

Abstract
Neuropsychiatric disorders have a complex genetic architecture. Human genetic population-based studies have identified numerous heritable sequence and structural genomic variants associated with susceptibility to neuropsychiatric disease. However, these germline variants do not fully account for disease risk. During brain development, progenitor cells undergo billions of cell divisions to generate the ~80 billion neurons in the brain. The failure to accurately repair DNA damage arising during replication, transcription, and cellular metabolism amid this dramatic cellular expansion can lead to somatic mutations. Somatic mutations that alter subsets of neuronal transcriptomes and proteomes can, in turn, affect cell proliferation and survival and lead to neurodevelopmental disorders. The long life span of individual neurons and the direct relationship between neural circuits and behavior suggest that somatic mutations in small populations of neurons can significantly affect individual neurodevelopment. The Brain Somatic Mosaicism Network has been founded to study somatic mosaicism both in neurotypical human brains and in the context of complex neuropsychiatric disorders.
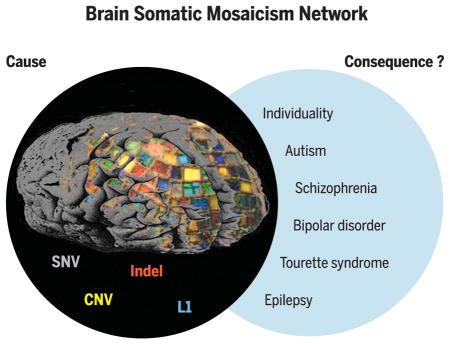
Graphical Abstract
Collectively, somatic SNVs, indels, structural variants (e.g., CNVs), and MEIs (e.g., L1 retrotransposition events) shape the genomic landscape of individual neurons. The Brain Somatic Mosaicism Network aims to systematically generate pioneering data on the types and frequencies of brain somatic mutations in both neurotypical individuals and those with neuropsychiatric disease. The resulting data will be shared as a large community resource.
The human body reaches a steady-state level of approximately 1014 cells in adulthood. Because DNA replication and DNA repair are imperfect processes (estimated at ~0.27 to 0.99 errors in ~109 nucleotides per cell division) (1), somatic cells within an individual must differ in the presence of single-nucleotide variants (SNVs) and/or small insertion/deletion (indel) mutations (2–4). In addition to SNVs and indels (5), subsets of neurons also harbor structural variants [which include large (>1 Mb) copy number variants (CNVs), inversions, translocations, and whole-chromosome gains or losses (6–10)] and smaller mobile genetic element insertions (MEIs) (11–16). Here, we define somatic mosaicism as the existence of different genomes within the cells of a monozygotic individual. Well-known examples of somatic mosaicism include ichthyosis with confetti and lines of Blaschko (4).
Healthy neuronal development requires that neural stem cells and progenitor cells (NPCs) undergo tens of billions of cell divisions, both before birth and during the first years of life, to generate the ~80 billion neurons in the fully developed human brain (17). Because neurons are among the longest-lived cells in the body, the accumulation of somatic mutations (i.e., SNVs, indels, structural variants, and MEIs) within NPCs, or perhaps postmitotic neurons (18), could influence neuronal development, complexity, and function (19, 20). Indeed, mounting evidence indicates that somatic mutations in small populations of neurons contribute to various neurodevelopmental disorders (Table 1).
Table 1. Mosaic mutations in genes and their associated signaling pathways and diseases.
Disease abbreviations: CLOVES, Congenital lipomatous overgrowth, vascular malformations, and epidermal nevi; FCD, focal cortical dysplasia; GPCR, G protein–coupled receptor; HME, hemimegalencephaly; MCAP, megalencephaly-capillary malformation-polymicrogyria syndrome; MPPH2, megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome-2; NF, neuro-fibromatosis; RALD, Ras-associated autoimmune leukoproliferative disorder; TSC, tuberous sclerosis complex. Mosaicism abbreviations: G, germline; S, somatic; OS, obligatory somatic; MS, milder somatic; SHS, second-hit somatic.
| Gene(s) | Signaling pathway(s) | Disease(s) | Cellular function(s) | Cancer(s) | Cancer role | Mosaicism |
|---|---|---|---|---|---|---|
| PIK3CA (100–104) | PI3K-AKT-mTOR | HME, mosaic overgrowth syndrome, type 2 segmental, CLOVES, MCAP | PI3K subunit, serine/threonine kinase | Cervical, various neoplasms, colorectal | Oncogene | OS |
| AKT1 (105) | PI3K-AKT-mTOR | Proteus syndrome | Serine/threonine kinase | Breast, ovarian, colorectal | Oncogene | OS |
| AKT2 (106) | PI3K-AKT-mTOR | Diabetes mellitus | Serine/threonine kinase | Ovarian, pancreatic, breast, colorectal, lung cancer | Oncogene | G/S |
| AKT3 (101, 103, 13, 107) | PI3K-AKT-mTOR | HME, MCAP, MPPH2 | Serine/threonine kinase | Melanoma, glioma, ovarian cancer | Oncogene | OS |
| MTOR (108) | PI3K-AKT-mTOR | FCD type II | Serine/threonine kinase | Carcinoma, glioblastoma, melanoma | Oncogene | OS |
| DEPDC5 (109, 110) | PI3K-AKT-mTOR | Epilepsy with FCD | mTORC1 repressor | Glioblastoma and ovarian tumors | Tumor suppressor | G/S |
| TSC1 (111, 112) | PI3K-AKT-mTOR | TSC | Negative regulator of mTORC1 | Renal angiomyolipomas | Tumor suppressor | SHS |
| TSC2 (111, 112) | PI3K-AKT-mTOR | TSC | Negative regulator of mTORC1 | Renal angiomyolipomas | Tumor suppressor | SHS |
| NRAS, BRAF, FGFR3, PIK3CA (113–118) | RAS, PI3K-AKT-mTOR | Congenital melanocytic, other nevi; seborrheic keratosis | Cell cycle regulation | (FGFR3) bladder, cervical, urothelial | Oncogene | G/S |
| NF2 (119) | RAS, PI3K-AKT-mTOR | NF type 2 | Negative regulator of Ras, mTOR pathways | Neurofibromas | Tumor suppressor | G/MS |
| NF1 (120–124) | RAS | NF type 1, Watson syndrome | Negative regulator of Ras pathway | Neurofibromas, leukemia | Tumor suppressor | SHS |
| BRAF, NRAS, KRAS (125) | RAS | Pyogenic granuloma | Cell cycle regulation | (KRAS) breast, colorectal, other; (NRAS) thyroid, melanoma, other; (BRAF) melanoma, colorectal | Oncogene | OS |
| HRAS, KRAS (126) | RAS | Schimmelpenning-Feuerstein-Mims syndrome | Cell cycle regulation | (KRAS) bladder, breast, colorectal, pancreatic, other; (HRAS) Colorectal, bladder, kidney, other | Oncogene | OS |
| KRAS (127, 128) | RAS | RALD | Cell cycle regulation | Breast, bladder, other | Oncogene | OS |
| GNAQ (129) | GPCR, MAPK | Sturge-Weber syndrome | G protein alpha subunit | Melanoma | Oncogene | OS |
| GNAQ, GNA11 (130) | GPCR, MAPK | Dermal melanocytosis and phakomatosis pigmentovascularis | G protein alpha subunit | Melanoma | Oncogene | OS |
| MAP3K3 (131) | MAPK | Verrucous venous malformation | Cell cycle regulation | Breast, colon, rectal cancers | Oncogene | OS |
| GNAS (132, 133) | GPCR | McCune-Albright syndrome | G protein alpha subunit | Adenomas, carcinomas, ovarian neoplasms | Oncogene | OS |
| JAK2 (134, 135) | JAK-STAT | Myelofibrosis, polycythemia vera, and essential thrombocythemia | Cell cycle regulation | Leukemia | Oncogene | SHS |
| SCN1A (136) | Sodium channel | Dravet syndrome | Neural excitation | – | – | G/MS |
| NLRP3 (137) | Caspase/inflammasome | CINCA syndrome | Inflammasome subunit | – | – | G/MS |
| PORCN (138) | Wnt | Focal dermal hypoplasia | O-acyltransferase | – | – | G/MS |
| PIGA (139) | Hematopoiesis | Paroxysmal nocturnal hemoglobinuria | ER protein processing | Leukemia | – | OS |
Genomic studies implicitly assume that every cell within an individual has the same genome. Family-based genetic studies, genome-wide association studies (GWAS), and exome sequencing analyses have identified numerous common, rare, and de novo germline SNVs and CNVs associated with an increased risk of autism spectrum disorder (ASD), schizophrenia, and bipolar disorder, but each variant only represents a minor component of population-level disease risk (21–24). In general, these approaches sequence the DNA from available clinical samples (e.g., peripheral blood) to interrogate an individual’s germline genome; they do not account for any additional disease risk brought about by somatic mutations that occur during brain development. To address this knowledge gap, the National Institute of Mental Health (NIMH) supported the formation of the Brain Somatic Mosaicism Network (BSMN). Notably, several outstanding reviews have recently discussed how somatic mutations within the brain may contribute to neurological disease [e.g., (2, 25, 26)]. Here, we build on these discussions and highlight how somatic mutations with in the brain may contribute to neuronal diversity. We also evaluate emerging genomic approaches to measure and validate somatic mosaicism and summarize BSMN efforts to generate a large publicly available resource to evaluate the contribution of somatic mosaicism to neuropsychiatric disease (Fig. 1).
Fig. 1. An overview of approaches employed by the BSMN.

The general approach of the BSMN is to identify mosaic variants in primary human brain tissue from large cohorts of neurotypical individuals and neuropsychiatric disease patients. The methods include bulk sequencing of tissues or sorted neurons (top), sequencing of single cells after whole-genome amplification (middle), or clonal expansion from single cells followed by bulk sequencing (bottom). Each method offers a trade-off between sensitivity and specificity.
Mechanisms of somatic mosaicism
DNA damage occurs constantly in every cell in our bodies, and many components of the DNA damage response are essential for neurodevelopment. Single-strand and double-strand DNA breaks, as well as base mutations, arise as a consequence of DNA replication, transcription, epigenetic modification, cellular respiration, and environmental stressors. If the resultant damage is not accurately repaired, DNA mutations can occur that can lead to somatic variation among neurons and other cell types.
The nonhomologous end-joining (NHEJ) pathway of DNA repair is required for neurodevelopment. Mice deficient in NHEJ proteins exhibit extensive NPC apoptosis and often die prenatally (27). Intriguingly, the embryonic lethality and NPC apoptosis phenotypes are rescued in a p53-null mouse background, suggesting that genotoxic stress contributes to lethality (28). Consistent with these data, compound heterozygous mutations in DNA damage response genes [e.g., ataxia telangiectasia mutated (ATM), ataxia telangiectasia-related (ATR), and ATR-interacting protein (ATRIP)] can lead to increased mutational loads, neurodevelopmental brain defects, and neuronal degeneration (29–31). More broadly, deficits in other DNA repair pathways, such as transcription-coupled repair, homologous recombination, and nucleotide excision repair, also can lead to human neurodevelopmental phenotypes (32, 33).
Defects in different DNA repair pathways are associated with distinct somatic mutation profiles. For example, SNVs and indels can arise from errors during base excision repair, nucleotide excision repair, and transcription-coupled repair (33). Moreover, the action of the apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like-3 (APOBEC3) family of cytosine deaminase proteins can lead to cytidine-to-uridine transition mutations on single-strand DNA that, upon replication, lead to guanosine-to-adenosine mutations on the opposing DNA strand (34). Errors made during DNA mismatch repair also can lead to either interspersed SNVs or indels within microsatellite repeat sequences, whereas errors made during double-strand break repair by homologous recombination, NHEJ, or alternative-NHEJ can lead to CNVs (35, 36).
Errors incurred during DNA replication or transcription also can lead to the formation of CNVs. Large, actively transcribed genes that undergo replication during late S-phase correspond to chromosomal fragile sites and are hot spots for the generation of genomic variants and translocations (37, 38). Because neuronal genes are overrepresented among the longest genes in the human genome, transcription may predispose these genes to somatic CNVs (39). Indeed, intragenic deletions within large, neuronally expressed genes (e.g., AUTS2, IMMP2L, NXRN1, and CNTNAP2) are associated with ASD, intellectual disability, and other neurodevelopmental disorders (40, 41). Thus, if individuals harbor somatic CNVs at these loci in many neurons or in neurons within specific functional brain regions, they may be susceptible to neurological disease.
Long interspersed element-1s (LINE-1s or L1s) can mobilize (i.e., retrotranspose) within the brain, leading to another form of somatic variation (42). Active L1s encode two proteins, ORF1p and ORF2p, which are required for retrotransposition. ORF2p contains endonuclease and reverse transcriptase activities that are needed to “copy-and-paste” L1 sequences into a new genomic location by a mechanism termed target-site primed reverse transcription (TPRT) (42, 43). In addition to canonical TPRT, L1s occasionally can integrate into endogenous DNA lesions (44). Moreover, recombination events that arise either during (15, 45–47) or after L1 retrotransposition (48) can lead to the formation of structural variants.
Somatic mutations in human disease
Mosaicism and structural brain abnormalities
One of the most common causes of medically refractory pediatric epilepsy is focal dysplasia of the cerebral cortex. Until recently, the basis of this disorder remained a medical mystery. Genetic studies of the most severe form of focal dysplasia, hemimegalencephaly, in which one entire cerebral hemisphere is enlarged in size, led to the identification of gain-of-function somatic mutations in the phosphatidylinositol-3-kinase (PI3K)–protein kinase B (Akt) and mammalian target of rapamycin (mTOR) signaling pathways (Table 1, Fig. 2). We now know that mutations in mTOR are the single largest contributor to focal dysplasia in pediatric epilepsy (49–51). Similarly, germline mutations in one allele of the TSC1 or TSC2 gene confer susceptibility to tuberous sclerosis, a disease characterized by facial and skin lesions, seizures, intellectual disability, cardiac and renal tumors, and cortical tubers (52). Because the Tsc1 and Tsc2 proteins are negative regulators of the mTOR-signaling pathway, a second somatically acquired mutation is required for disease onset. Somatic mutations that mildly activate the mTOR-signaling pathway also cause symmetrical overgrowth syndromes such as megalencephaly-capillary malformation syndrome, megalencephaly, and certain forms of polymicrogyria (49–51). Common to all of these phenotypes is the presence of hypertrophic neural-like “balloon” cells, which carry the somatic mutation yet fail to transform to a malignant cell type (52).
Fig. 2. An example of brain somatic mosaicism that leads to a focal overgrowth condition.

(A) Axial brain magnetic resonance imaging (MRI) of focal overgrowth of one hemisphere (arrows) from a 2-month-old child with intractable epilepsy and intellectual disability. MRI showed poor differentiation between the gray and white matter with dysplasia of the cortical gyri and sulci (arrows). (B) Brain mapping using high-resolution MRI or functional imaging such as positron emission tomography (PET), together with electrocorticography to fine-map specific epileptic foci, is followed by surgical resection of diseased brain tissue. (C) Histological analysis with hematoxylin/eosin showing characteristic balloon cells (arrows) consisting of large nuclei, distinct nucleoli, and glassy eosinophilic cytoplasm. (D) Immunostained section for phospho-S6 (green), as evidence of increased mTOR pathway activation. Arrows highlight large dysplastic cell showing strongest immunosignal. Scale bar, 50 μm. Bulk tissue sequencing showed somatic activating mutation in the MTOR gene c.6644C>T leading to p.S2215F in 15% of brain cells from the diseased hemisphere. After surgery, the patient showed clinical improvement.
Somatic mutations that inappropriately activate Ras signaling or related signaling pathways can likewise confer proliferation and survival phenotypes to subsets of cells and cause neurological disease. For example, a gain-of-function somatic mutation in GNAQ, encoding G protein subunit alpha q, can lead to Sturge-Weber syndrome, a disease characterized by vascular anomaly in the brain, glaucoma, seizures, stroke, and intellectual disability (53). The same GNAQ mutation, occurring in a different somatic cell type later in development, can cause uveal melanoma (54). Because mutations in certain neurodevelopmental disorders (e.g., neurofibromatosis, tuberous sclerosis, Proteus syndrome, and other neurocutaneous disorders) either activate proto-oncogenes or inactivate tumor suppressor genes, it is not surprising that similar mutations in non-neuronal cell types manifest as cancers. Intriguingly, postmitotic neurons are rarely the source of brain tumors, suggesting that postmitotic neurons may have safeguards that ensure against dedifferentiation and further proliferation.
Relative to germline mutations, somatic mutations can lead to milder cases of heritable neurodevelopmental disorders. For example, somatic mutations in genes involved in neuronal migration are estimated to represent 5 to 10% of de novo mutations and are detected more frequently in patients with unexplained brain malformations when studied with sensitive high-throughput sequencing methods (55). Moreover, somatic mutations within the LIS1 or DCX genes can lead to gross disruptions of neuronal migration, whereas germline mutations in LIS1 or DCX result in lissencephaly (56, 57). Results from several experiments also suggest that somatic mutations that lead to a reduction of gene copy number in migrating neurons can lead to cell-autonomous defects in neuronal migration, with severe epilepsy and intellectual disability as a consequence (56, 57).
ASD and other common neuropsychiatric diseases
Genetic approaches have not yet fully explained the etiology of ASD, bipolar disorder, schizophrenia, or Tourette syndrome. Although gene-by-gene and gene-by-environment interactions could, in principle, account for additional disease risk, somatic mosaicism is another potential mechanism that warrants exploration as a contributor to neuropsychiatric diseases (58).
De novo SNVs and CNVs, particularly loss-of-function mutations, are significant contributors to ASD risk (21, 59–62). In addition to de novo germline mutations, a substantial number of de novo somatic mutations (i.e., ~5.4% of de novo events) are detected in the blood of ASD patients and are enriched in ASD probands (22). Somatic mosaic mutations also have been identified throughout postmortem ASD brains or, in some instances, in more localized areas in ASD brains (59). Evidence of continuous, widespread cortical mismigration, as seen in some mutant mice, has not been reported in the postmortem ASD brain (63, 64). However, NPCs from a subset of ASD patients with enlarged brain volumes are inherently more proliferative and display abnormal neurogenesis when compared to controls (65, 66). Other ASD patients have focal cortical abnormalities, including disorganized neurons and lamina, polymicrogyria, and other local surface malformations (67). Thus, in addition to specific mutations, additional cell cycles may further affect somatic mutational loads in patients.
Prenatal challenges to the immune system in animals (i.e., maternal immune activation) (68) can also lead to many features like those present in ASD brains. Maternal immune activation leads to increased cellular proliferation, brain size, and ASD-like behaviors in animal models (69–72). Intriguingly, an elevated prevalence of MEIs was observed in a primate model of maternal immune activation (73). Elevated MEI levels likewise are observed in schizophrenia (73) and Rett syndrome patients (74), suggesting that somatic MEI burden may play a role in the etiology of some neurodevelopmental and neuropsychiatric diseases.
Methods to detect somatic mutations
The difficulty in detecting a somatic mutation depends on its frequency within a cell population. Whereas mutations affecting a large fraction (e.g., 50%) of cells are readily detected in bulk tissue sequencing experiments and generally result in high-confidence calls, mutations affecting one or a few cells are unlikely to be detected with bulk tissue sequencing approaches. The identification and validation of rare somatic mutations requires sequencing DNA derived from small pools of cells, single cells, or clonally reprogrammed cells followed by robust computational data analyses (Fig. 1).
Bulk tissue approaches
Whole-genome sequencing (WGS) or whole-exome sequencing (WES) of DNA derived from bulk brain tissue allows a straightforward approach to discovering somatic mosaicism (26). WGS and WES minimize sequencing artifacts that can confound downstream analyses and, in the case of WGS, provide an opportunity for identifying a wide range of structural rearrangements, including inversions and translocations. However, WGS and WES using standard sequencing depths have reduced statistical power to detect mutations that occur at low frequencies (i.e., <10% of cells in a population at 30 to 100x coverage). Although increasing sequence coverage allows detection of somatic variants at lower frequencies, it quickly becomes cost prohibitive. Moreover, WGS and WES do not provide information on how somatic variants are distributed across individual cell lineages within a bulk tissue sample.
Sorted-pools approaches
Fluorescence-activated cell or nuclei sorting (FACS/FANS) can be used to isolate specific neural populations (e.g., NeuN+ neurons versus NeuN− cells or cortical inhibitory interneurons versus excitatory principal neurons). Analysis of sorted nuclei populations (e.g., 5000 or 500,000 cells) from specific brain regions increases the power to detect somatic mosaicism that arises in one lineage, because these genomes are no longer diluted by genomes derived from other lineages. Independent pools of sorted nuclei can then be subjected to RNA sequencing (RNA-seq) and quantitative reverse transcription polymerase chain reaction (qRT-PCR) to confirm cell type–specific gene expression profiles (75). In addition to increasing the power for detecting a somatic mutation, cell sorting before DNA extraction could yield information about the embryological origin and developmental trajectory of somatic variation across the brain. Large pools of sorted cells can yield enough DNA for the direct examination of somatic variants by WGS or WES. However, smaller pool sizes will only generate small amounts of DNA; thus, they are best suited for generating PCR amplicon libraries (e.g., as used in MEI detection and other targeted sequencing) or for subsequent whole-genome amplification (WGA).
Single-cell approaches
WGA can be used to analyze the genomes of single neurons (26). The spectrum of mutations identified from the genomes of single neurons can then be compared to germline variants in bulk tissue data derived from a non-neuronal control (e.g., brain dural fibroblasts or heart) to identify candidate somatic mutations (5). WGA approaches already are used in pre-implantation genetic screening of embryos (76, 77) and include (i) degenerate-oligonucleotide-primed PCR (DOP-PCR), (ii) multiple displacement amplification (MDA), and (iii) multiple annealing and looping-based amplification (MALBAC). Each method has its advantages and drawbacks. In general, DOP-PCR provides coverage evenly across the genome, which facilitates the detection of large CNVs and chromosomal aneuploidies. However, DOP-PCR has a higher read duplication rate, lower mapping rate, and lower recovery rate when compared with MDA and MALBAC (78) and is cost prohibitive for SNV, indel, and MEI detection. By comparison, MDA yields a high rate of artificial chimeric DNA molecules that can lead to false-positive calls in downstream analyses (79), whereas MALBAC exhibits reduced coverage of certain genomic regions (14, 16, 80), especially those rich in repetitive sequences (78). Considerable advances have recently been made in detecting SNVs (81, 82), CNVs (83), and MEIs (16) in WGA samples; however, best practices necessitate evaluating each WGA approach for the detection of specific types of somatic mosaicism.
Clonal expansion of single cells using human-induced pluripotent stem cell (hiPSC) technology or somatic cell nuclear transfer (SCNT) provides a biological alternative to WGA (80, 84). Any variant uniformly identified in the clonal line, but not in controls, represents a candidate somatic mutation that requires confirmation in the tissue of origin. In contrast, mutations introduced during cell culture will be present in a lower frequency of cells within a clonal cell line and can be discriminated from bona fide somatic mutations in downstream computational analysis. Although the clonal isolation and expansion of primary human neural stem and progenitor cells is possible, the analysis of human neuronal genomes using clonal reprogramming has several limitations. Foremost among these is the availability of live human neurons. Moreover, neither clonal reprogramming nor SCNT have been reported using human neurons; SCNT is further limited by the expense and availability of human oocytes. Finally, reprogramming approaches currently are only successful in ~10% of cells; thus, any neurons harboring highly aberrant genomes may be refractory to reprogramming. Despite these caveats, clonal reprogramming of human neurons is theoretically possible. In addition, it is noteworthy that mouse neurons reprogrammed by SCNT contain genomic rearrangements (e.g., kataegis and chromothripsis) that would be very challenging to validate using current WGA approaches (84).
Computational methods for mutation detection
WGS and WES have been used successfully to detect somatic SNVs in family-based studies of Mendelian disease and large-scale sequencing studies of human patient cohorts (2). To identify SNVs, most computational approaches compare call sets generated from an affected sample to those generated from a matched healthy/unaffected sample and/or a control population. These comparisons allow the identification and subsequent exclusion of germline polymorphisms from downstream analyses; however, care must be taken to ensure that any candidate somatic mutations are not germline variants that were missed in the matched control. In general, variant callers initially developed to detect mutations in cancer offer higher sensitivity for detecting mosaic SNVs when compared with standard approaches used to detect germline variants (85, 86).
Somatic CNVs can be detected by identifying deviations either from the expected depth of sequence or in the expected distances between paired-end sequencing reads. Similarly, inversions can be identified through differences in the orientations of paired-end sequencing reads. Numerous approaches have been developed to identify CNVs from WGS (7, 87–89), and most can be applied directly to identify somatic mutations. For example, recent studies using WGA in conjunction with WGS have identified megabase-scale de novo CNVs in human and mouse neurons based on differences in read-depth across genomic bins (6–9). CNVs are more difficult to identify using WES due to the biases encountered during the capture of target exons (90).
Somatic MEIs can be detected from bulk tissue, PCR amplicons generated from sorted-cell fractions, or single-cell WGA DNA using split-read and paired-end information (e.g., one paired-end read may map to the reference genome, whereas another may map to a MEI) (91, 92). Detecting low-frequency MEIs with fewer supporting reads requires careful bioinformatic analyses that can distinguish signal from noise, followed by experimental validation with orthogonal methods (14, 93). The analysis of single-cell data remains challenging due to the presence of chimeras generated during WGA (14, 16, 94); thus, care must be taken in calling MEIs.
Validation of somatic mutations
It is essential to validate all candidate somatic mutations. False-positive calls can arise from DNA sequencing errors, contamination with germline variants, chimeric molecules generated during single-cell WGA, PCR-induced nucleotide substitutions, and the failure to amplify certain genomic regions. False-negative calls are dependent on the allele frequency of the somatic mutation within the sample, the type of mutation, and the method of detection. Orthologous experimental methods are required to eliminate false-positives and to calibrate the confidence of detection for different types of somatic mutations. Validation experiments can then be performed on either the tissue of origin or amplified material used to discover the variant. The first approach represents a biological validation, which establishes the presence of a variant call in unamplified DNA from the source sample. The second approach represents a technical validation, which establishes the presence/absence of variant calls in the DNA source material used for discovery.
Biological/primary validation in the tissue of origin
Validation on unamplified DNA from the tissue of origin provides confirmation that a candidate call is a genuine somatic variant and rules out the possibility that it corresponds to a DNA amplification artifact or a mutation that occurred during clonal expansion. Biological validation requires a variant to be present in multiple cells in the tissue of origin at a frequency above experimental detection limits. As such, the failure to validate a variant in the tissue of origin does not necessarily represent a false call. For example, only ~50% of CNVs manifested in hiPSC clones could be directly confirmed in the primary fibroblast cells used to derive hiPSCs (80).
Somatic variants can be confirmed in unamplified cell source material by (i) targeted DNA capture followed by high-coverage (>100x) DNA resequencing, (ii) high-coverage sequencing of multiplexed PCR amplicons, and (iii) droplet digital PCR (ddPCR). These approaches vary in throughput and sensitivity. Targeted DNA capture and resequencing can require the creation of several thousand custom oligonucleotides designed to capture the genomic DNA either including or surrounding the putative variants. The captured DNA then is subjected to high-coverage paired-end DNA sequencing, yielding a typical sensitivity of variant detection in greater than 1% of cells. Amplicon sequencing involves PCR amplification of candidate loci followed by high-coverage paired-end DNA sequencing, yielding a typical sensitivity of variant detection in greater than 0.1% of cells. Finally, ddPCR involves partitioning a DNA sample into large numbers of individual droplets that generally contain one copy of template DNA. PCR takes place within these droplets, leading to the production of a fluorescent readout, either through the use of an intercalating dye or a fluorescent oligomer probe, to indicate the presence or absence of the PCR target of interest. Subsequent quantification of the fluorescent droplets allows a determination of the number of copies of the target locus present in the sample, yielding a typical sensitivity of variant detection in greater than 0.001% of cells (95). Although extremely sensitive, ddPCR requires the optimization of primers, probes, and amplification conditions, which is time-consuming and limits throughput.
The goal when employing biological validation procedures is to detect putative somatic variants and to assess, as precisely as possible, the frequency of each variant in that tissue of origin. Biological validation can (i) determine whether certain individuals in the population are more prone to somatic variation than others, (ii) investigate whether different areas of the brain and/or specific brain cell types have varying amounts and types of particular forms of somatic variation, (iii) assess whether developmental timing contributes to somatic variation, and (iv) reveal whether somatic variations increase as a function of the number of cell divisions and/or a function of age in postmitotic neurons.
Technical validation on source/amplified material
If a somatic variant is only present in a single cell, it will be impossible to validate in bulk tissue. Likewise, a variant present in very few cells may be difficult to validate in the tissue of origin. Thus, technical validation in the source DNA used to discover a putative variant can be used to determine whether a call is true or false. Technical validation typically employs PCR, qPCR, and Sanger sequencing of the locus in the DNA source material (e.g., WGA DNA or DNA from a clonal cell population). Multiple true/false verdicts form the basis for estimating false-discovery and false-negative rates in the resultant call sets.
Present understanding of the prevalence of somatic mutation in neurotypical individuals
Recent studies revealed that mosaic neuronal genomes are the rule, rather than the exception; every neuron probably has a different genome than the neurons with which it forms synapses. Not unexpectedly, SNVs are the most prevalent somatic mutations. A “triple calling” strategy was used to identify and validate clonal SNVs in MDA-amplified DNA from single neurons isolated from a neurotypical brain, leading to estimates of ~1000 to 1500 SNVs per neuronal genome (5). By comparison to human cortical neurons, a SCNT experiment in reprogrammed mouse olfactory neurons detected hundreds of SNVs per neuron and a lower proportion of C-to-T transition mutations (84). Although the divergent SNV rates between these two studies may arise from technical differences (as discussed above), both approaches establish that SNVs represent an important form of somatic mutation in both human and mouse neurons.
Brain somatic CNVs initially were identified by comparing the sequences of bulk DNA derived from multicellular samples of different brain regions to the sequences of DNA derived from somatic tissues (96, 97). The first single-cell study of neuronal CNVs analyzed 110 human frontal cortex neurons and found that 13 to 41% of the neurons contained at least one megabase-scale de novo CNV (6). Additional studies, which analyzed fewer neuronal genomes, confirmed that de novo CNVs occur in at least 10% of neurons (7, 8). CNVs can be shared by multiple neurons and inherited in a clonal manner (8). Furthermore, megabase-scale CNVs typically alter the copy number of 10 or more genes in individual neurons. In addition to expression-level differences that can accompany gene copy number changes, mosaic neuronal CNVs also are expected to reveal or abate pernicious alleles on a neuron-by-neuron basis in every individual.
L1 retrotransposon insertions alter the transcriptional regulation of genes in myriad ways (42). Initial studies used engineered L1s containing a retrotransposition indicator cassette to discover MEI activity in mouse brain (98) and in human NPCs in vitro (99). Studies of MDA-amplified NeuN-positive nuclei isolated from a neurotypical human brain, followed by L1-transposon profiling (13) or WGS (15, 16), have since suggested that 0.2 to 1 L1 insertion occur per neuronal genome. Another report, which employed MALBAC WGA in conjunction with L1 capture technology (RC-seq), reported an average of 13 L1 insertions in every neuronal genome (11), although a subsequent study suggested a high false-positive rate in these data (14). By comparison, SCNT experiments in mouse olfactory neurons reported ≤1.3 MEI per neuronal genome (84). An extrapolation of these data indicates that potentially billions of neurons in the neurotypical brain contain de novo MEIs. Additional studies are required to determine whether L1s retrotranspose at varying rates in different brain regions, in different individuals, or preferentially insert into expressed genes, and whether other mobile elements [e.g., Alu retrotransposons (42)] also contribute to intra-individual neuronal genetic diversity.
Generation of a community resource
The BSMN will generate comprehensive maps of somatic genomic variation in neurotypical and diseased human brains, including a prioritized call set of confirmed somatic variants (Box 1) that may contribute to neuropsychiatric disease and epilepsy. Functional validation experiments will be performed using CRISPR/Cas9-mediated genome engineering, hiPSC-based neurogenesis, and mosaic mouse models generated by in utero electroporation (Fig. 3). The BSMN is initially determining concordance among disparate sequencing and bioinformatic approaches by performing a “common experiment” in which pulverized tissue from one neurotypical individual in the Lieber brain repository has been distributed to all of the working groups for independent assessment of mosaicism.
Box 1. Criteria used to prioritize somatic variants for functional characterization.
Absence from the germ line
We will focus on variants with a definitive somatic origin.
Recurrence and frequency of somatic variation at the locus of interest
We will prioritize loci at which somatic variations, across all types, recur in multiple disease samples but not in control samples.
Mutation severity
Highly deleterious variations will be prioritized for likely functional importance.
Intersection with known disease loci and biochemical pathways
Taking advantage of data on germline variations in brain disorders, we will prioritize loci that have been previously implicated in disease.
Intersection with brain expression and epigenomic data
Taking advantage of large, publicly funded consortia of human brain spatiatemporal expression data (e.g., BrainSpan) and epigenomic data (e.g., PsychENCODE and Roadmap Epigenomics), we will select genes that are expressed in brain regions associated with brain disorders and noncoding loci with potential regulatory function.
Fig. 3. A potential strategy to determine functional consequences of mosaic variants.

In utero electroporation (IUE) transfects a subpopulation of cortical neurons within a local area and will be combined with genome editing to generate mosaic mouse models for functional analysis. For example, a red fluorescent construct (CAG-TdTom) is shown labeling a transfected subset of neurons, shown in the context of a coronal brain section in which nuclei are stained blue with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar, 500 μm.
The BSMN will generate an estimated 10,000 sequencing data sets that comprise >600 terabytes of data and facilitate data-sharing through the BSMN Knowledge Portal (www.synapse.org/bsmn) and the NIMH Data Archive (https://data-archive.nimh.nih.gov). Coordinated analyses with data derived from some of the same brain samples by the CommonMind (www.synapse.org/cmc) and PsychENCODE (www.synapse.org/pec) initiatives may elucidate the effect of somatic mosaicism on tissue-wide gene expression. Data generated though the BSMN initiative will be released to the broader research community on an ongoing basis through a controlled-access mechanism that follows NIH policies and regulatory requirements.
Supplementary Material
Acknowledgments
We thank T. Insel for initiating this project, L. Bingaman for ongoing administrative assistance, and N. Leff and M. L. Gage for copyediting assistance. J.M.K acknowledges support provided by the Pew Biomedical Scholars Award. J.V.M. is an inventor on patent application 6150160, held by the John Hopkins University and the Trustees of the University of Pennsylvania, which covers the compositions and methods of use of mammalian retrotransposons. We also acknowledge the support of NIH R01 MH100914, Genomic mosaicism in developing human brain (F.M.V.). Some figures use images from the Servier Medical Art PowerPoint Image Bank. All the work was supported by U01MH106883, U01MH106874, U01MH106893, U01MH106892, U01MH106882, U01MH106876, U01MH1068898, U01MH106891, and U01MH106884. We regret that space constraints limit the number of references and apologize to many colleagues whose very valuable contributions to this field are not cited.
Footnotes
www.sciencemag.org/content/356/6336/eaal1641/suppl/DC1
Brain Somatic Mosaicism Network Listing
REFERENCES AND NOTES
- 1.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freed D, Stevens EL, Pevsner J. Somatic mosaicism in the human genome. Genes (Basel) 2014;5:1064–1094. doi: 10.3390/genes5041064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank SA. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: Mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci USA. 2010;107(suppl 1):1725–1730. doi: 10.1073/pnas.0909343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lupski JR. Genome mosaicism—One human, multiple genomes. Science. 2013;341:358–359. doi: 10.1126/science.1239503. [DOI] [PubMed] [Google Scholar]
- 5.Lodato MA, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–98. doi: 10.1126/science.aab1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConnell MJ, et al. Mosaic copy number variation in human neurons. Science. 2013;342:632–637. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knouse KA, Wu J, Amon A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 2016;26:376–384. doi: 10.1101/gr.198937.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai X, et al. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Reports. 2014;8:1280–1289. doi: 10.1016/j.celrep.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rehen SK, et al. Constitutional aneuploidy in the normal human brain. J Neurosci. 2005;25:2176–2180. doi: 10.1523/JNEUROSCI.4560-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yurov YB, et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLOS ONE. 2007;2:e558. doi: 10.1371/journal.pone.0000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Upton KR, et al. Ubiquitous L1 mosaicism in hippocampal neurons. Cell. 2015;161:228–239. doi: 10.1016/j.cell.2015.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baillie JK, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011;479:534–537. doi: 10.1038/nature10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evrony GD, et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151:483–496. doi: 10.1016/j.cell.2012.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evrony GD, Lee E, Park PJ, Walsh CA. Resolving rates of mutation in the brain using single-neuron genomics. eLife. 2016;5:e12966. doi: 10.7554/eLife.12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erwin JA, et al. L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat Neurosci. 2016;19:1583–1591. doi: 10.1038/nn.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evrony GD, et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron. 2015;85:49–59. doi: 10.1016/j.neuron.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lui JH, Hansen DV, Kriegstein AR. Development and evolution of the human neocortex. Cell. 2011;146:18–36. doi: 10.1016/j.cell.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macia A, et al. Engineered LINE-1 retrotransposition in nondividing human neurons. Genome Res. 2017;27:335–348. doi: 10.1101/gr.206805.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muotri AR, Gage FH. Generation of neuronal variability and complexity. Nature. 2006;441:1087–1093. doi: 10.1038/nature04959. [DOI] [PubMed] [Google Scholar]
- 20.Bushman DM, Chun J. The genomically mosaic brain: Aneuploidy and more in neural diversity and disease. Semin Cell Dev Biol. 2013;24:357–369. doi: 10.1016/j.semcdb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freed D, Pevsner J. The contribution of mosaic variants to autism spectrum disorder. PLOS Genet. 2016;12:e1006245. doi: 10.1371/journal.pgen.1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malhotra D, Sebat J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–1241. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341:1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee JH. Somatic mutations in disorders with disrupted brain connectivity. Exp Mol Med. 2016;48:e239. doi: 10.1038/emm.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekiguchi JM, et al. Nonhomologous end-joining proteins are required for V(D)J recombination, normal growth, and neurogenesis. Cold Spring Harb Symp Quant Biol. 1999;64:169–181. doi: 10.1101/sqb.1999.64.169. [DOI] [PubMed] [Google Scholar]
- 28.Frank KM, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/S1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 29.McConnell MJ, et al. Failed clearance of aneuploid embryonic neural progenitor cells leads to excess aneuploidy in the Atm-deficient but not the Trp53-deficient adult cerebral cortex. J Neurosci. 2004;24:8090–8096. doi: 10.1523/JNEUROSCI.2263-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coufal NG, et al. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci USA. 2011;108:20382–20387. doi: 10.1073/pnas.1100273108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iourov IY, Vorsanova SG, Liehr T, Kolotii AD, Yurov YB. Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum Mol Genet. 2009;18:2656–2669. doi: 10.1093/hmg/ddp207. [DOI] [PubMed] [Google Scholar]
- 32.Shen J, et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet. 2010;42:245–249. doi: 10.1038/ng.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224–238. doi: 10.1038/nrg.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 35.Arlt MF, Wilson TE, Glover TW. Replication stress and mechanisms of CNV formation. Curr Opin Genet Dev. 2012;22:204–210. doi: 10.1016/j.gde.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei PC, et al. Long neural genes harbor recurrent DNA break clusters in neural stem/progenitor cells. Cell. 2016;164:644–655. doi: 10.1016/j.cell.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson TE, et al. Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 2015;25:189–200. doi: 10.1101/gr.177121.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.King IF, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zylka MJ, Simon JM, Philpot BD. Gene length matters in neurons. Neuron. 2015;86:353–355. doi: 10.1016/j.neuron.2015.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabel HW, et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522:89–93. doi: 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richardson SR, et al. The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr. 2015;3:MDNA3-0061-2014. doi: 10.1128/microbiolspec.MDNA3-0061-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 44.Morrish TA, et al. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet. 2002;31:159–165. doi: 10.1038/ng898. [DOI] [PubMed] [Google Scholar]
- 45.Gilbert N, Lutz S, Morrish TA, Moran JV. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol. 2005;25:7780–7795. doi: 10.1128/MCB.25.17.7780-7795.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Symer DE, et al. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/S0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- 47.Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/S0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 48.Chen JM, Stenson PD, Cooper DN, Férec C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum Genet. 2005;117:411–427. doi: 10.1007/s00439-005-1321-0. [DOI] [PubMed] [Google Scholar]
- 49.Mirzaa GM, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 2016;73:836–845. doi: 10.1001/jamaneurol.2016.0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mirzaa GM, et al. Characterisation of mutations of the phosphoinositide-3-kinase regulatory subunit, PIK3R2, in perisylvian polymicrogyria: A next-generation sequencing study. Lancet Neurol. 2015;14:1182–1195. doi: 10.1016/S1474-4422(15)00278-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rivière JB, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henske EP, JóŸwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035. doi: 10.1038/nrdp.2016.35. [DOI] [PubMed] [Google Scholar]
- 53.Shirley MD, et al. Sturge-Weber syndrome and port-stains caused by somatic mutation in. GNAQ N Engl J Med. 2013;368:1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Raamsdonk CD, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jamuar SS, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733–743. doi: 10.1056/NEJMoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sicca F, et al. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology. 2003;61:1042–1046. doi: 10.1212/WNL.61.8.1042. [DOI] [PubMed] [Google Scholar]
- 57.Gleeson JG. Classical lissencephaly and double cortex (subcortical band heterotopia): LIS1 and doublecortin. Curr Opin Neurol. 2000;13:121–125. doi: 10.1097/00019052-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Insel TR. Brain somatic mutations: The dark matter of psychiatric genetics? Mol Psychiatry. 2014;19:156–158. doi: 10.1038/mp.2013.168. [DOI] [PubMed] [Google Scholar]
- 59.D’Gama AM, et al. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron. 2015;88:910–917. doi: 10.1016/j.neuron.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hutsler JJ, Casanova MF. Review: Cortical construction in autism spectrum disorder: Columns, connectivity and the subplate. Neuropathol Appl Neurobiol. 2016;42:115–134. doi: 10.1111/nan.12227. [DOI] [PubMed] [Google Scholar]
- 64.Donovan AP, Basson MA. The neuroanatomy of autism - A developmental perspective. J Anat. 2017;230:4–15. doi: 10.1111/joa.12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marchetto MC, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mariani J, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell. 2015;162:375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stoner R, et al. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014;370:1209–1219. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi GB, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–939. doi: 10.1126/science.aad0314. doi:3310.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith SE, Elliott RM, Anderson MP. Maternal immune activation increases neonatal mouse cortex thickness and cell density. J Neuroimmune Pharmacol. 2012;7:529–532. doi: 10.1007/s11481-012-9372-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26:607–616. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soumiya H, Fukumitsu H, Furukawa S. Prenatal immune challenge compromises development of upper-layer but not deeper-layer neurons of the mouse cerebral cortex. J Neurosci Res. 2011;89:1342–1350. doi: 10.1002/jnr.22636. [DOI] [PubMed] [Google Scholar]
- 73.Bundo M, et al. Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron. 2014;81:306–313. doi: 10.1016/j.neuron.2013.10.053. [DOI] [PubMed] [Google Scholar]
- 74.Muotri AR, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468:443–446. doi: 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lake BB, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vermeesch JR, Voet T, Devriendt K. Prenatal and pre-implantation genetic diagnosis. Nat Rev Genet. 2016;17:643–656. doi: 10.1038/nrg.2016.97. [DOI] [PubMed] [Google Scholar]
- 77.Yan L, et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next-generation sequencing with linkage analyses. Proc Natl Acad Sci USA. 2015;112:15964–15969. doi: 10.1073/pnas.1523297113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hou Y, et al. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. Gigascience. 2015;4:37. doi: 10.1186/s13742-015-0068-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lasken RS, Stockwell TB. Mechanism of chimera formation during the multiple displacement amplification reaction. BMC Biotechnol. 2007;7:19. doi: 10.1186/1472-6750-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abyzov A, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492:438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roth A, et al. Clonal genotype and population structure inference from single-cell tumor sequencing. Nat Methods. 2016;13:573–576. doi: 10.1038/nmeth.3867. [DOI] [PubMed] [Google Scholar]
- 82.Zafar H, Wang Y, Nakhleh L, Navin N, Chen K. Monovar: Single-nucleotide variant detection in single cells. Nat Methods. 2016;13:505–507. doi: 10.1038/nmeth.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garvin T, et al. Interactive analysis and assessment of single-cell copy-number variations. Nat Methods. 2015;12:1058–1060. doi: 10.1038/nmeth.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hazen JL, et al. The complete genome sequences, unique mutational spectra, and developmental potency of adult neurons revealed by cloning. Neuron. 2016;89:1223–1236. doi: 10.1016/j.neuron.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koboldt DC, et al. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao X, Emery SB, Myers B, Kidd JM, Mills RE. Resolving complex structural genomic rearrangements using a randomized approach. Genome Biol. 2016;17:126. doi: 10.1186/s13059-016-0993-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xi R, Lee S, Xia Y, Kim T-M, Park PJ. Copy number analysis of whole-genome data using BIC-seq2 and its application to detection of cancer susceptibility variants. Nucleic Acids Res. 2016;44:6274–6286. doi: 10.1093/nar/gkw491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol. 2014;15:R84. doi: 10.1186/gb-2014-15-6-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Meynert AM, Ansari M, FitzPatrick DR, Taylor MS. Variant detection sensitivity and biases in whole genome and exome sequencing. BMC Bioinformatics. 2014;15:247. doi: 10.1186/1471-2105-15-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee E, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337:967–971. doi: 10.1126/science.1222077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stewart C, et al. A comprehensive map of mobile element insertion polymorphisms in humans. PLOS Genet. 2011;7:e1002236. doi: 10.1371/journal.pgen.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Richardson SR, Morell S, Faulkner GJ. L1 retrotransposons and somatic mosaicism in the brain. Annu Rev Genet. 2014;48:1–27. doi: 10.1146/annurev-genet-120213-092412. [DOI] [PubMed] [Google Scholar]
- 94.Zhang CZ, et al. Calibrating genomic and allelic coverage bias in single-cell sequencing. Nat Commun. 2015;6:6822. doi: 10.1038/ncomms7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Piotrowski A, et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 97.O’Huallachain M, Karczewski KJ, Weissman SM, Urban AE, Snyder MP. Extensive genetic variation in somatic human tissues. Proc Natl Acad Sci USA. 2012;109:18018–18023. doi: 10.1073/pnas.1213736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muotri AR, et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 99.Coufal NG, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kurek KC, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee JH, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lindhurst MJ, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928–933. doi: 10.1038/ng.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rivière JB, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 105.Lindhurst MJ, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hussain K, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Poduri A, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lim JS, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. doi: 10.1038/nm.3824. [DOI] [PubMed] [Google Scholar]
- 109.Bar-Peled L, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baulac S, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol. 2015;77:675–683. doi: 10.1002/ana.24368. [DOI] [PubMed] [Google Scholar]
- 111.European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-Z. [DOI] [PubMed] [Google Scholar]
- 112.van Slegtenhorst M, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 113.Bourdeaut F, et al. Mosaicism for oncogenic G12D KRAS mutation associated with epidermal nevus, polycystic kidneys and rhabdomyosarcoma. J Med Genet. 2010;47:859–862. doi: 10.1136/jmg.2009.075374. [DOI] [PubMed] [Google Scholar]
- 114.Gantner S, et al. Absence of BRAF and HRAS mutations in eruptive Spitz naevi. Br J Dermatol. 2011;164:873–877. doi: 10.1111/j.1365-2133.2011.10210.x. [DOI] [PubMed] [Google Scholar]
- 115.Hafner C, et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc Natl Acad Sci USA. 2007;104:13450–13454. doi: 10.1073/pnas.0705218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hafner C, et al. Mosaicism of activating FGFR3 mutations in human skin causes epidermal nevi. J Clin Invest. 2006;116:2201–2207. doi: 10.1172/JCI28163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Papp T, et al. Mutational analysis of the N-ras, p53, p16INK4a, CDK4, and MC1R genes in human congenital melanocytic naevi. J Med Genet. 1999;36:610–614. [PMC free article] [PubMed] [Google Scholar]
- 118.Pollock PM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- 119.Rouleau GA, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neurofibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 120.Cawthon RM, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62:193–201. doi: 10.1016/0092-8674(90)90253-B. [DOI] [PubMed] [Google Scholar]
- 121.Garcia-Linares C, et al. Dissecting loss of heterozygosity (LOH) in neurofibromatosis type 1-associated neurofibromas: Importance of copy neutral LOH. Hum Mutat. 2011;32:78–90. doi: 10.1002/humu.21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Messiaen L, et al. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1) Hum Mutat. 2011;32:213–219. doi: 10.1002/humu.21418. [DOI] [PubMed] [Google Scholar]
- 123.Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33–40. doi: 10.1093/oxfordjournals.aje.a010118. [DOI] [PubMed] [Google Scholar]
- 124.Wallace MR, et al. Type 1 neurofibromatosis gene: Identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–186. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 125.Groesser L, et al. BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J Invest Dermatol. 2016;136:481–486. doi: 10.1038/JID.2015.376. [DOI] [PubMed] [Google Scholar]
- 126.Groesser L, et al. KRAS, HRAS and egfr mutations in sporadic sebaceous gland hyperplasia. Acta Derm Venereol. 2016;96:737–741. doi: 10.2340/00015555-2351. [DOI] [PubMed] [Google Scholar]
- 127.Niemela JE, et al. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117:2883–2886. doi: 10.1182/blood-2010-07-295501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Takagi M, et al. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2011;117:2887–2890. doi: 10.1182/blood-2010-08-301515. [DOI] [PubMed] [Google Scholar]
- 129.Shirley MD, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thomas AC, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136:770–778. doi: 10.1016/j.jid.2015.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Couto JA, et al. A somatic MAP3K3 mutation is associated with verrucous venous malformation. Am J Hum Genet. 2015;96:480–486. doi: 10.1016/j.ajhg.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Patten JL, et al. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. N Engl J Med. 1990;322:1412–1419. doi: 10.1056/NEJM199005173222002. [DOI] [PubMed] [Google Scholar]
- 133.Schwindinger WF, Francomano CA, Levine MA. Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc Natl Acad Sci USA. 1992;89:5152–5156. doi: 10.1073/pnas.89.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Baxter EJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)74230-6. [DOI] [PubMed] [Google Scholar]
- 135.Kralovics R, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 136.Depienne C, et al. Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. J Med Genet. 2010;47:404–410. doi: 10.1136/jmg.2009.074328. [DOI] [PubMed] [Google Scholar]
- 137.Tanaka N, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: Results of an International Multicenter Collaborative Study. Arthritis Rheum. 2011;63:3625–3632. doi: 10.1002/art.30512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Grzeschik KH, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet. 2007;39:833–835. doi: 10.1038/ng2052. [DOI] [PubMed] [Google Scholar]
- 139.Takeda J, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73:703–711. doi: 10.1016/0092-8674(93)90250-T. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.