

Abstract
Fucosyltransferase VII (FUT7) is one of a1,3-fucosyltransferases family that catalyzes the final fucosylation step in the synthesis of Lewis antigens and generates a unique glycosylated product sialyl Lewis X (sLeX). sLeX can serve as ligands for E- or P-selectin expressed on the cell surface and results in cancer metastasis and angiogenesis. However, the molecular biological mechanisms of FUT7 elevation in neoplastic cells are still largely unknown. In this study, we examined the impact of FUT7 on cell proliferation and migration in A549 cells by colony formation assay, cell cycle assay, gelatin zymography, wound-healing assay, transwell invasion assay and Western blot. In addition, we identified that FUT7 activated EGFR/AKT/mTOR signal pathway that correlated with sLeX augmentation. In conclusion, FUT7 overexpression augments sLeX synthesis to trigger cell proliferation via the activation of EGFR/AKT/mTOR signaling pathway, which indicated that FUT7 may be a potential therapeutic target for epithelial cancers with a high expression of FUT7 and sLeX.
Keywords: fucosyltransferase VII, epidermal growth factor receptor, lung cancer, proliferation, signal pathway
Introduction
Lung cancer is a high malignant carcinoma that has been reported to be the leading cause of cancer death in the US.1 Despite the advanced diagnostic techniques for early detection of lung cancer, the prognosis of lung cancer in patients is still unsatisfactory. The mortality remains high even among patients with early-stage lung cancer.2 Because of the limited knowledge of lung cancer, the therapeutic technology still needs further development. Therefore, the identification of molecules related to the progression of lung cancer will facilitate the development of therapeutic strategies for lung cancer.
It has been reported that overexpression or mutation of EGFR induced excessive proliferation of cells in lung cancer.3,4 Activation of EGFR signaling promotes cell proliferation, migration and adhesion via stimulating the downstream signal pathways, such as RAS/BRAF/MEK/ERK, PI3K/AKT/mTOR, JAK/STAT and PLC/PKC.5,6 EGF and TGF-α secreted by tumor cells combining with abnormal expression of EGFR activate the downstream signaling pathway that results in tumor malignant behavior and poor prognosis.7 In view of the important role of EGFR in the progression of lung cancer, the targeted therapy has become a new therapeutic method and achieved positive outcomes. EGFR-targeting drugs are widely used for clinical application, yet with unsatisfactory therapeutic effect. Therefore, searching for an effective EGFR-sensitizing target may enhance the therapeutic effect of EGFR-targeted therapy and improve the prognosis of lung cancer patients.
Glycosylation is a common post-translational regulator of eukaryotic gene expression. The synthesis of carbohydrate antigens is catalyzed by glycosyltransferases. a1,3-Fucosyltransferases (a1,3-FUTs) catalyze the final fucosylation step in the synthesis of Lewis antigens, which are a series of sugar chains derived from Galβ1-3/1-4GlcNac backbone.8 Six members of this family (FUT3, 4, 5, 6, 7 and 9) have already been cloned and identified.9–12 Each enzyme has a specific acceptor-substrate binding pattern and generates a unique range of glycosylated products. Lewis antigens are mainly located at the outer chains of glycolipids and the O- and N-linked sugar chains on the cell surface of glycoproteins.13 Fucosyltransferase VII (FUT7) catalyzes the synthesis of sialyl Lewis X (sLeX; NeuNAcα2-3Galβ1-4[Fucα1-3]GlcNAc), which is its only product.14 sLeX can serve as ligand for E- or P-selectin expressed on the cell surface and mediates the adhesion of malignant cells that result in cancer metastasis and angiogenesis.15
Ogawa et al16 first showed that FUT7 related to metastasis and remained a statistically significant prognostic factor in lung cancer. In early meta-analysis, we showed that sLeX overexpression was associated with tumor metastasis, recurrence and overall survival in cancer patients and played an important role in cancer prognosis.17 Farhan et al have shown that anti-Lewis Y antibody inhibited the EGF-stimulated EGFR signaling in A431 cells and prevented cell peoliferation.18 Downregulation of FUT3 and FUT6 caused inhibition of colon carcinoma cell proliferation.32 However, the regulation of FUT7 expression and its function in the proliferation of non-small lung cancer cells are largely unknown. The present study further identifies the correlation between FUT7 and EGFR in lung cancer cells.
Materials and methods
Reagents
Kinase inhibitor LY294002 was purchased from Beyotime Institute of Biotechnology (Shanghai, China). CCK-8 kit was purchased from Dojindo Molecular Technologies (Kumamoto, Japan). FUT7 expression construct and the blank vector were designed and synthesized by GenePharma (Shanghai, China). anti-AKT, anti-EGFR, anti-mTOR and anti-PCNA antibodies were purchased from Proteintech (Wuhan, China); anti-pEGFR, anti-pAKT (ser473) and anti-pmTOR (ser2448) antibodies were purchased from Bioworld Technology, Inc (St Louis Park, MN, USA); anti-FUT7 monoclonal antibody was purchased from Sigma-Aldrich Co. (St Louis, MO, USA) and anti-sLeX antibody was purchased from Merck Millipore (Billerica, MA, USA).
Cell culture and transfection
FUT7 overexpression plasmid pIRES2-EGFP-FUT7 (GenePharma) was constructed following the manufacturer’s instructions. The human lung cancer cell line (A549) was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). A549 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Hyclone, Logan, Utah, USA) supplemented with 10% fetal bovine serum, 100 IU/mL penicillin and 100 μg/mL streptomycin at 37°C with 5% CO2. pIRES2-EGFP-FUT7 was transiently transfected into A549 cells using 0.1 μg (low dose, FUT7 cDNA-1) or 0.4 μg (high dose, FUT7 cDNA-2) of plasmids in the presence of 2 μL of Lipofectamine reagent per manufacturer’s instructions.
Colony formation assay
Cells with different treatments were seeded in six-well plates at an initial cell density of 200 cells/well. Colonies were allowed to form for 2 weeks and stained with 0.05% crystal violet after methanol fixation.
Analysis of cell cycle
Cells were cultured in six-well plates and allowed to grow to 75%–80% confluency. Nonadherent cells were removed by gentle washing with PBS. Cells were trypsinized, collected and washed twice with PBS. Cell pellets were resuspended in 0.5 mL of PBS and fixed in 4.5 mL of 70% ethanol overnight. Cells were collected by centrifugation, and the pellets were resuspended in 0.2 mg/mL of propidium iodide (PI) containing 0.1% Triton X-100 and 0.1 mg/mL RNase A. The cell suspension was incubated in the dark for 30 min at room temperature and subsequently analyzed using FACScan flow cytometer for DNA content. The percentage of cells in different phases of the cell cycle was sorted using a ModFit 5.2 computer program.
Western blot
Cells were lyzed using radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-Hcl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium dexoycholate, 0.1% sodium dodecyl sulfate [SDS]) with protease inhibitors (EMD Biosciences, San Diego, CA, USA). After sonication for 2 min at 4°C and centrifugation for 10 min at 4°C, the supernatant was taken as the total cell lysate. Protein concentration was quantified using the Bradford method. Equal amounts of total protein (50 μg) were analyzed by SDS polyacrylamide gel electrophoresis (SDS-PAGE). Following transfer to the nitrocellulose (NC) membrane, the blot was probed with primary antibodies as indicated and then incubated with horseradish peroxidase (HRP)-labeled secondary antibodies for visualization using enhanced chemiluminescence reagents (Thermo Fisher Scientific, Waltham, MA, USA). The images were obtained by the Bio-Rad Imaging System.
Gelatin zymography
Cells were grown in six-well plates. The supernatants after the different treatments were collected. The supernatant protein (40 μg/lane) was loaded and electrophoresed on 8% SDS-PAGE copolymerized with 1% gelatin. After electrophoresis, the gels were washed five times in 2.5% Triton X-100 (20 min each) and two times in buffer without Triton X-100 to remove Triton X-100 and then incubated in 50 mmol/L Tris-Cl (pH 7.6) and 5 mmol/L CaCl2 (18 h, 37°C). The gels were stained with 0.1% Coomassie Blue R-250 and destained in 10% isopropanol and 10% acetic acid in H2O. MMP2 and MMP9 levels were detected as transparent bands on the blue background of a Coomassie Blue-stained gel.
Immunoprecipitation
Cells were lyzed in RIPA buffer (Beyotime Institute of Biotechnology), and 500 μg of protein was incubated with an anti-EGFR antibody for 1 h at 4°C. Antibodies were precipitated using Protein G-coated Dynabeads (Thermo Fisher Scientific), and bound protein was eluted in Laemmli buffer (Thermo Fisher Scientific) for 10 min at 70°C.
Wound-healing assay
Cells were seeded in six-well plates for 24 h. The monolayers were scratched with a 200 μL pipette tip and washed with media without serum to remove the detached cells. The wounded areas were observed and imaged using a microscope.
Transwell invasion assay
For invasion assays, cells (1×105/well) were placed into the upper chamber with the Matrigel-coated membrane, which was diluted with a serum-free culture medium. Cells were suspended in 200 μL of serum-free Roswell Park Memorial Institute (RPMI)-1640 medium and seeded into the upper chamber. In the lower chamber, 800 μL of the RPMI-1640 medium supplemented with 10% fetal bovine serum was added. After incubation for 24 h at 37°C and 5% CO2, the chambers were fixed with 100% methanol for 20 min and stained with 0.1% crystal violet for 15 min. Images were captured with an Olympus BX83 fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Cell viability assay
Cell proliferation inhibition was evaluated using the CCK-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. A549 cells (6×103/90 μL/well) were plated into 96-well plates in triplicate and cultured for 24 h before the onset of treatment. Then, cells were treated with LY294002 (10, 20, 30, 40, 50 and 60 μM) for 24 h in 96-well plates with 2,000 cells/well. Then, 20 μL of CCK-8 reagent was added to each well for 1 h, followed by light absorbance measurement at a wavelength of 450 nm.
Immunocytochemistry
Cells were seeded on glass coverslips and then fixed with PBS containing 4% formaldehyde for 10 min and permeabilized with 0.2% Triton X-100 in PBS for 10 min at room temperature. After washing with PBS, endogenous peroxidase activity was suppressed by 3% H2O2 and blocked with goat serum or 5% bovine serum albumin (BSA). Diluted primary monoclonal rabbit antibodies against sLeX (dilute 1:100) were added and incubated at 4°C overnight. As secondary reagents, biotin-labeled anti-IgG and an avidin–biotin HRP complex were used Zhongshan jinqiao (Beijing, China) followed by staining with chromogen diaminobenzidine (Zhongshan) until a brown color developed. Slides were counterstained with Mayer’s hematoxylin and differentiated in a solution containing 1% hydrochloric acid and 99% ethanol. Sections were dehydrated, and a transparent cover slip was added to enable observation by microscopy. All the immunohistochemical photographs were analyzed using Image-Pro Plus (IPP; Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
Results are expressed as the mean ± standard error of the mean (SEM) of at least three independent experiments. The statistical significance of differences among groups was analyzed by one-way analysis of variance (ANOVA) and the Student–Newman–Keuls (SNK) q test using SPSS 17.0 software (SPSS Inc., Chicago, IL, USA). Differences were considered to indicate a statistically significant result with a P-value <0.05.
Results
FUT7 induces sLeX expression in A549 cells
To examine the relationship between FUT7 and sLeX, two FUT7 cDNA constructs (cDNA-1 and cDNA-2) and the blank vector as control transfected A549 cells. As shown in Figure 1A, when FUT7 was overexpressed by FUT7 cDNA constructs, FUT7 levels were increased, concomitant with a notable increase in sLeX, indicating that FUT7 was a factor for promoting sLeX expression. In Figure 1B, we used immunocytochemistry to detect sLeX expression in different FUT7 expression A549 cells. It showed similar result as Western blot.
Figure 1.
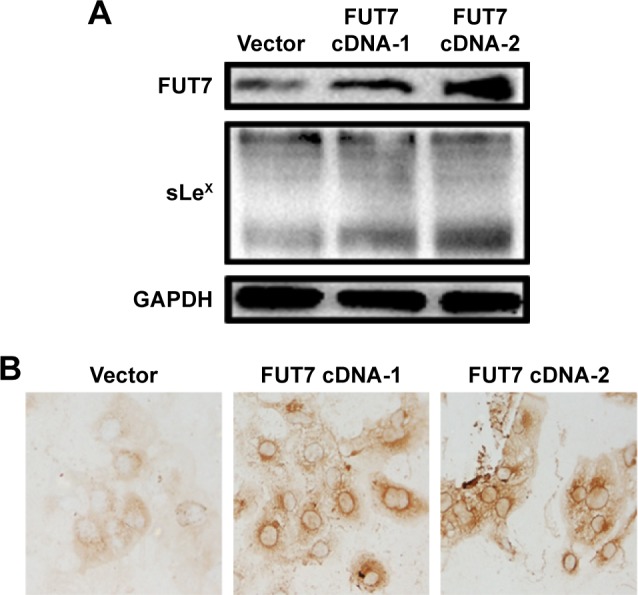
Overexpression of FUT7 increased the sLeX expression.
Notes: (A) Western blot analysis was used to assess the protein levels in A549 cells of forced expression of FUT7. (B) The expression of sLeX induced by overexpression of FUT7 was tested by immunocytochemistry.
Overexpression of FUT7 promotes cell growth
We measured cell proliferation utilizing colony formation assay (Figure 2A) and found that overexpression of FUT7 significantly increased the number of formed colonies. The impact of FUT7 overexpression on cell proliferation was further determined by cell cycle analysis (Figure 2B). Flow cytometry with PI-stained cells showed that A549-vector control cells were presented in G0/G1 (82.46%±1.89%), S (17.01%±2.04%) and G2/M (0.53%±0.01%) phases. While in A549-FUT7 cDNA cells, S phase fraction was increased (36.62%±1.48%, P<0.01) and G0/G1 fraction was decreased (0.00%±0.00% for A549-FUT7 cDNA cells, P<0.01). To further evaluate the impact of FUT7 overexpression on cell proliferation, PCNA expression was detected by Western blot (Figure 2C). A significant increase in PCNA expression was observed in A549-FUT7 cDNA cells.
Figure 2.

Overexpression of FUT7 stimulated the colony formation and cell proliferation in A549 cells.
Notes: (A) The colony formation assay was conducted with A549-vector and A549-FUT7 cDNA cells. Colonies formed during 10 days of culture were visualized by crystal violet staining. The number of colonies formed in each group was calculated from three individual experiments. ***P<0.001. (B) Cell cycle analysis – A549-vector and A549-FUT7 cDNA cells were stained for DNA by PI; the stained cells were analyzed by FACScan flow cytometer using a ModFit 5.2 computer program. (C) Western blot to detect PCNA expression – total protein from the whole-cell lysate of A549-vector and A549-FUT7 cDNA cells was subjected to Western blot analysis using PCNA antibody.
Abbreviation: PI, propidium iodide.
Overexpression of FUT7 promotes cell migration and invasion in A549 cells
We observed the migratory capacity of the cells through wound healing assay or scratch assay (Figure 3A). We found that the scratch was almost empty after 24 h in the A549-vector group, while the cells in the A549-FUT7 cDNA group showed higher migratory ability, as they filled over half of the scratch within 24 h. As the wound healing assay provides only semi-quantitative results, we proceeded with a transwell migration assay to evaluate the cell migratory capacity more precisely. The cells in the A549-FUT7 group showed a significant higher migratory ability compared with the cells in the A549-vector group (P<0.05; Figure 3B). We then emphasized the result by using the gelatin zymography assay and found that the activities of MMP2 and MMP9 were both increased in the A549-FUT7 cDNA group (Figure 3C).
Figure 3.

Overexpression of FUT7 promoted invasion and migration in A549 cells.
Notes: (A) Cell migration was detected by wound-healing assay. (B) Cell invasion was examined by transwell (Matrigel-coated membrane). (C) Analysis of MMP2 and MMP9 activity by gelatin zymography. *P<0.05.
FUT7 promotes proliferation via the EGFR/AKT/mTOR pathway in A549 cells by inducing sLeX generation
To elucidate the molecular mechanism underlying the augmented proliferation by FUT7, the EGFR/AKT/mTOR pathway was specifically analyzed in the present study. Using gene overexpression combined with anti-sLeX antibody blocking, it was found that FUT7 overexpression increased the activity of EGFR, AKT and mTOR as reflected by the phosphorylated form of EGFR, AKT and mTOR (Figure 4A). In addition, anti-sLeX antibody inhibited the activation of EGFR/AKT/mTOR signaling pathway. In addition, we used immunoprecipitation to detect the interaction between sLeX and EGFR; it was found that there is sLeX modification on EGFR, and the expression increased in FUT7 overexpression cells (Figure 4B). To further evaluate the involvement of EGFR/AKT/mTOR signaling pathway in FUT7 overexpession-induced activation on cell proliferation, we used LY294002 to inhibit AKT activation. To determine the working concentration of LY294002, CCK-8 assay was performed to evaluate the inhibitory rate of LY294002 on A549 cells. As shown in Figure 5A, A549 cells with LY294002 treatment displayed an increased inhibitory rate; the inhibition concentration of 10% of LY294002 against A549 cells was 10±0.29 μM. Our result showed that inactivation of AKT signaling significantly inhibits the cell proliferation induced by FUT7 overexpression. Increases in PCNA expression was seen in A549-FUT7 cDNA cells, while the inhibited expression of PCNA was observed in A549-FUT7 cDNA cells combined with LY294002 treatment (Figure 5B). The number of colonies in A549-FUT7 cDNA cells combined with LY294002 treatment was significantly decreased compared with those in A549-FUT7 cDNA cells (Figure 5C). The Akt activity was assessed by Western blot with a phospho-specific antibody against phosphorylated Ser473, which has been reported to be constitutively active in A549 cells under normal growth conditions.31 We found that the LY294002 treatment combined with FUT7 overexpression led to a marked decrease in the level of phosphorylated Akt compared with that of the A549-FUT7 cDNA cells (Figure 5B).
Figure 4.
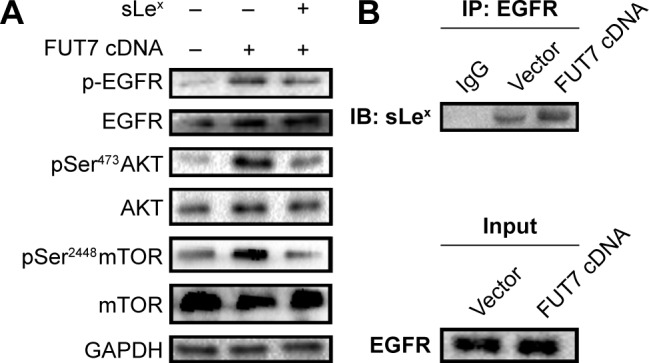
The impact of FUT7 overexpression and anti-sLeX antibody blocking on EGFR/Akt/mTOR signaling activation.
Notes: (A) P-EGFR, EGFR, p-Akt, Akt, p-mTOR and mTOR were detected by Western blot. (B) Immunoblot after immunoprecipitation of EGFR from cell lysates using an anti-sLeX antibody coupled to magnetic beads.
Figure 5.

Involvement of Akt/mTOR signaling pathway in FUT7 overexpression promoted cell growth.
Notes: (A) Inhibitory effects of LY294002 on A549 cells were tested by CCK-8 assay. (B) The expression of p-Akt, Akt and PCNA after treatment of FUT7 cDNA and LY294002 alone or in combination. (C) The inhibitory effect of LY294002 on colony formation of A549 cells was tested by colony formation assay. The number of colonies formed in each group was calculated from three individual experiments. **P<0.01.
Discussion
The carbohydrate moieties of cell surface glycoconjugates have been shown to play important roles in many biological processes. The multiple linkages, branching patterns and terminal modifications in these glycans are the main causes for their functional diversity. Over the last decades, important discoveries have been made in the field of glycoscience. It has been well studied that the neoplastic processes result in fundamental changes in the glycosylation patterns of cell surface glycoconjugates.19 Lewis antigens are a group of oligosaccharide residues constituting the end of various carbohydrate chains of glycoproteins and glycolipids protruding from the outer surface of mammalian cell membranes.20 Sialylated Lewis antigens sLeX are potentially involved in many cell adhesion events because they act as specific E-selectin ligands.21 The alteration in cell surface sLeX oligosaccharide expression has been reported to be correlated with many kinds of cancers. Our previous meta-analysis study has shown that sLeX overexpression is associated with tumor metastasis, recurrence and overall survival in cancer patients and plays an important role in cancer prognosis.17 Komatsu et al22 reported that serum sLeX concentration was an independent predictive factor for skip metastasis in non-small cell lung cancer (NSCLC). Therefore, exploring the mechanism of sLeX antigen synthesis is important for developing effective sLeX targeting therapeutic strategy. Biosynthesis of sLeX requires addition of fucose to N-acetyllactosamine by enzymatic catalysis.23 Among these enzymes, FUT7 was thought to participate in the biosynthesis of sLeX.24 In the present study, we established FUT7-overexpressed cell colonies. We found that FUT7 overexpression significantly augmented the sLeX antigen synthesis compared with that of the mock control cells (Figure 1).
It has been reported that abnormal expression of FUT7 is closely related to poor prognosis in lung cancer.16 In the study of Wang et al,25 the growth of hepatocarcinoma cells was significantly stimulated via the cyclin-dependent kinase inhibitor p27Kip1 by transfecting of FUT7 cDNA. Laubli et al26 have found that deficiency of induced endogenous L-selectin ligands caused by a low expression of FUT7 led to a low incidence of metastasis of hematogenous carcinoma in mice. We evaluated the effect of FUT7 expression on cell growth and migration using human adenocarcinoma A549 cells. We found that FUT7 overexpression significantly increased the cell growth. Cellular proliferation was detected by using flow cytometry. FUT7 overexpression enhanced the DNA replication through the cell cycle arrest at the S phase (Figure 2). It has been shown that overexpression of FUT7 enhanced the colorectal carcinoma metastases via promoting the carbohydration of glycoprotein CD24.27 In the present study, we represented that migration and invasion capability of A549 cells were enhanced after the transfection of FUT7 cDNA (Figure 3). These results suggested that FUT7 plays a critical role in cell growth and in its capability of migration and invasion.
The underlying mechanism of sLeX regulating tumor progression still needs further exploration. It has been studied that overexpression of FUT7 led to activation of the InR pathway, which is due to the high expression of sLeX on the glycans of cell-surface receptors.33 Liu et al28 revealed that using FUT7 antisense oligodeoxynucleotide leads to reduced expression of both FUT7 and sLeX and thereby inhibited the adhesion of embryo cells to endometrium. Using in vivo models, the adhesion rate was significantly reduced when the sLeX antigen on the surface of endometrial cells was blocked by the anti-sLeX antibody.29 The dysregulation of EGFR signaling has been well documented to contribute to the progression of NSCLC. The AKT signaling pathways are downstream of EGFR and deregulated via genetic and epigenetic mechanisms in many human cancers and play a pivotal role in oncogenesis.30 It has been revealed that anti-Lewis Y antibody (Lewis Y antigen is catalyzed by fut4, a member of fucosyltransferase gene family) effectively blocked the EGF-stimulated phosphorylation of MAPK through inactivating the EGFR signaling in A431 cells and prevented cell proliferation.18 Based on these studies, we hypothesized that blocking the sLeX antigen with anti-sLeX antibody may inhibit the EGFR/Akt/mTOR signaling pathway. In the present study, we verified our hypothesis by using PI3K/Akt inhibitor LY294002 and anti-sLeX antibody. Meantime, the immunoprecipitation result showed that EGFR glycoprotein is modified by sLeX that may participate in the phosphorylation of EGFR (Figures 4 and 5), but the underlying mechanism between the sLeX modification of EGFR and EGFR activation still needs further study. Our results indicated that anti-sLeX antibody inhibited tumorigenesis via inactivation of the EGFR/Akt/mTOR signaling pathway. FUT7 overexpression promotes tumorigenesis in NSCLC through activation EGFR/Akt/mTOR signaling pathway by augmenting the sLeX synthesis.
Conclusion
FUT7 played an important role in cell growth and proliferation via promoting the sLeX antigen synthesis and EGFR/AKT/mTOR signal pathway activation in the A549 cell line, which indicated that FUT7 may be a potential therapeutic target for epithelial cancers with a high expression of FUT7 and sLeX.
Acknowledgments
This study was supported by a grant from the National Natural Science Foundation of China (no 30870719).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Siegel R, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Nesbitt JC, Putnam JJ, Walsh GL, Roth JA, Mountain CF. Survival in early-stage non-small cell lung cancer. Ann Thorac Surg. 1995;60(2):466–472. doi: 10.1016/0003-4975(95)00169-l. [DOI] [PubMed] [Google Scholar]
- 3.Damstrup L, Wandahl Pedersen M, Bastholm L, Elling F, Skovgaard Poulsen H. Epidermal growth factor receptor mutation type III transfected into a small cell lung cancer cell line is predominantly localized at the cell surface and enhances the malignant phenotype. Int J Cancer. 2002;97(1):7–14. doi: 10.1002/ijc.1572. [DOI] [PubMed] [Google Scholar]
- 4.Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5(5):390–401. doi: 10.1016/j.apsb.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 6.Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):15–31. doi: 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh ET, Shepherd FA, Tsao MS. Co-expression of epidermal growth factor receptor and transforming growth factor-alpha is independent of ras mutations in lung adenocarcinoma. Lung Cancer. 2000;29(2):151–157. doi: 10.1016/s0169-5002(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 8.Javaud C, Dupuy F, Maftah A, Julien R, Petit JM. The fucosyltransferase gene family: an amazing summary of the underlying mechanisms of gene evolution. Genetica. 2003;118(2–3):157–170. [PubMed] [Google Scholar]
- 9.Natsuka S, Gersten KM, Zenita K, Kannagi R, Lowe JB. Molecular cloning of a cDNA encoding a novel human leukocyte alpha-1,3-fucosyltransferase capable of synthesizing the sialyl Lewis X determinant. J Biol Chem. 1994;269(32):16789–16794. [PubMed] [Google Scholar]
- 10.Kaneko M, Kudo T, Iwasaki H, et al. Alpha1,3-fucosyltransferase IX (Fuc-TIX) is very highly conserved between human and mouse; molecular cloning, characterization and tissue distribution of human Fuc-TIX. FEBS Lett. 1999;452(3):237–242. doi: 10.1016/s0014-5793(99)00640-7. [DOI] [PubMed] [Google Scholar]
- 11.Niemela R, Natunen J, Majuri ML, et al. Complementary acceptor and site specificities of Fuc-TIV and Fuc-TVII allow effective biosynthesis of sialyl-TriLex and related polylactosamines present on glycoprotein counterreceptors of selectins. J Biol Chem. 1998;273(7):4021–4026. doi: 10.1074/jbc.273.7.4021. [DOI] [PubMed] [Google Scholar]
- 12.Allahverdian S, Wojcik KR, Dorscheid DR. Airway epithelial wound repair: role of carbohydrate sialyl LewisX. Am J Physiol Lung Cell Mol Physiol. 2006;291(4):828–836. doi: 10.1152/ajplung.00120.2006. [DOI] [PubMed] [Google Scholar]
- 13.Varki A. Selectin ligands. Proc Natl Acad Sci U S A. 1994;91(16):7390–7397. doi: 10.1073/pnas.91.16.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasaki K, Kurata K, Funayama K. Expression cloning of a novel α1,3-fucosyltransferase that is involved in biosynthesis of the sialyl Lewis X carbohydrate determinants in leukocytes. J Biol Chem. 1994;269(20):14730–14737. [PubMed] [Google Scholar]
- 15.Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004;95(5):377–384. doi: 10.1111/j.1349-7006.2004.tb03219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogawa JI, Inoue H, Koide S. Expression of alpha-1,3-fucosyltransferase type IV and VII genes is related to poor prognosis in lung cancer. Cancer Res. 1996;56(2):325–329. [PubMed] [Google Scholar]
- 17.Liang JX, Liang Y, Gao W. Clinicopathological and prognostic significance of sialyl Lewis X overexpression in patients with cancer: a meta-analysis. Onco Targets Ther. 2016;9:3113–3125. doi: 10.2147/OTT.S102389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farhan H, Schuster C, Klinger M, et al. Inhibition of xenograft tumor growth and downregulation of ErbB receptors by an antibody directed against Lewis-Y antigen. J Pharmacol Exp Ther. 2006;319(3):1459–1466. doi: 10.1124/jpet.106.107318. [DOI] [PubMed] [Google Scholar]
- 19.Padler-Karavani V. Glycan microarray reveal the sweet side of cancer vaccines. Cell Chem Biol. 2016;23(12):1446–1447. doi: 10.1016/j.chembiol.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marionneau S, Cailleau-Thomas A, Rocher J, et al. ABH Lewis histo-blood group antigens, a model for the meaning of oligosaccharide diversity in the face of a changing world. Biochimie. 2001;83(7):565–573. doi: 10.1016/s0300-9084(01)01321-9. [DOI] [PubMed] [Google Scholar]
- 21.Läubli H, Borsig L. Selectins promote tumor metastasis. Semin Cancer Biol. 2010;20(3):169–177. doi: 10.1016/j.semcancer.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Komatsu H, Mizuguchi S, Izumi N, et al. Sialyl Lewis X as a predictor of skip N2 metastasis in clinical stage IA non-small cell lung cancer. World J Surg Oncol. 2013;11:309. doi: 10.1186/1477-7819-11-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes EH, Ostrander GK, Hakomori S. Biosynthesis of the sialyl-Lex determinant carried by type 2 chain glycosphingolipids (IV3NeuAcIII3-FucnLc4, VI3NeuAcV3FucnLc6, and 3NeuAcIII3V3Fuc2nLc6) in human lung carcinoma PC9 cells. J Biol Chem. 1986;261(8):3737–3743. [PubMed] [Google Scholar]
- 24.Kitagawa H, Paulson JC. Cloning of a novel a 2,3-sialyltransferase that sialylates glycoprotein and glycolipid carbohydrate groups. J Biol Chem. 1994;269(2):1394–1401. [PubMed] [Google Scholar]
- 25.Wang QY, Guo P, Duan LL, Shen ZH, Chen HL. alpha-1,3-Fucosyltransferase-VII stimulates the growth of hepatocarcinoma cells via the cyclin-dependent kinase inhibitor p27Kip1. Cell Mol Life Sci. 2005;62(2):171–178. doi: 10.1007/s00018-004-4349-8. [DOI] [PubMed] [Google Scholar]
- 26.Laubli H, Stevenson JL, Varki A, Varki NM, Borsig L. L-selectin facilitation of metastasis involves temporal induction of FUT7-dependent ligands at sites of tumor cell arrest. Cancer Res. 2006;66(3):1536–1542. doi: 10.1158/0008-5472.CAN-05-3121. [DOI] [PubMed] [Google Scholar]
- 27.Li W, Zhang W, Luo J, et al. Alpha 1,3 fucosyltransferase VII plays a role in colorectal carcinoma metastases by promoting the carbohydration of glycoprotein CD24. Oncol Rep. 2010;23(6):1609–1617. doi: 10.3892/or_00000802. [DOI] [PubMed] [Google Scholar]
- 28.Liu S, Zhang Y, Liu Y, Qin H, Wang X, Yan Q. FUT7 antisense sequence inhibits the expression of FUT7/sLeX and adhesion between embryonic and uterine cells. IUBMB Life. 2008;60(7):461–466. doi: 10.1002/iub.62. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Liu S, Liu Y, Wang Z, Wang X, Yan Q. Overexpression of fucosyltransferase VII (FUT7) promotes embryo adhesion and implantation. Fertil Steril. 2009;91(3):908–914. doi: 10.1016/j.fertnstert.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 30.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88(4):435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 31.Toulany M, Kehlbach R, Florczak U, et al. Targeting of Akt1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Ther. 2008;7(7):1772–1781. doi: 10.1158/1535-7163.MCT-07-2200. [DOI] [PubMed] [Google Scholar]
- 32.Hiller KM, Mayben JP, Bendt KM, et al. Transfection of alpha(1,3) fucosyltransferase antisense sequences impairs the proliferative and tumorigenic ability of human colon carcinoma cells. Mol Carcinog. 2000;27(4):280–288. [PubMed] [Google Scholar]
- 33.Wang QY, Zhang Y, Chen HJ, Shen ZH, Chen HL. Alpha 1,3-fucosyltransferase-VII regulates the signaling molecules of the insulin receptor pathway. FEBS J. 2007;274(2):526–538. doi: 10.1111/j.1742-4658.2006.05599.x. [DOI] [PubMed] [Google Scholar]