

Abstract
Recent evidence indicates that inflammation may significantly contribute to the pathogenesis of Alzheimer’s disease (AD). Since the apo A-I mimetic peptide D-4F has been shown to inhibit atherosclerotic lesion formation and regress already existing lesions (in the presence of pravastatin) and the peptide also decreases brain arteriole inflammation, we undertook a study to evaluate the efficacy of oral D-4F co-administered with pravastatin on cognitive function and amyloid β (Aβ) burden in the hippocampus of APPSwe-PS1ΔE9 mice. Three groups of male mice were administered D-4F and pravastatin, Scrambled D-4F (ScD-4F, a control peptide) and pravastatin in drinking water, while drinking water alone served as control. The escape latency in the Morris Water Maze test was significantly shorter for the D-4F+statin administered animals compared to the other two groups. While the hippocampal region of the brain was covered with 4.2±0.5 and 3.8±0.6% of Aβ load in the control and ScD-4F+statin administered groups, in the D-4F+statin administered group Aβ load was only 1.6±0.1%. Furthermore, there was a significant decrease in the number of activated microglia (p<0.05 vs the other two groups) and activated astrocytes (p<0.05 vs control) upon oral D-4F+statin treatment. Inflammatory markers TNFα and IL-1β levels were decreased significantly in the D-4F+statin group compared to the other two groups (for IL-1β p<0.01 vs the other two groups and for TNF-α p<0.001 vs control) and the expression of MCP-1 were also less in D-4F+statin administered group compared to the other two groups. These results suggest that the apo A-I mimetic peptide inhibits amyloid β deposition and improves cognitive function via exerting anti-inflammatory properties in the brain.
Keywords: Apo A-I, A-I mimetic, HDL, Amphipathic peptides, Amyloid β
Alzheimer’s disease (AD) is the world’s leading cause of dementia and affects 12 million people worldwide. AD is a neurodegenerative disorder and is characterized by progressive cognitive impairment (Hebert et al., 2003). Epidemiological and clinical studies suggest that inflammation and oxidative stress are implicated in AD (Akiyama et al., 2000; McGeer and McGeer, 2003; Christen, 2000; Pratico and Delanty, 2000). However, the role of inflammation in the pathogenesis of AD is not clearly understood. Activated microglia release a variety of pro-inflammatory molecules which result in the exacerbation of the disease process and contribute to neuronal death (Yates et al, 2000). On the other hand, activated microglia can possibly take up and degrade amyloid β (Aβ) (Boche and Nicoll, 2008). Mouse models of AD also suggest that AD may have an inflammatory component contributing to the pathogenesis of this disease (Heneka et al., 2005; Kunjathoor et al., 2004). Anti-inflammatory agents have been used as a therapy for AD (In t’ Veld et al., 2001; Zandi et al., 2002).
Hypercholesterolemia is also an important risk factor in the development of AD. Emerging evidence suggests that high dietary cholesterol increases Aβ accumulation and accelerates AD-related pathology (Li et al., 2003; Refolo et al., 2000). High density lipoproteins (HDL) and apolipoprotein A-I (apo A-I), the major protein component of HDL, possess several important protective functions including facilitating reverse cholesterol transport (Barter and Rye, 2006) and thereby is inversely correlated to the development of atherosclerosis. Apo A-I has also been shown to possess anti-oxidant and anti-inflammatory properties (Navab et al., 2005a; Barter and Rye, 2006). Apo A-I mimetic peptides were developed based on the presence of lipid-associating amphipathic α-helical domains in apo A-I (Segrest et al., 1992, Anantharamaiah et al., 2007). It has been shown that oral administration of D-4F, an apo A-I mimetic peptide synthesized from D-amino acids, remains intact in the circulation, significantly enhances HDL protective capacity, decreases LDL-induced monocyte chemotactic activity, and inhibits the formation of atherosclerotic plaques in young apo E null mice (Navab et al., 2002). In addition, it has been shown that D-4F decreases brain arteriole inflammation and improves cognitive performance in low density lipoprotein (LDL) receptor-null mice on Western diet (Buga et al, 2006). More interestingly, D-4F in the presence of pravastatin regressed already existing atherosclerotic lesions in older apo E null mice (Navab et al., 2005b). The mechanism of action of this peptide has been shown to be through modulation of inflammatory properties of circulating lipoproteins, particularly HDL.
Therefore, in the present study, we tested the hypothesis that oral administration of D-4F co-administered with pravastatin (at a dose at which this statin alone does not inhibit Aβ deposition (Chauhan et al., 2004)), would decrease Aβ deposition in the brain and improve cognitive performance in a mouse model of AD. We show that in the APPswe-PS1ΔE9 transgenic mouse model, the peptide D-4F, but not the control scrambled peptide ScD-4F, in the presence of pravastatin significantly inhibits Aβ deposition and improves cognitive performance. This is associated with decreased numbers of activated microglia and reactive astrocytes in the hippocampal region and a concomitant decrease in pro-inflammatory cytokine levels in the brain.
Materials and methods
Materials
D-4F (Ac-DWFKAFYDKVAEKFKEAF-NH2, synthesized using D-amino acids) and ScD-4F (Ac-DWFAKDYFKKAFVEEFAK-NH2), a control peptide with the same D-amino acids but arranged in a sequence that does not promote the formation of class A amphipathic helix, (thus rendering the peptide inactive) were synthesized as previously described (Datta et al., 2004).
Mice
Male APPswe-PS1ΔE9 mice (Jankowsky et al., 2004) were purchased from Jackson Laboratories, Bar Harbor, ME- Strain name B6C3-Tg(APPswe,PSEN1ΔE9)85Dbo/J; stock number 004462). A breeding colony was established by breeding male APPswe-PS1ΔE9-mice with female B6C3F1/J (Jackson Laboratories, Bar Harbor, ME). The animals were genotyped for the presence of transgene by PCR amplification of genomic DNA extracted from 1cm tail clippings. Three groups of 4–5 month old male APPswe-PS1ΔE9 were used for the study. The first group received water and served as control. The second group was treated with ScD-4F (200 μg/ml) and pravastatin (10 μg/ml) in their drinking water. The third group received the peptide D-4F (200 μg/ml) and pravastatin (10 μg/ml) in their drinking water. The water intake was monitored. The treatment period was for 3 months. Mice were housed under standard conditions in conventional cages and given standard rodent diet and water ad libitum. All animal procedures used for this study were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Morris Water Maze (MWM)
Spatial memory was evaluated by the MWM test as described earlier (Li et al., 2003). Briefly, the mice were tested in a water maze consisting of a round pool (diameter, 112 cm) filled with water (22 °C) to a height of 31 cm. An escape platform (8×8 cm) was submerged 1 cm below the water surface in the NorthEast (NE) quadrant. The acquisition of the spatial task consisted of placing the mouse in the water next to and facing the wall successively in north (N), east (E), south (S), and west (W) positions. In each trial, the mouse was allowed to swim until it found the hidden platform or until 60 s had elapsed, at which point the mouse was guided to the platform. The time required to find the hidden platform is referred to as escape latency. Shorter escape latency values represent improved cognitive function. The escape latency was recorded by the SMART system (SD instruments, San Diego, CA) for four trials daily for 5 days. The day after completion of the acquisition phase, a “probe trial” was also conducted by removing the platform and placing the mouse facing N side. The number of times the mouse crossed over the area where the platform was previously hidden and the amount of time spent in the target quadrant was counted in a single 1 min trial. The probe trial was followed by four visible platform trials, escape latencies were measured in an identical manner to the place learning task for four trials.
Brain tissue preparation
Mice were anesthetized with ketamine/xylazine, and blood was collected by cardiac puncture. After whole-body perfusion with 0.9% saline, brains were harvested. The brains were cut sagittally into left and right hemispheres. The right hemisphere was fixed in buffered formaldehyde for histological analysis. The hippocampal region was dissected out of the left hemisphere and was snap frozen in liquid nitrogen and stored at −80 °C until biochemical analysis.
Plasma total cholesterol levels were determined colorimetrically using commercial reagents (Infinity cholesterol reagent, Sigma, St. Louis, MO).
Enzyme-linked immunosorbent assay
Two or three hippocampi from each group were pooled. The hippocampi were homogenized with 15 volumes of ice-cold tris-buffered saline (TBS; pH 7.4) with protease inhibitors (Sigmafast protease inhibitor tablets, Sigma). The sample was centrifuged at 20,000 g for 20 min. at 4 °C. Supernatant (TBS extract) was transferred to a new tube and stored at −80 °C until analyzed. The pellet was washed with 50 μl of cold TBS. 400 μl of 5 M guanidine hydrochloride (GuHCl) containing complete protease inhibitor was added to the pellet. The sample was vortexed and incubated at room temperature for 4 h. Homogenate was spun at 20,000 rpm for 20 min. at 4 °C. The supernatant (guanidine extract) was transferred to new tubes and stored at −80 °C until analyzed. Total protein level in soluble and insoluble fractions was assayed using the BCA (bicinchoninic acid) protein assay reagent method (Pierce). The levels of Aβ40 and Aβ42 in the TBS soluble and guanidine soluble fractions were determined with Aβ40 and Aβ42 specific enzyme-linked immunosorbent assay kits (BioSource International, Camarillo, CA) using the manufacturer’s protocol. Interleukin -1β (IL-1β) and Tumor necrosis factor-α (TNF-α) in the TBS soluble fractions were measured using commercially available ELISA kits (R&D systems, Minneapolis, MN) according to manufacturer’s protocol. The values were expressed as amount per total protein.
Western blot analysis
50 μg of protein was separated on 4–20% SDS polyacrylamide gel under reducing conditions. Proteins were transferred to a nitrocellulose (Whatman) membrane. The membrane was blocked with 5% dry milk in 0.1% Tween 20/PBS for 1 h. and then incubated with rabbit polyclonal antibody CT695 against the C terminus of APP (1:1000; Invitrogen) overnight at 4 °C. After washing, blot was incubated with corresponding HRP-labeled secondary antibody (1:1000). Blot was stripped and labeled with mouse anti-tubulin monoclonal antibody (1:2000, Sigma) following the same procedure as above.
Immunohistochemistry
Formaldehyde-fixed and paraffin embedded tissues were sectioned at 5 μm and the sections were used for immunohistochemical analysis. The primary antibody used for assessing amyloid load was 6E10 (a monoclonal antibody raised against peptides 1–16 of Aβ, Covance, Dedham, MA) and visualized using Vectastain ABC kit (Vector laboratories, Burlingame, CA). Activated microglia were detected with rabbit polyclonal antibody to ionized calcium binding adaptor molecule-1 (IBA-1; Wako, 1:250). In brief, endogenous peroxidase was quenched using 0.3% H2O2 in absolute methanol for 10 min. Antigen retrieval was accomplished by steaming the sections for 30 min. Sections were allowed to cool to room temperature before they were washed with PBS. Blocking was done using 5% goat serum. Activated astrocytes were detected using rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP; Sigma, 1:100). The sections were quenched using 3% H2O2 in double distilled water for 10 min. Antigen retrieval was performed by immersing the sections in citrate buffer (pH6.0) and keeping in a boiling water bath for 5 min. After the sections were cooled to room temperature, they were blocked using commercially available Ultra V blocker (Labvision corp., CA) as per the manufacturer’s instructions. Microglia and astrocytes were visualized using Vectastain rabbit IgG kit (Vector laboratories, Burlingame, CA) and Extra avidin Peroxidase kit (Sigma, St. Louis, MO) respectively. Monocyte chemoattractant protein-1 (MCP-1) was detected using goat polyclonal antibody to MCP-1 (M-18) (Santa Cruz; 1:100). After endogenous peroxidase quenching, antigen retrieval was done by dipping the sections in formic acid for 1 min. MCP-1 was visualized using Vectastain ABC kit (Vector Laboratories, Burlingame, CA).
Microscopic image analysis
Image analysis was performed on two coronal sections per brain containing the caudal hippocampus. The hippocampal area was digitalized using Nikon Eclipse E600 microscope with camera, and the images were converted to grayscale using the Paint Shop Pro 7 program. To analyze amyloid depositions, activated microglial and astrocyte burden, the area in the hippocampus covered by Aβ (i.e. amyloid burden), IBA-1 positive area (i.e. activated microglia) and GFAP positive area (i.e. reactive astrocytes) were measured through the hippocampus using ScionImage (NIH) program. Burden was defined as the percentage of stained surface of total hippocampal area. For MCP-1 positive plaques, the plaque number is reported as the total number of plaques counted in the hippocampal sections.
Statistical analysis
Data were expressed as mean±SEM. Comparison of treatment groups were performed by Student’s t-test, and analysis of variance (ANOVA) with repeated measures. SigmaStat software (SPSS Science, Chicago, IL) was used for all statistical analysis. P<0.05 was considered statistically significant.
Results
Administration of peptides with statin did not show any changes in total plasma cholesterol levels
Male mice were used for these studies because they are shown to have lower Aβ plaque numbers compared to female mice of the same age (Burgess et al., 2006). Thus, to avoid potential confounding gender effects and see the effect of the drug clearly, we selected only young male mice. There was no difference in water intake and body weight among the three groups. Total plasma cholesterol levels were similar in the three groups (control: 97.9±6.6 mg/dl, ScD-4F+statin: 108.9±6.1 mg/dl, and D-4F+statin: 106.8±7.6 mg/dl). Thus, the small dose of pravastatin used did not exert any effect on plasma cholesterol levels.
D-4F+statin but not ScD-4F+statin improves cognitive function
Three months after the initiation of treatment the three groups of mice were subjected to behavioral testing. Two parameters were measured: 1) spatial memory was analyzed by determining escape latency in the MWM test. 2) Improvement in memory retention was determined during the probe trial by counting time spent in the target quadrant by mice and the number of times mice crossed over the platform where it was previously placed. In all of these parameters tested mice that received D-4F+statin showed significant improvement (Figs. 1A and B). ScD-4F+statin group mice behaved similarly to the control mice that received water only. These results demonstrate that despite the presence of statin, the control peptide had no effect in that this group of mice behaved similarly to the water-control group. These results indicate that D-4F was responsible for improvement in cognitive function. Since it is known that pravastatin can act peripherally as an anti-inflammatory agent, we administered mice with pravastatin alone (10 μg/ml) in their drinking water. When we performed MWM test, mice treated with pravastatin alone were not better than control mice either in finding the platform or during probe trial (data not shown). Hence, we excluded this group from further studies.
Fig. 1.
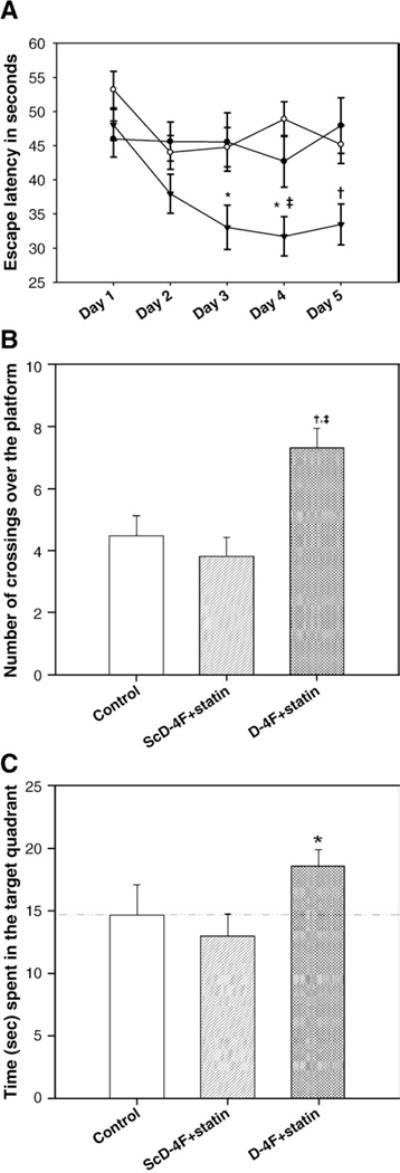
Water maze and probe trial performance. (A) Escape latency in seconds. Each point represents the average of 4 daily trials. D-4F+statin-treated mice (filled triangle, n=21) found the hidden platform significantly more quickly compared to control (filled circle, n=17) and the control peptide+statin-treated groups (open circle, n=15) from day 3 of trial period (*p<0.05 vs control and ScD-4F+statin group; ‡ p<0.001vs ScD-4F+statin; †p<0.01 vs control and ScD-4F+statin). (B) The graph shows the number of crossings over the previously hidden platform area in the probe trial. D-4F+statin-treated group crossed the platform area significantly more often compared to control (†, p <0.01) and control peptide + statin-treated group (‡, p<0.001). (C) The bar graph shows the average of four probe trials and represents the percent of probe trial time spent in target quadrant for each group. The dashed line indicates the chance level (25%) of performance. D-4F+statin-treated mice spent significantly more time in the target quadrant compared to ScD-4F+statin-treated group (p<0.05). Error bars indicate SEM.
Oral D-4F+statin significantly reduced amyloid β load in the hippocampal region of the brain
Sections of the brain were subjected to immunohistochemistry for Aβ deposition. As shown in Fig. 2, while Aβ deposition was found in the hippocampus to an equal extent in the control and ScD-4F+ statin group, the D-4F+statin group demonstrated significantly less Aβ deposition (p<0.001 vs control group and p<0.01 vs ScD-4F+statin group), in agreement with the observation that this group of mice also exhibited improved cognitive function. Two to three hippocampi from each group were pooled and homogenized. TBS (soluble) and GuHCl (insoluble) extracts were subjected to ELISA for Aβ40 and Aβ42 levels. Results showed that while there was no difference in soluble Aβ levels between the three groups, administration of D-4F+statin reduced insoluble total Aβ levels significantly compared to the control group (Table 1).
Fig. 2.
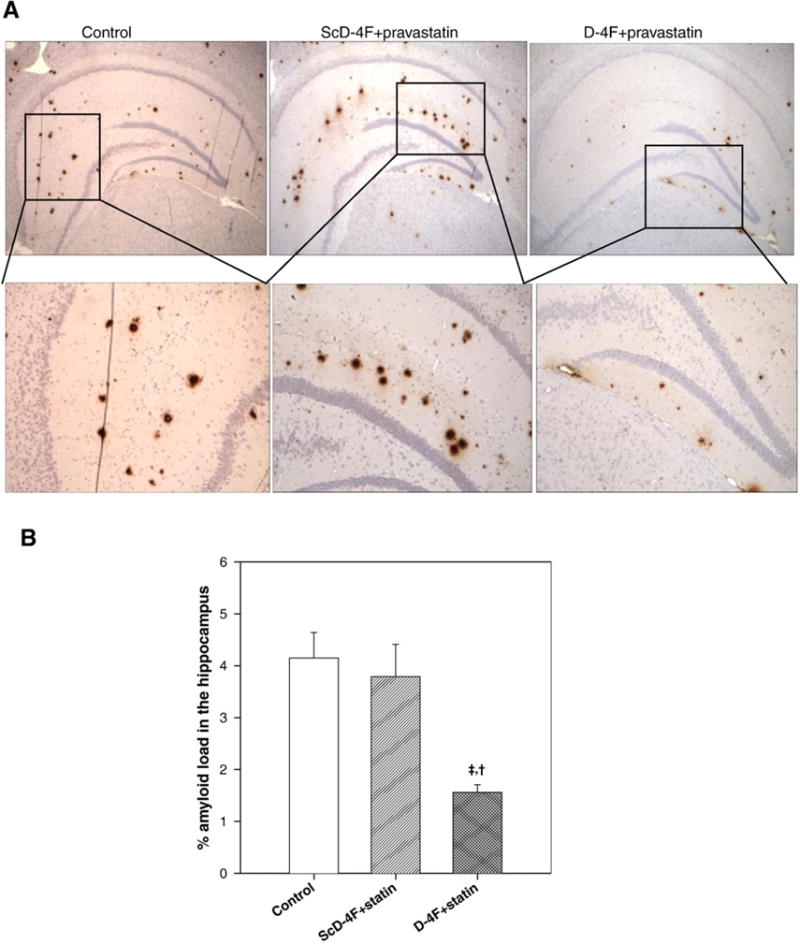
Aβ load in the hippocampus. (A) Representative photomicrographs from different treatment groups. Amyloid load was analyzed using 6E10 antibody. (B) Quantification of the percent amyloid load in the hippocampus, showing significant reduction in D-4F+statin-treated group (n=21) compared to either control (n=17), (‡, p<0.001) or ScD-4F+statin-treated group (n=15) (†, p<0.01).
Table 1.
Total Aβ in the hippocampi measured by ELISA.
| Control | ScD-4F+statin | D-4F+statin | |
|---|---|---|---|
| Aβ (TBS soluble) | 387.6±91.5 | 384.5±40.3 | 331.0±50.6 |
| Aβ (GuHCI soluble) | 5540.0±619.4 | 5745.6±757.9 | 4125.6±286.1* |
Two to three hippocampi from each group were pooled for each data point. Aβ40 and Aβ42 levels in the TBS (soluble) and GuHCl (insoluble fractions) were analyzed using ELISA. Amount of Aβ40 and Aβ42 in TBS and guanidine soluble fraction was summed, normalized to protein content and expressed as pg/mg protein. D-4F+statin-treated group had significantly decreased insoluble Aβ levels compared to control group (*p<0.05). Control, n=5, ScD-4F+statin, n=5, and D-4F+statin, n=6. Error bars indicate SEM.
D-4F+statin inhibits activated microglia in hippocampus
Microglia, the mononuclear phagocytes of the brain, accumulate in and near senile plaques in AD patients and in animal models of AD (McGeer et al., 1987, Dickson, 1999). Aβ can activate microglia to produce cytokines and neurotoxins, hence promoting neurodegeneration (Coraci et al., 2002). Microglial activation was analyzed by measuring the percent of IBA-1 stained area in the hippocampal sections from all three groups (Fig. 3A). Numbers of activated microglia were reduced to approximately 50% in the D-4F+statin group compared to the other two groups (Fig. 3B; p<0.05 vs control and ScD-4F groups). ScD-4F+statin administration had no effect on the levels of activated microglia compared to the control group.
Fig. 3.
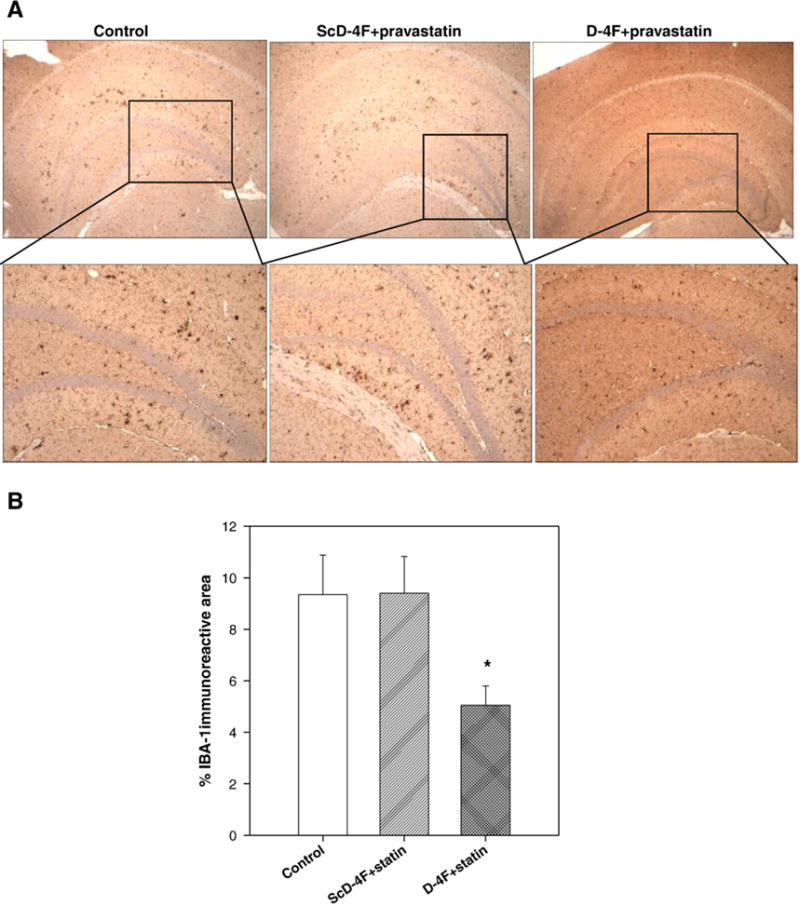
Activated microglias are inhibited by treatment with D-4F+statin. (A) Representative photomicrographs of activated microglia using IBA-1 antibody. (B) Quantification of percent activated microglial cells in the hippocampus demonstrated significant reduction in D-4F+statin-treated group (*p<0.05 vs control and ScD-4F+statin-treated group,). n=16 (control), 13 (ScD-4F+statin), and 19 (D-4F+statin). Error bars indicate SEM.
Treatment of mice with oral D-4F+statin reduces activated astrocyte levels in the hippocampal region
To determine if the anti-inflammatory peptide D-4F would have any effect on the level of activation of astrocytes, the sections were stained with GFAP, an astrocyte marker that is elevated in inflammatory conditions and is increased in amyloid-forming APP transgenic mice (Irizarry et al., 1997). While ScD-4F+statin showed levels of astrocytes similar to that found in the hippocampal region of control mice (Fig. 4), oral D-4F+statin significantly reduced the number of activated astrocytes (p<0.05 vs control).
Fig. 4.
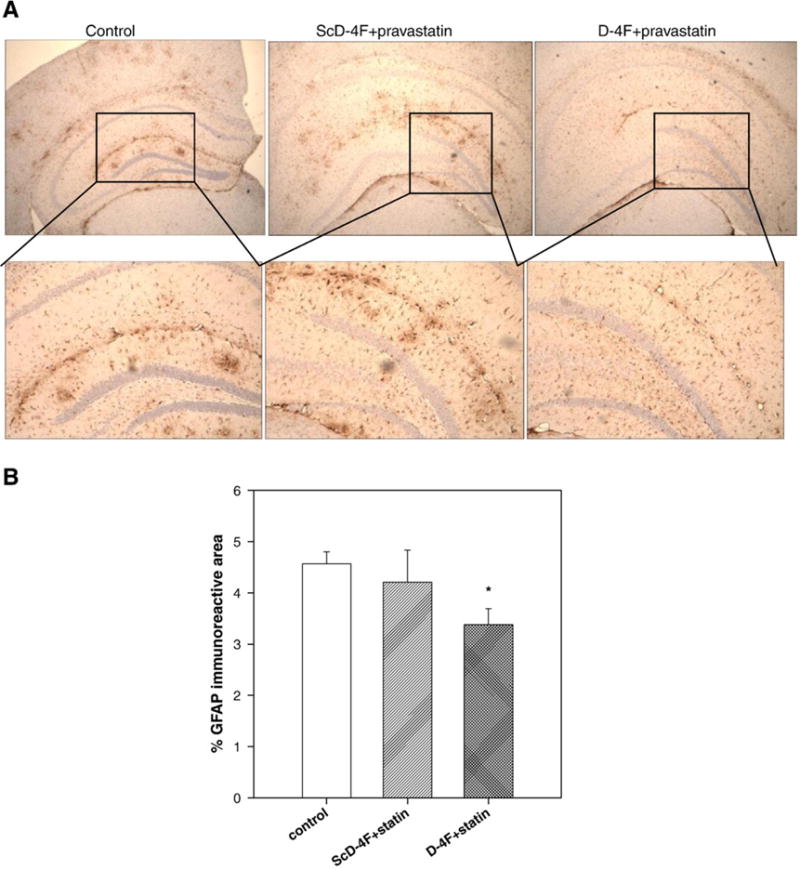
D-4F+statin treatment ameliorated glial activation. (A) Micrographs demonstrated that, compared with hippocampus of control treated group, D-4F+statin treatment reduced glial fibrillary acidic protein (GFAP) (activated astrocytes). (B) Quantitation of percentage GFAP staining in hippocampus demonstrated significant attenuation of activated gliosis by treatment with D-4F+statin (*vs control, p<0.05) n=15 (control), 12 (ScD-4F+statin), and 17 (D-4F+statin). Error bars indicate SEM.
D-4F+statin but not ScD-4F+statin inhibit pro-inflammatory markers in the hippocampal region of AD mouse model
Since activated microglias are involved in the release of pro-inflammatory cytokines, we measured levels of TNF-α and IL-1β in the hippocampi. Furthermore, age-related elevations of IL-1β in rodents have been implicated in age-related memory loss and defective long term potentiation (LTP) (Murray and Lynch, 1998). Two to three hippocampi from each group were pooled and homogenized, and TBS soluble extracts were assayed for TNF-α and IL-1β using ELISA. There was no significant difference in the levels of TNF-α and IL-1β between the control and control peptide administered group (n=5 pooled samples in each group). However, as shown in Fig. 5A, administration of D-4F with statin (n=7 pooled samples) caused a significant decrease in TNF-α (p<0.01 vs control) and IL-1β (p<0.01 vs control and ScD-4F+statin groups) levels.
Fig. 5.
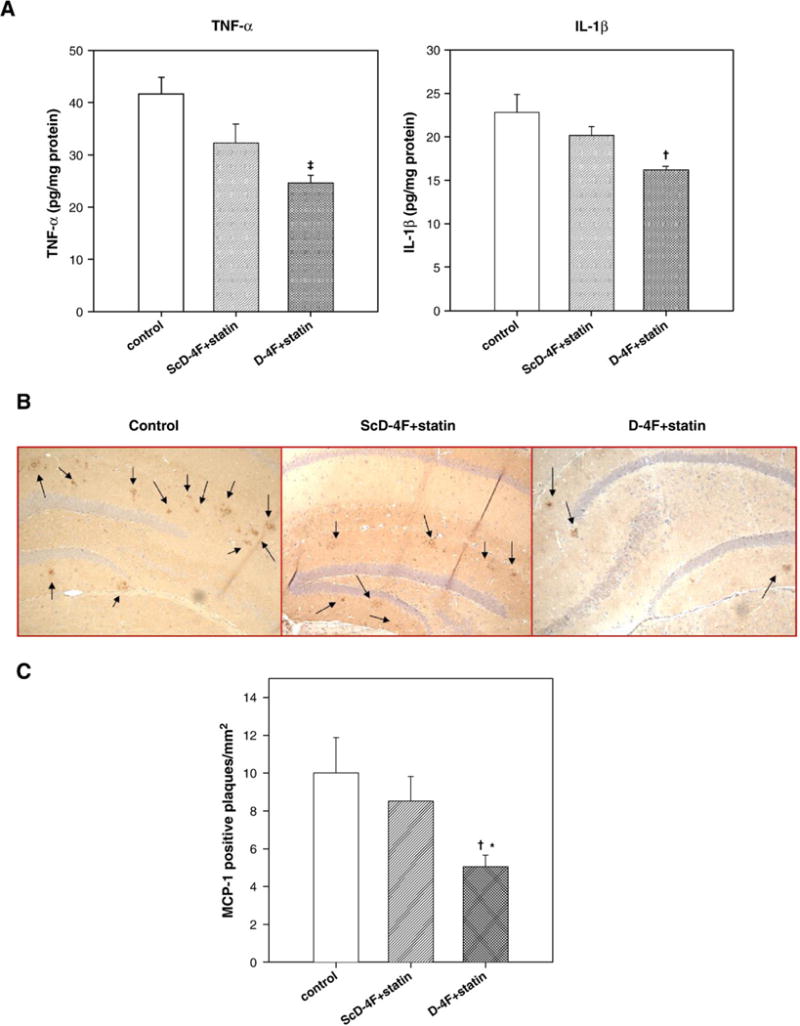
Oral D-4F+statin treatment inhibits pro-inflammatory markers in the brain. (A) Hippocampi from 2–3 mice were pooled in each group for each data point. The TBS soluble fractions were analyzed for pro-inflammatory markers, IL-1β and TNF-α using ELISA. Amount of IL-1β and TNF-α were normalized to protein content. (n=5 in control and ScD-4F+statin, and n=7 in D-4F+statin). There was significant reduction in TNF-α levels in D-4F+statin-treated group compared to control (‡, p<0.001), and in IL-1β levels compared to both control and ScD-4F+statin-treated groups (p<0.01). (B) Micrographs showing immunohistochemistry for monocyte chemoattractant protein-1 (MCP-1) in the hippocampus. (C) Bar graph showing number of MCP-1 plaques. Control and ScD-4F+statin-treated mice had significantly more plaque-associated MCP-1 levels compared to D-4F+statin group. (†p<0.01 vs control and *p<0.05 vs ScD-4F+statin) n=10 (control), 8 (ScD-4F+statin), and 12 (D-4F+statin). Error bars indicate SEM.
D4F+statin significantly reduce plaque-associated MCP-1 expression
MCP-1 is shown to be present in senile plaques and reactive microglia (Ishizuka et al., 1997) in AD brains. Astrocytes have been reported to be the major source of MCP-1 (Ransohoff et al., 1993). Thus, we determined MCP-1 levels in treated and untreated mice. As shown in Figs. 5B and C, oral D-4F+statin significantly reduced the levels of MCP-1 compared to the other two groups (p<0.01 vs control, and p<0.05 vs ScD-4F+statin).
Oral administration of D-4F+statin had no effect on APP processing
Aβ is produced from APP (Vassar et al., 1999) and readily aggregates to form insoluble, high-molecular-mass amyloid structure (Shankar et al., 2008). APP is cleaved by BACE1 enzyme at the N-terminal region, producing membrane-bound C-terminal fragments (CTFs). To determine whether the decrease in Aβ load observed in D-4F+statin-treated mice was due to its influence on Aβ generation from APP, we determined levels of FL-APP (full-length APP) and CTFs by Western blotting. Quantitation of the bands showed that there was no difference in APP processing between the three groups (Fig. 6). Thus suggesting that decrease in Aβ load in D-4F+statin treatment was not due to its effect on APP processing.
Fig. 6.
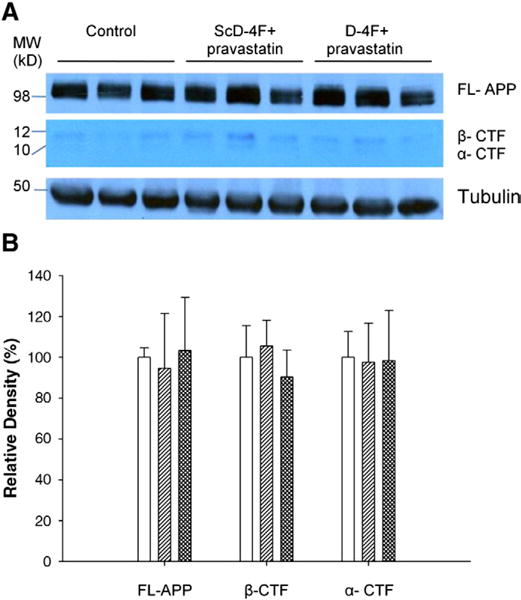
Effect of D-4F+statin on APP processing in APP/PS1dE9 mice: (A) Levels of FL-APP (full-length APP) and APP C-terminal fragments in hippocampus were visualized by Western blotting. (B) Quantitative analysis of FL-APP and APP-CTF’s. Tubulinwas used as a loading control. Results are normalized to loading control and values are expressed as change in the values from control mice (set to 100%). There was no difference in APP processing between control (open bar), ScD-4F+statin (single hatched bar) and D-4F+statin-treated mice (double hatched bar). Error bars indicate SEM.
Discussion
These data demonstrate for the first time that oral treatment of APPswe-PS1ΔE9 mice with an apo A-I mimetic peptide improves cognitive function and significantly reduces Aβ deposition in the hippocampal region. Furthermore, this is accompanied by decreased numbers of activated microglia and astrocytes, which are responsible for producing cytokines, oxidized lipids, and other pro-inflammatory molecules. In agreement with earlier observations (Navab et al., 2005b), our results also show that neither oral administration of D-4F nor pravastatin at the dose used showed any changes in plasma cholesterol levels.
While it is possible that D-4F alone would inhibit Aβ deposition in this mouse model (experiments are underway), to enhance the effectiveness of the peptide (as demonstrated by Navab et al., 2005b) we used it in combination with pravastatin. To analyze the effect of small amount of pravastatin used with the peptide D-4F, we fed a scrambled control peptide to mice in presence of the same amount of pravastatin. The ScD-4F+statin treatment had no effect on cognition, Aβ deposition, and inflammatory markers, therefore it appears that the improved cognition and decreased Aβ load and glial activation in mice administered with D-4F+statin group were due to the action of peptide D-4F.
It has been shown in humans with coronary heart disease (CHD) that oral administration of D-4F produces low plasma levels of D-4F (Bloedon et al., 2008). However, this is highly effective in converting pro-inflammatory HDL into an anti-inflammatory form (Bloedon et al., 2008). IL-1β and TNF-α are two major pro-inflammatory cytokines produced by microglia during CNS inflammation (Kim and Joh, 2006). These cytokines have been shown to be involved in the development of central nervous system (CNS) inflammation through the disruption of the blood brain barrier (BBB) and the induction of adhesion molecules and chemokines from astrocytes and endothelial cells, which facilitate the infiltration of leukocytes into the CNS (Sedgwick et al., 2000). Hickman et al. (2008) have used the same mouse model as we have used in this study and shown that, as these mice age, there is increased expression of IL-1β and TNF-α, showing that microglia in aged AD mice retain their pro-inflammatory response in the presence of continued Aβ deposition. Furthermore TNF-α and, to a lesser extent IL-1β, down regulate the expression of microglia scavenger receptors SRA and CD36, and reduce the uptake of Aβ in murine microglia. Oral D-4F reduced the number of activated microglia (Fig. 3) and this may be responsible for the reduction of inflammatory cytokines and decreased Aβ load.
Treatment with D-4F+statin lowered insoluble Aβ levels by about 25% compared to control animals. Although the level of soluble Aβ was not significantly different between the three groups, it should be noted that D-4F+statin-treated mice had lower levels compared to the other two groups. It has been shown that in the same mouse model as we have used that sucrose feeding led to worsening of cognitive deficits, increased amyloid load, increased insoluble Aβ levels without any change in the soluble Aβ levels (Cao et al., 2007). However, we observed a significant reduction in Aβ plaque burden. Although these results support the hypothesis that these reductions were caused by inhibition of the inflammatory process by the anti-inflammatory peptide, the other possibility could be that the peptide may have some direct effect on APP metabolism. However, our results showed that oral administration of D-4F+statin had no effect on the expression level of the APP transgene. Also, the levels of α- and β-CTF of APP, determined by secretase activities, were not altered. These results suggest that the production of Aβ from APP was not affected significantly by treatment with D-4F+statin.
D-4F has been shown to be highly effective in decreasing brain arteriole inflammation and improving cognitive functions in LDL receptor-null mice on Western diet (Buga et al., 2006). Results from previous studies also indicate that the peptide D-4F exerts potent anti-inflammatory and anti-oxidative effects in a number of inflammatory situations even when present in small quantities in vivo (Navab et al., 2005b). Decreased levels of superoxide anion that result from the administration of 4F or D-4F have been shown to result in improved vasodilatations and reduced oxidative stress, myocardial inflammation, and angiogenic potential in a mouse model of scleroderma (Weihrauch et al., 2007). D-4F has been shown to bind to oxidized lipids with high affinity (Van Lenten et al., 2008). In addition, D-4F has been shown to inhibit LPS-mediated inflammation and superoxide production (Gupta et al., 2005). It has been suggested and supported by experiments that D-4F, even when present in nanomolar quantities, avidly binds oxidized lipids and inhibits cytokine formation. While we have shown that levels of cytokines are significantly decreased upon oral D-4F treatment, presently we do not know if this peptide enters brain and interacts locally with markers of oxidative stress caused by Aβ deposition, or if it modulates lipoproteins to inhibit LDL oxidation and/or improve HDL function.
MCP-1 is a member of the CC chemokine family and promotes infiltration of macrophages into tumors. It plays an important role in the regulation of repair processes and cellular interactions in the central nervous system and is expressed by many cell types including astrocytes, microglia and axotomized neurons (Ubogu et al., 2006). Levels of IL-6, IL-8 and MCP-1 are shown to be higher in AD cases compared to controls, and MCP-1 was identified as a reliable predictor of disease, suggesting a major role for this cytokine in detrimental neuroinflammatory processes (Sokolova et al., 2008). In our studies, we found that administration of D-4F+statin significantly inhibited levels of MCP-1 in the hippocampus providing further evidence for the anti-inflammatory effect of this peptide in this AD mouse model.
In summary, we have shown that D-4F in the presence of a small amount of pravastatin improves cognitive function and reduces Aβ deposition. This occurs with a concomitant decrease in cytokines and activated microglia and astrocytes. These results, along with a large number of previous investigations, support the idea that anti-inflammatory apo A-I mimetics may also be a potential drug for Alzheimer’s disease.
Acknowledgments
This work was supported in part by Institute for the Study of Aging (S.P.H), NHLBI PO1 HL 34343 (D.W.G) and NIA AG 031846 (L.L). The behavioral studies were performed at Behavioral Assessment core, Dept. of Neurobiology, UAB which is supported by P30 NS47466.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharamaiah GM, Mishra VK, Garber DW, Datta G, Handattu SP, Palgunachari MN, Chaddha M, Navab M, Reddy ST, Segrest JP, Fogelman AM. Structural requirements for antioxidative and anti-inflammatory properties of apolipoprotein A-I mimetic peptides. J Lipid Res. 2007;48:1915–1923. doi: 10.1194/jlr.R700010-JLR200. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Rye KA. Relationship between the concentration and anti-atherogenic activity of high-density lipoproteins. Curr Opin Lipidol. 2006;17:399–403. doi: 10.1097/01.mol.0000236365.40969.af. [DOI] [PubMed] [Google Scholar]
- Bloedon LT, Dunbar R, Duffy D, Pinell-Salles P, Norris R, DeGroot BJ, Movva R, Navab M, Fogelman AM, Rader DJ. Safety, pharmacokinetics, and pharmacodynamics of oral apo A-I mimetic peptide D-4F in high-risk cardiovascular patients. J Lipid Res. 2008;49:1344–1352. doi: 10.1194/jlr.P800003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boche D, Nicoll JA. The role of immune system in clearance of Aβ from the brain. Brain Pathol. 2008;18:267–278. doi: 10.1111/j.1750-3639.2008.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buga GM, Frank JS, Mottino GA, Hendizadeh A, Hakhamian A, Tillisch JH, Reddy ST, Navab M, Anantharamaiah GM, Ignarro LJ, Fogelman AM. D-4F decreases brain arteriole inflammation and improves cognitive performance in LDL receptor-null mice on a Western diet. J Lipid Res. 2006;47:2148–2160. doi: 10.1194/jlr.M600214-JLR200. [DOI] [PubMed] [Google Scholar]
- Burgess BL, McIssac SA, Naus KE, Chan JY, Tansley GH, Yang J, Miao F, Ross CJ, van Eck M, Hayden MR, van Nostrand W, George-Hyslop P, Westaway D, Wellington CL. Elevated plasma triglyceride levels precede amyloid deposition in Alzheimer’s disease mouse models with abundant A beta in plasma. Neurobiol Dis. 2006;24:114–127. doi: 10.1016/j.nbd.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Cao D, Lu H, Lewis TL, Li L. Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer’s disease. J Bio Chem. 2007;282:36275–36282. doi: 10.1074/jbc.M703561200. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ, Feinstein DL. Effects of lovastatin and pravastatin on amyloid processing and inflammatory response in TgCRND8 brain. Neurochem Res. 2004;29:1897–1911. doi: 10.1023/b:nere.0000042217.90204.8d. [DOI] [PubMed] [Google Scholar]
- Christen Y. Oxidative stress and Alzheimer’s disease. Am J Clin Nutr. 2000;71:621s–629s. doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta G, Epand RF, Epand RM, Chaddha M, Kirksey MA, Garber DW, Lund-Katz S, Phillips MC, Hama S, Navab M, Fogelman AM, Palgunachari MN, Segrest JP, Anantharamaiah GM. Aromatic residue position on the nonpolar face of class A amphipathic helical peptides determines biological activity. J Biol Chem. 2004;279:26509–26517. doi: 10.1074/jbc.M314276200. [DOI] [PubMed] [Google Scholar]
- Dickson DW. Microglia in Alzheimer’s disease and transgenic models. How close the fit? Am J Pathol. 1999;154:1627–1631. doi: 10.1016/S0002-9440(10)65416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta H, Dai L, Datta G, Garber DW, Grenett H, Li Y, Mishra VK, Palgunachari MN, Handattu S, Gianturco SH, Bradley WA, Anantharamaiah GM, White CR. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein A-I mimetic peptide. Circ Res. 2005;97:236–243. doi: 10.1161/01.RES.0000176530.66400.48. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennet DA, Evans DA. Alzheimer’s disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O’Bannion K, Klockgether T, Van Leuven F, Landreth G. Acute treatment with the PPARg agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta 1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPswe transgenic mice develop age-related A beta deposits and neutrophile abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin Neurosci. 1997;51:135–138. doi: 10.1111/j.1440-1819.1997.tb02375.x. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific g secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in pathogenesis of Parkinson’s disease. Exp Mol Med. 2006;38:333–347. doi: 10.1038/emm.2006.40. [DOI] [PubMed] [Google Scholar]
- Kunjathoor VV, Tseng AA, Medeiros LA, Khan T, Moore KJ. Beta-amyloid promotes accumulation of lipid peroxides by inhibiting CD36 mediated clearance of oxidized lipoproteins. J Neuroinflammation. 2004;1:23–35. doi: 10.1186/1742-2094-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Cao D, Garber DW, Kim H, Fukuchi K. Association of aortic atherosclerosis with cerebral beta amyloidosis and learning defects in a mouse model of Alzheimer’s disease. Am J Pathol. 2003;163:2155–2164. doi: 10.1016/s0002-9440(10)63572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Inflammatory processes in Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. 2003. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patints with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin 1beta is a common trigger for age- and stress induced impairments in long-term potentiations. J Neurosci. 1998;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Hama S, Garber DW, Chaddha M, Hough G, Lallone R, Fogelman AM. Oral administration of an apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–292. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Fogelman AM. The role of high-density lipoprotein in inflammation. Trends Cardiovasc Med. 2005a;15:158–161. doi: 10.1016/j.tcm.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Hama S, Hough G, Reddy ST, Frank JS, Garber DW, Handattu S, Fogelman AM. D-4F and statins synergize to render HDL anti-inflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005b;25:1426–1432. doi: 10.1161/01.ATV.0000167412.98221.1a. [DOI] [PubMed] [Google Scholar]
- Pratico D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer’s disease. Am J Med. 2000;109:577–585. doi: 10.1016/s0002-9343(00)00547-7. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. 1993;7:592–600. doi: 10.1096/fasebj.7.6.8472896. [DOI] [PubMed] [Google Scholar]
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurthi K, Duff K, Papolla MA. Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Riminton DS, Cyster JG, Korner H. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol Today. 2000;21:110–113. doi: 10.1016/s0167-5699(99)01573-x. [DOI] [PubMed] [Google Scholar]
- Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in exchangeable apolipoproteins: a review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, et al. Amyloid-β protein dimmers isolated directly from Alzheimer’s brain impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova A, Hill MD, Rahimi F, Warden LA, Halliday GM, Shepherd CE. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00188.x. Electronic publication ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubogu E, Cossoy M, Ransohoff RM. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol Sci. 2006;27:48–55. doi: 10.1016/j.tips.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Van Lenten BJ, Wagner AC, Jung CL, Ruchala P, Waring AJ, Lehrer RI, Watson AD, Hama S, Navab M, Anantharamaiah GM, Fogelman AM. Anti-inflammatory apo A-I mimetic peptides bind oxidized lipids with much higher affinity than human apo A-I. J Lipid Res. 2008;49:2302–2311. doi: 10.1194/jlr.M800075-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Kahn S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the trans-membrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Weihrauch D, Xu H, Shi Y, Wang J, Brien J, Jones DW, Kaul S, Komorowski RA, Csuka ME, Oldham KT, Pritchard KA. Effects of D-4F on vasodilation, oxidative stress, angiostatin, myocardial inflammation, and angiogenic potential in tight-skin mice. Am J Physiol Heart Circ Physiol. 2007;293:H1432–H1441. doi: 10.1152/ajpheart.00038.2007. 2007. [DOI] [PubMed] [Google Scholar]
- Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokines production by THP-1 cells and murine microglia. J Neurochem. 2000;74:1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x. [DOI] [PubMed] [Google Scholar]
- Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59:880–886. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]