Synergi Fusion-RP, 4.0 μm, 4.6 mm × 150 mm); mobile phase isocratic 50:50 0.1% TFA in H2O:MeOH; detector, Waters 2996; injector, automated injector; detection wavelength, 254 nm; flow rate, 1.0 mL/min. Sample diluent 1:5 DMSO:MeOH. Compounds from the virtual screen predicted to bind (3–12, and compounds in Fig. S1), and 3b–3d, were purchased from Ambinter and fully characterized. All synthesized compounds and purchased analogs of 3 were > 95% pure except 3d, which was 93% pure, and 3l, which was 87% pure.
General Procedure for the Synthesis of the Dihydropyridopyrimidine Derivatives
Tetronic acid, cyclopentane-1,3-dione or 1-methylpyrrolidine-2.4-dione (0.50 mmol), the benzaldehyde (0.50 mmol), and the uracil (0.50 mmol) were added to a dry microwave vial (5 mL) and glacial AcOH (2 mL) was added. The vial was placed under N2 for 1 min and sealed. The vial was placed in an oil bath at 110 °C and stirred for 6 h, or until the solution solidified. The solution was allowed to cool to room temperature. AcOH was evaporated under N2 and the solid was washed with DCM (3 × 2 mL) to remove excess AcOH. The remaining solid was purified by washing with MeOH (8 × 1 mL) and dried to afford the dihydropyridopyrimidine.
1-Ethyl-5-(p-tolyl)-5,9-dihydrofuro[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3)
Yield (119 mg, 70%) as a white solid; m.p. 318–319 °C dec.; with a purity of 98%. 1H NMR (400 MHz, DMSO-d6) δ 11.07 (s, 1H), 10.12 (s, 1H), 7.12–7.10 (d, J = 8.0 Hz, 2H), 7.05–7.03 (d, J = 8.0 Hz, 2H), 4.97–4.93 (d, J = 16.5 Hz, 1H), 4.88–4.84 (d, J = 16.5 Hz, 1H), 4.63 (s, 1H), 3.98–3.87 (m, 2H), 2.22 (s, 3H), 1.21–1.17 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 161.6, 156.1, 149.8, 145.2, 142.0, 135.3, 128.5, 127.5, 102.3, 90.1, 65.8, 37.0, 33.8, 20.6, 13.7. HRMS: calcd for C18H17NaN3O4 [M + Na]+, 362.1111; found 362.1109.
5-(4-Methoxyphenyl)-1-methyl-5,9-dihydrofuro[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3a)
Yield (136 mg, 80%) as a white solid; m.p. 306–307 °C dec.; with a purity of 98%. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 1H), 10.15 (s, 1H), 7.17–7.15 (d, J = 8.3 Hz, 2H), 6.80–6.78 (d, J = 8.3 Hz, 2H), 4.96–4.91 (d, J = 16.5 Hz, 1H), 4.86–4.81 (d, J = 16.5 Hz, 1H), 4.61 (s, 1H), 3.69 (s, 3H), 3.35 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.0, 161.6, 157.8, 155.9, 150.1, 146.0, 137.2, 128.7, 113.3, 102.5, 90.0, 65.8, 55.0, 33.4, 29.1. HRMS: calcd for C17H15NaN3O5 [M + Na]+, 364.0904; found 364.0904.
1-Methyl-5-phenyl-8,9-dihydrofuro[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,5H)-trione (3b)
White solid; m.p. 308–309 °C dec.; with a purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 11.07 (s, 1H), 10.19 (s, 1H), 7.28–7.25 (m, 4H), 7.17–7.12 (m, 1H), 4.95 (d, J=16.6 Hz, 1H), 4.85 (d, J=16.7 Hz, 1H), 4.67 (s, 1H), 3.36 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 161.6, 156.2, 150.1, 146.2, 144.8, 127.9, 127.7, 126.3, 102.3, 89.7, 65.8, 34.3, 29.1. HRMS: calcd for C16H13N3NaO4 [M + Na]+, 334.0798; found 334.0798.
1-Ethyl-5-phenyl-8,9-dihydrofuro[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,5H)-trione (3c)
White solid; m.p. 305–306 °C dec.; with a purity 98%. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.15 (s, 1H), 7.27–7.21 (m, 4H), 7.18–7.12 (m, 1H), 4.96 (d, J=16.6 Hz, 1H), 4.87 (d, J=16.6 Hz, 1H), 4.67 (s, 1H), 4.02–3.85 (m, 2H), 1.19 (t, J=7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 161.6, 156.2, 149.8, 145.3, 144.8, 128.0, 127.6, 126.3, 102.2, 89.9, 65.8, 37.0, 34.2, 13.7. HRMS: calcd for C17H15N3NaO4 [M + Na]+, 348.0955; found 348.0955.
5-Phenyl-1-propyl-8,9-dihydrofuro[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,5H)-trione (3d)
White solid; m.p. 300–301 °C dec.; with a purity of 95%. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 10.07 (s, 1H), 7.28–7.21 (m, 4H), 7.18–7.12 (m, 1H), 4.97 (d, J=16.5 Hz, 1H), 4.85 (d, J=16.5 Hz, 1H). 4.67 (s, 1H), 3.85 (t, J=7.4 Hz, 2H), 1.66–1.55 (m, 2H), 0.91 (t, J=7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 171.0, 161.5, 156.3, 150.0, 145.5, 144.9, 128.0, 127.6, 126.3, 102.2, 89.9, 65.9, 42.9, 34.2, 21.2, 10.7. HRMS: calcd for C18H17N3NaO4 [M + Na]+, 362.1111; found 362.1111.
1,3-Dimethyl-5-(p-tolyl)-5,9-dihydrofuro[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3e)
Yield (115 mg, 68%) as white solid; m.p. 294–296 °C dec.; with a purity of 99%. 1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 7.14–7.12 (d, J = 7.2 Hz, 2H), 7.04–7.02 (d, J = 7.2 Hz, 2H), 4.96–4.82 (m, 2H), 4.65 (s, 1H), 3.42 (s, 3H), 3.08 (s, 3H), 2.22 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 160.8, 155.7, 150.4, 144.8, 142.0, 135.3, 128.4, 127.7, 102.5, 89.3, 65.7, 34.5, 30.1, 27.6, 20.6. HRMS: calcd for C18H17NaN3O4 [M + Na]+, 362.1111; found 362.1111.
5-(4-Methoxyphenyl)-1,3-dimethyl-5,9-dihydrofuro[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3f)
Yield (133 mg, 74%) as a white solid; m.p. 329–330 °C dec.; with a purity of 98%. 1H NMR (400 MHz, DMSO-d6) δ 10.21 (s, 1H), 7.17–7.15 (d, J = 8.6 Hz, 2H), 6.79–6.77 (d, J = 8.6 Hz, 2H), 4.97–4.93 (d, J = 16.6 Hz, 1H), 4.86–4.82 (d, J = 16.6 Hz, 1H), 4.64 (s, 1H), 3.69 (s, 3H), 3.42 (s, 3H), 3.08 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 160.8, 157.8, 155.7, 150.4, 144.7, 137.1, 128.7, 113.3, 102.6, 89.5, 65.7, 55.0, 34.0, 30.2, 27.6. HRMS: calcd for C18H17NaN3O5 [M + Na]+, 378.1060; found 378.1060.
5-(4-Chlorophenyl)-1-ethyl-5,9-dihydrofuro[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3g)
Yield (146 mg, 81%) as a white solid; m.p. 298–300 °C dec.; with a purity of 98%. 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 10.19 (s, 1H), 7.31–7.29 (d, J = 8.7 Hz, 2H), 7.27–7.25 (d, J = 8.7 Hz, 2H), 4.98–4.94 (d, J = 16.6 Hz, 1H), 4.90–4.86 (d, J = 16.6 Hz, 1H), 4.67 (s, 1H), 4.00–3.84 (m, 2H), 1.21–1.17 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 161.6, 156.5, 149.8, 145.5, 143.7, 130.9, 129.6, 127.9, 101.7, 89.5, 65.9, 37.1, 34.0,13.7. HRMS: calcd for C17H14ClNaN3O4 [M + Na]+, 382.0565; found 382.0565.
1-Ethyl-5-(4-(trifluoromethyl)phenyl)-5,9-dihydrofuro[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3h) Yield (140 mg, 71%) as a white solid; m.p
297–298 °C dec.; with a purity of 99%. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 10.24 (s, 1H), 7.62–7.60 (d, J = 8.1 Hz, 2H), 7.49–7.47 (d, J = 8.1 Hz, 2H), 4.99–4.95 (d, J = 16.7 Hz, 1H), 4.91–4.87 (d, J = 16.7 Hz, 1H), 4.78 (s, 1H), 4.02–3.85 (m, 2H), 1.22–1.19 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.8, 161.6, 156.8, 149.8, 149.2, 145.7, 128.6, 127.0, 124.89, 124.85, 124.82, 124.4, 101.4, 89.2, 66.0, 37.2, 34.6, 13.7. HRMS: calcd for C18H14F3NaN3O4 [M + Na]+, 416.0829; found 416.0829.
1-Cyclopropyl-5-(p-tolyl)-5,9-dihydrofuro[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3i)
Yield (125 mg, 71%) as a white solid; m.p. 280–281 °C dec.; with a purity of 96%. 1H NMR (400 MHz, DMSO-d6) δ 10.91 (s, 1H), 9.97 (s, 1H), 7.12–7.10 (d, J = 8.0 Hz, 2H), 7.04–7.02 (d, J = 8.0 Hz, 2H), 4.97–4.93 (d, J = 16.7 Hz, 1H), 4.88–4.84 (d, J = 16.7 Hz, 1H), 4.60 (s, 1H), 2.74–2.69 (m, 1H), 2.22 (s, 3H), 1.24–1.14 (m, 2H), 0.97–0.89 (m, 1H), 0.89–0.81 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 171.0, 161.9, 156.0, 150.1, 146.9, 141.9, 135.3, 128.5, 127.5, 102.2, 90.0, 66.0, 33.8, 24.5, 20.6, 10.1, 10.0. HRMS: calcd for C19H17NaN3O4 [M + Na]+, 374.1111; found 374.1110.
5-(p-Tolyl)-1-(2,2,2-trifluoroethyl)-5,9-dihydrofuro[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3j)
Yield (21 mg, 11%) as a white solid; m.p. 305–306 °C dec.; with a purity of 99%. 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H), 10.34 (s, 1H), 7.11–7.09 (d, J = 8.1 Hz, 2H), 7.06–7.04 (d, J = 8.1 Hz, 2H), 5.03–4.99 (d, J = 16.5 Hz, 1H), 4.88–4.75 (m, 3H), 4.64 (s, 1H), 2.23 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.7, 161.3, 155.6, 149.7, 144.8, 141.7, 135.6, 128.6, 127.4, 125.2, 122.4, 102.6, 90.9, 65.6, 33.8, 20.6. HRMS: calcd for C18H14F3NaN3O4 [M + Na]+, 416.0829; found 416.0829.
1-Ethyl-5-(p-tolyl)-5,7,8,9-tetrahydro-1H-cyclopenta[5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (3k)
Yield (66.0 mg, 59%) as a white solid; m.p. 308–310 °C dec.; with a purity of 99%. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H), 9.92 (s, 1H), 7.07–7.05 (d, J = 8.0 Hz, 2H), 7.00–6.98 (d, J = 8.0 Hz, 2H), 4.60 (s, 1H), 4.06–3.94 (m, 2H), 2.76–2.64 (m, 2H), 2.30–2.27 (m, 2H), 2.20 (s, 3H), 1.20–1.17 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 201.1, 164.1, 161.5, 149.9, 145.3, 142.6, 134.9, 128.4, 127.4, 117.3, 90.7, 36.8, 33.7, 33.6, 24.6, 20.6, 13.8. HRMS: calcd for C19H19N3O3 [M + Na]+, 360.1319; found 360.1319.
1-Ethyl-7-methyl-5-(p-tolyl)-5,7,8,9-tetrahydro-1H-pyrrolo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (3l)
Yield (88 mg, 50%) as a white solid; m.p. 293–295 °C dec.; with a purity of 87%. 1H NMR (400 MHz, DMSO-d6) δ 10.93 (s, 1H), 9.70 (s, 1H), 7.09–7.07 (d, J = 8.0 Hz, 2H), 7.01–6.99 (d, J = 8.0 Hz, 2H), 4.62 (s, 1H), 4.12–3.87 (m, 4H), 2.80 (s, 3H), 2.21 (s, 3H), 1.19–1.15 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 168.9, 161.7, 149.8, 146.4, 145.7, 142.9, 134.9, 128.3, 127.4, 109.3, 89.2, 49.4, 36.8, 34.0, 28.3, 20.6, 13.6. HRMS: calcd for C19H20N4O3 [M + Na]+, 375.1428; found 375.1428.
1-Ethyl-3-methyl-5-(p-tolyl)-5,9-dihydrofuro[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3m)
Yield (118.2 mg, 67%) as a white solid; m.p. 299–300 °C dec.; with a purity of 99%. 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1H), 7.13–7.11 (d, J = 7.9 Hz, 2H), 7.04–7.02 (d, J = 7.9 Hz, 2H), 4.98–4.94 (d, J = 16.5 Hz, 1H), 4.89–4.85 (d, J = 16.5 Hz, 1H), 4.66 (s, 1H), 4.06–3.92 (m, 2H), 3.08 (s, 3H), 2.22 (s, 3H), 1.23–1.20 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 160.8, 155.9, 150.1, 143.9, 141.9, 135.4, 128.5, 127.6, 102.4, 89.6, 65.8, 38.1, 34.4, 27.5, 20.6, 13.6. HRMS: calcd for C19H19NaN3O4 [M + Na]+, 376.1268; found 376.1260.
1-Ethyl-5-(p-tolyl)furo[3′,4′:5,6]pyrido[2,3-d]pyrimidine-2,4,6(1H,3H,8H)-trione (3n)
Compound 3 (22.0 mg, 0.07 mmol, 1.0 equiv) and chloranil (32.1 mg, 0.13 mmol, 2.0 equiv) were added to a dry microwave vial and DMSO (3 mL) was added. The vial was capped. The reaction was stirred at room temperature for 2 h. DMSO was then evaporated under N2. Purification by semi-automated silica gel chromatography (4:1 hexanes:EtOAc), yielded a white solid (19.0 mg, 87%); m.p. 278–279 °C dec.; with a purity of 99%. 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 7.20–7.18 (d, J = 8.0 Hz, 2H), 7.15–7.13 (d, J = 8.0 Hz, 2H), 5.39 (s, 2H), 4.29–4.27 (q, J = 6.9 Hz, 2H), 2.38 (s, 3H), 1.27–1.23 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 166.7, 159.7, 156.3, 154.3, 149.7, 137.3, 130.4, 127.8, 127.7, 112.2, 109.4, 68.9, 37.4, 21.0, 12.8. HRMS: calcd for C18H15NaN3O4 [M + Na]+, 360.0955; found 360.0948.
General Procedure for the Synthesis of the Dihydropyridopyrimidine Lactam Derivatives
Ethyl 7-(Chloromethyl)-1-ethyl-2,4-dioxo-5-(p-tolyl)-1,2,3,4,5,8-hexahydropyrido[2,3-d]pyrimidine-6-carboxylate (S1)
6-Amino-1-ethylpyrimidine-2,4(1H,3H)-dione (12.90 mmol) and ethyl 4-chloro-2-(4-methylbenzylidene)-3-oxobutanoate (6.45 mmol), which were synthesized according to the reported procedures,50,51 were placed into a round-bottom flask (250 mL). Magnesium sulfate was added and the mixture was heated to 45 °C for 48 h under a nitrogen atmosphere. After filtration, the solvent was removed in vacuo and the residue was purified by flash column chromatography on silica gel (40 g column, DCM + 1.5% MeOH) to furnish S1 as white solid (364 mg, 14%); m.p. 266–267 °C dec.; with a purity of 96%. 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H), 8.99 (s, 1H), 7.07 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 8.0 Hz, 2H), 5.17 (d, J = 11.0 Hz, 1H), 4.88 (s, 1H), 4.72 (d, J = 11.0 Hz, 1H), 4.07 (m, 3H), 3.96 - 3.86 (m, 1H), 2.20 (s, 3H), 1.16 (q, J = 7.0 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 165.2, 161.3, 149.8, 144.0, 143.2, 142.7, 135.5, 128.7, 127.1, 107.1, 89.8, 60.2, 48.6, 36.4, 35.7, 20.6, 13.9, 13.5. HRMS: calcd for C20H22ClN3NaO4 [M + Na]+, 426.1191; found 426.1199.
Compound S1 (0.05 mmol) and the substituted primary amine (0.05 mmol) were added into a dry microwave tube (2 mL) containing ethanol (1 mL). The mixture was heated to 150 °C for 1 h in a microwave reactor. The mixture was purified by flash column chromatography on silica gel (12 g column, DCM + 2.5% MeOH) to yield the target compound.
7-Allyl-1-ethyl-5-(p-tolyl)-5,7,8,9-tetrahydro-1H-pyrrolo[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (3o)
Yield (6.92 mg, 31%) as a yellow solid; m.p. 246–247 °C dec.; with a purity of 93%. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.89 (s, 1H), 7.15 (d, J = 7.9 Hz, 2H), 7.00 (d, J = 7.8 Hz, 2H), 5.78 (ddd, J = 15.3, 9.9, 4.8 Hz, 1H), 5.28 (d, J = 16.6 Hz, 1H), 5.09 (d, J = 16.6 Hz, 1H), 5.04 (d, J = 19.1 Hz, 1H), 4.93 (d, J = 19.1 Hz, 1H), 4.89 (s, 1H), 4.01 (q, J = 6.8 Hz, 2H), 3.86 (s, 2H), 2.20 (s, 3H), 1.09 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.4, 172.8, 162.0, 155.3, 150.9, 143.7, 134.8, 133.6, 128.3, 127.1, 115.9, 93.4, 90.0, 75.1, 43.3, 35.7, 32.3, 20.6, 14.0. HRMS: calcd for C21H23N4O3 [M + H]+, 379.1765; found 379.1771.
7-Benzyl-1-ethyl-5-(p-tolyl)-5,7,8,9-tetrahydro-1H-pyrrolo[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (3p)
Yield (9.01 mg, 35%) as a yellow solid; m.p. 223–224 °C dec.; with a purity of 97%. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 9.18 (s, 1H), 7.26 (d, J = 6.8 Hz, 3H), 7.17 (d, J = 7.9 Hz, 2H), 7.08 (d, J = 6.1 Hz, 2H), 7.02 (d, J = 7.9 Hz, 2H), 5.28 (d, J = 16.6 Hz, 1H), 5.12 (d, J = 16.6 Hz, 1H), 4.91 (s, 1H), 4.45 (d, J = 5.7 Hz, 2H), 4.02 (q, J = 6.8, 2H), 2.23 (s, 3H), 1.09 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.6, 172.8, 162.1, 155.3, 150.9, 143.7, 137.4, 134.9, 128.4, 128.4, 127.3, 127.2, 126.8, 93.5, 90.0, 75.2, 44.5, 35.7, 32.4, 20.6, 14.0. HRMS: calcd for C25H25N4O3 [M + H]+, 429.1921; found 429.1929.
1-Ethyl-7-phenethyl-5-(p-tolyl)-5,7,8,9-tetrahydro-1H-pyrrolo[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (3q)
Yield (6.61 mg, 25%) as a yellow solid; m.p. 252–253 °C dec.; with a purity of 93%. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.76 (s, 1H), 7.17 (s, 3H), 7.12 (d, J = 7.8 Hz, 2H), 7.01 (d, J = 7.8 Hz, 4H), 5.13 (d, J = 16.5 Hz, 1H), 5.00 (d, J = 16.5 Hz, 1H), 4.85 (s, 1H), 4.00 (dd, J = 14.6, 7.5 Hz, 2H), 3.46 (dd, J = 12.2, 6.1 Hz, 2H), 2.77 - 2.68 (m, 2H), 2.23 (s, 3H), 1.08 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.1, 172.8, 162.1, 155.4, 150.9, 143.7, 138.3, 134.8, 128.7, 128.4, 128.2, 127.1, 126.2, 93.4, 89.9, 75.0, 43.0, 35.7, 35.2, 32.3, 20.6, 14.0. HRMS: calcd for C26H27N4O3 [M + H]+, 443.2078; found 443.2068.
1-Ethyl-7-(4-methoxyphenethyl)-5-(p-tolyl)-5,7,8,9-tetrahydro-1H-pyrrolo[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (3r)
Yield (5.93 mg, 22%) as a yellow solid; m.p. 222–223 °C dec.; with a purity of 96%. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.73 (s, 1H), 7.12 (d, J = 7.9 Hz, 2H), 7.02 (d, J = 7.9 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 6.73 (d, J = 8.4 Hz, 2H), 5.17 (d, J = 16.5 Hz, 1H), 5.05 - 4.98 (d, J = 16.5 Hz, 1H), 4.85 (s, 1H), 4.00 (q, J = 6.7 Hz, 2H), 3.70 (s, 3H), 3.41 (dd, J = 13.6, 6.8 Hz, 2H), 2.70 - 2.62 (m, 2H), 2.23 (s, 3H), 1.08 (t, J = 6.7 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.0, 172.8, 162.0, 157.7, 155.4, 150.9, 143.7, 134.8, 130.2, 129.7, 128.4, 127.1, 113.6, 93.4, 89.8, 75.1, 54.9, 43.2, 35.7, 34.2, 32.3, 20.6, 14.0. HRMS: calcd for C27H29N4O4 [M + H]+, 473.2183; found 473.2180.
1-Ethyl-7-(4-hydroxyphenethyl)-5-(p-tolyl)-5,7,8,9-tetrahydro-1H-pyrrolo[3',4':5,6]pyrido[2,3-d]pyrimidine-2,4,6(3H)-trione (3s)
Yield (5.94 mg, 22%) as a yellow solid; m.p. 261–262 °C dec.; with a purity of 92%. 1H NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 9.16 (s, 1H), 8.73 (s, 1H), 7.12 (d, J = 7.9 Hz, 2H), 7.01 (d, J = 7.9 Hz, 2H), 6.79 (d, J = 8.2 Hz, 2H), 6.57 (d, J = 8.2 Hz, 2H), 5.15 (d, J = 16.4 Hz, 1H), 5.01 (d, J = 16.4 Hz, 1H), 4.86 (s, 1H), 4.00 (q, J = 6.8 Hz, 2H), 3.41 - 3.33 (m, 2H), 2.59 (m, 2H), 2.23 (s, 3H), 1.08 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 175.0, 172.8, 162.0, 155.7, 155.4, 150.9, 143.7, 134.8, 129.6, 128.3, 127.1, 115.0, 93.4, 89.8, 75.0, 43.4, 35.6, 34.4, 32.3, 20.6, 14.0. HRMS: calcd for C26H27N4O4 [M + H]+, 459.2027; found 459.2038.
Fluorescence anisotropy
All fluorescence anisotropy experiments were carried out in 50 mM Tris, 100 mM NaCl, pH = 7.4 on 384-well plates (Corning 4511). BrdT solutions also contained 3 mM DTT. Solutions for all experiments except those testing other detergents also contained 4 mM CHAPS. Other additives were present as described below. Stock solutions of fluorescent tracer BI-BODIPY in DMSO (25 μM) were diluted to 0.025 μM for these experiments. Plates were read on a Tecan Infinity 500 with an excitation wavelength at 485 nm and emission at 535 nm. For direct binding experiments, the protein was serially diluted across the plate, and the resulting anisotropy values were fit using Equation 1 in GraphPad Prism 6. In this equation, b and c are the maximum and minimum anisotropy values, respectively, a is the concentration of fluorescent tracer, x is the concentration of protein, and y is the observed anisotropy value.
| (Equation 1) |
The protein concentration for competition experiments was determined from the protein concentration in direct binding experiments at which the fluorescent tracer is 80% bound. Stock solutions of inhibitor molecules, which are 50 mM in DMSO, were serially diluted from 250 μM to subnanomolar concentrations, keeping the concentration of protein, BI-BODIPY, and other components constant. The DMSO concentration was held constant at 0.5% (v/v). The anisotropy values were then fit using GraphPad Prism’s log(inhibitor) vs. response (four parameters) function. The IC50 values thus obtained were converted to Ki values using a variant of the Cheng-Prusoff equation, where the Ki is the indirectly determined dissociation constant of the competitor ligand (Equation 2).52 Reported errors were determined from three or more independent experiments. Direct binding with BI-BODIPY and competition experiments with positive control 12 were carried out prior to each set of competition experiments to assess protein quality and assay stability.
| (Equation 2) |
Differential Scanning Fluorimetry
DSF experiments were carried out using a CFX384 Touch™ Real-Time PCR Detection System (Bio-Rad). The assay solution was composed of 10 μM BrdT(1) in a buffer of 50 mM HEPES (pH = 7.5) and 100 mM NaCl with a 5x concentration of SYPRO Orange (Life Technologies) as a fluorescent reporter. Compounds were tested in an 8-point dose response assay at concentrations ranging from 1250 to 9 μM (final DMSO concentration of 2.5 %); experiments were performed in duplicate with average values reported. Protein denaturation was measured from 25 to 75 °C after a 5 min incubation at 25 °C, in 0.2 °C increments with a dwell time of 5 seconds (ramping rate of 2.4 °C per min). The fluorescent response of the protein unfolding was recorded and the peak of the negative first derivative of the resulting melting curve was used to identify the apparent melting temperature (Tm) of the samples; calculations were performed by the Bio-Rad CFX Manager software. The change in melting temperature (ΔTm) was determined by subtracting the Tm of the DMSO control (2.5 % final concentration) from the Tm of the compound treated samples. Additional controls include a constant concentration of compound 2 at 100 μM and an 8-point dose response of 2 from 300 to 0.1 μM, included on each plate. A Z’ was calculated for each plate using 2 and DMSO controls; plates with a Z’ value less than 0.5 were excluded from analysis.
Virtual screen
A high throughput virtual screen (HTVS) was performed using the BrdT(1) X-ray crystal structure with (+)-JQ-1 (PDB code: 4FLP), using ~ 6,000,000 compounds from the ZINC clean drug-like molecules set.53 The crystal structure (4FLP) was retrieved from the Protein Data Bank, and downloaded into Schrodinger Maestro 9.3. The Protein Preparation Wizard in the Schrödinger suite was used to process the BrdT(1) structure. The protein structure was prepared by assigning bond orders, adding hydrogens, creating disulfide bonds and optimizing H-bonding networks with PROPKA. Missing side chains for some amino acids were found and added using Prime version 3.1. Energy minimization with root mean square deviation value of 0.30 was then applied using the Schrodinger OPLS_2005 force field. Compound 2 was removed and a receptor grid was generated. Subsets of ~100,000 molecules of the ZINC clean drug-like commercially available molecules set were then docked using Glide 5.8.54,55 The ZINC clean drug-like commercially available molecules set consists of molecules with molecular weight <500 g/mol, logP < 5, fewer than 5 H-bond donors, fewer than 10 H bond acceptors, fewer than 7 rotatable bonds, and polar surface area less than 150 Å2, and molecules with reactive groups, thiols and aldehydes were removed.56,57 The ligands were docked in low precision subsets of ~100,000. The best scoring 0.4% of each subset were retained and combined into one file, and then re-ordered. After re-ordering, the top 6,000 molecules were then re-docked using standard precision. The top 4,000 were re-docked with standard precision, with no significant change. The best 200 compounds were then examined, and high molecular weight compounds, PAINS, and phenols were removed. Twenty currently available compounds including two examples from the best scoring scaffold were ordered from a commercial source.
PrOF NMR
19F NMR spectra were acquired on a 500 MHz Bruker Avance III HD equipped with a 5 mM Prodigy TCI cryoprobe. Samples contained 40–50 μM 5FW-BrdT or 5FW-Brd4 in 50 mM Tris, 100 mM NaCl, pH = 7.4 with 5% D2O. 5FW-BrdT samples also contained 3 mM DTT. Experimental parameters included a 90° flip angle, a relaxation delay of 0.7 seconds, an acquisition time of 0.1 seconds, and 500 transients. Spectra are referenced to TFA at −76.55 ppm. Small molecules were titrated in from concentrated DMSO stocks. DMSO concentration was kept below 1% because of its potential for bromodomain binding. Kd values were obtained using a one-site binding equation accounting for ligand depletion.
Brd4(1) and BrdT(1) AlphaScreen™
Assays were performed with minor modifications from the manufacturer’s protocol (Perkin Elmer, USA). All reagents were diluted in AlphaScreen™ buffer (50 mM HEPES, 150 mM NaCl, 0.01% v/v Tween-20, 0.1% w/v BSA, pH = 7.4). After addition of Alpha beads to master solutions, all subsequent steps were performed under low light conditions. A 2x solution of components with final concentrations of His-Brd4(1) or His-BrdT(1) at 0.020 μM, Ni-coated Acceptor Bead at 10 μg/ml, and biotinylated compound 2 at 0.010 μM were added in 10 μL to 384-well plates (AlphaPlate-384, PerkinElmer) using an EL406 liquid handler (Biotek, USA). Plates were spun down at 1000 rpm. A 10-point 1: serial dilution of compounds in DMSO was prepared at 200x the final concentration. 100 nL of compound from these stock plates were added by pin transfer using a Janus Workstation (PerkinElmer). A 2x solution of streptavidin-coated donor beads with a final concentration of 10 μg/ml was added in a 10 μL volume. The plates were spun down again at 1000 rpm and sealed with foil to prevent light exposure and evaporation. The plates were then incubated at room temperature for 1 h and read on an Envision 2104 (PerkinElmer) using the manufacturer’s protocol. IC50 values were calculated using a 4-parameter logistic curve in Prism 6 (GraphPad Software, USA) after normalization to DMSO-treated negative control wells (0.2% DMSO, v/v).
AlphaScreen™
Assays were performed by Reaction Biology using an acetylated Histone4 (1-21), against Brd4(1) with a constant DMSO concentration of 1% (v/v). Compound 2 was used as a positive control.
Protein crystallography
Reagents for crystallographic experiments were purchased from Hampton Research. Brd4(1) was purified as described previously.27 Crystals of Brd4(1) were grown by vapor-diffusion in hanging drops using 0.2 M (NH4)2SO4, 0.1 M Tris/HCl (pH = 8.5) and 25 % (w/v) PEG 3350 as precipitant supplemented with 5 mM compound 1 and 10% (v/v) DMSO. Crystals were harvested in cryoprotectant (3 parts precipitant including 0.5 mM compound 1 and 1 part 100% (v/v) ethylene glycol) and flash frozen in a stream of nitrogen gas. X-ray diffraction data were recorded at −180 °C at the beamline 22-ID, SER-CAT, Advanced Photon Source, Argonne National Laboratories. Data were reduced and scaled with XDS.58 PHENIX59 was employed for phasing and refinement, and model building was performed using Coot.60 The structure was solved by molecular replacement using Phaser61 with the monomer of PDB entry 4O7A as the search model. An initial model of the inhibitor was generated using MarvinSketch (ChemAxon, Cambridge, MA) with ligand restraints from eLBOW of the PHENIX suite. All structures were validated by MolProbity62 and phenix.model_vs_data63. Figures were prepared using PyMOL (Schrödinger, LLC). Atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession code 5KDH.
Cell Viability Assays
MM1.S cells were purchased from ATCC and maintained in RPMI-1640 medium (Life Technologies) containing 10% fetal bovine serum. Cells were grown and maintained at 37 °C in a humidified atmosphere containing 5% CO2. Antibodies for c-MYC (#5605) and p21Cip1 (#2946) were purchased from Cell Signaling. Vinculin antibody (#V9131) was from Sigma-Aldrich. Peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch, West Grove, PA. MM1.S cells were seeded in 96-well plates at approximately 20,000 cells per well and incubated with drugs or vehicle (0.1% DMSO) for 72 h, with 6 replicates per drug concentration. After drug treatment, half volume of MTT reagent (Calbiochem) (2 mg/ml filtered PBS) was added to each well, followed by an incubation for 3 h at 37 °C. The supernatant was then aspirated and replaced with 100 μl DMSO to solubilize the formazan crystals. Plates were placed on an orbital shaker for 5 min, and absorbance at 540 nm was determined with a SpectraMax 340PC microplate reader (Molecular Devices) with SoftMax Pro 5.2 software. The data were analyzed with Prism 6 (GraphPad) to determine IC50 values.
Immunoblotting
Cells were seeded in 6-well plates (2 × 105 adherent or 1 × 106 suspension cells per well) and incubated for 6 h with increasing inhibitor concentrations. Cells were harvested by centrifugation at 300 g for 5 min and resuspended in CelLytic M Cell Lysis Reagent (Sigma-Aldrich) containing Halt Protease Inhibitor Cocktail and Halt Phosphatase Inhibitor Cocktail (Thermo Scientific) and 5 mM EDTA at 4 °C. Protein concentration was determined with Bio-Rad Protein Assay Reagent (Hercules, CA) and samples were diluted with 1/3 volume 4X SDS sample buffer and heated at 95 °C for 5 min. Samples were subjected to 10 or 12.5% SDS-PAGE and transferred to PVDF or nitrocellulose membranes. Western blots were developed with the appropriate pairs of primary and secondary antibodies and signals were visualized using HyGLO Chemiluminescent reagent (Denville Scientific).
Supplementary Material
Acknowledgments
This work was supported by the Contraception Research Branch, Eunice Kennedy Shriver National Institute of Child Health and Human Development: 5 U01 HD076542 (GIG and ES) and NICHD: HHSN275201300017C (GIG and ES), NIH 1U01HD76508 (JEB and JQ) and the NSF CAREER award (CHE-1352019, WCP). These studies were supported by the University of Minnesota through the Vince and McKnight Endowed Chairs (G.I.G) We thank the Southeast Regional Collaborative Access Team (SER-CAT, University of Georgia) for assistance with synchrotron data collection at Argonne National Laboratory. We also thank the Moffitt Chemical Biology Core for use of the protein crystallography facility (NCI grant P30-CA076292). BI-BODIPY was a generous gift from Wei Zhang at the University of Massachusetts, Boston.
ABBREVIATIONS
- BET
bromodomain and extra-terminal
- BrdT(1)
BrdT domain 1
- BrdT(2)
BrdT domain 2
- Brd4(1)
Brd4 domain 1
- DSF
differential scanning fluorimetry
- PrOF NMR
protein-observed fluorine NMR
Footnotes
Supporting Information. A listing of the contents of each file supplied as Supporting Information should be included. For instructions on what should be included in the Supporting Information as well as how to prepare this material for publications, refer to the journal’s Instructions for Authors.
The following files are available free of charge.
Additional data and methods (PDF)
List of compounds (CSV)
Accession Codes
Atomic coordinates of Brd4 bound to compound 1 (PDB code 5KDH) has been deposited with the Protein Data Bank.
Authors will release atomic coordinates and experimental data upon article publication.
References
- 1.Matzuk MM, McKeown MR, Filippakopoulos P, Li Q, Ma L, Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp S, Bradner JE. Small-Molecule Inhibition of BRDT for Male Contraception Cell. 2012;150:673–684. doi: 10.1016/j.cell.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prinjha RK, Witherington J, Lee K. Place Your BETs: the Therapeutic Potential of Bromodomains. Trends Pharmacol Sci. 2012;33:146–153. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filippakopoulos P, Knapp S. Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation. Nat Rev Drug Discovery. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Guo J, Wu Y, Zhou Q. The BET Bromodomain Inhibitor JQ1 Activates HIV Latency Through Antagonizing Brd4 Inhibition of Tat-Transactivation. Nucleic Acids Res. 2013;41:277–287. doi: 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, Bradner JE, McKinsey TA. BET Acetyl-Lysine Binding Proteins Control Pathological Cardiac Hypertrophy. J Mol Cell Cardiol. 2013;63:175–179. doi: 10.1016/j.yjmcc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, Alaiti MA, Bullard J, Alazem K, Margulies KB, Cappola TP, Lemieux M, Plutzky J, Bradner JE, Haldar SM. BET Bromodomains Mediate Transcriptional Pause Release in Heart Failure. Cell. 2013;154:569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung C-W, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of Inflammation by a Synthetic Histone Mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective Inhibition of BET Bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korb E, Herre M, Zucker-Scharff I, Darnell RB, Allis CD. BET Protein Brd4 Activates Transcription in Neurons and BET Inhibitor Jq1 Blocks Memory in Mice. Nat Neurosci. 2015;18:1464–1473. doi: 10.1038/nn.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fong CY, Gilan O, Lam EYN, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, Lee S C-W, Carpenter C, Gregory R, Ramsay RG, Lane SW, Abdel-Wahab O, Kouzarides T, Johnstone RW, Dawson S-J, Huntly BJP, Prinjha RK, Papenfuss AT, Dawson MA. BET Inhibitor Resistance Emerges From Leukaemia Stem Cells. Nature. 2015;525:538–542. doi: 10.1038/nature14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Syeda SS, Jakkaraj S, Georg GI. Scalable Syntheses of the BET Bromodomain Inhibitor JQ1. Tetrahedron Lett. 2015;56:3354–3457. doi: 10.1016/j.tetlet.2015.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ran X, Zhao Y, Liu L, Bai L, Yang C-Y, Zhou B, Meagher JL, Chinnaswamy K, Stuckey JA, Wang S. Structure-Based Design of Γ-Carboline Analogues as Potent and Specific BET Bromodomain Inhibitors. J Med Chem. 2015;58:4927–4939. doi: 10.1021/acs.jmedchem.5b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hay D, Fedorov O, Filippakopoulos P, Martin S, Philpott M, Picaud S, Hewings DS, Uttakar S, Heightman TD, Conway SJ, Knapp S, Brennan PE. The Design and Synthesis of 5- and 6-Isoxazolylbenzimidazoles as Selective Inhibitors of the BET Bromodomains. MedChemComm. 2013;4:140–144. doi: 10.1039/C2MD20189E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirguet O, Gosmini R, Toum J, Clément CA, Barnathan M, Brusq J-M, Mordaunt JE, Grimes RM, Crowe M, Pineau O, Ajakane M, Daugan A, Jeffrey P, Cutler L, Haynes AC, Smithers NN, Chung C-W, Bamborough P, Uings IJ, Lewis A, Witherington J, Parr N, Prinjha RK, Nicodeme E. Discovery of Epigenetic Regulator I-BET762: Lead Optimization to Afford a Clinical Candidate Inhibitor of the BET Bromodomains. J Med Chem. 2013;56:7501–7515. doi: 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- 16.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, Muller S, Brennan PE, Knapp S, Filippakopoulos P. RVX-208, an Inhibitor of BET Transcriptional Regulators with Selectivity for the Second Bromodomain. Proc Natl Acad Sci USA. 2013;110:19754–19759. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang G, Smith SG, Zhou M-M. Discovery of Chemical Inhibitors of Human Bromodomains. Chem Rev. 2015;115:11625–11688. doi: 10.1021/acs.chemrev.5b00205. [DOI] [PubMed] [Google Scholar]
- 18.McKeown MR, Shaw DL, Fu H, Liu S, Xu X, Marineau JJ, Huang Y, Zhang X, Buckley DL, Kadam A, Zhang Z, Blacklow SC, Qi J, Zhang W, Bradner JE. Biased Multicomponent Reactions to Develop Novel Bromodomain Inhibitors. J Med Chem. 2014;57:9019–9027. doi: 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Despite our attempts to remove all PAINS compounds, a rhodanine was tested (see structure in Figure S1). No measurable binding was detected for this compound in our fluorescence anisotropy assay.
- 20.Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR. BI 6727, a Polo-Like Kinase Inhibitor with Improved Pharmacokinetic Profile and Broad Antitumor Activity. Clin Cancer Res. 2009;15:3094–3102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- 21.Urick AK, Hawk LML, Cassel MK, Mishra NK, Liu S, Adhikari N, Zhang W, Santos Dos CO, Hall JL, Pomerantz WC. Dual Screening of BPTF and Brd4 Using Protein-Observed Fluorine NMR Uncovers New Bromodomain Probe Molecules. ACS Chem Biol. 2015;10:2246–2256. doi: 10.1021/acschembio.5b00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Kwiatkowski N, Zeng H, Lim SM, Gray NS, Zhang W, Yang PL. Leveraging Kinase Inhibitors to Develop Small Molecule Tools for Imaging Kinases by Fluorescence Microscopy. Mol BioSyst. 2012;8:2523–2524. doi: 10.1039/c2mb25099c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Philpott M, Yang J, Tumber T, Fedorov O, Uttarkar S, Filippakopoulos P, Picaud S, Keates T, Felletar I, Ciulli A, Knapp S, Heightman TD. Bromodomain-Peptide Displacement Assays for Interactome Mapping and Inhibitor Discovery. Mol BioSyst. 2011;7:2899–2908. doi: 10.1039/c1mb05099k. [DOI] [PubMed] [Google Scholar]
- 24.Zhao L, Wang Y, Cao D, Chen T, Wang Q, Li Y, Xu Y, Zhang N, Wang X, Chen D, Chen L, Chen Y-L, Xia G, Shi Z, Liu Y-C, Lin Y, Miao Z, Shen J, Xiong B. Fragment-Based Drug Discovery of 2-Thiazolidinones as BRD4 Inhibitors: 2. Structure-Based Optimization. J Med Chem. 2015;58:1281–1297. doi: 10.1021/jm501504k. [DOI] [PubMed] [Google Scholar]
- 25.Mirguet O, Lamotte Y, Chung C-W, Bamborough P, Delannée D, Bouillot A, Gellibert F, Krysa G, Lewis A, Witherington J, Huet P, Dudit Y, Trottet L, Nicodeme E. Naphthyridines as Novel BET Family Bromodomain Inhibitors. ChemMedChem. 2014;9:580–589. doi: 10.1002/cmdc.201300259. [DOI] [PubMed] [Google Scholar]
- 26.Chung C-W, Dean AW, Woolven JM, Bamborough P. Fragment-Based Discovery of Bromodomain Inhibitors Part 1: Inhibitor Binding Modes and Implications for Lead Discovery. J Med Chem. 2012;55:576–586. doi: 10.1021/jm201320w. [DOI] [PubMed] [Google Scholar]
- 27.Ember SWJ, Zhu J-Y, Olesen SH, Martin MP, Becker A, Berndt N, Georg GI, Schönbrunn E. Acetyl-Lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem Biol. 2014;9:1160–1171. doi: 10.1021/cb500072z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pomerantz WC, Wang N, Lipinski AK, Wang R, Cierpicki T, Mapp AK. Profiling the Dynamic Interfaces of Fluorinated Transcription Complexes for Ligand Discovery and Characterization. ACS Chem Biol. 2012;7:1345–1350. doi: 10.1021/cb3002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mishra NK, Urick AK, Ember SWJ, Schönbrunn E, Pomerantz WC. Fluorinated Aromatic Amino Acids Are Sensitive 19F NMR Probes for Bromodomain-Ligand Interactions. ACS Chem Biol. 2014;9:2755–2760. doi: 10.1021/cb5007344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gee CT, Koleski EJ, Pomerantz WCK. Fragment Screening and Druggability Assessment for the CBP/P300 KIX Domain Through Protein-Observed 19F NMR Spectroscopy. Angew Chem Int Ed Engl. 2015;54:3735–3739. doi: 10.1002/anie.201411658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ge X, MacRaild CA, Devine SM, Debono CO, Wang G, Scammells PJ, Scanlon MJ, Anders RF, Foley M, Norton RS. Ligand-Induced Conformational Change of Plasmodium Falciparum AMA1 Detected Using 19F NMR. J Med hem. 2014;57:6419–6427. doi: 10.1021/jm500390g. [DOI] [PubMed] [Google Scholar]
- 32.Gee CT, Arntson KE, Urick AK, Mishra NK, Hawk LML, Wisniewski AJ, Pomerantz WCK. Protein-Observed 19F NMR for Fragment Screening, Affinity, Quantification, and Druggability Assessment. Nat Protoc. 2015;11:1414–1427. doi: 10.1038/nprot.2016.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gee CT, Arntson KE, Urick AK, Mishra NK, Hawk LML, Wisniewski AJ, Pomerantz WCK. Protein-Observed (19)F-NMR for Fragment Screening, Affinity Quantification and Druggability Assessment. Nat Protoc. 2016;11:1414–1427. doi: 10.1038/nprot.2016.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fielding L. NMR Methods for the Determination of Protein-Ligand Dissociation Constants. Curr Top Med Chem. 2003;3:39–53. doi: 10.2174/1568026033392705. [DOI] [PubMed] [Google Scholar]
- 35.Tanifum EA, Kots AY, Choi B-K, Murad F, Gilbertson SR. Novel Pyridopyrimidine Derivatives as Inhibitors of Stable Toxin a (STa) Induced cGMP Synthesis. Bioorg Med Chem Lett. 2009;19:3067–3071. doi: 10.1016/j.bmcl.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 36.Magedov IV, Frolova L, Manpadi M, Bhoga UD, Tang H, Evdokimov NM, George O, Georgiou KH, Renner S, Getlik M, Kinnibrugh TL, Fernandes MA, Van slambrouck S, Steelant WFA, Shuster CB, Rogelj S, van Otterlo WAL, Kornienko A. Anticancer Properties of an Important Drug Lead Podophyllotoxin Can Be Efficiently Mimicked by Diverse Heterocyclic Scaffolds Accessible via One-Step Synthesis. J Med Chem. 2011;54:4234–4246. doi: 10.1021/jm200410r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evdokimov NM, Van slambrouck S, Heffeter P, Tu L, Le Calvé B, Lamoral-Theys D, Hooten CJ, Uglinskii PY, Rogelj S, Kiss R, Steelant WFA, Berger W, Yang JJ, Bologa CG, Kornienko A, Magedov IV. Structural Simplification of Bioactive Natural Products with Multicomponent Synthesis. 3. Fused Uracil-Containing Heterocycles as Novel Topoisomerase-Targeting Agents. J Med Chem. 2011;54:2012–2021. doi: 10.1021/jm1009428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamal A, Suresh P, Mallareddy A, Kumar BA, Reddy PV, Raju P, Tamboli JR, Shaik TB, Jain N, Kalivendi SV. Synthesis of a New 4-Aza-2,3-Didehydropodophyllotoxin Analogues as Potent Cytotoxic and Antimitotic Agents. Bioorg Med Chem. 2011;19:2349–2358. doi: 10.1016/j.bmc.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 39.Ioan P, Carosati E, Micucci M, Cruciani G, Broccatelli F, Zhorov SB, Chiarini A, Budriesi R. 1,4-Dihydropyridine Scaffold in Medicinal Chemistry, the Story So Far and Perspectives (Part 1): Action in Ion Channels and GPCRs. Curr Med Chem. 2011;18:4901–4922. doi: 10.2174/092986711797535173. [DOI] [PubMed] [Google Scholar]
- 40.Hantzsch A. Condensationsprodukte Aus Aldehydammoniak Und Ketonartigen Verbindungen. Chem Ber. 1881;14:1637–1638. [Google Scholar]
- 41.Chaikuad A, Petros AM, Fedorov O, Xu J, Knapp S. Structure-Based Approaches Towards Identification of Fragments for the Low-Druggability ATAD2 Bromodomain. MedChemComm. 2014;5:1843–1848. [Google Scholar]
- 42.Martin MP, Olesen SH, Georg GI, Schönbrunn E. Cyclin-Dependent Kinase Inhibitor Dinaciclib Interacts with the Acetyl-Lysine Recognition Site of Bromodomains. ACS Chem Biol. 2013;8:2360–2365. doi: 10.1021/cb4003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang X. Fluorescence Polarization Competition Assay: the Range of Resolvable Inhibitor Potency Is Limited by the Affinity of the Fluorescent Ligand. J Biomol Screening. 2003;8:34–38. doi: 10.1177/1087057102239666. [DOI] [PubMed] [Google Scholar]
- 44.Ciceri P, Muller S, O’Mahony A, Fedorov O, Filippakopoulos P, Hunt JP, Lasater EA, Pallares G, Picaud S, Wells C, Martin S, Wodicka LM, Shah NP, Treiber DK, Knapp S. Dual Kinase-Bromodomain Inhibitors for Rationally Designed Polypharmacology. Nat Chem Biol. 2014;10:305–312. doi: 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka M, Roberts JM, Seo H-S, Souza A, Paulk J, Scott TG, DeAngelo SL, Dhe-Paganon S, Bradner JE. Design and Characterization of Bivalent BET Inhibitors. Nat Chem Biol. 2016;12:1089–1096. doi: 10.1038/nchembio.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Faelth-Savitski M, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, den Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I, Crews CM. Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, Zeng L, Mujtaba S, Ohlmeyer M, Zhou M-M. Structure-Guided Design of Potent Diazobenzene Inhibitors for the BET Bromodomains. J Med Chem. 2013;56:9251–9264. doi: 10.1021/jm401334s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baud MGJ, Lin-Shiao E, Cardote T, Tallant C, Pschibul A, Chan K-H, Zengerle M, Garcia JR, Kwan TT-L, Ferguson FM, Ciulli A. Chemical Biology a Bump-and-Hole Approach to Engineer Controlled Selectivity of BET Bromodomain Chemical Probes. Science. 2014;346:638–641. doi: 10.1126/science.1249830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raux B, Voitovich Y, Derviaux C, Lugari A, Rebuffet E, Milhas S, Priet S, Roux T, Trinquet E, Guillemot J-C, Knapp S, Brunel J-M, Fedorov AY, Collette Y, Roche P, Betzi S, Combes S, Morelli X. Exploring Selective Inhibition of the First Bromodomain of the Human Bromodomain and Extra-Terminal Domain (BET) Proteins. J Med Chem. 2016;59:1634–1641. doi: 10.1021/acs.jmedchem.5b01708. [DOI] [PubMed] [Google Scholar]
- 50.Elzein E, Kalla RV, Li X, Perry T, Gimbel A, Zeng D, Lustig D, Leung K, Zablocki J. Discovery of a Novel A2B Adenosine Receptor Antagonist as a Clinical Candidate for Chronic Inflammatory Airway Diseases. J Med Chem. 2008;51:2267–2278. doi: 10.1021/jm7014815. [DOI] [PubMed] [Google Scholar]
- 51.Devasthale P, Wang Y, Wang W, Fevig J, Feng J, Wang A, Harrity T, Egan D, Morgan N, Cap M, Fura A, Klei HE, Kish K, Weigelt C, Sun L, Levesque P, Moulin F, Li Y-X, Zahler R, Kirby MS, Hamann LG. Optimization of Activity, Selectivity, and Liability Profiles in 5-Oxopyrrolopyridine DPP4 Inhibitors Leading to Clinical Candidate (Sa)-2-(3-(Aminomethyl)-4-(2,4-Dichlorophenyl)-2-Methyl-5-Oxo-5H-Pyrrolo[3,4-B]Pyridin-6(7H)-Yl)-N,N-Dimethylacetamide (BMS-767778) J Med Chem. 2013;56:7343–7357. doi: 10.1021/jm4008906. [DOI] [PubMed] [Google Scholar]
- 52.Kenakin TP. Pharmacologic Analysis of Drug-Receptor Interaction. Raven; New York: 1993. [Google Scholar]
- 53.Shoichet BK, Irwin JJ. ZINC − a Free Database of Commercially Available Compounds for Virtual Screening. J Chem Inf Model. 2004;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 55.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 56.Lipinski CA. Drug-Like Properties and the Causes of Poor Solubility and Poor Permeability. J Pharmacol Toxicol Methods. 2000;44:235–249. doi: 10.1016/s1056-8719(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 57.Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 58.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Crystallogr Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser Crystallographic Software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Afonine PV, Grosse-Kunstleve RW, Chen VB, Headd JJ, Moriarty NW, Richardson JS, Richardson DC, Urzhumtsev A, Zwart PH, Adams PD. Phenix.Model_vs_Data: a High-Level Tool for the Calculation of Crystallographic Model and Data Statistics. J Appl Crystallogr. 2010;43:669–676. doi: 10.1107/S0021889810015608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.