

Abstract
It is becoming increasingly impractical to indefinitely store raw sequencing data for later processing in an uncompressed state. In this paper, we describe a scalable compressive framework, Read-Quality-Sparsifier (RQS), which substantially outperforms the compression ratio and speed of other de novo quality score compression methods while maintaining SNP-calling accuracy. Surprisingly, RQS also improves the SNP-calling accuracy on a gold-standard, real-life sequencing dataset (NA12878) using a k-mer density profile constructed from 77 other individuals from the 1000 Genomes Project. This improvement in downstream accuracy emerges from the observation that quality score values within NGS datasets are inherently encoded in the k-mer landscape of the genomic sequences. To our knowledge, RQS is the first scalable sequence based quality compression method that can efficiently compress quality scores of terabyte-sized and larger sequencing datasets.
Availability: An implementation of our method, RQS, is available for download at: http://rqs.csail.mit.edu/
Keywords: RQS, quality score, sparsification, compression, accuracy, variant calling
1 Introduction
In the past two decades, genomic sequencing capabilities have increased exponentially, outstripping advances in computing power and storage [1, 2]. Capitalizing on this deluge of data is heavily dependent on the ability to efficiently store, process and extract meaningful biological insights from sequencing datasets, which is becoming correspondingly more difficult as more data is generated.
Early studies on compressing NGS datasets have mainly focused on compressing sequence data itself, aiming to leverage the inherent redundancy present in read sequences to reduce the space needed for storing ‘raw’ reads [3–7]. Furthermore, Loh et al. [8] demonstrated that it is possible to further exploit this redundancy through the use of succinct data structures that allow us to operate directly on the compressed data, saving CPU time as well as space.
That said, the Phred quality scores encoding the “base-calling confidence” take up more than twice the space on disk as the read sequence itself (∼ 2.3x − 2.8x for Illumina reads). Furthermore, it is more challenging to compress the quality scores [9], as they not only have a larger alphabet size (ranging from 63 to 94 characters depending on the sequencing technology), but also have limited repetitive patterns as well as little direct correlation with the bases sequenced.
Computational methods for NGS quality compression fall into two main camps: lossless compression methods that include general-purpose text-compressors, such as GZIP, BZIP2 or 7zip, as well as methods specialized to exploit the local similarity of quality values for further compression [7, 10, 11]; and lossy compression methods that aim to achieve further compression by sacrificing the ability to reconstruct the original quality values [9, 12, 13].
A recent area of investigation in quality score compression is exploiting sequence read information within NGS datasets in order to boost the compression of quality scores. Most of these methods need to compute expensive whole-genome alignments of the NGS read dataset, then use additional position and coverage information obtained from the alignments to compress quality values [4, 6, 14]. However, it has been shown that alignment-agnostic methods can also utilize sequence data to achieve better compression of quality values [15], but again require costly operations (e.g. BWT) to be run on the entire read dataset. Thus far neither approach has been able to truly address the scalability problem of quality score compression for terabyte-sized or larger NGS datasets.
In this paper, we introduce a highly efficient, scalable, and alignment-free k-mer based algorithm, “Read-Quality-Sparsifier” (RQS), which sparsifies quality score values by smoothing a large fraction of quality score values based on the k-mer neighborhood of their corresponding positions in the read sequences. In particular, RQS constructs a comprehensive database, or dictionary, of commonly occurring k-mers throughout a population-sized read dataset. Once this database is constructed it can be used to compress any given read dataset by identifying k-mers within each read that have a small Hamming distance from the database; assuming that any divergent base in a k-mer likely corresponds to a SNP or machine error, we preserve quality scores for probable variant locations and discard the rest.
Our “coarse” representation of quality scores leads to great savings in storage. Throwing away this much information significantly improves the compression ratios, allowing us on average to store quality scores at roughly 0.4 bits per value (from the original size of 6 – 7 bits). The scalability of our method arises from the fact that the k-mer database needs to be constructed only once for any given species, and the quality sparsification stage is very efficient. As a surprising result, our quality sparsification method not only significantly outperforms other de novo quality compression methods based on efficiency and compression ratio, but is also able to improve downstream variant calling accuracy. The improvement in downstream SNP-calling accuracy of the compressed dataset emerges from the fact that base calling confidences within NGS datasets are inherently encoded in the k-mer landscape of genomic sequences. Notably, supported by experimental results on annotated real data, our study demonstrates that k-mer density profiles of read sequences are more informative on average than ∼95% of the quality score information.
We validate RQS on real NGS exome and genome read datasets taken from 1000 Genomes Project Phase 1 [16]. Specifically, we demonstrate the superior compression ratio and efficiency of our method on Illumina read sequences of 77 British individuals (see Appendix B), comparing the variant call accuracy to other de novo quality compression methods. We also give preliminary results showing that, compared to a ground truth genotype annotation of NA12878, RQS compression achieves better downstream SNP-call accuracy compared to the uncompressed quality values.
While lossy compression of quality scores has not been widely adopted by biologists due to loss of precision [6], our RQS method remediates this effect by improving the accuracy of downstream genotyping applications. It does so by capitalizing on the k-mer landscape of a read dataset. The usefulness of k-mer frequencies for inferring knowledge about the error content of a read sequence has been studied [17–19]—in fact, many sequence-correction and assembly methods directly or indirectly make use of this phenomenon [20–23]; however, to our knowledge, RQS is the first such method to traverse the k-mer landscape for quality score compression, thereby improving efficiency, compression-ratio, and accuracy.
2 Methods
At a high level, RQS is divided into two separate stages (Figure 1). In the first preprocessing stage, we generate a dictionary, D, of all k-mers that appear with high multiplicity in a representative collection (corpus) of reads. In the second sparsification stage, we look at the k-mers in a read. k-mers that are close to our dictionary (as measured by Hamming distance) have nearly all of their quality scores discarded. RQS keeps only the low quality scores for bases where the k-mer differs from the dictionary.
Fig. 1. Preprocessing.
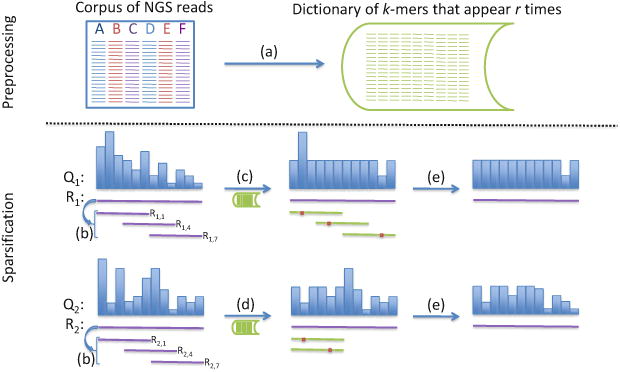
(a) A dictionary of k-mers that appear at least r times in a corpus of NGS reads is generated. Sparsification. R is a read sequence and Q is a corresponding quality score string. Ri, j is a k-mer within Ri starting at position j. (b) We choose multiple k-mers to cover the read sequence. If a particular k-mer is within Hamming distance 1 of a k-mer in the dictionary, then we can sparsify the quality scores that correspond to the positions in the k-mer. (c) R1,1,R1,4, and R1,7 are exactly distance 1 from the dictionary. We mark the locations where they do not match the dictionary k-mer and smooth all the other quality scores. Note that although R1,4 has a mismatch, that location is still smoothed because the location is covered by R1,1, which does not have a corresponding mismatch at the same position in the read. (d) Only R2,1 is within Hamming distance 1 of the dictionary. However, it has two Hamming neighbors in the dictionary. We only smooth the quality scores where there is concord among all Hamming neighbors and R2,1. Neither R2,4 nor R2,7 contribute because they are too far away from the dictionary. (e) Last, we smooth all quality scores above a threshold.
More precisely, with NGS read data, we are given a corpus C of reads with depth coverage t of some consensus sequence G. We will assume an independent accuracy rate of p for each base call, and let q = 1 − p be the variation rate (whether caused by machine error or by a SNP). As usual, we identify reverse complements together. Let γk be the multiset of all k-mers that appear in a read γ, counting multiplicity. Similarly, let Gk and Ck be respectively the multiset of all k-mers that appear in G and the reads of C.
Let Δ (x, y) be the Hamming distance between two k-mers x and y, and let Δ(x, D) be the minimum Hamming distance from x to any k-mer in D. Then we generate a dictionary of all k-mers that appear at least r times in Ck, which approximates Gk for r, k and p sufficiently large. These “good” k-mers are then used to identify high confidence base calls in reads: if a k-mer is within Hamming distance 1 from D, we assign high confidence to all locations where there is concordance among it and all its Hamming neighbors in D. Each read can be covered by overlapping k-mers, allowing us to identify high confidence base calls in that case as well.
The sparsification procedure then consists of two separate steps. First, we discard quality values for all high confidence base calls. Then, we discard quality values for all base calls above some threshold value 𝒬. In our implementation, for later downstream analysis, we replace all discarded values with 𝒬.
Below we present pseudocode for a simplified version of RQS (an efficient implementation is described in appendix A). DICT(C, k, r) (alg. 1) takes the corpus of reads C and returns a list D of all k-mers that appear at least r times. Using Hamming distance from D, MARK_KMER(x, D) (alg. 2) generates a boolean vector marking each position in a k-mer x that corresponds to a high-confidence call. MARK_READ(γ, D) (alg. 3) then repeatedly calls MARK_KMER to generate the vector of high-confidence calls in read γ. SPARSIFY_RQ(γ, Q, D, 𝒬) (alg. 4) calls MARK_READ to locate high-confidence calls and then discards both the corresponding quality scores and quality scores above a cut-off threshold 𝒬.
| Algorithm 1. DICT(C, k, r): Generates a dictionary of all k-mers that appear at least r times in a corpus C of reads |
| Input: C, k, r |
| Output: D |
| D = {} |
| A = [0, … , 0] ∈ ℕ4k |
| for x ∈ Ck do |
| A[x]+ = 1 |
| for x ∈ [4k] do |
| if A[x] ≥ r then |
| D.append(x) |
| return D |
| Algorithm 2. MARK_KMER(x, D): Marks high confidence locations in a k-mer x using a dictionary D |
| Input: x, D |
| Output: M |
| if Δ(x, D) > 1 then |
| M = [F, … , F] ∈ {T, F}k |
| else |
| M = [T, … , T] ∈ {T, F}k |
| for y ∈ D s.t. Δ(x, y) = 1 do |
| for i ∈ [k] do |
| if xi ≠ yi, then M[i] = F |
| return M |
| Algorithm 3. MARK_READ(γ, D): Marks high confidence calls in read γ using dictionary D |
| Input: γ, D |
| Output: ℳ |
| Let xa be the k-mer in γ starting at a. |
| Cover γ by k-mers {xa1,… , xan}. |
| for i ∈ [n] do |
| Mi = MARK_KMER(xai, D) |
| M̄i = [F,…, F] ∈ {T, F} length(γ) |
| for j ∈ [k] do |
| ℳ = M̄1 OR … OR M̄n |
| return ℳ |
| Algorithm 4. SPARSIFY_RQ (γ, Q, D, 𝒬): Sparsifies the quality vector Q associated with read γ using a dictionary D; discarded qualities replaced with 𝒬 |
| Input: γ, Q, D, 𝒬 |
| Output: Q′ |
| Q′ = Q |
| ℳ = MARK_READ(γ, D) |
| for i ∈ length(γ) do |
| if (Qi > 𝒬) OR (ℳi = T) then |
| Q′i = 𝒬 |
| return Q′ |
2.1 Theoretical Guarantees
For completeness, we present an analysis of the composition of D, showing that under certain conditions, D ≈ Gk. Let Σ = {A, C, G, T} ≅ ℤ/4ℤ be the alphabet, and let G ∈ ΣN be the consensus sequence—note, we will assume that we are not given G. Let the multiset counting multiplicity of all k-length substrings of G be denoted by Gk.
We construct an idealized variation model combining both machine error in the base call and the presence of SNPs. Let p ∈ [0, 1] and q = 1 − p. Let the random variable σ : [0, 1] → Σ be defined by
| (1) |
Thus ∀l ∈ Σ, l + σ = l with probability p. This is to say that a base is read correctly with probability p and incorrectly with probability q.
Given x ∈ Σk, define xi as the ith letter (base) of x. For all x ∈ Σk, define independently the Σk-valued random variables Rx by ∀i, , where σi,… ,σk are i.i.d. copies of σ. Thus, Rx can be thought of as a read of x, including machine errors and SNPs. Let Ĝk ≡ {Rx|x ∈ Gk}. Ĝk thus corresponds to a version of Gk with noise.
We are given t independent noisy copies Ĝk,1,…, Ĝk, t of Gk, but with a low error rate q > 0. This assumption corresponds to being given reasonably accurate reads covering the target genome t times and counting all k-mers. We want to recover a dictionary Dr approximating Gk (without multiplicity) from the collection Ḡk = Ĝk,1,…, Ĝk, t. Let Ck be the multiset defined as the disjoint union of Ĝk,1, … , Ĝk, t. We will construct the dictionary by simply taking all k-mers that appear in Ck at least r times, where r is an adjustable parameter. Intuitively, this process should work because provided the variation rate q is small, Rx = x often so there will be many exact copies of x in Ck if x ∈ Gk; however, then Rx = y only rarely for x ∈ Gk and y ∉ Gk, so there will not be many copies of y in Ck.
Let Δ(x, y) be the Hamming distance between x, y ∈ Σk. Let Δ(x, Gk) = miny∈Gk Δ (x, y). Let α(x) = |{i : ∈ Ĝk, i}|, the number of times x appears in Ck. Let us denote the i.i.d. copies of Rx in Ĝk,1,…, Ĝk, t by Rx,1, … , Rx, t. Then for x ∈ Gk,
| (2) |
which is just the chance that a noise-free version of x was stored in Ĝk, j. Let 1x ∈ Ĝk, j be an indicator variable for the event {x ∈ Ĝk, j}. Then
| (3) |
Furthermore, by applying a Chernoff bound, for any δ1 > 0,
| (4) |
Recall that we defined our dictionary Dr ≡ {x ∈ Ck|α(x) ≥ r}, which consists of all members of Ck with multiplicity at least r. Then,
| (5) |
where unique(Gk) is the k-mer set obtained by discarding multiplicity of k-mers in Gk. So long as tpk is reasonably large and (1 – δ1) is not very big, most of Gk is expected to fall in Dtpk(1−δ1).
We also want to be able to say that most elements not in Gk do not fall in Dr. For simplicity of analysis, let us assume that all Gk is well-separated and sparse in Σk (NB: this assumption does not hold for repetitive regions in the genome) so that for x, y ∈ Gk, ℙ(Rx = Ry|x ≠ y) is negligible. Then we can separately consider for each x ∈ Gk, the number of collisions among Rx,1,…, Rx, t. . If kq < 1, the probability mass decreases and is spread thinner for higher d, so it is sufficient to bound collisions conditional upon all the probability mass staying within Hamming distance 1. Note that this assumption also implies that |unique(Gk)| = |Gk|.
By symmetry, under these conditions, since there are 3k possible positions for Rx to go. Let 1Rx=y be an indicator variable. Then for every y,
| (6) |
By again applying Chernoff, for any δ2 > 0,
| (7) |
Putting it all together,
| (8) |
Thus, equation 8 shows that as read multiplicity r increases Dr contains exponentially fewer k-mers that are not in Gk. Additionally, equation 5 shows that if r is small compared tpk, Dr contains nearly all of Gk. Whether or not there exists a value of r that makes Dr sufficiently close to Gk for our purposes is of course dependent on the exact parameters k, q, and t. However, because the simplifying assumptions we made do not perfectly reflect real data, instead of attempting to compute r, we swept over different values of r in our results section. These bounds do however show that as coverage t grows, there are parameters for which Dr asymptotically approaches Gk. As we demonstrate in the results, we are close enough to that regime for accurate and effective compression.
3 Results and Discussion
RQS performs impressively in terms of both compression rates and effects on downstream variant calling. In the first experiments, we demonstrate that RQS' performance is superior to existing methods in a more careful analysis of effects on downstream variant calling on chromosome 21 using the gold standard of NA12878. Most interestingly, RQS improves downstream variant calling, despite throwing away most of the quality scores. The second experiment demonstrates RQS' ability to successfully scale to the whole human genome by using a sampling algorithm for generating the dictionary.
Datasets
We generated our dictionary from a subset of the reads from the genomes of 77 British individuals with data taken from the 1000 Genomes Project, Phase 1 (see Appendix B for details). Read lengths ranged from 50-110bp, and there was a total depth coverage of 460 across all 77 individuals. Variant calling was performed using samtools [24], and BZIP2 was used to further compress the sparsified quality scores. For chromosome 21 analyses, we first filtered the reads in our corpus by those mapping to chromosome 21 using BWA [25] in combination with GATK [26]. For the whole human genome, we again used the same 77 British individuals and considered all reads mapping to any chromosome using a sampling approach.
Parameters
We chose k = 32 for two reasons. Importantly, 32-mers can be stored efficiently in 64-bit numbers, facilitating both ease of implementation and runtime. Additionally, our theoretical results were dependent on k-mers from our corpus being sparse; thus, k needs to be sufficiently large. We chose 𝒬 = 40 as 40 was close to the average quality score in the corpus.
Choice of read multiplicity r for inclusion in the dictionary is highly dependent on read depth of the corpus. Additionally, our theoretical guarantees assume random distribution of k-mers in the corpus, which is not necessarily true for repetitive regions of the human genome. Thus, we used several different values of r in our experiments. For our last analysis though, we sweep over k-mer multiplicity r = 25, 50, 100, 200, 350, 550 to provide some guidance as to the trade-offs involved.
3.1 RQS Out-Performs Existing Compression Methods
Here we show that our method offers a reduction in size over state-of-the-art lossless algorithms currently available and performs at least as well as lossy algorithms we have encountered in the literature. We measured the performance of several different compressors on the quality scores of HG02215, chromosome 21. General purpose lossless text compressors naturally have perfect fidelity for downstream variant calling, but also are unable on their own to achieve compression levels better than roughly 3.6 bits per quality score, which is only just over a 50% reduction in space. Of the three general purpose compressors we used, 7zip (PPMd) outperformed both BZIP2 and GZIP.
Read-Quality-Sparsifier, as implemented in this paper, only makes use of redundancy by replacing scores with the constant threshold value 𝒬, and so must be paired with one of the general purpose compressors to compress the modified scores. Surprisingly, the relative efficacy of compression after preprocessing with RQS with r = 50 (see Section 3.4 for choice of read multiplicity parameter) did not match that of the original scores; indeed, RQS + BZIP2 was by far the most effective combination, using only 0.2540 bits per quality value for storage.
In addition to the usual general purpose compressors, we also compared our compressive framework to QualComp [27], which features a tuning parameter to specify the number of bits needed for quality scores per read. We chose to sweep the QualComp parameter, bits per read, to match RQS' compression level and accuracy. As displayed in Table 1, RQS performs considerably better on accuracy than QualComp when a comparable compression level was chosen. Indeed, for QualComp to match the F-measure of RQS, QualComp needed over 6 times the space.
Table 1.
Relative compression rates of different compressors on the HG02215, chromosome 21 dataset. For each method, best results with respect to F-measure are bolded. Note that QualComp has its own quality storage format, whereas for Janin et al, we used 7zip (PPMd) to postprocess the smoothed quality scores (as this gave the best results for their method).
| Method | Size | Bits/Q | Precision | Recall | F-measure |
|---|---|---|---|---|---|
| Uncompressed | 273 MiB | 8.0000 | 1 | 1 | 1 |
| GZIP | 143 MiB | 4.1923 | 1 | 1 | 1 |
| BZIP2 | 133 MiB | 3.8791 | 1 | 1 | 1 |
| 7zip (PPMd) | 124 MiB | 3.6269 | 1 | 1 | 1 |
| RQS (r = 50) + GZIP | 14 MiB | 0.3825 | 0.9867 | 0.9963 | 0.9914 |
| RQS (r = 50) + BZIP2 | 8.7 MiB | 0.2540 | 0.9867 | 0.9963 | 0.9914 |
| RQS (r = 50) + 7zip (PPMd) | 11 MiB | 0.2935 | 0.9867 | 0.9963 | 0.9914 |
| QualComp (25 bits/read) | 9.4 MiB | 0.2747 | 0.8988 | 0.9934 | 0.9436 |
| QualComp (50 bits/read) | 19 MiB | 0.5494 | 0.9329 | 0.9943 | 0.9625 |
| QualComp (100 bits/read) | 38 MiB | 1.0988 | 0.9746 | 0.9949 | 0.9846 |
| QualComp (150 bits/read) | 57 MiB | 1.6482 | 0.9874 | 0.9957 | 0.9914 |
| Janin et al (2013) (c = 0, s = 1, r = 29) | 3.4 MiB | 0.0987 | 0.9279 | 0.9754 | 0.9509 |
| Janin et al (2013) (c = 1, s = 1, r = 29) | 4.4 MiB | 0.1284 | 0.9283 | 0.9751 | 0.9510 |
| Janin et al (2013) (c = 1, s = 5, r = 29) | 43 MiB | 1.2402 | 0.9903 | 0.9887 | 0.9894 |
| Janin et al (2013) (c = 0, s = 5, r = 29) | 43 MiB | 1.2416 | 0.9902 | 0.9887 | 0.9894 |
| Janin et al (2013) (c = 0, s = 10, r = 29) | 90 MiB | 2.6336 | 0.9953 | 0.9944 | 0.9948 |
Lastly, we compared against the method of Janin et al, 2013 [15], which uses a computation-intensive Burrows-Wheeler transform and least-common-prefixes table to smooth high-confidence reads. To ensure comparability, we display in Table 1 best-in-class parameters taken from those used in their paper. Although one set of parameters did have slightly higher accuracy than RQS, it came at the cost of needing over 10 times the disk space. Even at 5 times less compression, their method was not as accurate at variant calling, as measured by the balanced F-measure.
3.2 Comparison of Effects on Genotyping Accuracy by RQS, QualComp, and Janin et al
In the previous section, we considered any differences from the genotype information obtained from the original uncompressed data to be errors. In this section we show preliminary results suggesting that the differences between the SNP-calls of the RQS-compressed data and uncompressed data do not necessarily indicate errors, but actually some of them are corrections of the false positive and false negative SNP-calls obtained using the uncompressed data, improving the overall area-under-curve (AUC) of the SNP-calls. To demonstrate this, we tested our compressive framework on NA12878 genome, an extensively studied individual within the 1000 Genomes Project, for which a high-quality trio-validated genotype annotation is available [28].
In order to compress the quality scores of NGS reads of NA12878 chromosome 21, we reused the dictionary generated from the set of 77 British individuals, of which NA12878 is not a member. Table 2 shows the read length, coverage and sequence type of the NGS datasets used for this experiment.
Table 2.
Depth-of-coverage and read length information for datasets used in Figure 2
| Dataset | Read depth-of coverage | Read length |
|---|---|---|
| NA12878, chromosome 21 (76bp) | 16.9 | 2×76bp |
| NA12878, chromosome 21 (36bp) | 22.5 | 2×36bp |
| NA12878, exome, chromosome 21 | 5.15 | 2×76bp |
For each read set, we used samtools to call SNPs over the uncompressed dataset and computed the ROC curve using annotated variant locations in chromosome 21. Then the same test was performed after compressing the quality scores. As it is demonstrated in Figure 2, even though the ROC curves occasionally cross, AUC values favor the RQS-compressed data. Because the exome dataset was much smaller, it was more tractable for multiple comparisons with other compression tools. Thus, for the exome dataset specifically, we also compare the ROC curves of Qualcomp and Janin et al. at comparable compression levels. Unlike the other methods, RQS does not decrease AUC when compared to that of the uncompressed SNP calls. Furthermore, it is also demonstrably faster.
Fig. 2.
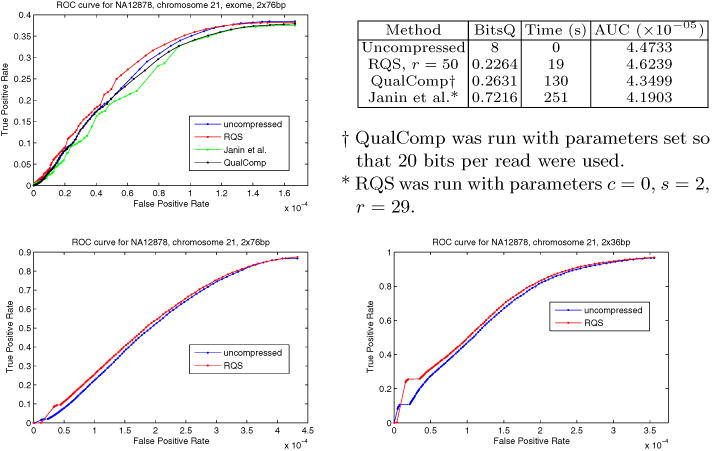
ROC curves of genotype calls made from the NA12878 dataset, both before and after compression, for exome reads of length 76bp, genome reads of length 36bp and 76bp. For the exome dataset (top), we also compared genotype call accuracies to QualComp and Janin et al. AUC values are included, integrated up to the largest of the maximum false positive rates to ensure comparability. To our knowledge, RQS is unique in improving accuracy, as measured by AUC, through compression. Note that the maximum true positive rate in the exome dataset is limited by the total fraction of variants covered by exome reads.
3.3 Compressing Large-Scale Whole-Genome Sequencing Datasets with RQS
We demonstrate that RQS scales to the whole genome using a probabilistic dictionary construction algorithm, as counting the exact number of appearances in the corpus of k-mers of a large whole-genome NGS dataset of 77 individuals is computation and memory intensive. Our sampling-based dictionary construction method identifies 32-mers that in expectation appear at least 500 times in the original corpus—by taking only 32-mers that appear at least 5 times in a randomly-chosen 1% of the reads.
Compressing the sampled reads using this method resulted in an average compression of 1.934 bits per quality score. Compressing all the reads—both sampled and unsampled—of HG02215 (one representative genome from the corpus) resulted in an average compression of 1.8406 bits/score. As is shown in the next section, these compression levels are better than the rates achieved with r = 350 or r = 550 when analyzing just chromosome 21 (Figure 3). These preliminary results indicate that we can effectively perform dictionary generation with a probabilistic sampling scheme and also larger (and potentially more redundant) genome sequences can facilitate better compression.
Fig. 3.
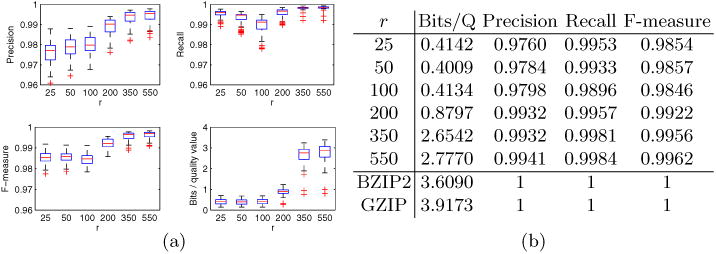
(a) Box plots of precision, recall, F-measure, and compression ratios across multiple values of read multiplicity r; each data point corresponds to one of the 77 genomes. Compression rates shown here include post-processing with BZIP2. (b) Table of averages for the same data shown in (a), along with compression levels of BZIP2 and GZIP.
3.4 Effect of Read Multiplicity on Compression and Accuracy
We have used several different values for read multiplicity r. Here, we apply RQS to the chromosome 21 reads in the corpus itself while sweeping over r to demonstrate the effect on compression and accuracy of variant calling; F-measure, precision, and recall are measured against the variant calls generated from the uncompressed data. Note that it is the ratio of read multiplicity to total depth coverage (here, 460) that matters, rather than absolute read multiplicity. However, since each read is independently compressed, once the dictionary is generated from the corpus, read coverage no longer affects the compression ratio.
As depicted in Figure 3, across multiple r values, RQS is able to maintain high fidelity with respect to the variants called in the uncompressed dataset. Furthermore, note that the loss of fidelity at r < 200 is not necessarily negative, as the improvements in accuracy shown in Figure 2 naturally require that the SNP-calls after compression differ from those for the uncompressed quality scores. However, we observe that compression rates become dramatically worse with r values higher than 200. This indicates that, for r > 200, our dictionary becomes too sparse to achieve high compression ratios.
4 Conclusions
Here we have shown that our RQS compressive framework capitalizes on the redundancies in the k-mer landscape of NGS read data to discard and sparsify nearly all quality scores, while at the same time enhancing compression ratio, speed, and downstream genotyping accuracy.
Acknowledgments
Y.W.Y. gratefully acknowledges support from the Fannie and John Hertz Foundation. This research was partially supported by the National Institutes of Health (NIH) R01GM108348. This content is solely the responsibility of the authors and does not reflect the official views of the NIH.
A Experimental Setup and Implementation of RQS
In the Results section, in order to perform an in-depth comparison between the performance of RQS and other compression methods on NGS reads from a large number of individuals, we reduced the domain of the genome to chromosome 21 (∼ 1.5% of whole genome in size).
In sections 3.1 and 3.2, read alignments were generated using BWA [25] in combination with GATK (Genome Analysis Toolkit) [26]. After chromosome 21 alignments were extracted, positional alignment information within mappings were removed before running RQS and other compression schemes.
In section 3.3, we used the reads aligned to the whole genome (instead of only chromosome 21), again removing all positional information from the alignments. Due to the large number of possible k-mers, it was not possible to fit the entire hash table into memory during dictionary generation. Though it is possible to compute exact k-mer frequencies using a parallelization approach, here we designed a less computation and memory intensive sampling-based dictionary construction method and demonstrated that the quality of the constructed dictionary is not affected.
In the RQS implementation, the dictionary generation from the corpus of reads was implemented using an unordered set data type in C++. For detecting all reads within Hamming distance 1 to the dictionary D; we stored D in a Boost multi-index hash table with 4 keys. Each key covers 24 out of 32 bases and each base of the 32-mer is covered exactly by 3 keys. Defining the key in this way allows us to aggressively prune most 32-mers within the dictionary, guaranteeing that each matching key implies a close match Δ(x, D) ≤ 8. If the total number of matches is < 96, we check the remaining nucleotides to verify that the matching 32-mers are within Hamming distance of 1. Otherwise, we enumerate all 96 neighboring 32-mers of the query 32-mer and check whether any of these exist in the dictionary.
During the sparsification step of our experiments, we chose 𝒬, the replacement quality value for discarded and smoothed positions, to be “40”. Although this is a user-defined parameter within our implementation, we selected this value as it was close to the average quality score value in our dataset.
After the sparsification step was completed, as a rough measure of entropy, we ran the quality scores through BZIP2 and recorded the number of bits per quality score required for storage. Although a production implementation would probably use a custom file format storing only a subset of the quality scores, using a modification of the standard SAM file format allowed for easy input into downstream analysis tools.
As a lossy compression method, there are two separate criteria upon which we can compare different methods: (1) the compression ratio and (2) the accuracy of downstream analysis. For the comparison between compression ratios, we fixed an F-measure threshold of 0.99 and searched for the best compression ratio of different tools across a range of input parameters, satisfying the minimum accuracy threshold. For fixed accuracy-level, RQS displayed superior compression rates compared to other methods (see Table 1). For the comparison between downstream variant call accuracies, we fixed a compression rate of 25× and searched for the best downstream variant call accuracy (with respect to the uncompressed data) of different tools across a range of parameters, satisfying minimum compression rate. For each fixed compression-level, RQS gave the most accurate F-measure values.
B Data Sources
Corpus from 1000 Genomes Project Phase 1:
HG00096 HG00100 HG00103 HG00106 HG00108 HG00111 HG00112 HG00114 HG00115 HG00116 HG00117 HG00118 HG00119 HG00120 HG00122 HG00123 HG00124 HG00125 HG00126 HG00127 HG00131 HG00133 HG00136 HG00137 HG00138 HG00139 HG00140 HG00141 HG00142 HG00143 HG00145 HG00146 HG00148 HG00149 HG00150 HG00151 HG00152 HG00154 HG00155 HG00156 HG00157 HG00158 HG00159 HG00160 HG00231 HG00232 HG00233 HG00236 HG00237 HG00239 HG00242 HG00243 HG00244 HG00245 HG00246 HG00247 HG00249 HG00250 HG00251 HG00252 HG00253 HG00254 HG00256 HG00257 HG00258 HG00259 HG00260 HG00261 HG00262 HG00263 HG00264 HG00265 HG01334 HG01789 HG01790 HG01791 HG02215
Exome reads for NA12878: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ ftp/phase1/data/NA12878/exome_alignment/NA12878.mapped.illumina. mosaik.CEU.exome.20110411.bam
Human Genome 18: ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/ technical/working/20101201_cg_NA12878/Homo_sapiens_assembly18.fasta
References
- 1.Berger B, Peng J, Singh M. Computational solutions for omics data. Nature Reviews Genetics. 2013;14:333–346. doi: 10.1038/nrg3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahn SD. On the future of genomic data. Science. 2011;331(6018):728–729. doi: 10.1126/science.1197891. [DOI] [PubMed] [Google Scholar]
- 3.Apostolico A, Lonardi S. Compression of biological sequences by greedy off-line textual substitution; Proceedings of the Data Compression Conference, DCC; 2000; pp. 143–152. IEEE 2000. [Google Scholar]
- 4.Kozanitis C, Saunders C, Kruglyak S, Bafna V, Varghese G. Compressing genomic sequence fragments using SlimGene. Journal of Computational Biology. 2011;18(3):401–413. doi: 10.1089/cmb.2010.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones DC, Ruzzo WL, Peng X, Katze MG. Compression of next-generation sequencing reads aided by highly efficient de novo assembly. Nucleic Acids Research. 2012;40(22):e171. doi: 10.1093/nar/gks754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fritz MHY, Leinonen R, Cochrane G, Birney E. Efficient storage of high throughput DNA sequencing data using reference-based compression. Genome Research. 2011;21:734–740. doi: 10.1101/gr.114819.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deorowicz S, Grabowski S. Compression of DNA sequence reads in FASTQ format. Bioinformatics. 2011;27(6):860–862. doi: 10.1093/bioinformatics/btr014. [DOI] [PubMed] [Google Scholar]
- 8.Loh PR, Baym M, Berger B. Compressive genomics. Nature Biotechnology. 2012;30:627–630. doi: 10.1038/nbt.2241. [DOI] [PubMed] [Google Scholar]
- 9.Bonfield JK, Mahoney MV. Compression of FASTQ and SAM format sequencing data. PloS one. 2013;8(3):e59190. doi: 10.1371/journal.pone.0059190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hach F, Numanagic I, Alkan C, Sahinalp SC. SCALCE: boosting sequence compression algorithms using locally consistent encoding. Bioinformatics. 2012;28(23):3051–3057. doi: 10.1093/bioinformatics/bts593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tembe W, Lowey J, Suh E. G-SQZ: compact encoding of genomic sequence and quality data. Bioinformatics. 2010;26(17):2192–2194. doi: 10.1093/bioinformatics/btq346. [DOI] [PubMed] [Google Scholar]
- 12.Popitsch N, von Haeseler A. NGC: lossless and lossy compression of aligned high-throughput sequencing data. Nucleic Acids Research. 2013;41(1):e27. doi: 10.1093/nar/gks939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan R, Anh VN, Asai K. Transformations for the compression of FASTQ quality scores of next-generation sequencing data. Bioinformatics. 2012;28(5):628–635. doi: 10.1093/bioinformatics/btr689. [DOI] [PubMed] [Google Scholar]
- 14.Christley S, Lu Y, Li C, Xie X. Human genomes as email attachments. Bioinformatics. 2009;25(2):274–275. doi: 10.1093/bioinformatics/btn582. [DOI] [PubMed] [Google Scholar]
- 15.Janin L, Rosone G, Cox AJ. Adaptive reference-free compression of sequence quality scores. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt257. [DOI] [PubMed] [Google Scholar]
- 16.Consortium TGP. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:1. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang X, Chockalingam SP, Aluru S. A survey of error-correction methods for next-generation sequencing. Briefings in Bioinformatics. 2013;14(1):56–66. doi: 10.1093/bib/bbs015. [DOI] [PubMed] [Google Scholar]
- 18.Melsted P, Pritchard JK. Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinformatics. 2011;12(1):333. doi: 10.1186/1471-2105-12-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27(6):764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelley DR, Schatz MC, Salzberg SL, et al. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11(11):116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Schröder J, Schmidt B. Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics. 2013;29(3):308–315. doi: 10.1093/bioinformatics/bts690. [DOI] [PubMed] [Google Scholar]
- 22.Ilie L, Molnar M. RACER: Rapid and accurate correction of errors in reads. Bioinformatics. 2013;29(19):2490–2493. doi: 10.1093/bioinformatics/btt407. [DOI] [PubMed] [Google Scholar]
- 23.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ochoa I, Asnani H, Bharadia D, Chowdhury M, Weissman T, Yona G. QualComp: a new lossy compressor for quality scores based on rate distortion theory. BMC Bioinformatics. 2013;14:187. doi: 10.1186/1471-2105-14-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Consortium TGP. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]