

ABSTRACT
A novel bi-partite fluorescence platform exploits the high affinity and selectivity of antibody scaffolds to capture and activate small-molecule fluorogens. In this report, we investigated the property of multi-selectivity activation by a single antibody against diverse cyanine family fluorogens. Our fluorescence screen identified three cell-impermeant fluorogens, each with unique emission spectra (blue, green and red) and nanomolar affinities. Most importantly, as a protein fusion tag to G-protein-coupled receptors, the antibody biosensor retained full activity – displaying bright fluorogen signals with minimal background on live cells. Because fluorogen-activating antibodies interact with their target ligands via non-covalent interactions, we were able to perform advanced multi-color detection strategies on live cells, previously difficult or impossible with conventional reporters. We found that by fine-tuning the concentrations of the different color fluorogen molecules in solution, a user may interchange the fluorescence signal (onset versus offset), execute real-time signal exchange via fluorogen competition, measure multi-channel fluorescence via co-labeling, and assess real-time cell surface receptor traffic via pulse-chase experiments. Thus, here we inform of an innovative reporter technology based on tri-color signal that allows user-defined fluorescence tuning in live-cell applications.
KEY WORDS: Antibody, Biosensor, FAP, Fluorescence, Fluorogen, scFv
Summary: We present a multi-color bipartite protein reporter for cell surface labeling of receptors. This permits innovative strategies of detection previously difficult or impossible with conventional fluorescent systems.
INTRODUCTION
Fluorescent protein reporters have dramatically transformed the probing methods for discerning cellular and molecular phenomena. In more detail, this fluorescence revolution commenced with the gene sequence identification and molecular cloning of the jellyfish Aequorea victoria fluorescent protein (Prasher et al., 1992; Shimomura et al., 1962), and was followed by the finding of fluorescent proteins in other animal models (Masuda et al., 2006; Matz et al., 1999; Shagin et al., 2004). Such isolated fluorescent proteins were often bioengineered as functional reporter tags for use in living cells – with features of improved thermal stabilities, multi-detection wavelengths, bipartite split-domains and environmental sensing probes, to highlight a few (Cabantous et al., 2005a,b; Kent et al., 2009; Sample et al., 2009; Shaner et al., 2004, 2005). Today, fluorescence biosensors form an indispensable arsenal for every sector of biological research – academia, industry and medicine. Accordingly, their application, developability and influence will further continue in this new century, with innovative technologies already emerging.
In the past decade, novel biosensing reporter approaches started to challenge the conventional paradigm of fluorescent proteins. That is, scientists started to explore bio-conjugate platforms where fluorescent modalities and protein scaffolds would interact to form stable complexes. Here, some researchers identified and developed protein scaffolds that form covalent interactions with small-molecule fluorescent ligands via chemical or enzymatic coupling mechanisms. As a result, such bipartite reporters offered enhanced spatial and temporal resolutions at the surface of cells and/or intracellular milieu (Chen et al., 2005; Fernández-Suárez et al., 2007; Gautier et al., 2008; Griffin et al., 1998; Hori et al., 2009; Keppler et al., 2002, 2004; Los et al., 2008; Luedtke et al., 2007). More advanced approaches utilized the capture of fluorogenic molecules, which are inherently non-fluorescent unless sterically restricted. The most successful of these to date are the fluorogen-activating proteins (FAPs), which utilize the high affinity and selectivity of antibodies to form stable non-covalent bonds with target fluorogens (Szent-Gyorgyi et al., 2008). Here, the antibody functions as a protein cage that sterically confines the small-molecule fluorogen, and, upon light excitation, the fluorogen emits fluorescence due to non-radiative energy decay and energy release. Incidentally, FAP technology also offers a malleable approach for altering fluorescence signals, primarily by modifying the chemical composition of the synthetic fluorogens in order to tune their binding affinities and/or spectra (Pham et al., 2015; Rastede et al., 2015; Saunders et al., 2013, 2014; Szent-Gyorgyi et al., 2010). Furthermore, FAP reporters have demonstrated a rapid advancement as tools for labeling targets at the surface of cells (Fig. S1), showing absence of intracellular background/noise and high cell-surface signal brightness that is comparable to (or greater) than conventional fluorescent proteins (Holleran et al., 2010; Saunders et al., 2012; Szent-Gyorgyi et al., 2008, 2010).
The majority of current fluorescent protein technologies show lack of multi-color detection and signal modulation. Some breakthroughs occurred in the covalent bio-conjugate field, where the same target ligand for capture may be chemically coupled with unique color fluorophores, a very similar approach to using commercially labeled antibodies for labeling cells (Chen et al., 2007; Kosaka et al., 2009; Vivero-Pol et al., 2005; Lee et al., 2010; Liu et al., 2014; Uttamapinant et al., 2010; Wombacher et al., 2010; Yao et al., 2012). Likewise, other groups have utilized bio-conjugate platforms based on tandem dye interactions that have resulted in fluorescence resonance energy transfer (FRET), a donor-acceptor approach that amplifies the Stokes shift of a molecule resulting in fluorescence emissions at longer wavelengths (Brun et al., 2009, 2011; Gallo et al., 2015; Pham et al., 2015; Rajapakse et al., 2010; Robers et al., 2009; Saunders et al., 2014; Yushchenko et al., 2012; Zürn et al., 2010). As a result, we find that current methods prove lacking in multi-color detection and real-time signal modulation. In this regard, FAP technology may prove better capable for generating multi-fluorescence detection from a single reporter, due to the non-covalent nature of the affinity interactions.
Recently, a group isolated a multi-selective single-chain variable fragment (scFv) with affinity activation for various cyanine family fluorogens with differing poly-methine group lengths (Özhalici-Ünal et al., 2008). In summary, this work showed that an scFv FAP may display binding promiscuity with different small-molecule fluorogen analogs. Inspired by this observation, we set out to screen a previously isolated scFv FAP for multi-fluorogen activation against a family of small-molecule variants. Thus, we explored fluorogens with assorted structural and electrostatic properties, with the aim to isolate fluorogens with diverse spectra.
Our affinity screen identified three cell-impermeant fluorogens with sub-micromolar affinities for the scFv scaffold. Most importantly, each fluorogen possesses a unique spectra that results in coverage of the entire visible spectrum from a single fluorescent reporter. As a result, we further explored advanced multi-color fluorescence detection at the surface of live cells. We investigated fluorescence signal manipulation via fluorogen removal and addition, multi-color target detection via co-labeling, and real-time fluorescence color-switch via affinity binding competition. To further highlight the potential of a multi-color scFv reporter, we tracked in real-time the cell surface internalization of a G-protein-coupled receptor via tri-color pulse-chase in live cells.
Thus, when compared to conventional systems, where signal is ever-present and monochromatic (such as fluorophore-labeled antibodies or fluorescent proteins), our findings show that a multi-selective affinity biosensor expands the opportunities of detection, resulting in user-defined spatial and temporal resolutions. In brief, here we report of a multi-color scFv FAP and demonstrate its importance for fluorescence detection on live cells.
RESULTS
Assessment of multi-fluorogen activation from scFv HL1.0.1-TO1
We set up a fluorescence-activation fluorogen screen using diverse cyanine family fluorogens against a previously isolated scFv, called HL1.0.1-TO1, with known specificity for a Thiazole Orange (TO) fluorogen derivative (Szent-Gyorgyi et al., 2008). In our screen, we included small-molecule analogs with variances in structural and physico-chemical properties. More specifically, we screened fluorogens with altered polymethine group lengths, atomic substitutions, electrostatic alterations, altered molecular polarities, and removal or addition of subgroups at distinct spatial locations. Incidentally, the molecular screening method utilized purified scFv protein in the presence of fluorogen, and was biased for high-affinity interactions with measurements at sub-micromolar concentrations of each moiety.
Fig. 1A compares normalized activation of TO and analog derivatives TO1, TO2 and TO1-2p. We observed improved fluorescence emission when a negative charged subgroup was present at the benzothiazole ring position, such as in TO versus TO1 or TO1-2p, while the same addition to the quinoline ring of TO substantially reduced fluorescence emission, such as for TO versus TO2. On the other hand, the addition of diethylene-glycol-diamine (neutral charge group) at the quinoline ring position did not affect fluorescence, such as for TO1 versus TO1-2p. Taken together, we observed that molecular electrostatics play a determining role for fluorogen–scFv interactions and their subsequent fluorescence activation.
Fig. 1.

A fluorescence activation screen using HL1.0.1-TO1 protein and diverse cyanine family fluorogens. All measurements were performed in PBS using triplicate samples with 500 nM fluorogen and purified HL1.0.1-TO protein. We included the excitation and emission maxima for the fluorogens that exhibit fluorescence activation, which is listed below the name of the corresponding fluorogen.
Fluorine atoms possess strong electron-withdrawing properties. As a result, fluorine atoms disrupt the electrical polarity of a fluorogen when present in the molecule. In our fluorescence screen, we observed diminished activation using different TO fluorogens modified with fluorine atoms at the benzothiazole ring (Fig. 1B). On the other hand, when measuring the same fluorine-modified fluorogens using an activating medium, such as 90% glycerol, the fluorescence output of the fluorogens remained comparable (Shank et al., 2009). This shows that the fluorogenic modifications themselves are not intrinsically disruptive. It also highlights that the charge polarity of fluorogens (whole-molecule electrostatics) prove important for directing the affinity interactions with the scFv FAP.
Previous groups have demonstrated that a twisted fluorogen conformation may be corrected into a planar geometry inside the binding pocket of a scFv, resulting in fluorescence emission upon excitation (Shank et al., 2013; Silva et al., 2007). As a result, we screened HL1.0.1-TO1 against twisted cyanine fluorogens αCN-TO and αCN-DIR. The data revealed that both fluorogens lack fluorescence activation (Fig. 1C,D). Incidentally, our results showed a lower signal emission profile for the αCN-DIR fluorogen in the presence of protein rather than when free in solution (observed multiple independent times – data not shown). Here, we hypothesize that αCN-DIR fluorogen loosely interacts with the scFv protein, but that its twisted geometry prevents the fluorogen from activating since a planar conformation is required. Incidentally, the lower observed signal may be attributed to the protein-binding pocket encapsulating and shielding the fluorogen from background activation when free in solution. A similar observation was previously identified when measuring the photo-insulating properties of scFv scaffolds and cognate fluorogens (Saurabh et al., 2015; Szent-Gyorgyi et al., 2013). Thus, here, we find that HL1.0.1-TO1 lacks the ability to stabilize a twisted conformation into a planar geometry for fluorescence activation.
In Fig. 1E–G, we show that extension of the methine-linker group resulted in decreased or loss of fluorescence signal — such as for PO-PRO1 versus PO-PRO3, TO-PRO1 versus TO-PRO5, and YO-PRO1 versus YO-PRO3. Thus, we speculate that the scFv affinity binding region proves restrictive in regards to spatial proximity between the heterocycles. This observation contrasts with a previously identified multi-selective scFv (Özhalici-Ünal et al., 2008) – with affinity activation for different fluorogens primarily based on poly-methine group length diversities. As a result, we find some properties are not universal across scFv FAPs, and that different antibody scaffolds possess unique affinity requirements in regards to methine group length extensions.
Additionally, we also screened tandem fluorogen molecules, such as TO-TO1 or YO-YO1 fluorogens, for affinity activation against HL1.0.1-TO1. For both molecules, we observed robust emission profiles in presence of the scFv (Fig. 1I). Thus, tandem molecules show fluorogenic activation, regardless of their increased molecular size footprint. As a result, since the scFv HL1.0.1-TO1 was previously isolated against a monomeric fluorogen, we suggest that only one tandem-half of the molecule may actually form the affinity complex rather than the whole-molecule tandem fluorogen. A similar observation was previously determined when using tandem pairings of fluorogen–fluorophore or fluorogen–polyethylene–biotin molecules (Pham et al., 2015; Szent-Gyorgyi et al., 2010; Vasilev et al., 2016). In these three studies, the fluorogen component shows affinity interaction with the scFv irrespective of its tandem fluorogen pair, where most likely both moieties display independent functional activities.
Determining multi-fluorogen activation from scFv HL1.0.1-TO1 and HL1-TO1 at the surface of cells
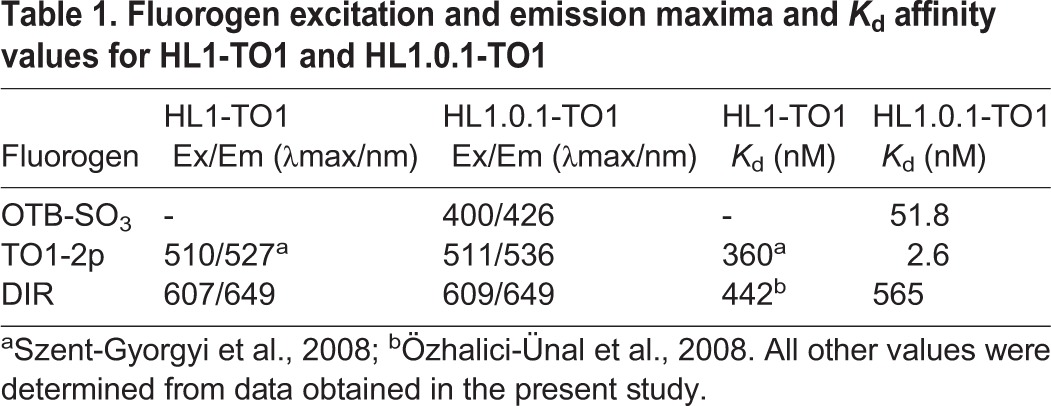
Based on our fluorescence validation screen, we selected three different spectra cell-impermeant fluorogens for further validation on live cells (Fig. S4): a Oxazole Thiazole Blue derivative (OTB-SO3) (Zanotti et al., 2011), a Thiazole Orange derivative (TO1-2p), and Dimethyl-Indole Red (DIR) (Constantin et al., 2008). In addition, we included in our assessment scFv HL1.0.1-TO1 and its parent scFv HL1-TO1 (a non-affinity matured version) (Szent-Gyorgyi et al., 2008). The reasoning behind this was to compare how affinity maturation may influence scFv–fluorogen multi-selectivity and activation, specifically in regards to fluorescence output and affinity values. As a result, Fig. 2A shows the fluorescence spectra of each fluorogen in the presence of surface displayed scFvs on yeast cells with measurements acquired at same Kd values for correct comparisons (see Table 1). Accordingly, we observed that all three fluorogens activated in the presence of scFv HL1.0.1-TO1 (as previously identified from our protein screen), while its parent scFv HL1-TO1 failed to activate OTB-SO3. Additionally, we also observed improved fluorescence signal from TO1-2p with HL1.0.1-TO1 versus HL1-TO1. On the other hand, HL1.0.1-TO1 showed a lower signal emission profile with DIR rather than HL1-TO1 (parent scFv) – likely due to the stricter methine group length restrictions for HL1.0.1-TO1, as previously identified in Fig. 1E–G. Taken together, affinity-matured scFv HL1.0.1-TO1 showed greater binding affinity for all fluorogens when compared to its parent, scFv HL1-TO1 (Table 1). Incidentally, affinity maturation may further explain why HL1.0.1-TO1 activated OTB-SO3 fluorogen while the parent scFv HL1-TO1 failed.
Fig. 2.
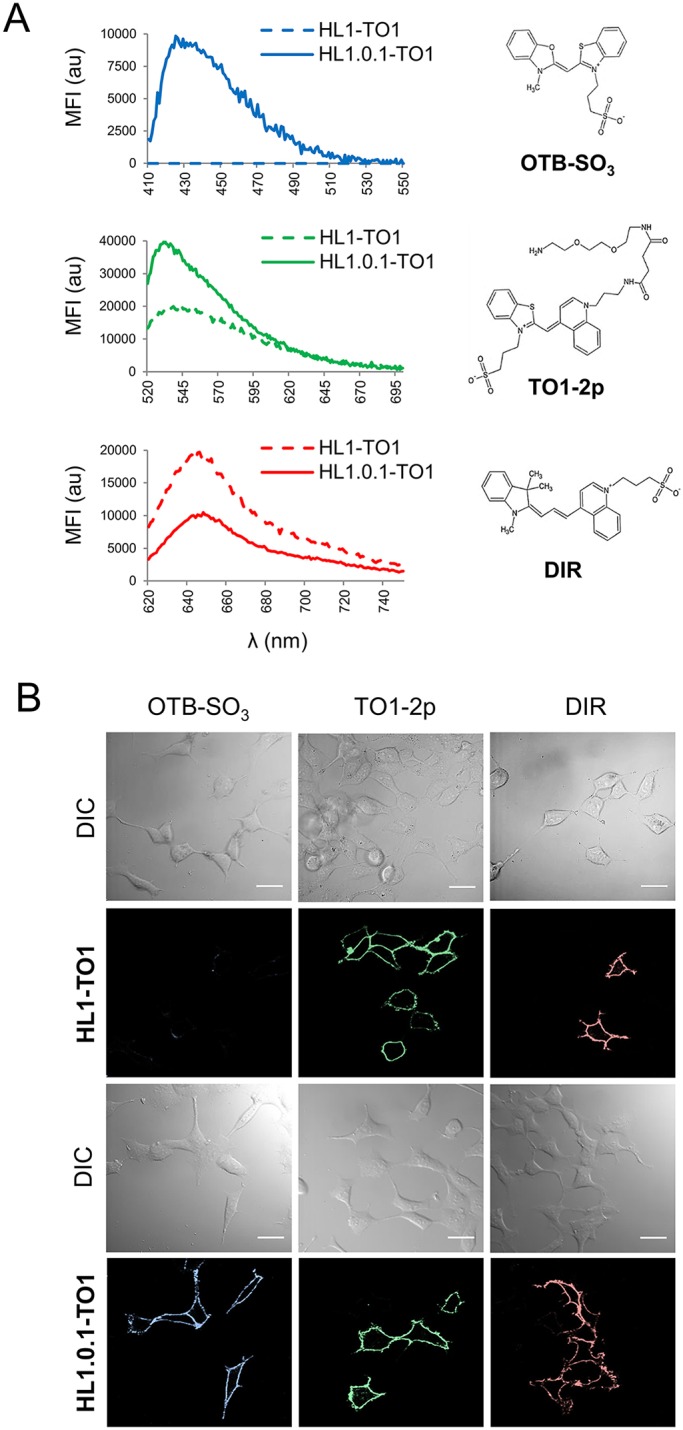
Comparison of HL1-TO1 or HL1.0.1-TO1 proteins expressed at the surface of live cells and screened for activation of different fluorogens. (A) Fluorescence emission spectra of surface displayed HL1-TO1 versus HL1.0.1-TO1 on yeast cells in presence of different fluorogens. Comparisons assessments were performed at 2- or 5-fold the Kd values of each fluorogen for its cognate scFv (see Table 1). (B) Micrographs of live mammalian cells expressing HL1.0.1-TO1 fused to adrenoreceptor β2 (ADRB2) at the surface. Cells were imaged in the presence of 100 nM of each fluorogen independently. Scale bars: 30 μm.
Table 1.
Fluorogen excitation and emission maxima and Kd affinity values for HL1-TO1 and HL1.0.1-TO1
Likewise, the yeast cell results also corresponded with additional mammalian cell validations utilizing scFv genetic fusions with adrenoreceptor-β2 (ADRB2) at the cell surface. Here, we observed abundant and distinct cell surface signal from HL1.0.1-TO1 with all three different color fluorogens independently (Fig. 2B), while the untransfected cells remained dark, indicative of low background/noise from the fluorogens in the medium. On the other hand, parent scFv HL1-TO1 lacked signal activation for OTB-SO3 fluorogen, and corresponded with our previous yeast cell assessments. Taken together, scFv HL1.0.1-TO1 demonstrates functional activity as a genetic reporter, showing opportunities of tri-color detection at the surface of live cells.
Evaluating fluorescence signal loss upon fluorogen removal from the medium
A property of scFv biosensors is their ability to form non-covalent complexes with their ligands. Hence, fluorogen removal from the cellular medium results in fluorescence signal loss due to the thermodynamically favorable exchange from bound to unbound fluorogen. After first labeling, for cells tagged with HL1.0.1-TO1 at the cell surface and then subjected to fluorogen removal, we observed signal loss for all fluorogens (Fig. 3A). Conversely, re-addition of each fluorogen resulted in rapid regain of initial fluorescence. Furthermore, some fluorogens exhibited more pronounced signal off-rates than others. That is, TO1-2p fluorogen proved the least susceptible to fluorescence signal loss, attributed to the high affinity of HL1.0.1-TO1 for this fluorogen (see Table 1). Conversely, OTB-SO3 showed the greatest loss of fluorescence activity for all three fluorogens (Fig. 3B). Note, this proved unexpected since DIR possesses the weakest affinity values for HL1.0.1-TO1 from all three fluorogens (Table 1). As a result, we asked whether photobleaching effects contributed to reduced fluorescence signal over time since fluorogens and fluorophores are susceptible over time exposure to irreversible photochemical alterations that may permanently reduce their ability to fluoresce. We subjected the same cells labeled with each fluorogen to multiple cycles of photobleaching using identical laser power settings as before. Data analysis showed OTB-SO3 to be the most susceptible fluorogen to photobleaching, while TO1-2p and DIR showed minor sensitivities (Fig. 3C). Accordingly, we repeated the fluorogen removal assay and measured fluorescence at only two time points instead of multiple time points (such as 15 s intervals for 10 consecutive minutes; see Materials and Methods for descriptive methods) in order to minimize the photobleaching time exposure. The data showed smaller OTB-SO3 signal loss than in the prior experiment (over the same time interval) (Fig. 3D). Interestingly, OTB-SO3 showed comparable values of fluorescence loss to that of DIR fluorogen, which may indicate similar ligand off-rates (k-off) for these fluorogens. In conclusion, scFv HL1.0.1-TO1 permits signal manipulation via fluorogen removal from cellular medium; however, photobleaching effects must be taken into consideration for correct assessments.
Fig. 3.

Assessing fluorescence signal loss at the surface of live mammalian cells after fluorogen removal from the cellular medium. All analyses were performed using cells expressing HL1.0.1-TO1 fused to ADRB2 at the cell surface. (A) Time-lapse micrographs from cells initially labeled with fluorogen, then washed and imaged for 10 min; at the last time point fluorogen was re-added. Each fluorogen time-series micrograph represents multiples images over time. Scale bar: 15 μm. (B) Bar graph summary of fluorescence intensities from fluorogen removal time-lapse images (n=5). (C) Bar graph summary of fluorescence intensities from photobleached time-lapse images in presence of fluorogen (n=5). (D) Bar graph summary of fluorescence intensities from images at only two time points after fluorogen removal (n=5). All assays were performed with 100 nM of each fluorogen. Results are mean±s.d.
Assessing fluorescence signal exchange via fluorogen competition
In multi-selective settings, different fluorogens compete for binding with the same scFv, where the labeling with different fluorogens may result in real-time color exchange. For this analysis, we excluded fluorogen competitions between TO1-2p and DIR due to potential FRET between these chromophores. We initially labeled the HL1.0.1-TO1-tagged cells with OTB-SO3 fluorogen and then added TO1-2p or DIR fluorogens to the cellular medium. Time-lapse image analysis showed that the majority of OTB-SO3 signal diminished after TO1-2p fluorogen addition (Fig. 4A). On the other hand, co-labeling was observed when DIR fluorogen was added to the medium (Fig. 4A). For both examples, the data correlates with HL1.0.1-TO1 fluorogen affinity values, where the high scFv affinity of TO1-2p competes for binding and displaces OTB-SO3 over time (Fig. 4B), while the weaker affinity DIR only partially displaces OTB-SO3, and after 10 min the majority of the OTB-SO3 signal remains (Fig. 4C). As above, in order to assess any contribution of photobleaching effects, we repeated the fluorogen competition assay and measured for fluorescence activation at only two time points instead of multiple time points. The results showed comparable signal loss for OTB-SO3 when TO1-2p was the competing fluorogen (Fig. 4D). On the other hand, we observed higher OTB-SO3 signal when DIR was the competing fluorogen (Fig. 4E), indicative of partial OTB-SO3 sensitivity to photobleaching (as previously measured). Thus, multi-selective scFvs may allow fluorescence signal exchange when presented with a second fluorogen; however, the extent of the color switch depends upon the binding affinities of each fluorogen.
Fig. 4.

Determining fluorescence signal exchange at the surface of live cell via two-fluorogen competition. All analyses were performed using mammalian cells expressing HL1.0.1-TO1 fused to ADRB2 at the cell surface. (A) Time-lapse micrographs from cells initially labeled with OTB-SO3 fluorogen, then presented in the medium with either TO1-2p or DIR fluorogens and immediately time-lapse imaged for 10 min. Note, we observed improved image resolutions at longer wavelength emissions due to reduced cellular background and higher laser excitation power settings. Each fluorogen time-series micrograph represents multiple images over time. Scale bar: 15 μm. (B) Bar graph summary of fluorescence intensities from fluorogen competition time-lapse images of OTB-SO3 vs TO1-2p. The TO1-2p signal was normalized to the fluorescence values for the final time point. (C) Bar graph summary of fluorescence intensities from fluorogen competition time-lapse images of OTB-SO3 vs. DIR. The DIR signal was normalized to the fluorescence values for the final time point. (D) Bar graph summary of OTB-SO3 fluorescence at only two time points after addition of TO1-2p in the medium. (E) Bar graph summary of OTB-SO3 fluorescence at only two time points after addition of DIR in the medium. All assays were performed in presence of 100 nM of each fluorogen, and each bar graph analysis was determined from n=5 images for each group. Results are mean±s.d.
Measuring multi-color surface receptor trafficking on live cells
The potential of multi-color activation from a single reporter allows for advanced strategies of cellular detection. In this regard, we transfected live mammalian cells with HL1.0.1-TO1 fused to ADRB2. Upon agonist molecule receptor stimulation, we measured the receptor trafficking from the cell surface towards cytoplasm using sequential pulse-chase in three colors. Here, the overall goal was to measure cell surface receptor traffic in real-time via successful removal of surface staining, but with maintenance of staining in the internalized receptors. Thus, we first labeled the cells with DIR – the fluorogen with the weakest affinity binding – and then followed by presenting the small-molecule receptor agonist isoproterenol. Post-internalization trafficking, the cells were washed and relabeled with OTB-SO3 fluorogen. The same process was repeated again and followed by labeling with TO1-2p. In this case, we were able to determine spatial and temporal locations of ADRB2 receptors using live cells in real-time (Fig. 5). Furthermore, we also noted that fluorogen removal via a wash-step and then followed by exchange resulted in faster relabeling of the cell surface receptors rather than direct competition (compare to Fig. 4). As a result, we were able to perform single reporter tri-color fluorescence imaging to track, in real time, the spatial and temporal location of cell surface receptors.
Fig. 5.

Imaging the real-time intracellular trafficking response of cell surface receptors via three-color fluorogen sequential labeling. Mammalian cells expressing HL1.0.1-TO1 fused to ADRB2 at the surface were initially labeled using 100 nM DIR in the medium and subsequently presented with an ADRB2 agonist ligand (isoproterenol). The cells were washed with PBS and labeled with 50 nM OTB-SO3 and the ADRB2 agonist. Then, the cells were washed with PBS and labeled with 500 nM TO1-2p. Each fluorogen time-series micrograph represents multiples images over time. Scale bar: 30 μm.
DISCUSSION
The properties of high affinity and specificity from single-chain variable fragments (scFvs) for target ligands generates highly stable and functional scFv–fluorogen biosensors. As demonstrated in this report, in certain cases a single scFv molecule may target multiple different fluorogens from the same chemical family. Our fluorescence screen showed a high percentage activation, of greater than 50%, with diverse fluorogen analogs against scFv HL1.0.1-TO1 (see Fig. 1 and Fig. S3). Accordingly, we propose that antibody multi-selectivity may prove more common in settings of small-molecule targets than with conventional antibody antigens, such as peptides or proteins. For the latter case, macromolecules possess a larger affinity interface that must be occluded from the surrounding solvent, and results in lower energetically favorable conditions for binding. Here, the proteins and peptides possess geometric conformations and structural features that restrict the number of possible spatial orientations for binding when compared to what is seen for small molecules. Also, macromolecules are subject to long-range affinity interactions (i.e. electrostatic, hydrogen bonds and hydrophobic interactions) that may indirectly influence protein affinity and the specificity interface (Janin, 1997; Schreiber and Fersht, 1995). Conversely, long-range interactions also negatively influence whole-protein affinity associations via charge repulsions and/or unfavorable dipole orientations (Lockless and Ranganathan, 1999; Tompa, 2002). By contrast, fluorogen molecules possess small and flexible scaffolds that offer a greater range of protein surface interactions leading to increased freedom of contact regions and geometrical orientations. Furthermore, small-molecules tend to bury within the affinity binding region, which fully encapsulates the fluorogen from bulk-solvent and increases the free-energy conditions (i.e. lower entropy) for affinity binding (Bogan and Thorn, 1998).
Our findings further show that, when comparing fluorogen analogs for scFv activation, altered molecular electrostatics reveal a propensity for affinity disruptions. That is, we detected diminished or absence of fluorescence signal when charged groups were replaced or whole-molecule electrostatic distributions were modified (see Fig. 1A,B). In this regard, our findings suggest that electrical forces largely influence the free-energy landscape for affinity binding of fluorogens, and minor disturbances at electrostatic interfaces lead in severe loss of or poor binding. This observation is reflective of protein–protein interaction models, where the principles of charge–charge and charge–dipole interactions prove critical for thermodynamic stability and association kinetics (Dill, 1990; Janin, 1997; Schreiber and Fersht, 1996). On the other hand, fluorogen structural modifications based on cyclic or methine group modifications proved less detrimental in fluorescence signal loss. For example, when comparing TO1-2p versus OTB-SO3 (smaller heterocycle group), or TO1-2p versus DIR (longer methine linker), in both cases we still observed scFv-mediated fluorogen activation, yet with reduced binding affinities (see Table 1). Taken together, our data hints that for antibody–small-molecule interactions, the scFv binding region allows greater malleability of fluorogens with structural rather than electrostatic fluorogens modifications.
Our experimental findings may be further explained using metadata studies based on affinity hot-spot regions that stabilize protein–protein interactions (Morrow and Zhang, 2012). More specifically, hot-spots show involvement in affinity steering of target molecules for docking, and show preferential enrichment of tyrosine, tryptophan and arginine residues (Bogan and Thorn, 1998; Lise et al., 2011). As a result, here we propose that polar and charged interactions (such as with arginine and others) must govern the free-energy landscape of the fluorogen affinity interface with the scFv, thereby guiding and orienting the small molecule. However, aromatic residues in the region of the protein that binds the fluorogen, such as tyrosine, tryptophan and others, may help stabilize the fluorogens via π-stacking, hydrogen bonding and hydrophobic interactions. This assertion also corresponds with multiple other protein–protein affinity models that describe an early affinity complex that is further stabilized by a later precise docking stage (Chakrabarti and Janin, 2002; Schreiber and Fersht, 1995, 1996).
In this report, we present a screening method for identifying multi-selective FAPs, and also we highlight novel approaches for microscopy detection based on multi-color fluorescence. More specifically, when compared to conventional methods, a bipartite non-covalent platform offers increased flexibility for target molecule detection – in regards to temporal and spatial fluorescence. That is, in contrast to ever-present signal, such as fluorescent antibodies or reporter proteins, fluorescence detection remains absent until the addition of a cognate fluorogen to the cellular medium. Conversely, here we observed that fluorogen removal from the cellular medium results in fluorescence loss, and is regulated by affinity off-rates (Fig. 3). Application-wise, a tunable signal detection reporter offers opportunities for advanced protein discovery strategies involving protein turnover at the cellular membrane, pulse-chase of vesicular traffic or post-cell-labeling assays that require the removal of signal (Bodor et al., 2012; Fuchs et al., 2010; Giepmans et al., 2006; Lippincott-Schwartz and Patterson, 2003; Mizukami et al., 2012). Furthermore, in order to increasingly accelerate the off-rates of signal detection using multi-selective FAPs, the future focus will be placed on isolating weak-affinity fluorogens. Here, two distinct engineering strategies may rapidly facilitate this: the site-directed mutagenesis of the protein scaffold or chemical alterations of fluorogen sub-groups (Rastede et al., 2015; Shank et al., 2009; Yates et al., 2013; Zanotti et al., 2011).
A multi-color and selective scFv FAP offers increased user freedom when compared to covalent-based bipartite reporters or standard fluorescent proteins. That is because a non-covalent affinity platform allows fluorescence signal manipulations via the kinetic tuning of fluorogen molecules in the surrounding medium. Here, a user may initially present a fluorogen to the cellular medium, and subsequently induce binding competition via addition of a different color fluorogen. As demonstrated in this report, such fluorescence color manipulation is contingent upon the fluorogen affinities for the scFv. Here, the higher affinity fluorogens compete with weaker affinity fluorogens to force a new equilibrium, resulting in rapid exchange of fluorescence signals (see Fig. 4). Incidentally, one may further accelerate color signal exchange via first removing the first fluorogen from the medium (wash-off) followed by subsequent presentation of a second color fluorogen (as demonstrated in Fig. 5). Accordingly, a user may perform measurements of real-time signal exchange using different color channels; this has direct application for cellular trafficking studies such as the downregulation and re-sensitization of cell surface receptors (Fisher et al., 2014; Wu et al., 2012, 2014). On the other hand, fluorogen competition using similar affinity fluorogens, results in multi-color co-labeling instead of color-switch. Here, co-labeling may prove critical for the detection of targets with high background/noise by the method of signal overlap measurements using two independent channels.
In summary, the data presented in this report advances the field of fluorescence reporters, and reveals novel strategies of detection that were previously problematic or impossible using conventional methods. Furthermore, the work presented here offers proof-of-concept examples with direct implications for live-cell detection strategies. Thus, here we seek to challenge the conventional paradigm and offer new directions in the field of fluorescent reporters.
MATERIALS AND METHODS
Plasmid constructions and fluorogen reagents
The surface expression of scFvs HL1.0.1-TO1 and HL1-TO1 (Fig. S2) fused at the N-terminal to adrenoreceptor β2 (ADRB2) on mammalian cells was performed using the pDisplaySacLac2 plasmid (Holleran et al., 2010). HL1.0.1-TO1 and HL1-TO1 gene inserts (Szent-Gyorgyi et al., 2008) were PCR amplified to include a 5′ SfiI restriction site sequence 5′-GGCCCAGCCGGCC-3′ and 3′ SfiI restriction site sequence 5′-GGCCGCAGGGGCC-3′, and each insert cloned into a SfiI enzyme-digested pDisplaySacLac2. Subsequently, a complete ORF and stop codon from the ADRB2 gene, encoding the b2AR receptor, was PCR amplified (template: fosmid WI2-2202O9/G248P86156H5, BACPAC Resources Center, Oakland, CA) to include 5′ and 3′ BsmI restriction sites using primer sequences 5′-GATCTGAATGCTATGGGGCAACCCGG-3′ and 5′-CCCACAGCATTCTACAGCAGTGAGTCATTTCTACTACAATT-3′, as previously shown (Fisher et al., 2010), and cloned into BsmI enzyme-digested pDisplaySacLac2. Fluorogens used in this report were provided as generous gifts from Dr. Armitage's and Dr. Schmidt's laboratories at Carnegie Mellon University, Pittsburgh, PA, USA, and the Molecular Biosensor and Imaging Center (MBIC) at Carnegie Mellon University, Pittsburgh, PA, USA.
Cell lines and culture conditions
We utilized a JAR200 yeast cell line (Mat a ura3-52, trp1, leu2δ200, his3δ200, pep4:HIS3, prbd1.6R, can1, GAL, GAL promoter-AGA1::URA3:G418r) for the surface display of HL1-TO1 or HL1.0.1-TO1 scFvs using methods previously described (Szent-Gyorgyi et al., 2008). Also, we utilized a HEK-293 mammalian cell line (ATCC, Manassas, VA) for all transient plasmid transfections using reagent TransIT®-LT1 (Mirus Bio, Madison, WI) according to the manufacturer's instructions. The mammalian cells were grown at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin.
Protein expression and purification
Purified HL1.0.1-TO1 scFv protein was obtained using a Rosetta-Gami E.coli strain (Novagen, Billerica, MA), where the cells were induced with 0.5 mm isopropyl-β-d-thiogalactopyranoside (RPIcorp, Prospect, IL), lysed and pelleted via high-speed centrifugation (30,000 g for 45 min). The supernatant was used in nickel-nitrilotriacetic acid chromatography (Ni-NTA; Thermo-Fisher, Waltham, MA) according to the manufacturer's instructions. The eluted fractions were purified via gel-filtration chromatography, then pooled and concentrated using centrifugal-filter units (EDM Millipore, Billerica, MA). The final scFv protein concentration was determined by spectroscopy (measured at 280 nm wavelength) and calculated using the Beer–Lambert equation. The protein samples were aliquoted and stored in phosphate-buffered saline (PBS) with 0.09% sodium azide at −20°C. All thawed samples were subsequently stored at 4°C for 1 month, and then discarded.
Optical spectroscopy assays
The samples were analyzed in an Infinite M1000 plate spectrometer (TECAN, Männedorf, Switzerland) in transparent, flat-bottom, 96-well microtiter plates (Corning, Corning, NY). For the fluorogen activation screen, the fluorescence measurements were performed in triplicate using 0.5 µM purified scFv protein and fluorogen in 200 µl of PBS. The emission spectra comparisons between HL1-TO1 and HL1.0.1-TO1 were determined using triplicate samples of 106 surface displayed scFv yeast cells in 200 µl of PBS, and corrected for background fluorescence using wild-type control cells. For correct comparisons, the fluorogens were presented at the same Kd concentration values, with 2-fold Kd for OTB-SO3 and DIR and 5-fold Kd for TO1-2p (see Table 1). All kinetic titration measurements were determined by using yeast with surface displayed HL1.0.1-TO1 in triplicate samples and fluorogen titrated at different concentrations, and corrected for background fluorescence using wild-type control cells. The data were fit to a one-site binding equation (Fig. S5) using Graph-pad Prism 5.0 (GraphPad Software, San Diego, CA), where x is the fluorogen concentration:
Fluorescence microscopy
The images were acquired with a Carl Zeiss LSM 510 Meta/UV DuoScan inverted confocal microscope using a 405 nm laser and a 430–480 nm band-pass filter for OTB-SO3 fluorogen, a 488 nm laser and a 505–550 nm band-pass filter for TO1-2p fluorogen, and a 561 nm laser and 575 nm LP band-pass filter for DIR fluorogen. Cells were imaged in PBS in 35-mm glass-bottom dishes (MatTek, Ashland, MA), and images analyzed with ImageJ software (http://rsb.info.nih.gov/ij/).
Cell surface imaging assays
HEK293 cells transiently expressing HL1.0.1-TO1 fused to ADRB2 were labeled with 100 nM OTB-SO3, TO1-2p or DIR fluorogens. For evaluating signal loss by fluorogen removal, the cells were presented with fluorogen in the medium and incubated for 10 min at room temperature. Subsequently, the cells were washed with PBS plus Ca2+ and Mg2+, and immediately imaged through time-lapse microscopy at 15 s intervals for 10 min. Next, the cells were presented again with fluorogen and imaged. For evaluating signal exchange via fluorogen competition, the cells were presented with OTB-SO3 fluorogen in the medium and incubated for 10 min. Subsequently, either TO1-2p or DIR fluorogens were added, and the cells were immediately imaged through time-lapse microscopy at 15 s intervals for 10 min. For evaluating receptor traffic via agonist stimulation, initially 100 nM of DIR fluorogen was added to the cellular medium and the cells imaged after 5 min incubation with the fluorogen. Next, a final concentration of 10 μM isoproterenol (Sigma-Aldrich, St Louis, MO) was added to the medium and the cells were incubated for 30 min at 37°C. Then, the cells were re-imaged. The medium was then removed, the cells gently washed with PBS plus Ca2+ and Mg2+, and medium re-added with 100 nM OTB-SO3 fluorogen. After this, the cells were re-imaged after incubation for 5 min with the fluorogen. Next, a final concentration of 10 μM isoproterenol was added to the medium and the cells were incubated for 30 min at 37°C, and subsequently the cells were re-imaged. Finally, the media was removed, the cells were gently washed with PBS plus Ca2+ and Mg2+, and cell medium was re-added with 100 nM TO1-2p fluorogen. Subsequently, after 5 min the cells were re-imaged.
Cellular fluorescence photobleaching assay
HEK293 cells transiently expressing HL1.0.1-TO1 fused to ADRB2 were imaged in presence of either 100 nM OTB-SO3, TO1-2p or DIR fluorogens. The cells were photobleached over four z-stack planes for a total of 20 cycles that consisted of 30 s iterations of 10% laser power (a 405 nm laser for OTB-SO3, a 488 nm laser for TO1-2p, or a 561 nm laser for DIR). Fluorescence microscopy acquisition was performed between each cycle, while maintaining the same settings across each fluorogen group. Image fluorescence values from each z-stack were converted into maximum intensity averages onto a single image. Subsequently, the fluorescence values for each image cycle were normalized to the initial image (pre-photobleaching) for analysis comparisons.
Acknowledgements
We would like to thank H. Teng for confocal microscopy assistance. Also, B. Schmidt, N. Shank, E. Rastede, G. Silva, and other MBIC chemists for the synthesis of most of the fluorogens used in this report. We are grateful to A. Dempsey and C. Szent-Gyorgyi for providing the HL1-TO1 and HL1.0.1-TO1 surface displayed yeast cell lines and yeast cell culture reagents.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: E.G.; Methodology: E.G.; Investigation: E.G.; Resources: J.J.; Writing - original draft: E.G.; Writing - review & editing: J.J.; Supervision: J.J.; Funding acquisition: J.J.
Funding
This work was supported by the National Institutes of Health (grant U54GM103529). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.202952.supplemental
References
- Bodor D. L., Rodríguez M. G., Moreno N., Jansen L. E. T., Bodor D. L., Rodríguez M. G., Moreno N. and Jansen L. E. T. (2012). Analysis of protein turnover by quantitative SNAP-based pulse-chase imaging. In Current Protocols in Cell Biology (eds J. Bonifacino, J. Harford, and K. Yamada), pp. 8.8.1-8.8.34. Hoboken, NJ, USA: John Wiley & Sons, Inc; 10.1002/0471143030.cb0808s55 [DOI] [PubMed] [Google Scholar]
- Bogan A. A. and Thorn K. S. (1998). Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280, 1-9. 10.1006/jmbi.1998.1843 [DOI] [PubMed] [Google Scholar]
- Brun M. A., Tan K.-T., Nakata E., Hinner M. J. and Johnsson K. (2009). Semisynthetic fluorescent sensor proteins based on self-labeling protein tags. J. Am. Chem. Soc. 131, 5873-5884. 10.1021/ja900149e [DOI] [PubMed] [Google Scholar]
- Brun M. A., Griss R., Reymond L., Tan K.-T., Piguet J., Peters R. J. R. W., Vogel H. and Johnsson K. (2011). Semisynthesis of fluorescent metabolite sensors on cell surfaces. J. Am. Chem. Soc. 133, 16235-16242. 10.1021/ja206915m [DOI] [PubMed] [Google Scholar]
- Cabantous S., Pédelacq J. D., Mark B. L., Naranjo C., Terwilliger T. C. and Waldo G. S. (2005a). Recent advances in GFP folding reporter and split-GFP solubility reporter technologies. Application to improving the folding and solubility of recalcitrant proteins from Mycobacterium tuberculosis. J. Struct. Funct. Genomics 6, 113-119. [DOI] [PubMed] [Google Scholar]
- Cabantous S., Terwilliger T. C. and Waldo G. S. (2005b). Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23, 102-107. 10.1038/nbt1044 [DOI] [PubMed] [Google Scholar]
- Chakrabarti P. and Janin J. (2002). Dissecting protein-protein recognition sites. Proteins Struct. Funct. Genet. 47, 334-343. 10.1002/prot.10085 [DOI] [PubMed] [Google Scholar]
- Chen I., Howarth M., Lin W. and Ting A. Y. (2005). Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2, 99-104. 10.1038/nmeth735 [DOI] [PubMed] [Google Scholar]
- Chen I., Choi Y.-A. and Ting A. Y. (2007). Phage display evolution of a peptide substrate for yeast biotin ligase and application to two-color quantum dot labeling of cell surface proteins. J. Am. Chem. Soc. 129, 6619-6625. 10.1021/ja071013g [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin T. P., Silva G. L., Robertson K. L., Hamilton T. P., Waggoner A. S., Armitage B. A. and Fague K. (2008). Synthesis of new fluorogenic cyanine dyes and incorporation into RNA fluoromodules. Org. Lett. 10, 1561-1564. 10.1021/ol702920e [DOI] [PubMed] [Google Scholar]
- Dill K. A. (1990). Dominant forces in protein folding. Biochemistry 29, 7133-7155. 10.1021/bi00483a001 [DOI] [PubMed] [Google Scholar]
- Fernández-Suárez M., Baruah H., Martínez-Hernández L., Xie K. T., Baskin J. M., Bertozzi C. R. and Ting A. Y. (2007). Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nat. Biotechnol. 25, 1483-1487. 10.1038/nbt1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher G. W., Adler S. A., Fuhrman M. H., Waggoner A. S., Bruchez M. P. and Jarvik J. W. (2010). Detection and quantification of β2AR internalization in living cells using FAP-based biosensor technology. J. Biomol. Screen. 15, 703-709. 10.1177/1087057110370892 [DOI] [PubMed] [Google Scholar]
- Fisher G. W., Fuhrman M. H., Adler S. A., Szent-Gyorgyi C., Waggoner A. S. and Jarvik J. W. (2014). Self-checking cell-based assays for GPCR desensitization and resensitization. J. Biomol. Screen. 19, 1220-1226. 10.1177/1087057114534299 [DOI] [PubMed] [Google Scholar]
- Fuchs J., Böhme S., Oswald F., Hedde P. N., Krause M., Wiedenmann J. and Nienhaus G. U. (2010). A photoactivatable marker protein for pulse-chase imaging with superresolution. Nat. Methods 7, 627-630. 10.1038/nmeth.1477 [DOI] [PubMed] [Google Scholar]
- Gallo E., Snyder A. C. and Jarvik J. W. (2015). Engineering tandem single-chain Fv as cell surface reporters with enhanced properties of fluorescence detection. Protein Eng. Des. Sel. 28, 327-337. 10.1093/protein/gzv016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier A., Juillerat A., Heinis C., Corrêa I. R., Kindermann M., Beaufils F. and Johnsson K. (2008). An engineered protein tag for multiprotein labeling in living cells. Chem. Biol. 15, 128-136. 10.1016/j.chembiol.2008.01.007 [DOI] [PubMed] [Google Scholar]
- Giepmans B. N. G., Adams S. R., Ellisman M. H. and Tsien R. Y. (2006). The fluorescent toolbox for assessing protein location and function. Science 312, 217-224. 10.1126/science.1124618 [DOI] [PubMed] [Google Scholar]
- Griffin B. A., Adams S. R. and Tsien R. Y. (1998). Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269-272. 10.1126/science.281.5374.269 [DOI] [PubMed] [Google Scholar]
- Holleran J., Brown D., Fuhrman M. H., Adler S. A., Fisher G. W. and Jarvik J. W. (2010). Fluorogen-activating proteins as biosensors of cell-surface proteins in living cells. Cytom. Part A 77, 776-782. 10.1002/cyto.a.20925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori Y., Ueno H., Mizukami S. and Kikuchi K. (2009). Photoactive yellow protein-based protein labeling system with turn-on fluorescence intensity. J. Am. Chem. Soc. 131, 16610-16611. 10.1021/ja904800k [DOI] [PubMed] [Google Scholar]
- Janin J. (1997). The kinetics of protein-protein recognition. Proteins Struct. Funct. Genet. 28, 153-161. 10.1002/(SICI)1097-0134(199706)28:2<153::AID-PROT4>3.0.CO;2-G [DOI] [PubMed] [Google Scholar]
- Kent K. P., Oltrogge L. M. and Boxer S. G. (2009). Synthetic control of green fluorescent protein. J. Am. Chem. Soc. 131, 15988-15989. 10.1021/ja906303f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H. and Johnsson K. (2002). A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86-89. 10.1038/nbt765 [DOI] [PubMed] [Google Scholar]
- Keppler A., Pick H., Arrivoli C., Vogel H. and Johnsson K. (2004). Labeling of fusion proteins with synthetic fluorophores in live cells. Proc. Natl. Acad. Sci. USA 101, 9955-9959. 10.1073/pnas.0401923101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N., Ogawa M., Choyke P. L., Karassina N., Corona C., McDougall M., Lynch D. T., Hoyt C. C., Levenson R. M., Los G. V. et al. (2009). In vivo stable tumor-specific painting in various colors using dehalogenase-based protein-tag fluorescent ligands. Bioconjug. Chem. 20, 1367-1374. 10.1021/bc9001344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-D., Lord S. J., Iwanaga S., Zhan K., Xie H., Williams J. C., Wang H., Bowman G. R., Goley E. D., Shapiro L. et al. (2010). Superresolution imaging of targeted proteins in fixed and living cells using photoactivatable organic fluorophores. J. Am. Chem. Soc. 132, 15099-15101. 10.1021/ja1044192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J. and Patterson G. H. (2003). Development and use of fluorescent protein markers in living cells. Science 300, 87-91. 10.1126/science.1082520 [DOI] [PubMed] [Google Scholar]
- Lise S., Buchan D., Pontil M. and Jones D. T. (2011). Predictions of hot spot residues at protein-protein interfaces using support vector machines. PLoS ONE 6, e16774 10.1371/journal.pone.0016774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. S., Nivón L. G., Richter F., Goldman P. J., Deerinck T. J., Yao J. Z., Richardson D., Phipps W. S., Ye A. Z., Ellisman M. H. et al. (2014). Computational design of a red fluorophore ligase for site-specific protein labeling in living cells. Proc. Natl. Acad. Sci. USA 111, E4551-E4559. 10.1073/pnas.1404736111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless S. W. and Ranganathan R. (1999). Evolutionarily conserved pathways of energetic connectivity in protein families. Science 286, 295-299. 10.1126/science.286.5438.295 [DOI] [PubMed] [Google Scholar]
- Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M. et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373-382. 10.1021/cb800025k [DOI] [PubMed] [Google Scholar]
- Luedtke N. W., Dexter R. J., Fried D. B. and Schepartz A. (2007). Surveying polypeptide and protein domain conformation and association with FlAsH and ReAsH. Nat. Chem. Biol. 3, 779-784. 10.1038/nchembio.2007.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H., Takenaka Y., Yamaguchi A., Nishikawa S. and Mizuno H. (2006). A novel yellowish-green fluorescent protein from the marine copepod, Chiridius poppei, and its use as a reporter protein in HeLa cells. Gene 372, 18-25. 10.1016/j.gene.2005.11.031 [DOI] [PubMed] [Google Scholar]
- Matz M. V., Fradkov A. F., Labas Y. A., Savitsky A. P., Zaraisky A. G., Markelov M. L. and Lukyanov S. A. (1999). Fluorescent proteins from nonbioluminescent Anthozoa species. Nat. Biotechnol. 17, 969-973. 10.1038/13657 [DOI] [PubMed] [Google Scholar]
- Mizukami S., Watanabe S., Akimoto Y. and Kikuchi K. (2012). No-wash protein labeling with designed fluorogenic probes and application to real-time pulse-chase analysis. J. Am. Chem. Soc. 134, 1623-1629. 10.1021/ja208290f [DOI] [PubMed] [Google Scholar]
- Morrow J. K. and Zhang S. (2012). Computational prediction of protein hot spot residues. Curr. Pharm. Des. 18, 1255-1265. 10.2174/138161212799436412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özhalici-Ünal H., Pow C. L., Marks S. A., Jesper L. D., Silva G. L., Shank N. I., Jones E. W., Burnette J. M., Berget P. B. and Armitage B. A. (2008). A rainbow of fluoromodules: a promiscuous scFv protein binds to and activates a diverse set of fluorogenic cyanine dyes. J. Am. Chem. Soc. 130, 12620-12621. 10.1021/ja805042p [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham H. H., Szent-Gyorgyi C., Brotherton W. L., Schmidt B. F., Zanotti K. J., Waggoner A. S. and Armitage B. A. (2015). Bichromophoric dyes for wavelength shifting of dye-protein fluoromodules. Org. Biomol. Chem. 13, 3699-3710. 10.1039/C4OB02522A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher D. C., Eckenrode V. K., Ward W. W., Prendergast F. G. and Cormier M. J. (1992). Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111, 229-233. 10.1016/0378-1119(92)90691-H [DOI] [PubMed] [Google Scholar]
- Rajapakse H. E., Gahlaut N., Mohandessi S., Yu D., Turner J. R. and Miller L. W. (2010). Time-resolved luminescence resonance energy transfer imaging of protein-protein interactions in living cells. Proc. Natl. Acad. Sci. USA 107, 13582-13587. 10.1073/pnas.1002025107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastede E. E., Tanha M., Yaron D., Watkins S. C., Waggoner A. S. and Armitage B. A. (2015). Spectral fine tuning of cyanine dyes: electron donor-acceptor substituted analogues of thiazole orange. Photochem. Photobiol. Sci. 14, 1703-1712. 10.1039/C5PP00117J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robers M., Pinson P., Leong L., Batchelor R. H., Gee K. R. and Machleidt T. (2009). Fluorescent labeling of proteins in living cells using the FKBP12 (F36V) tag. Cytom. Part A 75A, 207-224. 10.1002/cyto.a.20649 [DOI] [PubMed] [Google Scholar]
- Sample V. and Newman R. H. and Zhang J. (2009). The structure and function of fluorescent proteins. Chem. Soc. Rev. 38, 2852-2864. 10.1039/b913033k [DOI] [PubMed] [Google Scholar]
- Saunders M. J., Szent-Gyorgyi C., Fisher G. W., Jarvik J. W., Bruchez M. P. and Waggoner A. S. (2012). Fluorogen activating proteins in flow cytometry for the study of surface molecules and receptors. Methods 57, 308-317. 10.1016/j.ymeth.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders M. J., Liu W., Szent-Gyorgyi C., Wen Y., Drennen Z., Waggoner A. S. and Meng W. S. (2013). Engineering fluorogen activating proteins into self-assembling materials. Bioconjug. Chem. 24, 803-810. 10.1021/bc300613h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders M. J., Block E., Sorkin A., Waggoner A. S. and Bruchez M. P. (2014). A bifunctional converter: fluorescein quenching scFv/Fluorogen activating protein for photostability and improved signal to noise in fluorescence experiments. Bioconjug. Chem. 25, 1556-1564. 10.1021/bc500273n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saurabh S., Zhang M., Mann V. R., Costello A. M. and Bruchez M. P. (2015). Kinetically tunable photostability of fluorogen-activating peptide-fluorogen complexes. Chemphyschem 16, 2974-2980. 10.1002/cphc.201500587 [DOI] [PubMed] [Google Scholar]
- Schreiber G. and Fersht A. R. (1995). Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248, 478-486. 10.1016/S0022-2836(95)80064-6 [DOI] [PubMed] [Google Scholar]
- Schreiber G. and Fersht A. R. (1996). Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 3, 427-431. 10.1038/nsb0596-427 [DOI] [PubMed] [Google Scholar]
- Shagin D. A., Barsova E. V., Yanushevich Y. G., Fradkov A. F., Lukyanov K. A., Labas Y. A., Semenova T. N., Ugalde J. A., Meyers A., Nunez J. M. et al. (2004). GFP-like proteins as ubiquitous metazoan superfamily: evolution of functional features and structural complexity. Mol. Biol. Evol. 21, 841-850. 10.1093/molbev/msh079 [DOI] [PubMed] [Google Scholar]
- Shaner N. C., Campbell R. E., Steinbach P. A., Giepmans B. N. G., Palmer A. E. and Tsien R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 22, 1567-1572. 10.1038/nbt1037 [DOI] [PubMed] [Google Scholar]
- Shaner N. C., Steinbach P. A. and Tsien R. Y. (2005). A guide to choosing fluorescent proteins. Nat. Methods 2, 905-909. 10.1038/nmeth819 [DOI] [PubMed] [Google Scholar]
- Shank N. I., Zanotti K. J., Lanni F., Berget P. B. and Armitage B. A. (2009). Enhanced photostability of genetically encodable fluoromodules based on fluorogenic cyanine dyes and a promiscuous protein partner. J. Am. Chem. Soc. 131, 12960-12969. 10.1021/ja9016864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank N. I., Pham H. H., Waggoner A. S. and Armitage B. A. (2013). Twisted cyanines: a non-planar fluorogenic dye with superior photostability and its use in a protein-based fluoromodule. J. Am. Chem. Soc. 135, 242-251. 10.1021/ja308629w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura O., Johnson F. H. and Saiga Y. (1962). Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J. Cell. Comp. Physiol. 59, 223-239. 10.1002/jcp.1030590302 [DOI] [PubMed] [Google Scholar]
- Silva G. L., Ediz V., Yaron D. and Armitage B. A. (2007). Experimental and computational investigation of unsymmetrical cyanine dyes: understanding torsionally responsive fluorogenic dyes. J. Am. Chem. Soc. 129, 5710-5718. 10.1021/ja070025z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C., Schmidt B. A. F., Schmidt B. A. F., Creeger Y., Fisher G. W., Zakel K. L., Adler S., Fitzpatrick J. A. J., Woolford C. A., Yan Q. et al. (2008). Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol. 26, 235-240. 10.1038/nbt1368 [DOI] [PubMed] [Google Scholar]
- Szent-Gyorgyi C., Schmidt B. F., Fitzpatrick J. A. J. and Bruchez M. P. (2010). Fluorogenic dendrons with multiple donor chromophores as bright genetically targeted and activated probes. J. Am. Chem. Soc. 132, 11103-11109. 10.1021/ja9099328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C., Stanfield R. L., Andreko S., Dempsey A., Ahmed M., Capek S., Waggoner A. S., Wilson I. A. and Bruchez M. P. (2013). Malachite green mediates homodimerization of antibody VL domains to form a fluorescent ternary complex with singular symmetric interfaces. J. Mol. Biol. 425, 4595-4613. 10.1016/j.jmb.2013.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. (2002). Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527-533. 10.1016/S0968-0004(02)02169-2 [DOI] [PubMed] [Google Scholar]
- Uttamapinant C., White K. A., Baruah H., Thompson S., Fernández-Suárez M., Puthenveetil S. and Ting A. Y. (2010). A fluorophore ligase for site-specific protein labeling inside living cells. Proc. Natl. Acad. Sci. USA 107, 10914-10919. 10.1073/pnas.0914067107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilev K. V., Gallo E., Shank N. and Jarvik J. W. (2016). Novel biosensor of membrane protein proximity based on fluorogen activated proteins. Comb. Chem. High Throughput Screen. 19, 392-399. 10.2174/1386207319666160408150320 [DOI] [PubMed] [Google Scholar]
- Vivero-Pol L., George N., Krumm H., Johnsson K. and Johnsson N. (2005). Multicolor imaging of cell surface proteins. J. Am. Chem. Soc. 127, 12770-12771. 10.1021/ja0533850 [DOI] [PubMed] [Google Scholar]
- Wombacher R., Heidbreder M., van de Linde S., Sheetz M. P., Heilemann M., Cornish V. W. and Sauer M. (2010). Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods 7, 717-719. 10.1038/nmeth.1489 [DOI] [PubMed] [Google Scholar]
- Wu Y., Tapia P. H., Fisher G. W., Simons P. C., Strouse J. J., Foutz T., Waggoner A. S., Jarvik J. and Sklar L. A. (2012). Discovery of regulators of receptor internalization with high-throughput flow cytometry. Mol. Pharmacol. 82, 645-657. 10.1124/mol.112.079897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Tapia P. H., Jarvik J., Waggoner A. S., Sklar L. A., Wu Y., Tapia P. H., Jarvik J., Waggoner A. S. and Sklar L. A. (2014). Real-time detection of protein trafficking with high-throughput flow cytometry (HTFC) and fluorogen-activating protein (FAP) base biosensor. In Current Protocols in Cytometry (ed. J. P. Robinson), pp. 9.43.1-9.43.11. Hoboken, NJ, USA: John Wiley & Sons, Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J. Z., Uttamapinant C., Poloukhtine A., Baskin J. M., Codelli J. A., Sletten E. M., Bertozzi C. R., Popik V. V. and Ting A. Y. (2012). Fluorophore targeting to cellular proteins via enzyme-mediated azide ligation and strain-promoted cycloaddition. J. Am. Chem. Soc. 134, 3720-3728. 10.1021/ja208090p [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates B. P., Peck M. A. and Berget P. B. (2013). Directed evolution of a fluorogen-activating single chain antibody for function and enhanced brightness in the cytoplasm. Mol. Biotechnol. 54, 829-841. 10.1007/s12033-012-9631-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushchenko D. A., Zhang M., Yan Q., Waggoner A. S. and Bruchez M. P. (2012). Genetically targetable and color-switching fluorescent probe. Chembiochem 13, 1564-1568. 10.1002/cbic.201200334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti K. J., Silva G. L., Creeger Y., Robertson K. L., Waggoner A. S., Berget P. B. and Armitage B. A. (2011). Blue fluorescent dye-protein complexes based on fluorogenic cyanine dyes and single chain antibody fragments. Org. Biomol. Chem. 9, 1012-1020. 10.1039/C0OB00444H [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zürn A., Klenk C., Zabel U., Reiner S., Lohse M. J. and Hoffmann C. (2010). Site-specific, orthogonal labeling of proteins in intact cells with two small biarsenical fluorophores. Bioconjug. Chem. 21, 853-859. 10.1021/bc900394j [DOI] [PubMed] [Google Scholar]