

Abstract
Purpose
A long natural history and a predominant osseous pattern of metastatic spread are impediments to the adoption of precision medicine in patients with prostate cancer. To establish the feasibility of clinical genomic profiling in this disease, we performed targeted deep sequencing of tumor and normal DNA from patients with locoregional, metastatic noncastrate, and metastatic castration-resistant prostate cancer.
Patients and Methods
Patients consented to genomic analysis of their tumor and germline DNA. A hybridization capture-based clinical assay was used to identify single-nucleotide variations, small insertions and deletions, copy number alterations, and structural rearrangements in more than 300 cancer-related genes in tumors and matched normal blood.
Results
We successfully sequenced 504 tumors from 451 patients with prostate cancer. Potentially actionable alterations were identified in DNA damage repair, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase pathways. Twenty-seven percent of patients harbored a germline or a somatic alteration in a DNA damage repair gene that may predict for response to poly (ADP-ribose) polymerase inhibition. Profiling of matched tumors from individual patients revealed that somatic TP53 and BRCA2 alterations arose early in tumors from patients who eventually developed metastatic disease. In contrast, comparative analysis across disease states revealed that APC alterations were enriched in metastatic tumors, whereas ATM alterations were specifically enriched in castration-resistant prostate cancer.
Conclusion
Through genomic profiling of prostate tumors that represent the disease clinical spectrum, we identified a high frequency of potentially actionable alterations and possible drivers of disease initiation, metastasis, and castration resistance. Our findings support the routine use of tumor and germline DNA profiling for patients with advanced prostate cancer for the purpose of guiding enrollment in targeted clinical trials and counseling families at increased risk of malignancy.
INTRODUCTION
Prostate cancer is a disease characterized by distinct clinical states with highly variable outcomes.1 Surgery and radiation therapy are potentially curative for patients with localized disease, whereas androgen-deprivation therapy (ADT) is effective but palliative for patients who develop metastases with a testosterone level in the noncastrate range (metastatic noncastrate prostate cancer).2 Metastatic noncastrate prostate cancer inevitably evolves into castration-resistant prostate cancer (mCRPC) —the lethal form of the disease.
Recent molecular profiling efforts, including the Stand Up to Cancer-PCF (SU2C-PCF) mCRPC project and The Cancer Genome Atlas (TCGA) primary localized prostate cancer study, have identified distinct molecular subsets of prostate cancer and potentially targetable alterations that occur somatically as well as in the germline.3-7 Of note, there are limited genomic data on metastatic noncastrate disease. Although molecular profiling is not yet considered a standard-of-care for patients with this disease, new evidence points to enhanced treatment response in specific molecular contexts, paving the way for therapy selection on the basis of tumor molecular characteristics. In particular, genomic alterations in genes that are involved in DNA damage repair (DDR) by homologous recombination may predict for increased sensitivity to poly-ADP ribose polymerase (PARP) inhibitors and platinum-based therapy.8,9 We sought to determine whether routine prospective genomic profiling in the clinical practice setting was feasible and informative for patients with prostate cancer, as well as to define the frequency of potential driver genomic alterations across the disease clinical spectrum.
PATIENTS AND METHODS
Patients and Samples
Patients with prostate cancer consented to an institutional review board–approved protocol for tumor genomic profiling using the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) sequencing assay. Specific consent was required for analysis of germline variants in an identifiable manner. After consent, either archival or new tumor samples were obtained and blood was drawn for germline DNA. Archival tumor samples were formalin fixed and paraffin embedded. New biopsies were obtained under radiographic guidance and were formalin fixed. Bone biopsies were nondecalcified. All tumors were reviewed by pathologists who specialize in genitourinary oncology for confirmation of malignant histology of prostatic origin.
Sequencing and Analysis
We used the MSK-IMPACT assay as previously described10,11 (Fig 1A). The assay was performed in a Clinical Laboratory Improvement Amendments–certified laboratory and designed to robustly identify single-nucleotide variations, small insertions and deletions (indels), somatic copy number alterations, and structural rearrangements in more than 300 cancer-related genes in formalin-fixed and paraffin-embedded tumors and matched normal blood.
Fig 1.
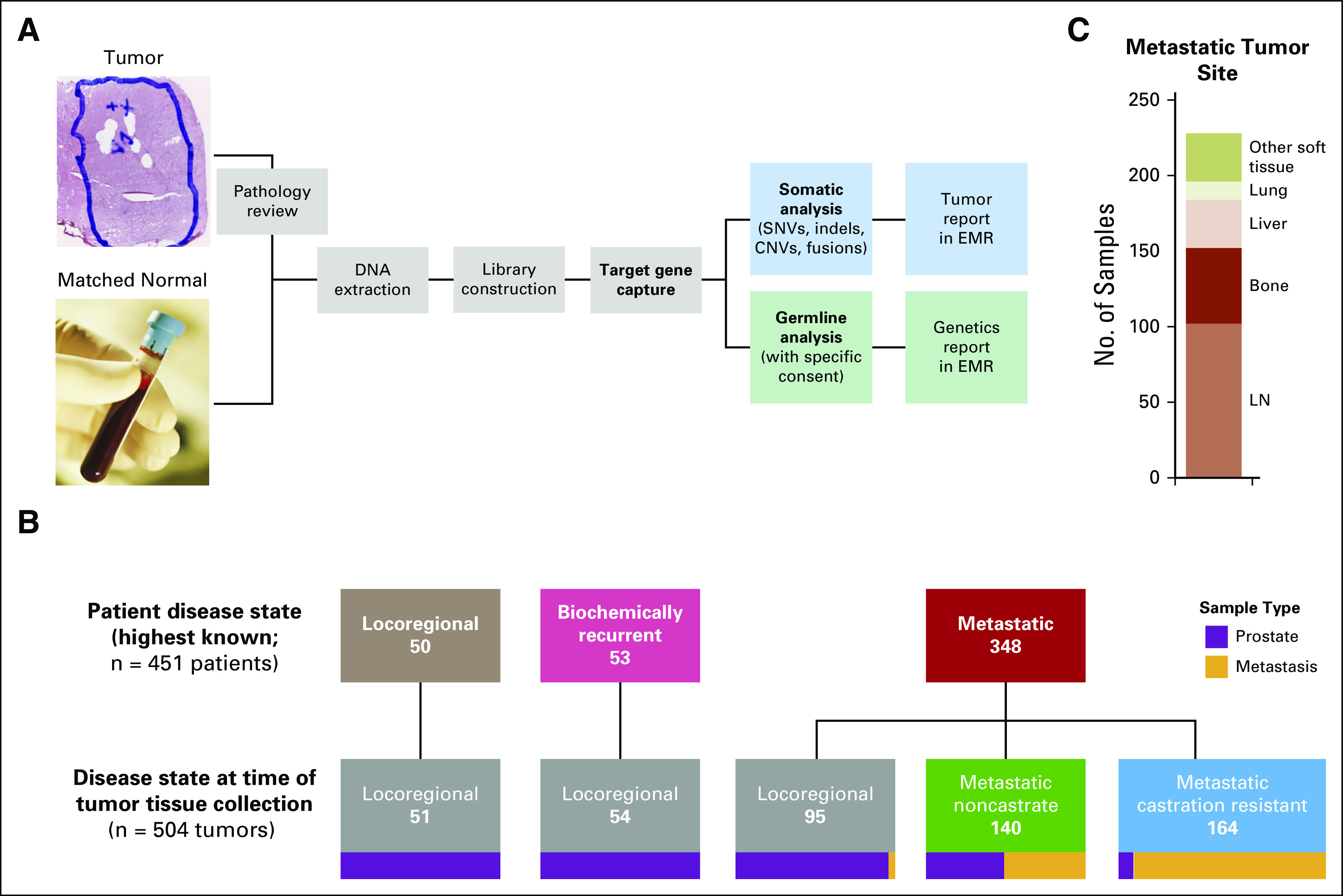
Clinical sequencing of tumors and germline for patients with prostate cancer. (A) MSK-IMPACT assay workflow. (B) Four hundred fifty-one patients underwent tumor profiling in the clinic. Their last known disease state when seen in the clinic is represented at the top. Their disease state at the time of tissue collection for the 504 tumors that were profiled is represented at bottom. Tumors that were profiled represented all three prostate cancer clinical disease states: locoregional, metastatic noncastrate, and metastatic castration resistant. Locoregional disease indicates disease without distant clinical or pathologic spread, including lymph node (LN) involvement in the pelvis only (TxN0/1). Sample type (prostate v metastasis) is represented at bottom. (C) Site of disease for metastatic tumors successfully profiled by MSK-IMPACT. CNV, copy number variation; EMR, electronic medical record, MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; SNV, single-nucleotide variation.
Somatic variant analysis was performed as described,10,11 with germline variants identified in matched blood samples filtered out in the somatic analysis process. Somatic findings were reported in the electronic medical record and anonymized and uploaded to cBioPortal for visualization and analysis.12-14 Clonality of mutations was estimated as cancer cell fraction15 and implemented in the FACETS algorithm.16
Beginning in May 2015, patients were given the option to consent to analysis of germline variants that were identified via sequencing of normal blood samples. Germline analysis of 76 known cancer-predisposing genes was performed as previously described.16a
Additional methods are provided in the Data Supplement.
RESULTS
Targeted DNA Sequencing of Tumor-Normal Pairs From Patients With Prostate Cancer
Using the MSK-IMPACT assay, we successfully profiled 504 tumors from 451 patients with prostate cancer who presented to the clinic (Fig 1A and Data Supplement). In total, 348 patients (77%) had metastatic prostate cancer, 53 (12%) had biochemical recurrence after definitive therapy, and 50 (11%) had locoregional disease (Fig 1B). The 504 tumors were either archival or newly acquired primary prostate or metastatic tumors of prostate origin, with 44 patients having more than one tumor profiled. Metastatic tumors that were successfully profiled were obtained from lymph node (45%), bone (22%), liver (14%), lung (5%), and other soft tissue sites (14%; Fig 1C). Disease state at the time of collection of the tumor is shown in Fig 1B. Unlike the TCGA and SU2C-PCF studies, tumors that were profiled represented all three prostate cancer clinical states: locoregional, metastatic noncastrate, and metastatic castration resistant.
We began with 746 biopsy/surgical samples to successfully sequence the 504 tumors reported above, with an overall success rate of 68% (Appendix Fig A1). The highest success rates were for prostate tumor samples that were obtained from diagnostic prostate needle biopsy, radical prostatectomy, or transurethral resection of prostate performed for palliation. For metastatic samples, success rates of ≥ 69% were observed for lymph node, liver, and other soft tissue samples, whereas bone and lung samples were more challenging (42% to 52% success rate).
Somatic and Germline Alterations Identified in the MSK-IMPACT Data Set
Somatic alterations in biologically relevant genes in prostate cancer were identified in all disease states and are shown in Fig 2, grouped in pathways that are potentially clinically actionable.
Fig 2.
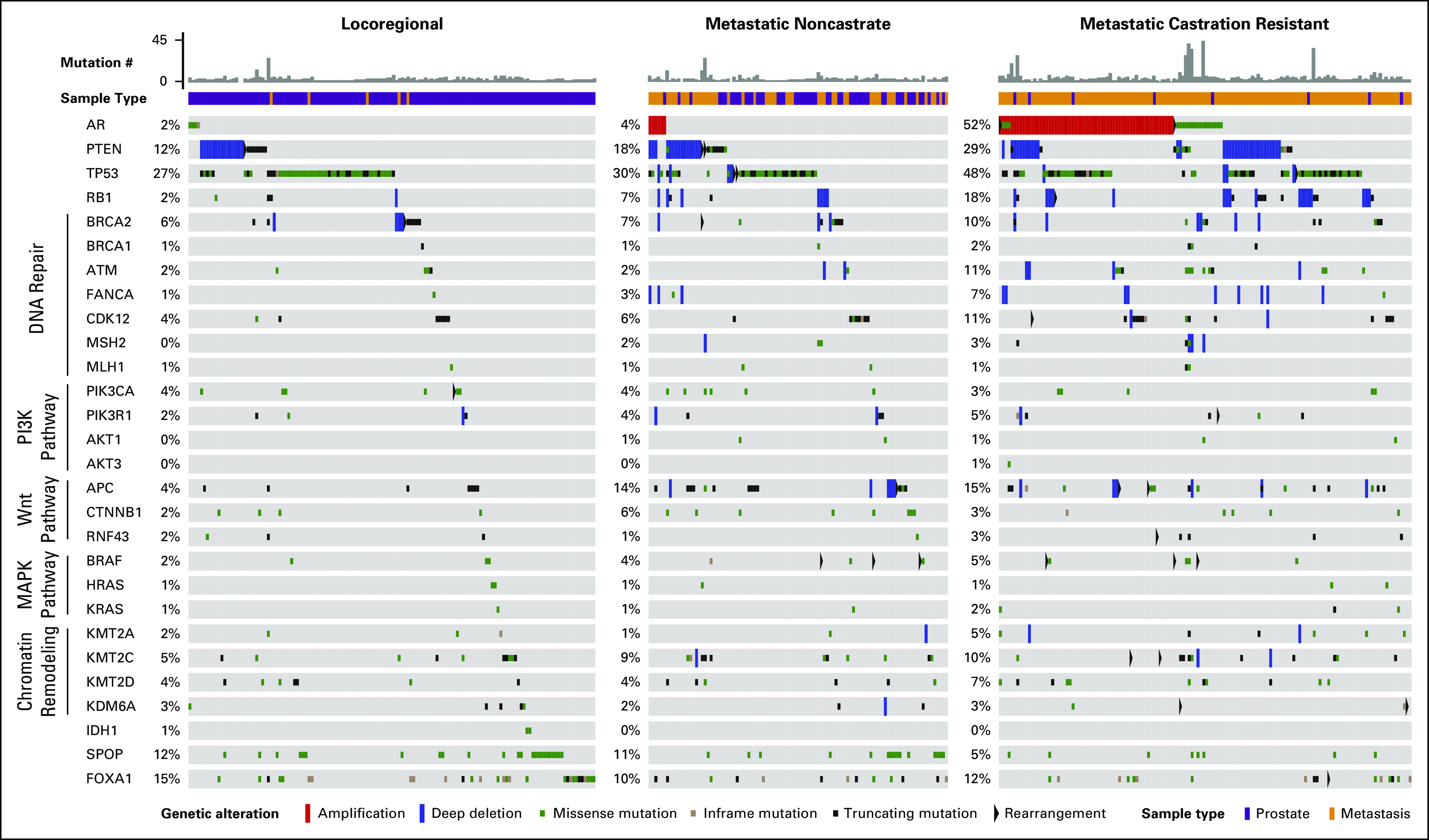
Selected genomic alterations across disease states in the MSK-IMPACT data set. Each column represents an individual patient with prostate cancer whose tumor was acquired in the disease state indicated at the top. Total number of nonsynonymous somatic mutations in the tumor is represented in histogram form. Sample type (prostate v metastasis) is also represented. Alterations in commonly affected genes in prostate cancer (eg, AR, PTEN, TP53, FOXA1) are shown in addition to genes in potentially actionable or biologically relevant pathways. The type of alteration (eg, copy number variation, rearrangement, mutation) is indicated in the bottom row. MAPK, mitogen-activated protein kinase; MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; PI3K, phosphatidylinositol-3-kinase.
Overall, the frequency of alterations in genes of interest in prostate cancer was similar for mCRPC tumors that were profiled in the MSK-IMPACT and SU2C-PCF mCRPC data sets4 (Appendix Fig A2) but demonstrated now in a clinical practice setting. However, notable differences were observed when comparing primary localized tumors in the MSK-IMPACT data set with those that were profiled in the prospective TCGA study, including a higher frequency of alterations in TP53 and FOXA1 in MSK-IMPACT tumors (Appendix Fig A3). This is most likely a result of the more aggressive nature of the primary localized tumors in the MSK-IMPACT cohort, which were predominantly obtained from patients who subsequently developed biochemically recurrent and metastatic disease (Fig 1B) relative to the prospectively acquired primary TCGA tumors (Appendix Fig A3).
In total, 24% of patients carried somatic alterations in the PI3K/AKT pathway, including in PTEN, PIK3CA, PIK3CB, PIK3R1, AKT1, and AKT3 (Appendix Fig A4). The majority of point mutations in PIK3CA, AKT1, and AKT3 were known activating hotspot mutations in those genes.11 In addition, 5% of patients harbored somatic alterations in mitogen-activated protein kinase pathway genes (Appendix Fig A5), including hotspot mutations in BRAF, HRAS, KRAS, and MAP2K1.
Fifteen percent of patients carried somatic alterations in the Wnt-β catenin pathway, including in APC, CTNNB1, and RNF43 (Appendix Fig A6). Consistent with the results of the SU2C-PCF study, 22% of patients harbored a somatic alteration in a gene that is involved in DDR by homologous recombination, including BRCA2, BRCA1, ATM, FANCA, RAD50, PALB2, and CDK1217-21 (Fig 3A and Appendix Fig A7).
Fig 3.
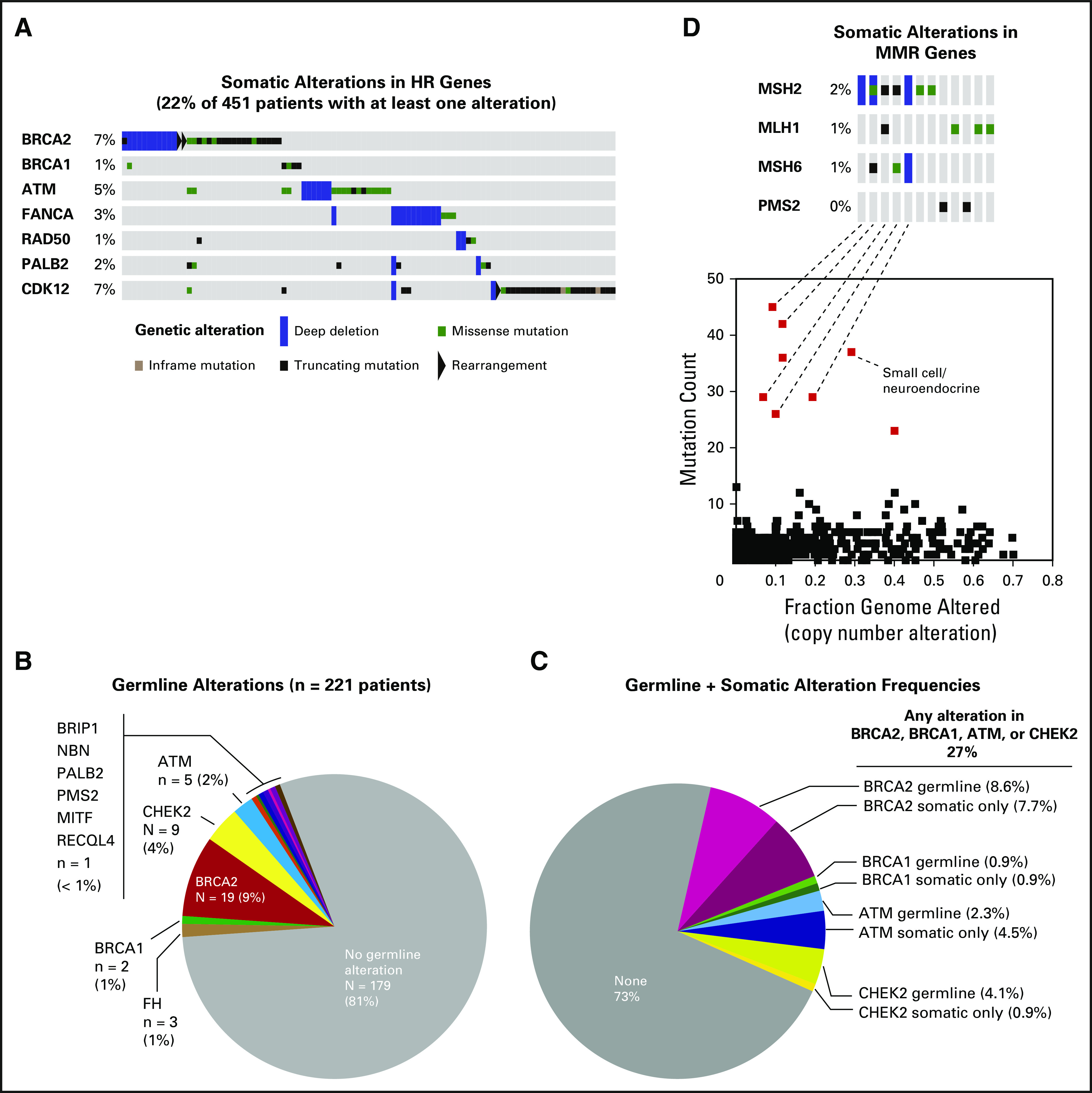
Somatic and germline alterations in DNA damage repair genes. (A) Oncoprint of somatic alterations in genes that are involved in DNA repair by homologous recombination (HR) for 451 patients. Twenty-two percent of patients harbor a tumor alteration in one of the listed genes. (B) Germline alterations for patients who consented to germline analysis (n = 221). In total, 19% of patients were found to have a germline pathogenic alteration. (C) Frequency of combined somatic and germline alterations in DNA repair genes BRCA2, BRCA1, ATM, and CHEK2 for patients who consented to germline analysis. Overall, 27% of patients had either a germline or a somatic-only alteration in one of these genes. (D) Oncoprint of somatic alterations in genes that are involved in DNA mismatch repair (MMR; top). Three percent of patients harbor a tumor alteration in one of these genes. The majority of tumors that display more than 20 somatic mutations (bottom) harbor a somatic alteration in an MMR gene.
A recent multi-institutional study identified a high frequency of DDR gene alterations in the germline of patients with advanced prostate cancer.22 Of patients in our data set, 221 underwent formal germline analysis, the first 124 of whom were included in the previously reported study.22 Of these 221 patients, 42 (19% of total) had a known or likely pathogenic germline mutation in BRCA2 (9% of total), CHEK2 (4%), ATM (2%), BRCA1 (1%), FH (1%), and PMS2, NBN, PALB2, and BRIP1 (< 1% each; Fig 3B and Appendix Table A1). Whereas germline DDR gene alterations may predict for response to PARP inhibition or platinum agents, somatic-only alterations in these genes without a germline event may still predict for drug response.9 Of the 221 patients who underwent germline analysis, 27% demonstrated alterations in BRCA2, BRCA1, ATM, or CHEK2, either in the germline or somatically (Fig 3C). Of note, germline analysis alone accounted for approximately one half of these patients only, which suggests that both germline and somatic analysis should be performed to identify patients with DDR gene deficiency.
In total, 3% of patients had tumors with somatic alterations in mismatch repair (MMR) genes MSH2, MLH1, PMS2, or MSH6. These tumors accounted for the majority of samples with the highest mutation counts on MSK-IMPACT profiling (Fig 3D), which were confirmed to be enriched for previously described MMR and microsatellite instability signatures23,24 (Appendix Fig A8). Identification of MMR-deficient prostate cancers may have immediate clinical applicability, given recent data that suggest sensitivity of such tumors to immune checkpoint blockade in colorectal cancer and other malignancies.25-27
Overall, 36% of patients were found to have a potentially actionable alteration by using the OncoKB annotation platform27a (Appendix Fig A9). This platform does not include non-BRCA/ATM germline alterations, missense alterations of unknown significance, and genes whose clinical significance is less clear, including CDK12 and FANCA. As genomic alterations undergo further characterization and new trials and drug targets emerge, the frequency of alterations that are defined as actionable may increase.
Comparative Analysis of Somatic Alterations Across Disease States
A unique aspect of this data set is that it includes genetic profiles of tumors that represent all three clinical states: locoregional, metastatic noncastrate, and metastatic castration resistant. The number of nonsynonymous mutations per tumor increased significantly from tumors in the locoregional disease state to those in the mCRPC disease state (1.74 v 4.02 mean mutations/megabase; P < .001), whereas tumors in the metastatic non–castration-resistant state had a mutation burden similar to locoregional tumors (2.08 mutations/megabase). Consistent with previous studies,3,4 we identified recurrent areas of copy number loss that involved chromosomes 6q, 8p, 13q, and 16q, and areas of copy number gain that involved chromosomes 1q, 3q, 7, 8q, and X (Fig 4). mCRPC tumors displayed the highest burden of copy number alterations, whereas those that represented locoregional disease displayed the lowest (Fig 4 and Appendix Fig A10).
Fig 4.
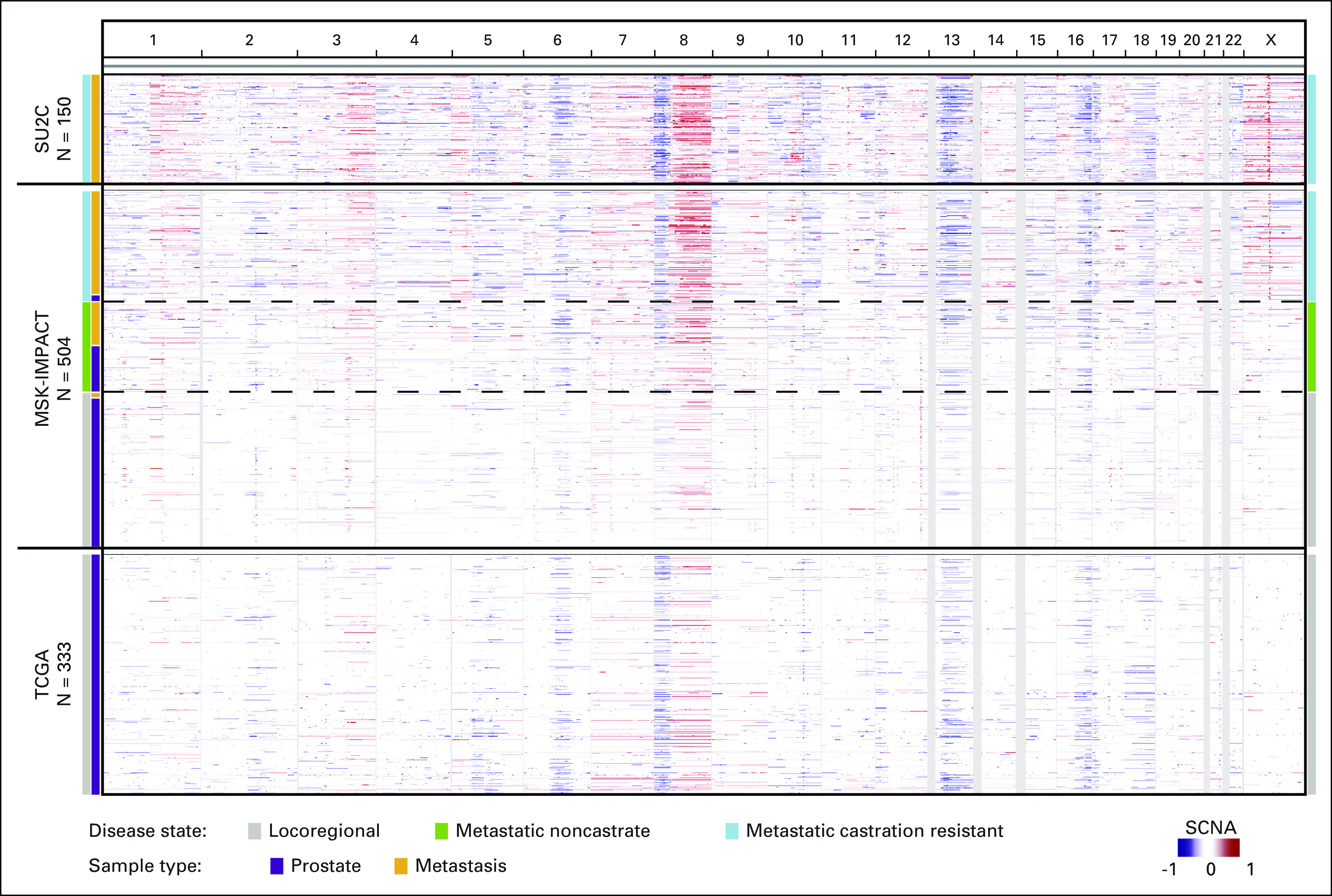
Cross-cohort comparative analysis of the pattern and degree of copy number alterations across the genome. Represented here are regions of amplification (red) or deletion (blue) with chromosomes listed horizontally (top) in the MSK-IMPACT, SU2C-PCF (metastatic castration-resistant prostate cancer [CRPC]), and TCGA (primary localized prostate cancer) data sets. MSK-IMPACT tumors are sorted by disease state at time of tissue acquisition, from locoregional (bottom, gray) to metastatic CRPC (top, blue). Whereas sequencing platforms differ between studies, the degree and pattern of copy number alteration were similar for tumors that were acquired in the same disease state. MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; SCNA, somatic copy number alterations; SU2C-PCF, Stand Up to Cancer-Prostate Cancer Foundation; TCGA, The Cancer Genome Atlas.
Aiming to identify possible genomic drivers of disease progression, we performed a selective enrichment analysis to identify genes that were more frequently altered in mCRPC compared with locoregional disease (Fig 5A). AR amplification/mutation was the most enriched alteration in mCRPC, as shown in previous studies.3,4 Other genes that were more commonly altered in mCRPC included TP53, RB1, PTEN, APC, ATM, FANCA, and CDK12.
Fig 5.
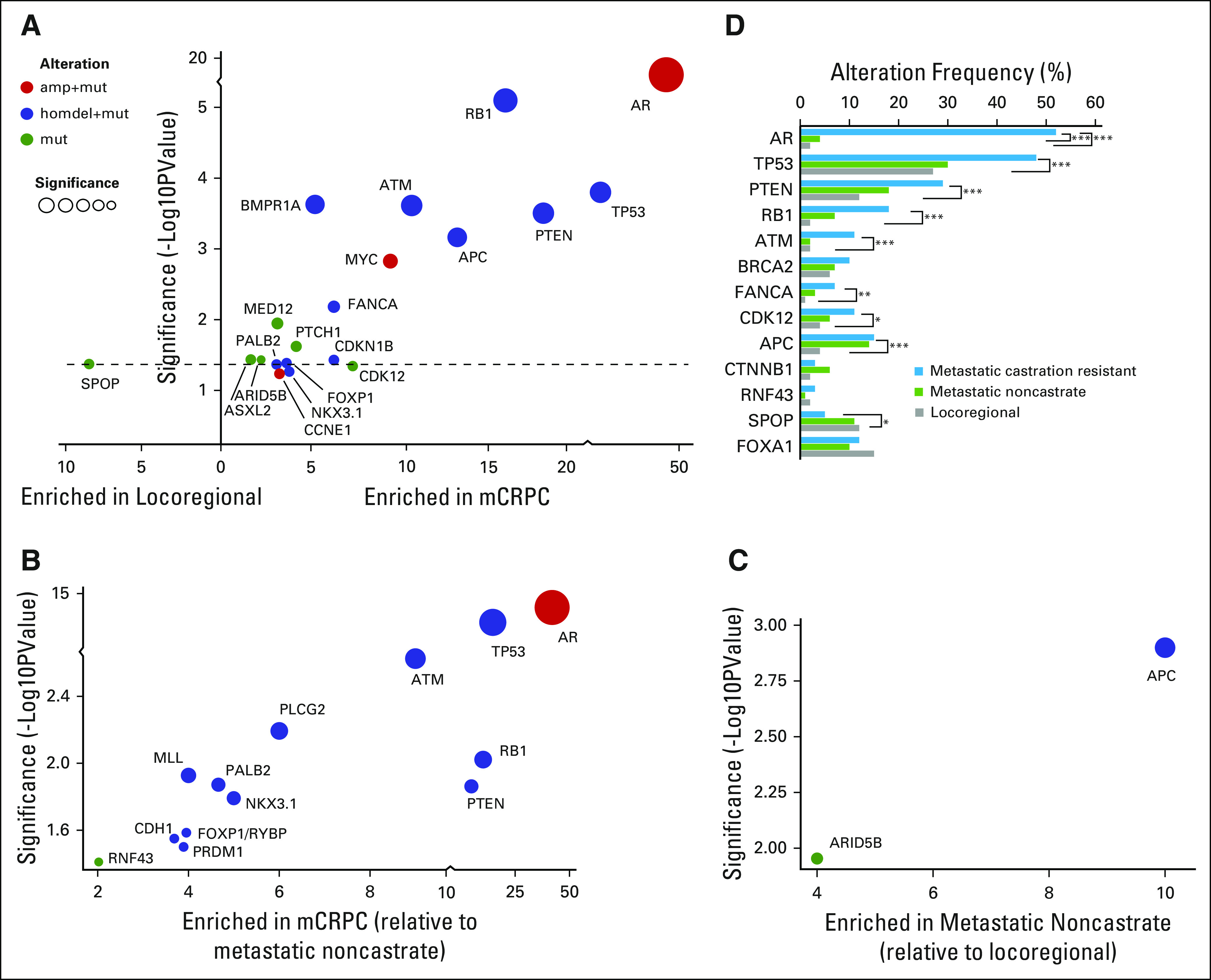
Enrichment of genomic alterations between disease states. (A) Enrichment of genomic alterations in tumors from patients with metastatic castration-resistant prostate cancer (mCRPC) versus locoregional disease. The level of enrichment is represented as difference in frequency between the two indicated classes (x-axis) and its significance (P value, y-axis). The type of alteration is represented by color. (B) Enrichment of genomic alterations in tumors from patients with metastatic CRPC versus metastatic noncastrate disease. (C) Enrichment of genomic alterations in tumors from patients with metastatic noncastrate disease versus locoregional disease. (D) Frequencies of alterations in select genes across disease states in the MSK-IMPACT data set. P values are represented (Fisher’s exact test). amp, amplification; homdel, homozygous deletion; mut, mutation.
We performed a similar analysis that compared alterations in mCRPC with metastatic noncastrate prostate cancer (Fig 5B). AR was again the most enriched gene in this analysis. The high enrichment of alterations in AR in mCRPC relative to both locoregional and metastatic non-castrate disease serves as a positive control, consistent with the known role of AR as a driver of castration resistance.28-31 Beyond frequent amplification of the gene, AR antiandrogen resistance mutations were identified in tumors from patients with mCRPC (Appendix Fig A11), including an F877L enzalutamide/ARN509 resistance mutation32,33 that was found in the tumor of a patient who experienced progression after 4 years of treatment on ARN509. Of note, a 4% alteration frequency in AR was identified in tumors from patients with metastatic noncastrate disease (Figs 2 and 5D). These were tumors that were exposed to ADT and were likely transitioning to a castration-resistant phenotype that had not yet manifested clinically. Four locoregional tumors were found to have mutations in AR, including an H875Y mutation that is known to confer resistance to flutamide29,34 in a prostatectomy sample from a patient who was treatment-naïve but who had received dutasteride for benign prostate enlargement.
In addition to AR, we again identified enrichment of alterations in TP53, RB1, PTEN, and ATM in mCRPC compared with metastatic noncastrate disease. Enrichment of these genes in mCRPC relative to both earlier disease states implicates them in the development of castration resistance, a finding that is of particular interest for ATM, a gene that is involved in DDR. FANCA and CDK12, two other DNA repair genes, did not show statistically significant enrichment in mCRPC compared with metastatic noncastrate disease as they did versus locoregional disease, although there is a trend that suggests a role in castration resistance as well (Fig 5D). When analysis was limited to metastatic tumors or lymph nodes only, similar trends for enrichment were observed, although statistical significance was not always reached because of smaller sample size (Appendix Fig A12).
Only two genes were enriched in metastatic noncastrate versus locoregional disease: APC and, to a lesser extent, ARID5A (Fig 5C). Enrichment of APC alterations in both metastatic states relative to locoregional disease (Fig 5D) implicates this gene in metastasis. Conversely, alterations in SPOP7,35 were enriched in locoregional disease—and possibly metastatic noncastrate disease, although this does not meet statistical significance—compared with mCRPC (Figs 5A and 5D), which suggests increased sensitivity of SPOP mutant tumors to ADT. These findings will require functional validation in the laboratory.
Matched Samples Identify Clonal Alterations in Prostate Cancer
In total, 44 patients had more than one tumor site profiled by MSK-IMPACT, including 16 with a matched primary localized tumor and a subsequent metastatic tumor. Tumors from the same patient that were acquired at a later time point typically had a higher mutation count (Fig 6A). Given the high frequency of TP53 alterations that were observed in primary localized tumors in this data set and prior reports of aggressive behavior of prostate tumors that harbor TP53 alterations,36-37a we sought to determine whether TP53 alterations were present in tumors early in their evolution or whether they were acquired later at disease progression. As shown in Fig 6B, TP53 alterations arose early in affected patients and were identified in all tumors from the same patient. TP53 mutations were clonal, including in cases in which both a primary localized tumor and a later metastasis were available (cancer cell fraction ≥ 0.9 in the later metastasis). Likewise, we found that somatic alterations in BRCA2 were present in matched tumors, which again suggested that somatic BRCA2 loss of function alterations occur early in tumorigenesis for affected patients. Conversely, alterations in AR did not occur early in matched samples (Fig 6B, patients P-0003597 and P-0004910), which is consistent with treatment-related changes that promote castration resistance. Of note, other potentially actionable alterations may arise later in disease evolution, as was the case for patient P-0002149 (Fig 6B), who acquired an activating PIK3CA E545K mutation (Appendix Fig A4B) in a recurrent tumor nearly 3 years after his radical prostatectomy.
Fig 6.
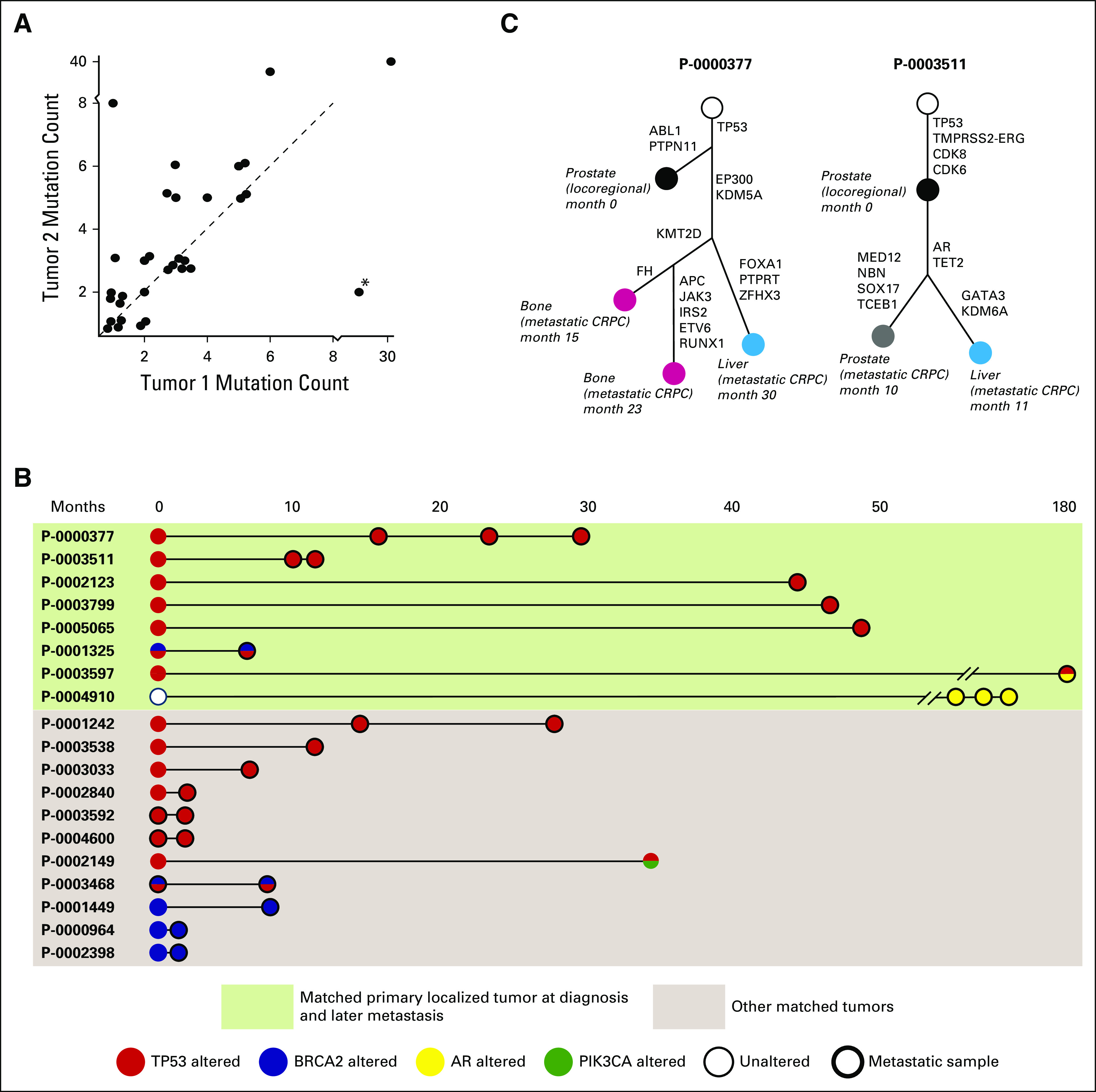
Somatic alterations identified in matched tumors from the same patients. (A) Somatic mutation count in pairs of matched tumors. The latter tumor (Tumor 2) tends to have a higher mutation burden. A notable exception (*) involves a bladder metastasis and a bone metastasis acquired 5 months later, where the patient had received salvage radiation to the pelvis, possibly explaining the higher mutation count in the earlier bladder tumor. (B) Somatic alterations in TP53 (red), BRCA2 (blue), AR (gold), and PIK3CA (green) in matched tumors in the data set, including localized primaries and later metastases (green box) and other matched tumors from the same patients. (C) Evolutionary analysis representing the acquisition of genomic alterations in sequential tumors obtained from two patients. Filled circles represent tumors sequenced by MSK-IMPACT, labeled with their sites, disease state, and relative dates of acquisition. CRPC, castration-resistant prostate cancer.
To confirm the above findings, we performed phylogenetic analysis on cases in which several matched tumors were available from the same patient (Fig 6C). Both phylogenetic trees shown reveal the early truncal nature of TP53 alterations. For patient P-0000377, alterations in EP300 and KDM5A—genes that are involved in epigenetic modulation—occurred truncally for metastatic tumors. An alteration in KMT2D was identified subclonally in metachronous metastases from bone obtained from separate sites, but not in a liver metastasis. For patient P-0003511, AR amplification was truncal in castration-resistant tumors, which is consistent with its well-characterized role in castration resistance.28 As the number of patients with prostate cancer who are profiled longitudinally throughout their clinical care increases, such findings may provide insight into clonal driver events that promote disease progression and site-specific metastasis.
DISCUSSION
Unlike previous prostate cancer genomic studies, we profiled tumors that represent the clinical spectrum of the disease, from locoregional to metastatic noncastrate and metastatic castration- resistant prostate cancer, which enabled comparisons of genomic landscape across disease states using a single assay. Whereas locoregional tumors in this data set typically represented more aggressive disease than TCGA, patients with such tumors are those in greatest need of new treatment approaches and profiling of their tumors may have particular clinical relevance.
An increase in copy number alterations and mutation frequency was evident in mCRPC compared with earlier disease states, as was an increased frequency of alterations in AR, TP53, RB1, and PTEN. SPOP mutations, however, were more frequent in the earlier disease states, which suggested better outcomes for patients with SPOP-mutant tumors, possibly through increased sensitivity to ADT. Importantly, the ability to compare alteration frequencies across three prostate cancer disease states can provide insight into genes that promote metastasis versus castration resistance. In this analysis, APC and ATM emerged as candidate genes of interest that may independently contribute to metastasis and castration resistance, respectively, pending functional validation in the laboratory. Other genes that emerged as being enriched in mCRPC, although not to the same extent, are FANCA and CDK12. These genes, like ATM, are involved in DDR, alluding to a possible role for DNA repair defects in the development of castration resistance.
We also found that TP53 alterations are early clonal events in matched tumor samples from individual patients. This suggests that TP53 alterations in localized tumors may predict for increased risk of progression to metastatic disease, which is consistent with recent reports of aggressive behavior of TP53-altered prostate cancers.37-39 As long-term outcomes from prospective primary prostate cancer data sets become available, it will be possible to validate this finding, guiding more aggressive treatment approaches early on for these patients.
Overall, we identified potentially actionable alterations, including hotspot activating alterations in genes that are known drug targets, consistent with findings of the SU2C mCRPC study, but this time in a prospective clinical practice setting. In allowing for separate somatic and germline analyses, our study showed that 27% of patients with advanced prostate cancer have a combination of either somatic or germline alterations in BRCA2, BRCA1, ATM, or CHEK2, and that 3% harbor an alteration in an MMR gene. These findings have immediate therapeutic relevance, given the recently reported sensitivity of these tumors to PARP inhibition9 or immune checkpoint blockade.25 The higher frequency of DNA repair alterations that were identified via integrative germline and somatic analyses strongly argues for performing both germline and somatic genomic analyses in all patients with advanced prostate cancer who will require systemic treatment, irrespective of screening on the basis of family history.
In summary, this study shows that a large genomic data set that represents the clinical spectrum of prostate cancer can provide mechanistic insight into possible genomic drivers of disease initiation, metastasis, and drug resistance. Our ability to profile metastatic tumors allowed us to detect the evolution of potential driver alterations in matched tumors from individual patients, identifying alterations in TP53 and BRCA2 as early events that may confer a more aggressive phenotype. Our study reveals that identifying actionable genomic alterations is feasible in the clinical practice setting for patients with prostate cancer, but several challenges remain. First, the availability of trials that target these alterations remains a limitation. Trials of PARP inhibitors for patients with prostate cancer with homologous recombination gene alterations are due to open shortly, and the findings of this study and others should prompt the development of multi-institutional molecularly guided studies for smaller subsets of patients with other molecular alterations. Second, a critical difficulty is in obtaining sufficient tumor material for sequencing, particularly for patients with disease that is restricted to bone. Circulating tumor DNA sequencing assays—currently under investigation in prostate cancer—may offer a solution to this problem. Overall, given the high frequency of potentially actionable alterations, early but compelling evidence of the clinical benefit of targeted therapy for patients with DNA repair gene-deficient prostate cancers, and the implication of germline findings for family members, our data argue for the routine use of germline and somatic genomic profiling assays as standard practice for all patients with advanced prostate cancer.
ACKNOWLEDGMENT
We thank Emily Waters, Vanessa Robinson, Amal Gulaid, Melanie Hullings, and Joseph Jang.
Appendix
Fig A1.
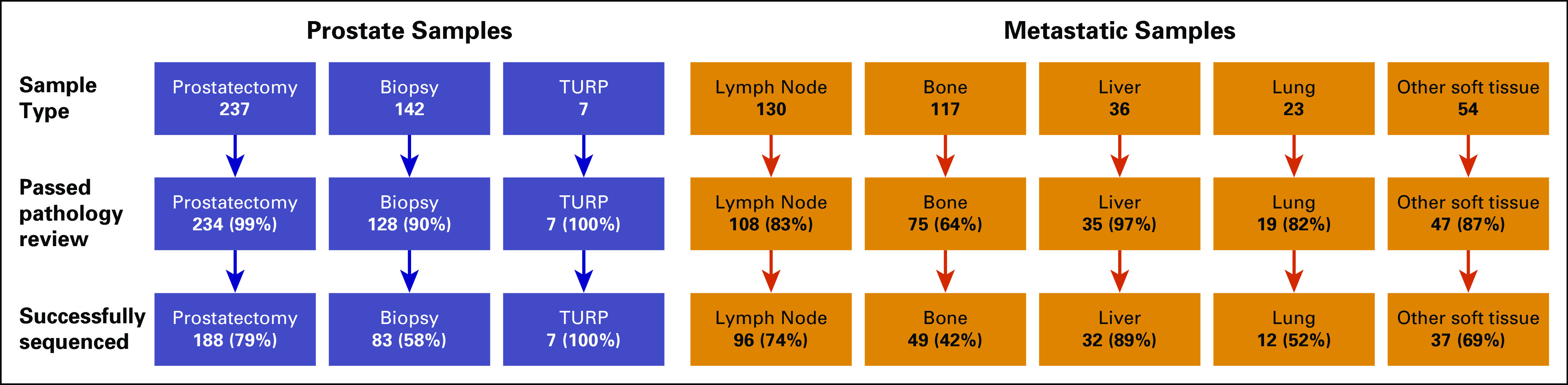
Tumor sequencing success rates. Sequencing success rate for prostate and metastatic samples (includes archived samples acquired for sequencing as well as fresh biopsies). Highest overall success rates were observed for prostate samples, with failures occurring primarily among older archived tumors (median age of failed primary sample: 41.2 months, median age of successful primary sample: 13.9 months). Lowest overall success rates were for bone and lung (42-52%).
Fig A2.

MSK-IMPACT versus SU2C-PCF dataset comparison. Frequencies of alterations in select genes in metastatic CRPC tumors from the MSK-IMPACT dataset (orange) versus the SU2C-PCF dataset (red). Overall, the frequencies of alterations in these genes of interest are similar in the two datasets. (*) Does not include germline alterations.
Fig A3.
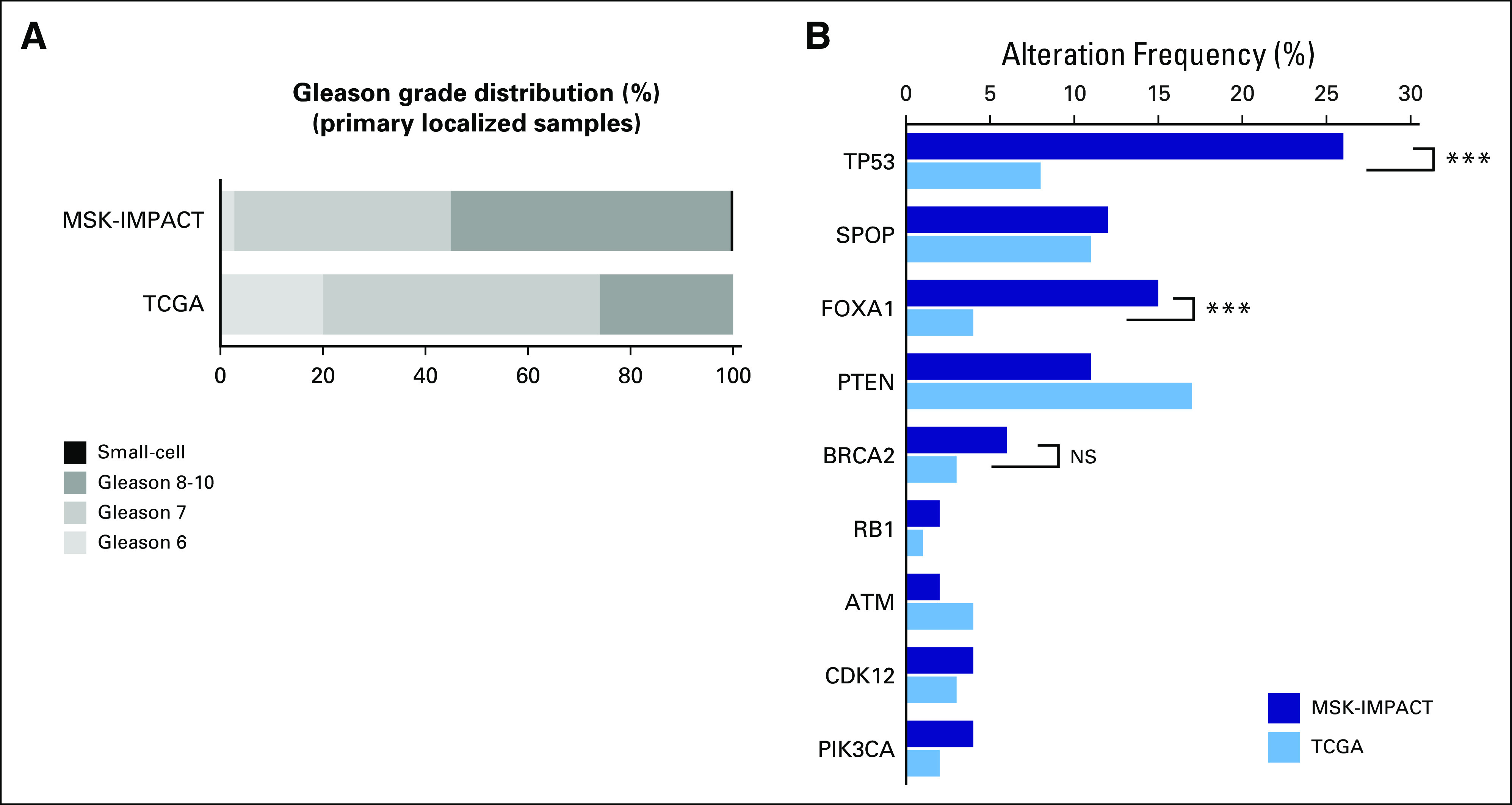
Comparison of primary localized samples from the MSK-IMPACT dataset to TCGA. (A) Gleason score comparison for TCGA and MSK-IMPACT primary localized tumors. More than 50% of primary localized tumors from the MSK-IMPACT dataset are Gleason 8-10 tumors, compared with approximately 25% in the TCGA dataset. (B) Frequencies of alterations in select genes in primary localized tumors from the MSK-IMPACT dataset (dark blue) versus the TCGA dataset (light blue). P-values are represented (Fisher’s exact test).
Fig A4.

Alterations in the PI3K pathway. (A) Oncoprint of somatic alterations in PI3K pathway genes. 24% of patients harbor a somatic alteration in one of the genes listed. (B) Mutations in PIK3CA, AKT1 and AKT3. Known activating hotspot mutations are labeled.
Fig A5.

Alterations in the MAP kinase pathway. (A) Oncoprint of somatic alterations in MAP kinase pathway genes. 5% of patients harbor a somatic alteration in one of the genes listed. (B) Missense mutations in MAP kinase pathway genes. Known activating hotspot mutations are labeled.
Fig A6.

Alterations in the Wnt-β catenin pathway. (A) Oncoprint of somatic alterations in Wnt-β catenin pathway genes. Fifteen percent of patients harbor a somatic alteration in one of the genes listed. (B) Mutations in APC, CTNNB1 and RNF43. APC alterations are primarily deletions and truncating mutations predicted to inactivate the protein. CTNNB1 alterations are primarily hotspot N-terminal missense mutations reported to prevent phosphorylation and degradation of the protein product β catenin.
Fig A7.

Somatic mutations identified in DNA damage repair genes. Shown here are missense, truncating and in-frame mutations in BRCA2, BRCA1, ATM and CDK12. The majority of somatic alterations in BRCA2 are predicted to result in a truncated version of the protein product or in loss of expression of the gene, while somatic alterations in ATM were primarily missense mutations occurring across ATM coding regions. Alterations in CDK12 were primarily truncation mutations, as previously reported in ovarian cancer.
FigA 8.

MMR/MSI mutation signatures in hypermutated tumors. K-means clustering of mutations/Mb (mut/Mb) across all 504 tumors identified two distinct clusters: cluster1 with Mutations/Mb10. The latter represents hypermutated tumors. Tumors in the hypermutated group correspond to the 8 patients with > 20 MSK-IMPACT mutations represented in Figure A5B. Mutational signature decomposition analysis for hypermutated tumors and control non-hypermutated tumors with >8mutation/Mb revealed a high contribution of MMR/MSI signatures to all hypermutated tumors, as measured by proportion of mutations attributed to a specific signature across 30 different mutational signatures.22,33
Fig A9.

Fraction of patients with actionable and oncogenic alterations per OncoKB database. The dataset was queried for actionable and oncogenic alterations using the OncoKB genomic alteration annotation tool (www.OncoKB.org), and frequencies are represented for the 451 patients in the study. Actionable alterations are ranked by level of evidence (Level 2B: Standard of care biomarker predictive of response to an FDA-approved drug in another indication, but not standard of care for this indication; Level 3A: Compelling clinical evidence supports the biomarker as being predictive of response to a drug in this indication, but neither biomarker nor drug are standard of care; Level 3B: Compelling clinical evidence supports the biomarker as being predictive of response to a drug in another indication, but neither biomarker nor drug are standard of care; Level 4: Compelling biological evidence supports the biomarker as being predictive of response to a drug, but neither biomarker nor drug are standard of care). Only the highest level actionable alteration is represented per patient.
Fig A10.

Copy number variations represented as fraction of the genome altered across disease states. Copy number alterations increase from tumors in the locoregional disease state to tumors in the metastatic non-castrate state to tumors in metastatic castrationresistant disease.
Fig A11.
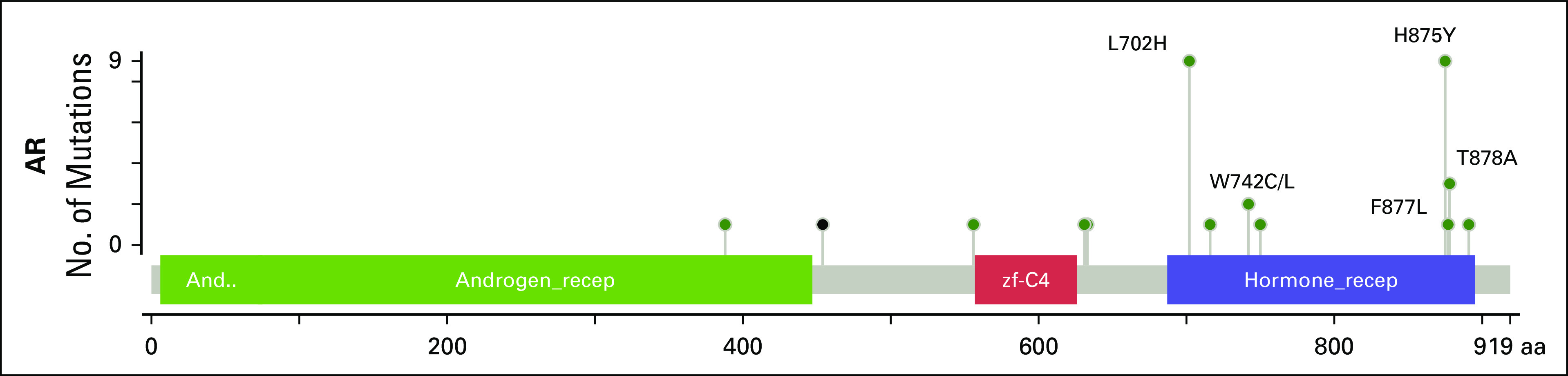
Mutations identified in AR. Missense mutations are represented as green dots, and an in-frame insertion as the single black dot. Well-characterized mutations in the ligand binding domain (blue box) are labeled.
Fig A12.

Frequency of alterations in select genes in metastatic castration-resistant prostate cancer (mCRPC) versus metastatic noncastrate tumors by site of biopsy. The genes listed showed a statistically significant enrichment in mCRPC when comparing all samples in the two sets (Figs. 5B and 5D). We compared alterations in these genes among (A) metastatic samples only (excluding prostate samples) and (B) lymph node samples only. Trends for enrichment in mCRPC still hold in most cases; however, statistical significance was often lost as a result of the lower number of samples included in these subsets. NS, not significant.
Table A1.
Germline Pathogenic Variants Identified in 42 of 221 Patients Who Underwent Germline Analysis

Footnotes
See accompanying editorial doi:https://doi.org/10.1200/PO.17.00064
Supported by Prostate Cancer Foundation Challenge and Young Investigator Awards (to W.A., H.I.S., N.A., and B.S.T.), the National Institutes of Health (National Cancer Institute Prostate SPORE Grant No. P50-CA92629 and National Cancer Institute Cancer Center Support Grant No. P30-CA008748), the Department of Defense Prostate Cancer Research Program (Grants No. PC071610 and PC121111), the David H. Koch Fund for Prostate Cancer Research, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, the Robertson Foundation (to N.S. and B.S.T.), and an American Cancer Society Research Scholar Award (to B.S.T.)
Presented in part at the 2015 ASCO Genitourinary Cancers Symposium, Orlando, FL, February 26-28, 2015, 2015 ASCO Annual Meeting, Chicago, IL, May 29-June 2, 2015, and the 2016 American Association of Cancer Research Annual Meeting, New Orleans, LA, April 16-20, 2016.
AUTHOR CONTRIBUTIONS
Conception and design: Wassim Abida, David M. Hyman, Maria E. Arcila, Mark E. Robson, Kenneth Offit, Philip W. Kantoff, David B. Solit, Charles L. Sawyers, Howard I. Scher
Administrative support: Ryan Brennan, Kristen Curtis, David M. Hyman, Kenneth Offit, David B. Solit, Howard I. Scher
Financial support: David B. Solit, Howard I. Scher
Provision of study materials or patients: Wassim Abida, Joshua Armenia, Daniel Danila, Michael Morris, Susan Slovin, Marc Ladanyi, Liying Zhang, Victor E. Reuter, David B. Solit, Howard I. Scher
Collection and assembly of data: Wassim Abida, Joshua Armenia, Ryan Brennan, Michael Walsh, David Barron, Daniel Danila, Michael Morris, Susan Slovin, Brigit McLaughlin, Kristen Curtis, David M. Hyman, Jeremy C. Durack, Stephen B. Solomon, Maria E. Arcila, Ahmet Zehir, Aijazuddin Syed, Mark E. Robson, Vijai Joseph, Ritika Kundra, Zachary J. Heins, Marc Ladanyi, Victor E. Reuter, David B. Solit, Michael F. Berger, Howard I. Scher
Data analysis and interpretation: Wassim Abida, Joshua Armenia, Anuradha Gopalan, Michael Walsh, Daniel Danila, Dana Rathkopf, Michael Morris, Susan Slovin, Jeremy C. Durack, Maria E. Arcila, Aijazuddin Syed, Jianjiong Gao, Debyani Chakravarty, Hebert Alberto Vargas, Mark E. Robson, Vijai Joseph, Mark T.A. Donoghue, Adam A. Abeshouse, Alexander V. Penson, Christopher Harris, Barry S. Taylor, Diana Mandelker, Liying Zhang, Philip W. Kantoff, David B. Solit, Michael F. Berger, Charles L. Sawyers, Nikolaus Schultz, Howard I. Scher
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Impact Clinical Decision Making
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
Wassim Abida
Honoraria: Caret Healthcare
Consulting or Advisory Role: Clovis Oncology
Research Funding: AstraZeneca, Zenith Epigenetics
Joshua Armenia
No relationship to disclose
Anuradha Gopalan
No relationship to disclose
Ryan Brennan
No relationship to disclose
Michael Walsh
No relationship to disclose
David Barron
No relationship to disclose
Daniel Danila
Honoraria: Astellas Scientific, Medical Affairs, Angle, Bayer, Xian-Janssen Pharmaceutical
Consulting or Advisory Role: Angle, Bayer
Research Funding: Prostate Cancer Foundation, Genentech, Janssen Research & Development (Inst)
Patents, Royalties, Other Intellectual Property: Gene expression profile associated with prostate cancer
Travel, Accommodations, Expenses: Cambridge Healthtech Institute, Prostate Cancer Foundation, Angle, Bayer, American Austrian Open Medical Institute, Global Technology Community, Xian-Janssen Pharmaceutical, Oncology Education
Dana Rathkopf
Consulting or Advisory Role: Janssen Oncology
Research Funding: Janssen Oncology (Inst), Medivation (Inst), Celgene (Inst), Takeda (Inst), Millennium Pharmaceuticals (Inst), Ferring (Inst), Novartis (Inst), Taiho Pharmaceutical (Inst), AstraZeneca (Inst), Genentech (Inst), TRACON Pharma (Inst)
Michael Morris
Consulting or Advisory Role: Astellas Pharma, Bayer, Endocyte
Research Funding: Bayer (Inst), Sanofi (Inst), Endocyte (Inst), Progenics (Inst)
Travel, Accommodations, Expenses: Bayer, Endocyte
Susan Slovin
Consulting or Advisory Role: Bayer
Brigit McLaughlin
No relationship to disclose
Kristen Curtis
No relationship to disclose
David M. Hyman
Consulting or Advisory Role: Atara Biotherapeutics, Chugai Pharma, CytomX Therapeutics
Research Funding: AstraZeneca, Puma Biotechnology
Jeremy C. Durack
Stock and Other Ownership Interests: Adient Medical
Consulting or Advisory Role: Adient Medical
Patents, Royalties, Other Intellectual Property: Patent disclosures, provisionals submitted for instrument to analyze biopsy specimens (unrelated to the small renal masses topic). Patent holder for device related to x-ray imaging data collection (unrelated to small renal masses topic), July 2008.
Other Relationship: Society of Interventional Radiology Foundation
Stephen B. Solomon
Leadership: Devicor Medical Products
Stock and Other Ownership Interests: Johnson & Johnson, Progenics, Aspire Bariatrics
Consulting or Advisory Role: GE Healthcare, AngioDynamics, Medtronic
Research Funding: GE Healthcare (Inst), AngioDynamics (Inst)
Patents, Royalties, Other Intellectual Property: GE Healthcare-patents pending (Inst), Aspire Bariatrics
Maria E. Arcila
Consulting or Advisory Role: AstraZeneca
Travel, Accommodations, Expenses: AstraZeneca, Invivoscribe, Raindance Technologies
Ahmet Zehir
No relationship to disclose
Aijazuddin Syed
No relationship to disclose
Jianjiong Gao
No relationship to disclose
Debyani Chakravarty
No relationship to disclose
Hebert Alberto Vargas
No relationship to disclose
Mark E. Robson
Honoraria: AstraZeneca
Consulting or Advisory Role: McKesson, AstraZeneca
Research Funding: AstraZeneca (Inst), AbbVie (Inst), Myriad Genetics (Inst), Biomarin (Inst), Medivation (Inst), Tesaro (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Vijai Joseph
Stock and Other Ownership Interests: Juno Therapeutics
Kenneth Offit
No relationship to disclose
Mark T.A. Donoghue
No relationship to disclose
Adam A. Abeshouse
No relationship to disclose
Ritika Kundra
No relationship to disclose
Zachary J. Heins
No relationship to disclose
Alexander V. Penson
No relationship to disclose
Christopher Harris
No relationship to disclose
Barry S. Taylor
No relationship to disclose
Marc Ladanyi
Honoraria: Merck (I)
Consulting or Advisory Role: NCCN/Boehringer Ingelheim Afatinib Targeted Therapy Advisory Committee
Consulting or Advisory Role: NCCN/AstraZeneca Tagrisso RFP Advisory Committee
Research Funding: LOXO Pharmaceuticals (Inst)
Diana Mandelker
No relationship to disclose
Liying Zhang
No relationship to disclose
Victor E. Reuter
No relationship to disclose
Philip W. Kantoff
Stock and Other Ownership Interests: Bellicum Pharmaceuticals, Metamark Genetics, Placon, Druggablity Technologies, Tarveda
Consulting or Advisory Role: Bavarian Nordic, Janssen, Millennium Pharmaceuticals, MorphoSys, Pfizer, Astellas Pharma, Bellicum Pharmaceuticals, BIND Biosciences, Endocyte, Metamark Genetics, Medivation, Merck, MTG Therapeutics, OncoCellMDX, Oncogenex, Sotio, Sanofi, Tokai Pharmaceuticals, Bayer, Genentech, Cristal Therapeutics, Ipsen, Omnitura, GTx, Tarveda, Druggablity Technologies, Progenity
Research Funding: Medivation (Inst), Sanofi (Inst), Oncogenex (Inst), Aragon Pharmaceuticals (Inst), Amgen (Inst), Astellas Pharma (Inst), Bayer (Inst), Bavarian Nordic (Inst), Dendreon (Inst), Exelixis (Inst), Janssen (Inst)
Patents, Royalties, Other Intellectual Property: Method for predicting the risk of prostate cancer morbidity and mortality, predicting and treating prostate cancer, methods for predicting likelihood of responding to treatment, chromosome copy number gain as a biomarker of urothelial carcinoma lethality, drug combinations to treat cancer, somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma (patent), Up-to-Date royalties, Wolters-Kluwer royalties
Expert Testimony: Sanofi, Janssen
Travel, Accommodations, Expenses: Sanofi, Janssen, BIND Biosciences, Bavarian Nordic, Millennium Pharmaceuticals
David B. Solit
Honoraria: Loxo, Pfizer
Consulting or Advisory Role: Pfizer, Loxo
Michael F. Berger
Consulting or Advisory Role: Cancer Genetics, Sequenom
Charles L. Sawyers
Leadership: Novartis
Stock and Other Ownership Interests: Novartis, Agios, Blueprint Medicines, BeiGene, ORIC Pharmaceuticals
Consulting or Advisory Role: Novartis, Blueprint Medicines, Agios, BeiGene, ORIC Pharmaceuticals
Patents, Royalties, Other Intellectual Property: Xtandi
Nikolaus Schultz
No relationship to disclose
Howard I. Scher
Leadership: Asterias Biotherapeutics
Consulting or Advisory Role: Ferring, WIRB-Copernicus Group, Medivation, Blue Earth Diagnostics, Med IQ, Roche, Elsevier, Merck, Clovis Oncology, Janssen Research & Development, Astellas Pharma, Sanofi
Research Funding: Janssen (Inst), Medivation (Inst), Innocrin Pharma (Inst), Illumina (Inst)
Travel, Accommodations, Expenses: Ferring, WIRB-Copernicus Group, Astellas Pharma, Roche, Sanofi, Janssen, Clovis
REFERENCES
- 1.Scher HI, Heller G. Clinical states in prostate cancer: Toward a dynamic model of disease progression. Urology. 2000;55:323–327. doi: 10.1016/s0090-4295(99)00471-9. [DOI] [PubMed] [Google Scholar]
- 2.National Comprehensive Cancer Network Prostate cancer (version 1.2015) https://www.nccn.org/patients/guidelines/prostate/files/assets/common/downloads/files/prostate.pdf [DOI] [PubMed]
- 3.Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [Erratum: Cell 162:454, 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grasso CS, Wu YM, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng HH, Pritchard CC, Boyd T, et al. Biallelic inactivation of BRCA2 in platinum-sensitive metastatic castration-resistant prostate cancer. Eur Urol. 2016;69:992–995. doi: 10.1016/j.eururo.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hyman DM, Solit DB, Arcila ME, et al: Precision medicine at Memorial Sloan Kettering Cancer Center: Clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today 20:1422-8, 2015. [DOI] [PMC free article] [PubMed]
- 11. Cheng DT, Mitchell T, Zehir A, et al: MSK-IMPACT: A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17:251-264, 2015. [DOI] [PMC free article] [PubMed]
- 12.cBioportal cBio cancer genomics portal. http://www.cbioportal.org/study?id=prad_mskcc_2017.
- 13.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGranahan N, Favero F, de Bruin EC, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra54. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a. Schrader KA, Cheng DT, Joseph V, et al: Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol 2:104-11, 2016. [DOI] [PMC free article] [PubMed]
- 17.Joshi PM, Sutor SL, Huntoon CJ, et al. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J Biol Chem. 2014;289:9247–9253. doi: 10.1074/jbc.M114.551143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17a. Chang MT, Asthana S, Gao SP, et al: Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 34:155-163, 2016. [DOI] [PMC free article] [PubMed]
- 18.Bajrami I, Frankum JR, Konde A, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–297. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buisson R, Dion-Côté AM, Coulombe Y, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17:1247–1254. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 21.Cremona CA, Behrens A. ATM signalling and cancer. Oncogene. 2014;33:3351–3360. doi: 10.1038/onc.2013.275. [DOI] [PubMed] [Google Scholar]
- 22.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [Erratum: Nature 502:258, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Deciphering signatures of mutational processes operative in human cancer. Cell Reports. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27a. Chakravarty D, Gao J, Phillips SM, et al: OnkoKB: A precision oncology knowledge base. JCO Precis Oncol doi: 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed]
- 28.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 29.Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 30.Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 31.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 32.Balbas MD, Evans MJ, Hosfield DJ, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joseph JD, Lu N, Qian J, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 34.Duff J, McEwan IJ. Mutation of histidine 874 in the androgen receptor ligand-binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Mol Endocrinol. 2005;19:2943–2954. doi: 10.1210/me.2005-0231. [DOI] [PubMed] [Google Scholar]
- 35.Boysen G, Barbieri CE, Prandi D, et al. SPOP mutation leads to genomic instability in prostate cancer eLife 4e09207, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aparicio AM, Shen L, Tapia EL, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res. 2016;22:1520–1530. doi: 10.1158/1078-0432.CCR-15-1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Z, Flesken-Nikitin A, Corney DC, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006;66:7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]
- 37a. Mu P, Zhang Z, Benelli M, et al: SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355:84-88, 2017. [DOI] [PMC free article] [PubMed]
- 38.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong MK, Macintyre G, Wedge DC, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605. doi: 10.1038/ncomms7605. [DOI] [PMC free article] [PubMed] [Google Scholar]