

Abstract
Background
Using whole genome sequence data might improve genomic prediction accuracy, when compared with high-density SNP arrays, and could lead to identification of casual mutations affecting complex traits. For some traits, the most accurate genomic predictions are achieved with non-linear Bayesian methods. However, as the number of variants and the size of the reference population increase, the computational time required to implement these Bayesian methods (typically with Monte Carlo Markov Chain sampling) becomes unfeasibly long.
Results
Here, we applied a new method, HyB_BR (for Hybrid BayesR), which implements a mixture model of normal distributions and hybridizes an Expectation-Maximization (EM) algorithm followed by Markov Chain Monte Carlo (MCMC) sampling, to genomic prediction in a large dairy cattle population with imputed whole genome sequence data. The imputed whole genome sequence data included 994,019 variant genotypes of 16,214 Holstein and Jersey bulls and cows. Traits included fat yield, milk volume, protein kg, fat% and protein% in milk, as well as fertility and heat tolerance. HyB_BR achieved genomic prediction accuracies as high as the full MCMC implementation of BayesR, both for predicting a validation set of Holstein and Jersey bulls (multi-breed prediction) and a validation set of Australian Red bulls (across-breed prediction). HyB_BR had a ten fold reduction in compute time, compared with the MCMC implementation of BayesR (48 hours versus 594 hours). We also demonstrate that in many cases HyB_BR identified sequence variants with a high posterior probability of affecting the milk production or fertility traits that were similar to those identified in BayesR. For heat tolerance, both HyB_BR and BayesR found variants in or close to promising candidate genes associated with this trait and not detected by previous studies.
Conclusions
The results demonstrate that HyB_BR is a feasible method for simultaneous genomic prediction and QTL mapping with whole genome sequence in large reference populations.
Background
Whole genome sequence data is available for an increasing number of species. In some cases enough individuals have been sequenced to serve as a reference panel for imputation of individuals that have been genotyped with SNP arrays to whole genome sequence variant genotypes. A good example of such a reference set is the 1000 bull genomes project which includes 234 bulls with whole-genome sequencing data and 28.3 million genotyped sequence variants [1]. Compared with dense SNP arrays, the advantage of using whole genome sequence data might potentially include more accurate genomic predictions within and across breeds [2–5], better persistence of accuracy of genomic predictions across generations, and more precise QTL mapping [5], all as a result of including the causal mutation genotypes in the data set.
As the resulting data sets will be extremely large (thousands of individuals with millions of imputed genotypes), the algorithms used to derive genomic predictions must be computationally efficient. Ideally, they should also implement a non-linear model at the level of the SNP effects, including the possibility of excluding some SNPs from the model, as such models have been demonstrated to give higher accuracies of genomic predictions for some traits with high-density genotype data [5, 6]. Although computationally efficient, GBLUP and BLUP do not satisfy the second criteria (they implement a linear model and all SNPs are always in the genomic prediction model). BayesR [7] is a flexible non-linear model, which assumes that SNP effects follow a mixture of four normal distributions (with zero variance, very small variance, small variance, and moderate variance). Compared with GBLUP, BayesR results in superior accuracy of genomic prediction for some traits [6, 8–12]. However, as Bayesian models are typically implemented with MCMC (Markov Chain Monte Carlo) sampling, application of BayesR with sequence data is currently not feasible.
Another advantage of non-linear models such as BayesR, is the application of QTL mapping [5, 6, 8, 13, 14]. Loh et al. [14] pointed out that Bayesian mixed-models with speed-up schemes (termed fastBayesB [15]) could improve the power of detecting genes associated with human diseases. There are several modified versions of Bayesian model implemented for the identification of causal mutations. Speed and Balding [13] developed an efficient approach termed multiBLUP (a mixture model of SNP effects, similar to nonlinear models), which was applied on the Welcome Trust Case Control Consortium (WTCCC) human disease data. Later, Kemper et al. [6] implemented a nonlinear model (BayesR) for mapping QTL to 250 kb windows in dairy cattle. Then, Moser et al. [8] applied a modified version of BayesR (updating the additive genetic variance in the MCMC chain instead of fixing it, as in the original BayesR) to WTCCC human disease data. Furthermore, MacLeod et al. [5] proposed the algorithm referred to as BayesRC, which is a modified version of BayesR incorporating biological prior information. All these studies have demonstrated that nonlinear models, which might exclude SNPs from the models with the assumptions of Bayesian mixture priors for SNP effects, could actually help to improve the precision of QTL mapping or association studies in human or dairy cattle.
To take advantage of the accuracy superiority of MCMC nonlinear models but improve their time-efficiency, a hybrid scheme (termed HyB_BR) was proposed by Wang et al. [16]. This scheme has three steps: 1) Implement the mixture model of BayesR, which had been demonstrated to be quite flexible for genomic prediction; 2) run an expectation-maximisation algorithm that estimates the parameters in the mixture model; 3) Using the solutions from the EM as starting points, run a limited number of MCMC iterations to improve the parameter estimates. The results of the Hybrid algorithm on 600 K SNP data in dairy cattle data and 300 K SNP data in human disease data from Welcome Trust Case Control Consortium (WTCCC) have demonstrated that the Hybrid algorithm performed as well as BayesR while requiring half of the running time demanded by MCMC iterations [16].
With the aim of investigating whether HyB_BR gave comparable accuracies to BayesR with MCMC for genomic prediction and precision of QTL mapping with whole genome sequence data, we implemented HyB_BR on a large subset of imputed whole-genome sequence data with 994,019 variants in 16,214 cattle. The genotype data came from the imputed sequence variants in or close to gene coding regions and some SNP from the 600 K Bovine HD SNP genotypes. The HyB_BR algorithm was evaluated on this data set with three criteria: 1) computational performance (speed) compared to a full MCMC implementation, 2) prediction accuracy for a range of complex traits with different genetic architecture. The traits included fat yield, milk yield, protein yield, fat percent, protein percent, fertility and heat tolerance and 3) the precision of HyB_BR for QTL mapping of milk production traits, fertility and heat tolerance.
Methods
High density and sequence genotypes
Two types of genomic data, 600 K Bovine HD SNP array, and imputed sequence data were used in this study. As described by Kemper et al. (2015) [6], 10,311 Holstein, 4738 Jersey and 249 Australian Red bulls and cows were genotyped with the Bovine SNP50 Array (Illumina, San Diego, CA). In addition, 1620 Holstein bulls and cows, 125 Jersey bulls, and 114 Australian Red bulls were genotyped with the 777 K bovine HD SNP panel. After quality control steps described by Erbe et al. (2012) [7], all genotypes were imputed to 632,003 SNP using Beagle 3.0 [17].
For the Sequence data set (termed SEQ), the sequences of 136 Holstein and 27 Jersey bulls from the 1000 Bulls Genome Project [1] were used as a reference set for imputation. All the animals described above with real or imputed 600 K SNP genotypes were imputed to whole genome sequence data using Beagle 3.0 software [18]. In total there were 2.785 million sequence variants imputed, including both SNPs and indels in either coding regions or putative regulatory regions flanking genes [5]. After quality control including minor allele frequency filtering and LD pruning by PLINK [19], there were 994,019 variants remaining including 370,259 markers from the 600 K SNP panel, and 623,760 sequence variants in gene coding regions or 5000 bp up- and down-stream of the gene start stop positions as detailed by MacLeod et al. (2016) [5].
Phenotypes
Protein, fat and milk yields and, fat and protein percent are key traits in dairy cattle breeding. Phenotypes that were pre-corrected for fixed effects (herd-year-season, and lactation number) were used in this study, these are known as trait deviations (TDs) and daughter trait deviations (DTDs) for cows and bulls respectively. TDs and DTDs are provided by DataGene (and its predecessor, the Australian Dairy Herd Improvement Scheme), which is the organisation responsible for providing genetic evaluations to the Australian dairy industry. (e.g. DataGene; http://datagene.com.au/index.php). A summary of the phenotypes is shown in Table 1. For milk production traits, there were 16,214 bulls and cows from Holstein and Jersey breeds as the reference set. Then, for the validation sets, Holstein and Jersey bulls were used to assess the accuracy of within-breed prediction. These bulls were the youngest cohorts (born after 2005) in the data set. As mentioned in [6], all the bulls of the validation set have more than 20 effective daughters. In addition, Australian Red bulls (a third breed; not included in the reference set) were included for the validation set to evaluate the performance of across-breed prediction. We implemented the calculation of Garrick et al. (2009) [20] to appropriately weight the phenotypes of bulls and cows as follows:
Table 1.
The number of animals in the reference sets and validation sets
| Traits | Reference sets | Validation sets | |||||
|---|---|---|---|---|---|---|---|
| Holstein | Jersey | Holstein Bulls | Jersey Bulls | Australian Red Bulls | |||
| Bulls | Cows | Bulls | Cows | ||||
| Milk production traits (FatY/MilkY/ProtY/Fat%/Protein%) | 3049 | 8478 | 770 | 3917 | 262 | 105 | 114 |
| Fertility | 2806 | 7838 | 716 | 3830 | 396 | 81 | 114 |
| Heat Tolerance traits (FatY_HT/MilkY_HT /ProtY_HT) | 2028 | 2037 | 476 | 1116 | 252 | 101 | - |
Milk production traits include fat yield (FatY), milk yield (MilkY), protein yield (ProteinY), fat percent (Fat%) and protein percent (Protein%); Heat tolerance traits are the decline of fat yield (FatY_HT), milk yield (MilkY_HT) and protein yield (ProtY_HT) under heat stress
where, h 2 is the heritability of the trait; t is the repeatability of the traits; d is the number of the daughter of each bulls; r is the number of records; c is the proportion of additive genetic variance not accounted for by the SNP [20]. To compare the prediction accuracy of GBLUP, BayesR and HyB_BR for multi-breeds and across-breed, the weight calculation is included in all three models.
In addition to milk production traits, fertility is another important complex trait. The DTD and TD that DataGene calculate and that was available to this study was calving interval (CI) which is the number of days between consecutive calving, For fertility, the number of bulls and cows in the reference set, i.e. with genotypes and fertility phenotypes was around 15,190. The validation set includes Holstein bulls (youngest cohort born after 2004) and Jersey bulls (youngest cohort born after 2005).
As weather becomes warmer and less predictable, there is growing interest in developing genomic breeding values for heat tolerance [21]. In Australian dairy genetics studies, heat tolerance is defined as the rate of the decline in production traits (e.g. fat, milk and protein yield) with increasing heat stress [21]. The rate of decline for each trait was estimated for each cow in the data set with a linear random regression of yield on daily temperature-humidity index (THI), when THI was above a threshold of 60 units [21–23]. The total number of animals with phenotypes for heat tolerance was 5657 and included Holstein and Jersey cows and bulls. The validation set for heat tolerance was a set of Holstein bulls and a set of Jersey bulls, Table 1. In contrast to the milk production and fertility phenotypes, heat tolerance is still under development and is not yet officially released as a breeding value in Australia.
The input parameters for HyB_BR were estimated from the data with ASReml4 [24] and included additive genetic variance, error variance, and additive polygenic variance (Table 2). Using the variances, the heritability is calculated based on the “narrow-sense” definition [25] as the ratio of additive genetic variance and the sum of additive genetic variance, error variance and additive polygenetic variance (). The heritability for milk production traits is consistent with the published results of Kemper et al. (2015) [6]. Compared with milk production traits, heritabilities for heat tolerance traits and fertility were lower. Across all the traits, the prediction accuracy is evaluated using the correlations between genomic estimated breeding value (GEBV) and DTD in the validation sets. The regression of DTD on GEBV in the validation sets was used to investigate if any of the methods resulted in biased predictions.
Table 2.
The genetic architecture of milk production traits and Fertility estimated by ASReml
| Additive genetic variance () | Additive polygenic variance () | Error variance () | Heritability (h 2) | |
|---|---|---|---|---|
| FatY | 118.594 | 48.689 | 234.326 | 0.421 |
| MilkY | 114.827e + 03 | 38.532e + 03 | 135.598e + 03 | 0.528 |
| ProtY | 72.488 | 36.072 | 140.417 | 0.443 |
| Fat% | 0.056 | 0.008 | 0.018 | 0.781 |
| Protein% | 0.012 | 0.003 | 0.003 | 0.818 |
| Fertility | 42.990 | 0.003e-01 | 340.287e-01 | 0.013 |
| FatY_HTa | 0.041 | 0.581e-07 | 0.571 | 0.072 |
| MilkY_HTa | 0.004 | 0.353e-06 | 0.035 | 0.091 |
| ProtY_HTa | 0.035 | 0.564e-07 | 0.561 | 0.059 |
aLabels the traits of Fat yield, milk yield, and protein yield under heat stress
Genomic prediction methods
GBLUP
GBLUP assumes all marker effects follow a normal distribution with the same additive genetic variance. The overall model of GBLUP is:
| 1 |
Where,
y = vector of n phenotypes.
β = vector of b fixed effects, following uninformative priors.
u = vector of q random genetic effects (q = number of animals) captured by the SNP, with . G is the q x q genomic similarity matrix between pairs of individuals constructed as described by [26]; is the additive genetic variance.
v = vector of q additive polygenic effects (q = number of animals), with ). A is the q × q pedigree-based relationship matrix, and is the additive polygenic variance.
e= vector of n residual errors. For cattle data, , the n × n diagonal matrix E is especially designed to evaluate the different contributions of the phenotype records from different sex to the error variance, de-regressing predicted breeding values and weighting information for genomic regression analyses [20].
X = n × b design matrix, allocating phenotypes y to fixed effects β. b is the number of fixed effects
W= n × q design matrix, which aims at allocating the q × 1 vector of polygenic effects to y.
S = n × q design matrix, allocating the q × 1 vector of genetic values to y.
BayesR
Compared with the common prior distributions of GBLUP, BayesR [7] assumes SNP effects are drawn from the mixture of four normal distributions. BayesR aims at estimating each SNP effects instead of estimating breeding values directly for each animal. Therefore, the genetic value u in the model (1) is substituted with Zg in the BayesR model. Briefly, the data model of BayesR can be written as:
| 2 |
Where,
g = m vector of SNP effects, , . Therefore, each SNP have four possible normal distributions: ), ), ), and ). Related to such mixture priors, there are two other parameters including b(i, k) and Pr.
b(i, k) = {0, 1}, which defines whether or nor SNP i follows normal distribution k (k = 1 , 2 , 3 , 4). Therefore, the prior distribution of each SNP i conditional on b(i, k) can be written as:
Pr= the vector of proportion parameter, which defines the proportion SNPs in each of four normal distributions. The prior of Pr is drawn from Dirichlet distribution Pr ~ Dirichlet(α), with α = [1, 1, 1, 1]. The conditional distribution of SNP effect on the proportion parameter Pr is:
Z is the standardised (for mean and variance) n × m genotype matrix.
To implement the BayesR model, and arrive at posterior estimates of parameters, Gibbs sampling has been used, as described by Kemper et al. (2015) [6]. On the sequence data, we use five independent replicate chains of the Gibbs sampling, and for each independent chain, there are 40,000 iterations, with the first 20,000 iterations discarded as burn in, as described by Kemper et al. (2015) (for 630 K SNP data).
HyB_BR
The HyB_BR model [16] incorporates the same assumption for SNP effects as BayesR, but serially hybridizes the expectation-maximization (EM) and MCMC to reduce large number of iterations required by MCMC. That is, HyB_BR first implements an EM algorithm to perform the Maximum A Posterior (MAP) estimation until converged. Then, to improve accuracy, a limited number of MCMC iterations are performed to improve parameter estimates [16].
As described in Wang et al. (2016) [16], the HyB_BR model for a SNP effect is:
| 3 |
Assumptions in the model are 1) each SNP effect gi follows the same prior assumption as BayesR with Z i being the standardized genotype for SNP i. 2) to correct the prediction errors generated by all other SNPs, HyB_BR introduces the genetic values u, whereby a correction based on the prediction error variance (PEV) is introduced to account for the effects of all the other SNP with a GBLUP model as detailed by Wang et al. [16]. Then under the model (3), the posterior distribution for all related parameter sets {g i , Pr , β , u , v, } are derived according to the theory: p(θ| y) ∝ f(y| θ)p(θ), where f(y| θ) is the likelihood function based on model (3) and p(θ) is the prior density function for the parameter sets θ. Based on the derived marginal posterior distribution p(θ| y), the expectation- maximization steps are implemented to estimate each parameter while “integrating out” the other parameters detailed by Wang et al. (2016). The process of the EM module is presented in pseudo code in Fig. 1.
Fig. 1.

The pseudo-code of the EM module
As shown in Fig. 1, the EM module begins by initializing all the input parameters including SNP effects (g), Proportion parameter (Pr), the variance for each SNP (), the fixed matrix (X), the pedigree based relationship matrix (A), the genomic relationship matrix (G), the error matrix (E), and index matrix for polygenic effects (W). Similar to emBayesR [27], the starting values of g and Pr are set as g = 0.01 and Pr = {0.5, 0.487, 0.01, 0.003}, while . The additive genetic variance , error variance , and polygenic variance are obtained from ASReml. Later, HyB_BR fixes the value of the additive genetic variance and additive polygenic variance (not updating them in later MCMC and EM iterations). The n×3 matrix X is a design matrix, allocating the phenotypes to fixed effects. In our case, matrix X is set up with first column being the mean, the second and third columns defining the breeds (Holstein and Jersey) and sex (bulls and cows) of the cattle. The pedigree relationship matrix, A, is built using the lower symmetrical matrix Ped detailed by Henderson [28]; while the genomic relationship matrix G is constructed using the equation G = Z s Z s′/n, Z s is the standardized Z matrix with . The diagonal error matrix E is constructed according to the equation defined by Garrick et al. [20] and described above for the phenotypes used in this study.
The EM steps require the time complexity O(mn). For the calculation of which is calculated prior to the EM steps, the required time is O(m 2 n). This calculation accounts for 40% of the total computational time of EM module. Since the calculation is independent for each SNP, we parallelize the operations by chromosomes, which reduce total running time by approximately 30%.
Once the EM has converged using the criterion ( with q the iteration number, the parameter estimates from the EM are used as starting points of parameter values in the MCMC iterations. The steps of MCMC iterations iteration were detailed by Kemper et al. (2015) [6]. Furthermore, Wang et al. (2016) [16] suggested a speed-up scheme to improve computational efficiency. The scheme is as follows. After 500 MCMC iterations, the SNPs with high probability in the distribution with zero variance will be excluded from the model. In other words, when P(i, 1) is greater than 0.90, the SNP effects will be set as zero. Previous investigation showed that 4000 MCMC iterations were required by HyB_BR for both 600 K SNP panel and imputed sequence data to maximize accuracy of genomic prediction across all the traits [16].
To compare the computational cost between BayesR and HyB_BR and how this changed with an increasing number of individuals in the reference set, we divided the data (Table 1) into three different referent sets (Ref1, Ref2, and Ref3) (with the number of sequence variants held constant). Ref1 had Holstein bulls only with 3049 bulls; Ref2 included Holstein bulls and cows with 12,527 animals; Ref3 had all the data (16,214 animals).
In all three reference sets, the speed advantage of HyB_BR compared with BayesR was investigated. Then the accuracy of genomic prediction from BayesR, HyB_BR and GBLUP was compared in the full data (including the sequence variants).
In addition, the precision of mapping QTL from the three methods was compared.
Results
Computational time comparison between GBLUP, BayesR and HyB_BR
For both 600 K and SEQ data sets, HyB_BR was more than 10 times faster than BayesR, Fig. 2. As the size of the data set increased (from Ref 1 to Ref3 or from 600 K to SEQ data), the computational time required for HyB_BR could be reduced by a greater and greater margin relative to BayesR. On 600 K data, HyB_BR had a similar compute time to GBLUP. For the SEQ data, HyB_BR was up to four fold faster than GBLUP.
Fig. 2.
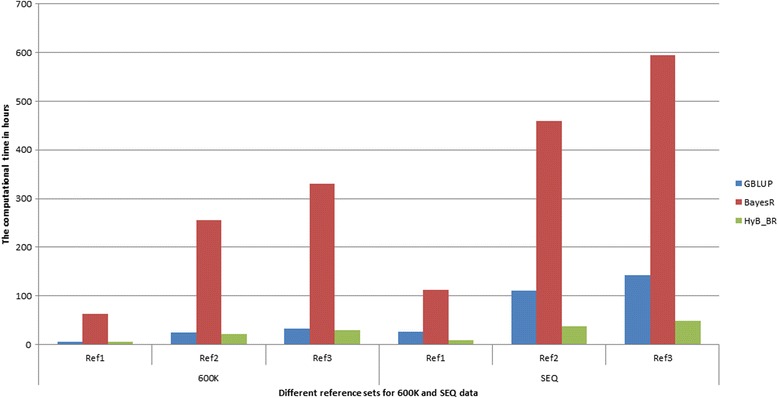
The computational time comparison between GBLUP, BayesR and HyB_BR on 600 K and SEQ data. Three reference sets (Ref1, Ref2 and Ref3) with the same number of variants (600 K or SEQ) are used here. Ref1 has Holstein bulls data with 3049 animals; Ref2 has Holstein bulls and cows data with 12,527 animals; Ref3 has Holstein and Jersey bulls and cows with 16,214 individuals
These timings were recorded on a server with Intel E5–2680 2.7GHz processors and 384GB of 1333 MHz RAM.
Accuracy of genomic prediction for GBLUP, BayesR, and hybrid with sequence data
Prediction accuracy for milk production traits and fertility
For the milk production and fertility traits, the combined Holstein and Jersey reference sets were used to predict three validation sets including Holstein bulls (Table 3), Jersey bulls (Table 3), and Australian Red bulls & cows (Table 4).
Table 3.
The multi-breed prediction accuracy and bias of GBLUP, BayesR, and HyB_BR on SEQ data related to Fat Yield, Milk Yield, Protein Yield, Fat%, Protein% and Fertility
| Holstein and Jersey reference to predict Holstein validation | |||||||||||||
| Fat Yield | Milk Yield | Protein Yield | Fat% | Protein% | Fertility | ||||||||
| Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | ||
| GBLUP | +Polya | 0.64 | 1.07 | 0.66 | 0.92 | 0.63 | 0.95 | 0.76 | 0.95 | 0.83 | 0.98 | 0.42 | 1.70 |
| -Polyb | 0.62 | 1.32 | 0.60 | 0.83 | 0.58 | 1.15 | 0.75 | 1.01 | 0.81 | 1.09 | 0.42 | 1.70 | |
| BayesR | +Polya | 0.65 | 1.27 | 0.69 | 0.91 | 0.68 | 1.04 | 0.81 | 1.01 | 0.83 | 0.99 | 0.42 | 1.32 |
| -Polyb | 0.63 | 1.17 | 0.67 | 0.85 | 0.65 | 0.91 | 0.80 | 1.01 | 0.82 | 0.96 | 0.42 | 1.32 | |
| HyB_BR | +Polya | 0.66 | 1.04 | 0.69 | 0.89 | 0.68 | 0.96 | 0.81 | 0.99 | 0.83 | 0.96 | 0.42 | 1.32 |
| -Polyb | 0.63 | 0.96 | 0.69 | 0.89 | 0.66 | 0.88 | 0.81 | 0.99 | 0.81 | 0.94 | 0.42 | 1.32 | |
| Holstein and Jersey reference to predict Jersey validation | |||||||||||||
| Fat Yield | Milk Yield | Protein Yield | Fat% | Protein% | Fertility | ||||||||
| Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | ||
| GBLUP | +Polya | 0.54 | 0.76 | 0.65 | 0.88 | 0.69 | 0.94 | 0.67 | 0.86 | 0.77 | 0.94 | 0.23 | 1.13 |
| -Polyb | 0.52 | 0.93 | 0.65 | 1.03 | 0.68 | 1.24 | 0.66 | 0.93 | 0.75 | 1.02 | 0.23 | 1.13 | |
| BayesR | +Polya | 0.57 | 0.88 | 0.70 | 0.96 | 0.72 | 1.22 | 0.77 | 0.97 | 0.77 | 0.89 | 0.23 | 1.03 |
| -Polyb | 0.52 | 0.73 | 0.68 | 0.87 | 0.67 | 1.02 | 0.76 | 0.95 | 0.77 | 0.87 | 0.23 | 1.02 | |
| HyB_BR | +Polya | 0.58 | 0.87 | 0.69 | 0.95 | 0.73 | 0.91 | 0.77 | 0.93 | 0.79 | 0.87 | 0.23 | 0.97 |
| -Polyb | 0.57 | 0.74 | 0.69 | 0.85 | 0.73 | 0.91 | 0.76 | 0.93 | 0.78 | 0.85 | 0.23 | 0.97 | |
The bulls and cows from two breeds of Holstein and Jersey are used as the reference set to predict Holstein bulls and Jersey bulls separately. aThe prediction accuracy when adding the polygenic term in the model; whilebis the prediction accuracy when leaving out the polygenic term from the model
Table 4.
The across breed prediction accuracy of GBLUP, BayesR, and HyB_BR on SEQ data related to Fat Yield, Milk Yield, Protein Yield, Fat%, Protein% and Fertility
| Across breeds prediction on Australian red bulls | ||||||||||||
| Fat Yield | Milk Yield | Protein Yield | Fat% | Protein% | Fertility | |||||||
| Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | |
| GBLUP | 0.13 | 0.58 | 0.21 | 0.59 | 0.15 | 0.71 | 0.39 | 0.61 | 0.50 | 1.32 | 0.22 | 0.96 |
| BayesR | 0.35 | 1.31 | 0.22 | 0.77 | 0.24 | 0.92 | 0.40 | 0.61 | 0.53 | 0.86 | 0.27 | 0.97 |
| HyB_BR | 0.28 | 0.74 | 0.36 | 0.70 | 0.26 | 0.74 | 0.47 | 0.66 | 0.53 | 0.88 | 0.27 | 0.95 |
| Across breeds prediction on Australian red cows | ||||||||||||
| Fat Yield | Milk Yield | Protein Yield | Fat% | Protein% | Fertility | |||||||
| Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | Acc. | Bias | |
| GBLUP | 0.15 | 0.77 | 0.11 | 0.37 | 0.12 | 0.57 | 0.31 | 0.92 | 0.34 | 1.09 | 0.07 | 0.61 |
| BayesR | 0.28 | 1.02 | 0.22 | 0.55 | 0.16 | 0.60 | 0.37 | 0.94 | 0.34 | 0.93 | 0.07 | 0.52 |
| HyB_BR | 0.25 | 0.88 | 0.23 | 0.54 | 0.16 | 0.59 | 0.37 | 0.91 | 0.34 | 0.91 | 0.07 | 0.57 |
The bulls and cows from two breeds of Holstein and Jersey are used as the reference set to predict Australian red bulls and cows
When predicting the Holstein validation bull data, BayesR and HyB_BR performed equally well. Compared with GBLUP, BayesR and HyB_BR had a small but consistent accuracy improvement for the milk production traits except protein%. For fat% trait, BayesR and HyB_BR gave a 5% improvement in accuracy compared with GBLUP. However, for protein% and fertility there was no difference between the methods. With the Jersey validation set, the accuracy superiority of HyB_BR and BayesR over GBLUP was greater; for example for fat percent, BayesR and HyB_BR gave a 10% higher accuracy than GBLUP. HyB_BR and BayesR also gave regression coefficients (DTD on GEBV) closer to one than GBLUP for most traits.
In addition, when incorporating the polygenic effects into the prediction model, a small but consistent accuracy improvement was observed for milk production traits, Table 3. However, for fertility, including the polygenic effects did not affect the prediction accuracy at all.
When predicting Australian red bulls and cows using the combined Holstein and Jersey reference set (across breed prediction), both HyB_BR and BayesR had a considerable accuracy advantage (up to 12% increase) over GBLUP for all the traits (Table 4). Compared with BayesR, HyB_BR performed equally, or better, in terms of accuracy for all traits except fat yield.
Accuracy of genomic prediction for heat tolerance
The accuracy of genomic prediction for the heat tolerance traits was similar for GBLUP, BayesR, and HyB_BR, Table 5. There were two exceptions when predicting the validation set of Jersey bulls: 1) for the fat yield trait associated heat tolerance, there was a 6% accuracy reduction for BayesR and HyB_BR in comparison with GBLUP; 2) For milk yield, a 9% increase in accuracy from BayesR and HyB_BR over that of GBLUP was observed. Given the small size of the validation populations, these differences were not statistically significantly different. HyB_BR and BayesR did give regression coefficients closer to one compared with GBLUP for all the traits.
Table 5.
The multi-breed prediction accuracy and bias of GBLUP, BayesR, and HyB_BR on SEQ data related to traits affected by heat tolerance
| Holstein and Jersey reference Prediction on Holstein bulls | ||||||
| Fat | Milk | Protein | ||||
| Acc. | Bias | Acc. | Bias | Acc. | Bias | |
| GBLUP | 0.35 | 1.47 | 0.24 | 0.84 | 0.32 | 1.24 |
| BayesR | 0.35 | 1.05 | 0.29 | 0.88 | 0.33 | 0.92 |
| HyB_BR | 0.35 | 1.05 | 0.28 | 0.86 | 0.33 | 1.01 |
| Holstein and Jersey reference Prediction on Jersey bulls | ||||||
| Fat | Milk | Protein | ||||
| Acc. | Bias | Acc. | Bias | Acc. | Bias | |
| GBLUP | 0.33 | 1.25 | 0.37 | 1.11 | 0.35 | 0.72 |
| BayesR | 0.27 | 0.89 | 0.46 | 0.89 | 0.35 | 0.76 |
| HyB_BR | 0.27 | 0.88 | 0.46 | 0.89 | 0.35 | 0.77 |
The bulls and cows from two breeds of Holstein and Jersey are used as the reference set to predict Holstein bulls and Jersey bulls separately
Compared with 600 K SNP panels, the impact of sequence data (SEQ) on the prediction accuracy of GBLUP, BayesR, and HyB_BR was dependent on the trait and validation population (Fig. 3). For the prediction of the validation sets of Holstein or Jersey bulls (which were closely related to the reference set), only a very small accuracy gain (1% ~ 2%) was observed from using sequence data compared to using the 600 K panel. However, when the validation set comprised of Australian Red bulls and cows, there was greater advantage of using the sequence data, provided BayesR or HyB_BR was used. For example, the accuracy using BayesR and HyB_BR with the sequence data was up to 13% higher than when the 600 K SNP panel was used. When using sequence data, GBLUP gave only a very limited increase (or even a reduction for Fat Yield trait).
Fig. 3.
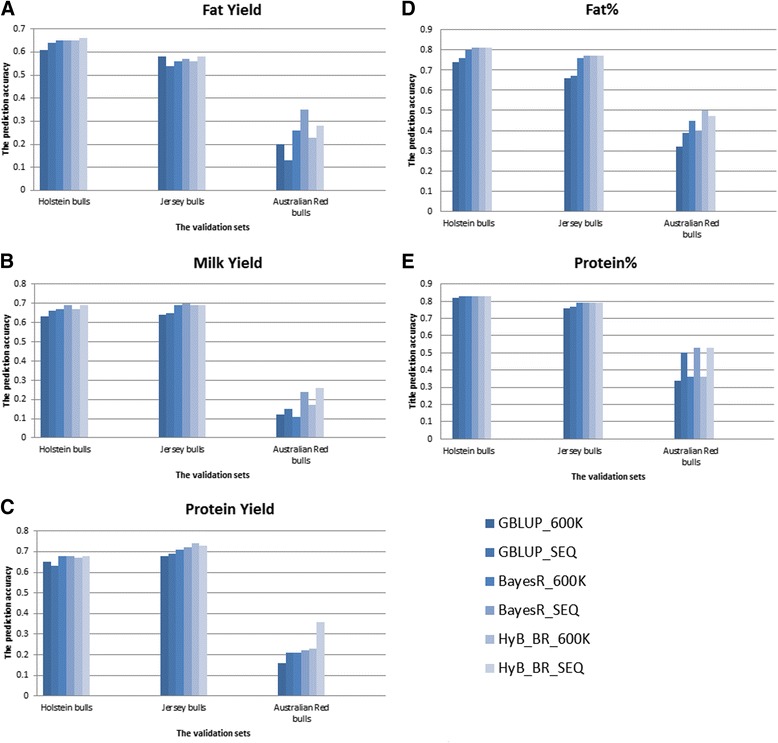
The prediction accuracy of GBLUP, BayesR, and HyB_BR on 600 K and SEQ data related to three milk production traits including Fat Yield (a), Milk Yield (b), Protein Yield (c), Fat Percent (d), and Protein Percent (e)
Inference of genetic architecture
To compare the genetic architecture of the traits using whole genome sequence data, the number of SNPs in each of four distributions (with the variance , , , or ) was investigated (Table 6). Across all the traits, BayesR and HyB_BR gave a similar proportion of SNP in the distribution with the largest variance . However, there was a difference in the proportion of SNPs in each of the four distributions, in that is HyB_BR systematically estimated more variants in the distributions with non-zero variances than BayesR.
Table 6.
The proportion of variants in each of four distributions (0, , , or ) estimated from BayesR (termed BR) and HyB_BR (termed HB)
| Fat Yield | Milk Yield | Protein Yield | Fat% | Protein% | Fertility | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BR (%) | HB (%) | BR (%) | HB (%) | BR (%) | HB (%) | BR (%) | HB (%) | BR (%) | HB (%) | BR (%) | HB (%) | |
| 0 | 99.484 | 99.015 | 99.542 | 99.242 | 99.494 | 99.224 | 99.660 | 99.380 | 99.577 | 99.171 | 99.515 | 99.298 |
| 0.513 | 0.958 | 0.449 | 0.717 | 0.501 | 0.725 | 0.333 | 0.612 | 0.405 | 0.799 | 0.453 | 0.676 | |
| 0.002 | 0.027 | 0.009 | 0.039 | 0.004 | 0.05 | 0.004 | 0.006 | 0.015 | 0.028 | 0.030 | 0.025 | |
| 0.001 | 0.001 | 0.001 | 0.002 | 0.001 | 0.001 | 0.002 | 0.002 | 0.002 | 0.002 | 0.001 | 0.002 | |
QTL mapping
For all the traits, estimated posterior possibilities from BayesR and HyB_BR were plotted across the whole genome locations of SNPs, Figs. 4, 5, 6, 7, 8, 9, 10 and 11. According to the posterior possibilities, the thresholds (the grey horizon lines in the figures; the probabilities above which there are the same number of SNPs as in the distribution with largest variance ) were set to highlight the top SNPs. Top variants with the highest posterior probability of being in the distribution with the largest variance from BayesR and HyB_BR were investigated.
Fig. 4.
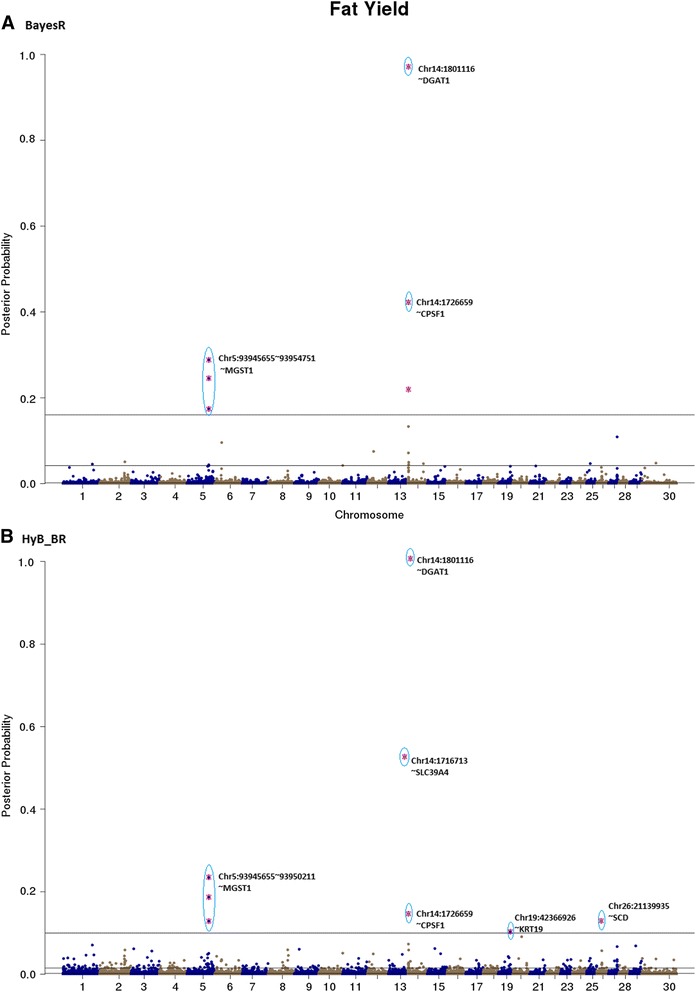
Posterior possibilities of all the variants on fat yield estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole genome. The top SNPs with highest posterior possibilities are labelled with blue circle
Fig. 5.
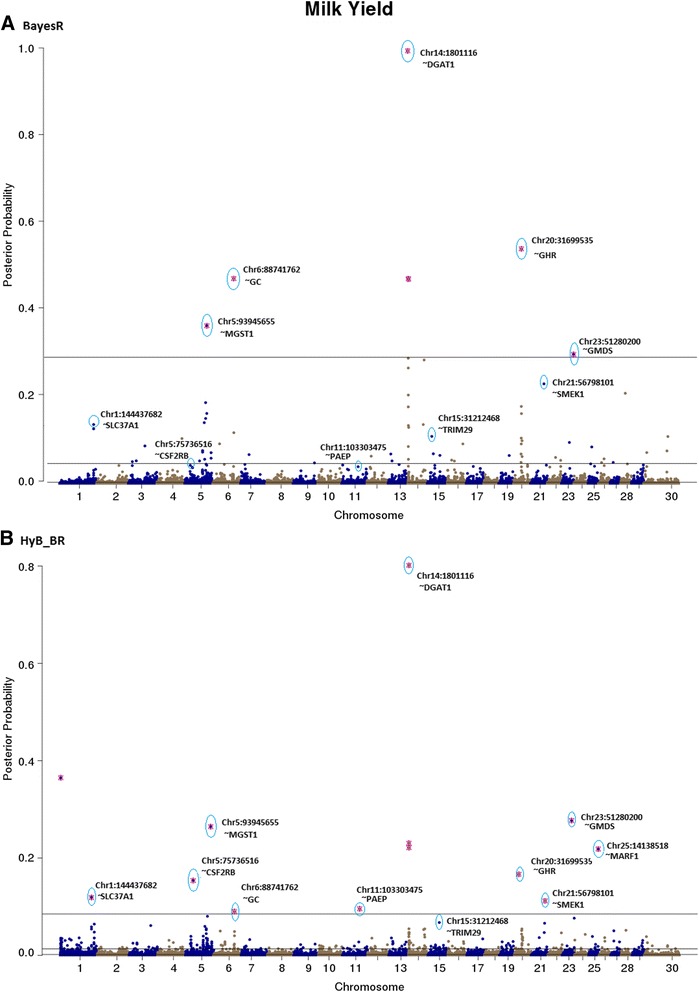
Posterior possibilities of all the variants for milk yield estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole genome. The top SNPs with highest posterior possibilities are labelled with blue circle
Fig. 6.
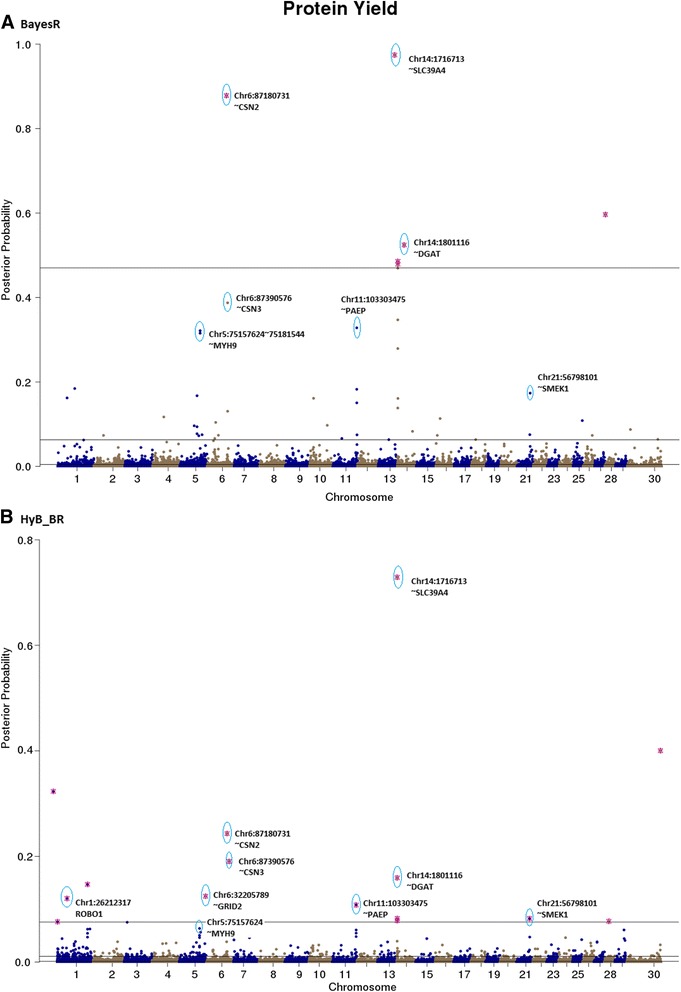
Posterior possibilities of all the variants for protein yield estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole chromosome genome. The top SNPs with highest posterior possibilities are labelled with blue circle
Fig. 7.
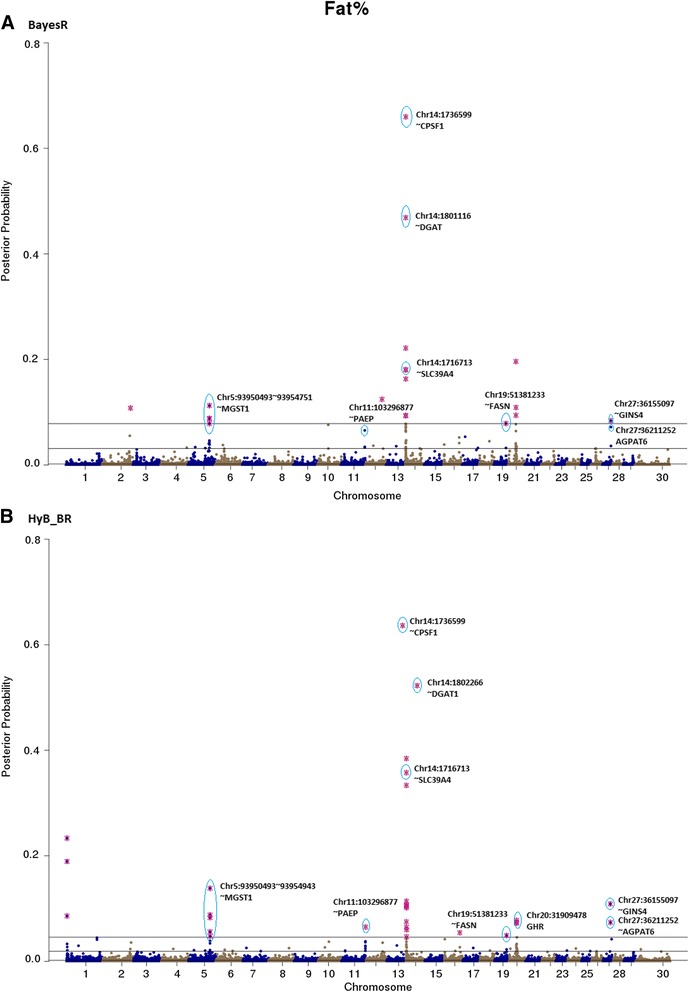
Posterior possibilities of all the variants for fat percent estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole genome. The top SNPs with highest posterior possibilities are labelled with blue circle
Fig. 8.
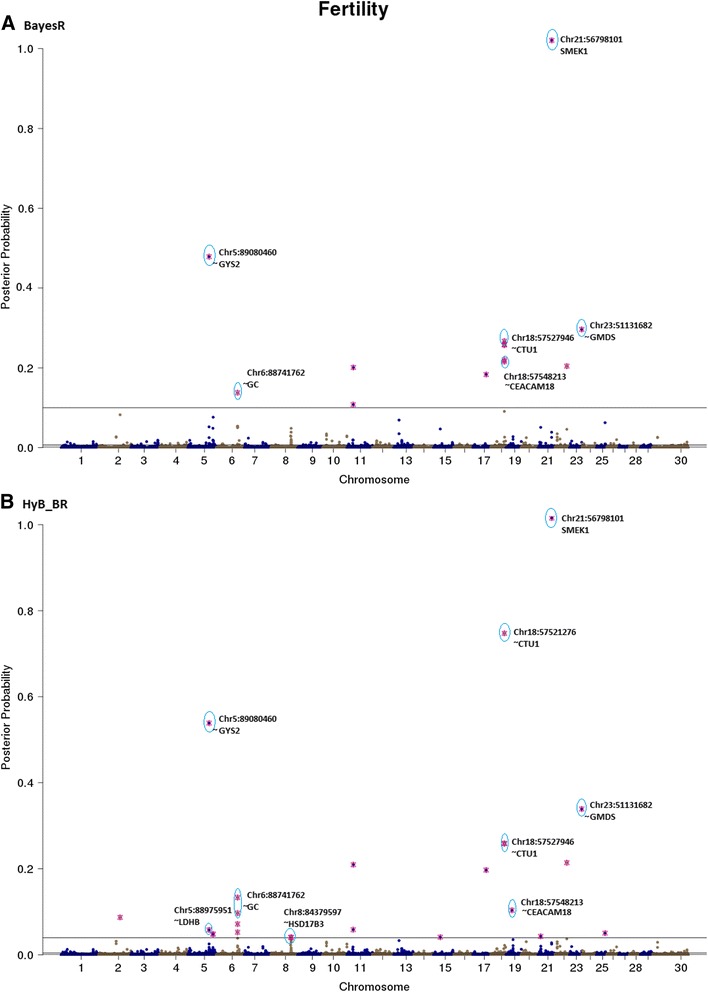
Posterior possibilities of all the variants on fertility estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole genome. The top SNPs with highest posterior possibilities are labelled with blue circle
Fig. 9.
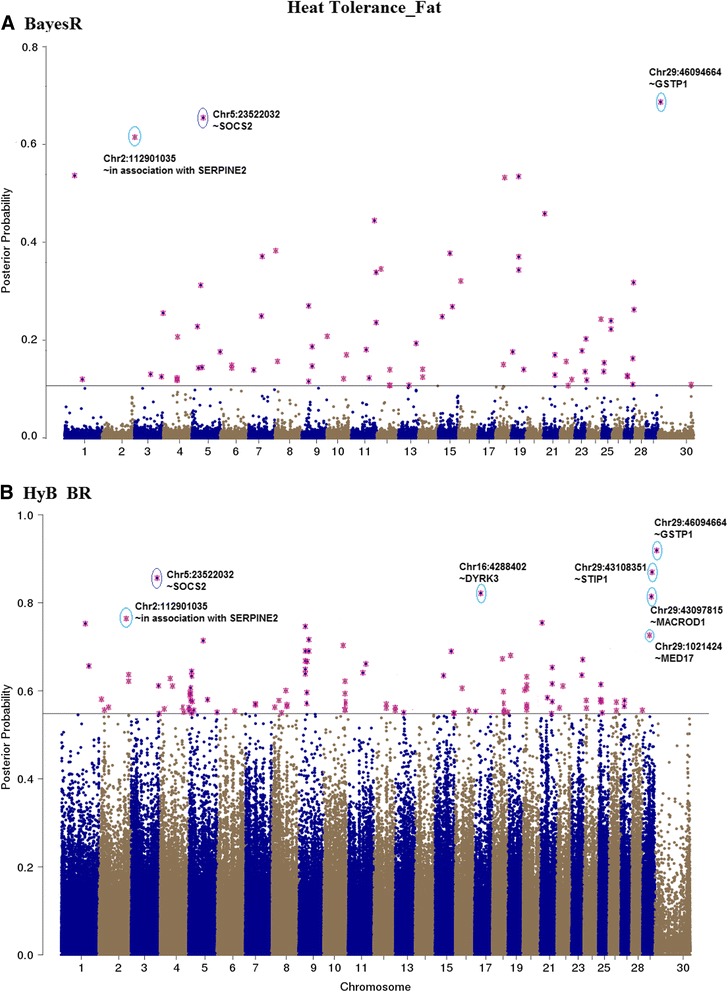
Mapping posterior probabilities of all the variants estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole chromosome related to Fat yield affected by heat tolerance. The top SNPs with highest posterior possibilities are labelled with blue circle
Fig. 10.
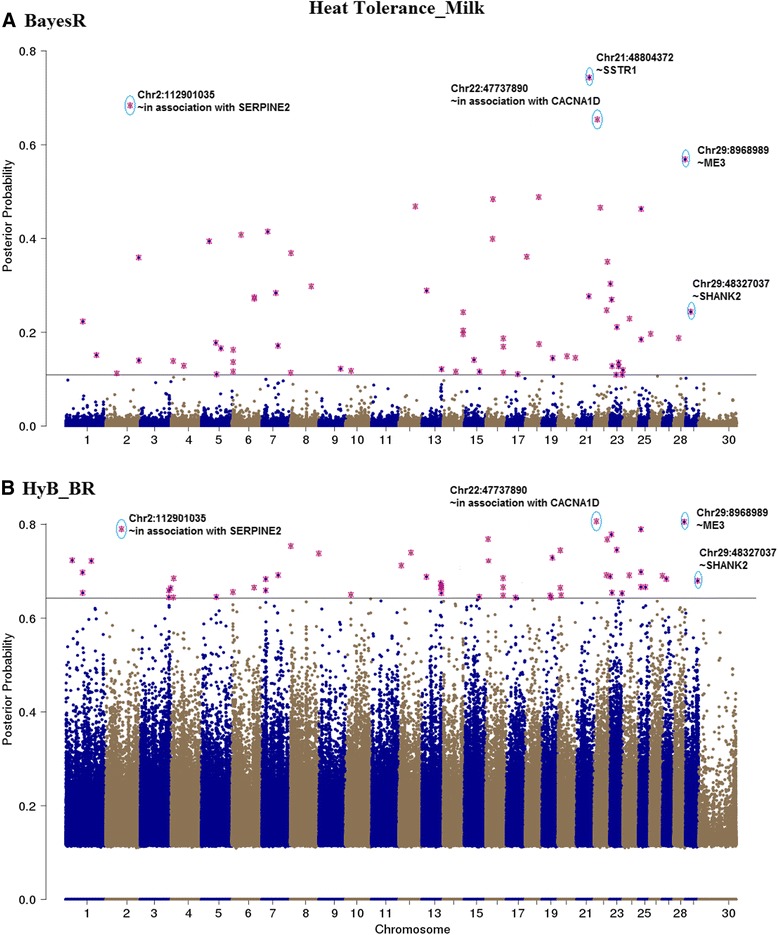
Mapping the posterior probabilities of all the variants estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole chromosome related to Milk yield affected by heat tolerance. The top SNPs with highest posterior possibilities are labelled with blue circle
Fig. 11.
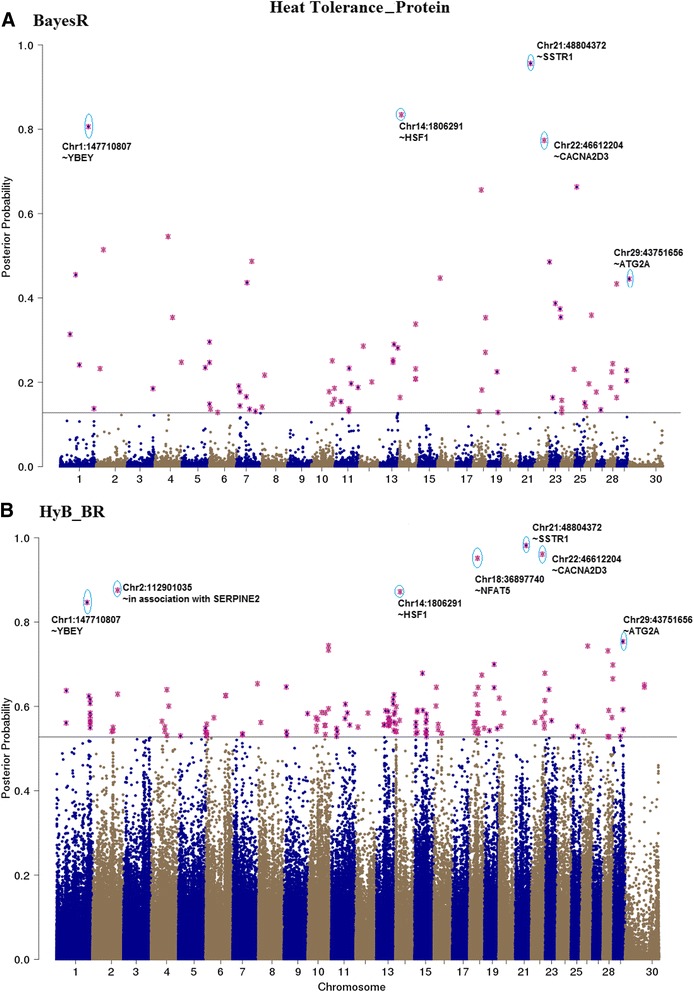
Mapping the posterior probabilities of all the variants estimated from BayesR (a) and HyB_BR (b) according to their positions (base pairs) across the whole chromosome related to protein yield affected by heat tolerance. The top SNPs with highest posterior possibilities are labelled with blue circle
QTL mapping for milk production traits
The top variants detected by both BayesR and HyB_BR (Table 7) were in, or close to, many previously described genes involved with milk production. For example, in Table 7, some well-known mutations impacting milk synthesis included DGAT1 [29–31], FASN [32], SCD [33], PAEP [34], AGPAT6 [35, 36], and CNS2/3 [5]. Notably, for the trait Fat% (Fig. 7), HyB_BR was able to find the real causal mutation in the DGAT1 gene, located at 1802266 bp of Chromosome 14, which has been reported by Grisart et al., 2004 [29]. In addition, HyB_BR could detect some novel potential causal mutations including in the genes GC (encoding the vitamin D binding protein, affecting milk yield), SMEK1 (regulating the Insulin/IGF pathway, indirectly impacting milk production and fertility) and MYH9 (myosin, heavy chain 9, non-muscle; impacting protein yield [5, 37, 38].
Table 7.
Known genes (impacting milk production traits and fertility) identified by HyB_BR using the variants with the largest variances [49–55]
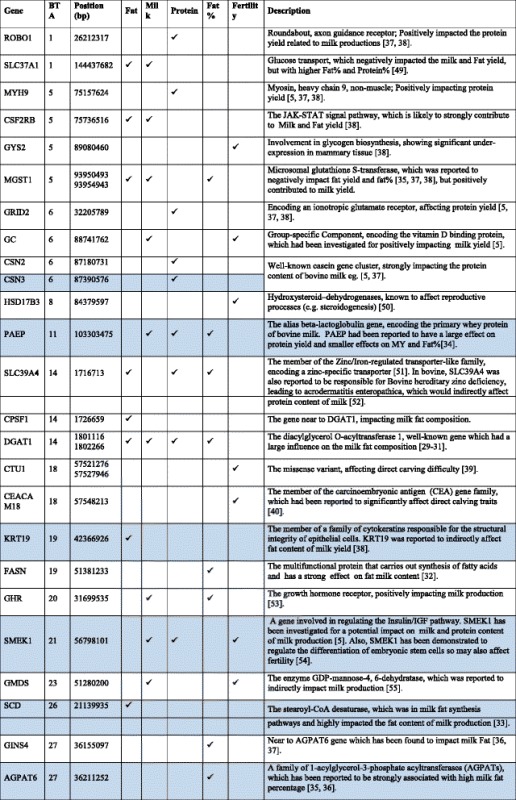
The blue bar highlights the genes that were not detected by BayesR in the proportion with the largest variances
QTL mapping for fertility
For fertility, a putative candidate gene located on Chromosome 18 including (around genes CTU1 and CEACAM18) was detected by BayesR and HyB_BR. These genes haveb previously been reported to be associated with calving traits [39, 40].
QTL mapping for heat tolerance traits
As there is a significant unfavourable correlation between milk production and heat tolerance, at least for the traits we have used for heat tolerance (decline in milk production with increasing heat stress) [21], mutations that affect milk production are also likely to affect heat tolerance. To avoid detecting just QTL with large effects on milk production, QTL mapping for heat tolerance traits was performed fitting fixed effects of the mutations in DGAT1, ROBO1, PAEP, and MGST1 (the mutations with largest effects on milk production, to ensure these mutations were not picked up again in the heat tolerance mapping) in the BayesR and HyB_BR models. The posterior possibilities of all the variants estimated by HyB_BR and BayesR were plotted across the whole genome sequence in Figs. 9, 10, and 11. Compared with BayesR, HyB_BR systematically detected more SNPs with small effects () while identifying fewer SNP with zero effects.
In total, we found fourteen novel variants (Table 8) in our study which have previously been associated with heat tolerance in humans or other species. YBEY [41, 42], located at BTA1 with the position 147,710,807 bp, has been reported to be important in the response of infection of Escherichia coli of human or other animals under heat-shock response. Variants in SERPINE2 and CACNA1D (close to the variants detected in our study, BTA2:112,901,035 and BTA22:47,737,890 respectively) have been reported to impact the sweating rate and respiration rate of dairy cattle [43]. DYRK3 (The dual specificity tyrosine-phosphorylation-regulated kinase 3), has been reported to affect respiration rate (breaths per minute) in dairy cattle [43]. HSF1, heat shock factor protein 1, coordinates stress-induced transcription in Human [44]. One single nucleotide polymorphism (SNP) in the 3′-untranslated region (g.4693G > T) of HSF1 has been reported to be in association with thermo tolerance in Chinese Holstein cattle [45]. STIP1, stress inducible protein 1, has been reported to be homologous to hsc70/hsp90 in human [46]. In mice, STIP1 could play a key role on in the ability of germ cells to survive in stress conditions including high temperatures [47]. Further investigation of the effect of these genes on heat tolerance is required.
Table 8.
Known genes interacting with heat stress
| Gene | BTA | Position | Traits | Description | ||
|---|---|---|---|---|---|---|
| Fat | Milk | Protein | ||||
| YBEY | 1 | 147,710,807 | ✓ | The translation-associated heat shock genes, playing key roles in the heat-shock response of E. coli under heat shock stress [41, 42]. | ||
| Unknown | 2 | 112,901,035 | ✓ | ✓ | ✓ | In association with the gene SERPINE2, which had been proven to impact the sweating rate of dairy cattle [43] |
| SOCS2 | 5 | 23,522,032 | ✓ | Suppressor of cytokine signalling 2, might be responsible for heat stress abatement during the dry period of dairy cattle [56]. | ||
| HSF1 | 14 | 1,806,291 | ✓ | Genes involved in the bovine heat stress response [45, 57]. | ||
| DYRK3 | 16 | 4,288,402 | ✓ | The dual specificity tyrosine-phosphorylation-regulated kinase 3, impacting Respiration rate (breaths per minute) in dairy cattle [43] | ||
| NFAT5 | 18 | 36,897,740 | ✓ | Nuclear factor of activated T cells, simulating transcription of Heat shock protein 70 [58]. | ||
| SSTR1 | 21 | 48,804,372 | ✓ | Somatostatin receptor 1, playing a role in heat stress sensing or communicating stress status between cells [59]. | ||
| CACNA2D3 | 22 | 46,612,204 | ✓ | Methylation of the Calcium Channel-Related Gene, showing impaired behavioural heat pain sensitivity in mice and human studies [60]. | ||
| MED17 | 29 | 1,021,424 | ✓ | The mediator mutant yeast, which was temperature-sensitive [61]. | ||
| ME3 | 29 | 8,968,989 | ✓ | Malic Enzyme 3, conferring heat-stable resistance to root-knot nematodes in plants [62]. | ||
| MACROD1 | 29 | 43,097,815 | ✓ | Heat shock protein 90 kDa alpha (cytosolic), class A member 1, which might be in association with PAR (had been proved to function heat shock response) [63, 64]. | ||
| STIP1 | 29 | 43,108,351 | ✓ | ✓ | Stress inducible protein 1, was homologous to the human heat shock cognate protein 70 (hsc70)/heat shock protein 90 (hsp90) [47]. | |
| GSTP1 | 29 | 46,094,664 | ✓ | Glutathione S-transferase Pi, which was reported to play a positive role under heat stress in controlling cellular toxicants and to alleviate the destructive effect on cattle [65]. | ||
| ATG2A | 29 | 43,751,656 | ✓ | Autophagy Related 2 Homolog A, which had been referred to as the Heat Stress-repressed target genes by Niskanen et al., 2015 [66]. | ||
All the listed genes are identified by HyB_BR using the variants with the largest variances
Discussion
In this paper, we have demonstrated that HyB_BR [16] could be efficiently implemented for simultaneous prediction of genomic estimated breeding values, inference of genetic architecture, and potential causal mutation discovery using whole-genome sequence data. As mentioned by Wang et al. (2016), HyB_BR was developed to overcome two challenges:
Long compute times are the main limitation of traditional MCMC Bayesian models applied to whole genome sequence data with very large data size. Therefore, an Expectation-Maximisation scheme was introduced to reduce number of iterations of MCMC.
Fast schemes (mainly including Iterative Conditional Expectation, and Expectation-Maximisation algorithms) implemented for Bayesian models have tended to reduce the accuracy compared with MCMC.
HyB_BR implements an EM algorithm to quickly converge for estimates of SNP effects and other parameters, followed by a limited number of MCMC iterations to optimise the posterior estimation for SNP effects. When applied to whole genome sequence data, our results indicated HyB_BR had similar accuracy of genomic prediction and precision of QTL mapping to BayesR implemented with full MCMC, but with 10 fold less computational time required. Furthermore, compared with the prediction accuracy on 600 K SNP panels, we have demonstrated that using sequence data improved the accuracy of genomic prediction for some of the traits, and particularly in multi-breed evaluations, if a breed was not included in the reference population.
The key improvement for computational efficiency was that HyB_BR reduced the iteration times. BayesR required a huge number of MCMC iterations, which was dependent on the size of the data. For example, on the whole genome sequence data with 16,214 animals and almost 1 million variants, 40,000 iterations with first 20,000 as burn-in were required. For each MCMC iteration, the basis operation times were O(mn 2). In comparison with BayesR, HyB_BR has the same number of basic operations. But after the EM converges (with very small number of iterations as demonstrated by Wang et al. (2015) [27]), HyB_BR implemented MCMC iterations with speed-up schemes, which could reduce the iteration number to 4000 iterations. The results from Fig. 2 provided the evidence that HyB_BR was up to 10 times faster than BayesR in the whole genome sequence data set.
In addition to the computational time, the prediction accuracy of HyB_BR for multi-breed prediction and across-breed prediction was very similar to BayesR for a range of traits with various genetic architectures, shown in Tables 3, 4 and 5. The accuracy advantage of HyB_BR and BayesR over GBLUP for across-breed prediction demonstrated the benefit of the non-linear Bayesian models. Also, the increase in accuracy using whole genome sequence data for across-breed prediction in comparison with using 600 K data, confirmed the results from [5].
For the genetic architecture identification of milk production traits, there was one notable difference between BayesR and HyB_BR: In comparison with BayesR, HyB_BR does not shrink variants with small effects () as strongly, the same is true for very small effects (), (Table 6). The same is true for the identification of causal mutations for heat tolerance, Figs. 9, 10 and 11. One explanation is that EM steps do not have enough power to shrink SNPs with small effects [27], which limits the following MCMC steps.
For the heat tolerance traits, there is relatively little literature reporting QTL for heat tolerance in cattle. Only one of the additive genetic variants (located at Chromosome 29 with the position 48,329,079 base pairs; close to FGF4) [48], later suggested to be SHANK2 by [43] has previously been reported. In Table 7, the gene SHANK2 was detected but not in the list of top causal mutations. However, both BayesR and HyB_BR did pick up mutations in or close to seven genes (e.g. YEBY, HSF1, MED17, ME3, STIP1, SERPINE2 and CACNA1D), which have been reported by previous studies to be involved in response to heat stress events in cattle (e.g. [45]), human, mice, or other species. In addition, HyB_BR also detected two other unknown variants. All these variants required the further investigation in regards to their function interacting between milk productions and heat tolerance.
The computational advantage of HyB_BR makes it attractive for implementation of genomic prediction in many applications. However, there are still two limitations: 1) the speed-up scheme of HyB_BR defines the fixed threshold for different traits and various densities of genomic data, which could hinder its flexibility for practical applications; 2) when the size of the data increases dramatically to 30 million variants on millions of animals, which is possible in the near future, HyB_BR is still not computationally efficient enough. Therefore, a flexible and more efficient speed-up scheme will play an important role to further improve the computational performance of HyB_BR.
Conclusion
A hybrid scheme of Expectation-Maximisation algorithm and MCMC sampling was implemented on whole-genome sequence data for simultaneous genomic prediction, inference of genetic architecture inference and causal mutation identification. The accuracy of HyB_BR for multi-breed and across breed prediction for all traits was very similar to the results from BayesR (implemented with full MCMC) while requiring only 1/10 of the total running time of BayesR. HyB_BR could identify some well-known mutations (e.g. DGAT1) with the highest posterior probability, which demonstrated the value of the method for QTL mapping of complex traits. The advantage of using sequence data and HyB_BR was greatest for multi-breed and across breed predictions.
Acknowledgements
The authors thank DataGene (Melbourne Australia) for contributing milk production and fertility phenotypes and Dr. Thuy Nguyen (Research Scientist in Department of Economic Development, Jobs, Transport and Resources) for providing the phenotype data of heat tolerance traits.
Funding
The research was funded by Dairy Futures CRC project and the project titled “MIRprofit: integrating very large genomic and milk mid-infrared data to improve profitability of dairy cows” funded by the Commonwealth of Australia.
Availability of data and materials
For 600 K SNP chips, we can provide meta-analysis data related to our paper which can be easily used to conduct the analysis by other researchers.
For the sequence data set, as detailed by [5], the 1000 Bull Genomes Project (Run 2&3) has published the sequences of 136 Holstein and 27 Jersey bulls that were used as our reference for sequence imputation. The list of 2.875 million sequence variants used for the analysis is available on request.
The HyB_BR compiled program is available for request for non-commercial research.
Authors’ contributions
BJH and Y-PPC supervised this project; TW developed HyB_BR algorithm, analysed the sequence data and drafted the manuscript. IMM provided help with the processing of the whole genome sequence data. JEP gave support to genomic prediction on heat tolerance data. MEG contributed the valuable ideas about the application of hybrid scheme to the whole genome sequence data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no completing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tingting Wang, Email: tingting.wang@ecodev.vic.gov.au.
Yi-Ping Phoebe Chen, Email: phoebe.chen@latrobe.edu.au.
Iona M. MacLeod, Email: iona.macleod@ecodev.vic.gov.au
Jennie E. Pryce, Email: jennie.pryce@ecodev.vic.gov.au
Michael E. Goddard, Email: Mike.Goddard@ecodev.vic.gov.au
Ben J. Hayes, Email: Ben.Hayes@ecodev.vic.gov.au
References
- 1.Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brondum RF, Liao X, Djari A, Rodriguez SC, Grohs C, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet. 2014;46(8):858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 2.Clark SA, Hickey JM, van der Werf JHJ. Different models of genetic variation and their effect on genomic evaluation. Genet Sel Evol. 2011;43(1):1–9. doi: 10.1186/1297-9686-43-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Druet T, Macleod IM, Hayes BJ. Toward genomic prediction from whole-genome sequence data: impact of sequencing design on genotype imputation and accuracy of predictions. Heredity. 2014;112(1):39–47. doi: 10.1038/hdy.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.IM ML, Hayes BJ, CJ VJ, Kemper KE, Haile-Mariam M, Bowman PJ, Schrooten C, Goddard ME. A Bayesian analysis to exploit imputed sequence variants for QTL discovery. In: Proceedings on 10th World Congress of Genetics Applied to Livestock Production: 2014. Vancouver, BC, Canada; 2014. p. 193.
- 5.MacLeod IM, Bowman PJ, Vander Jagt CJ, Haile-Mariam M, Kemper KE, Chamberlain AJ, Schrooten C, Hayes BJ, Goddard ME. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genomics. 2016;17(1):1–21. doi: 10.1186/s12864-016-2443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kemper KE, Reich CM, Bowman PJ, Vander Jagt CJ, Chamberlain AJ, Mason BA, Hayes BJ, Goddard ME. Improved precision of QTL mapping using a nonlinear Bayesian method in a multi-breed population leads to greater accuracy of across-breed genomic predictions. Genet Sel Evol. 2015;47:29. doi: 10.1186/s12711-014-0074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erbe M, Hayes BJ, Matukumalli LK, Goswami S, Bowman PJ, Reich CM, Mason BA, Goddard ME. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J Dairy Sci. 2012;95(7):4114–4129. doi: 10.3168/jds.2011-5019. [DOI] [PubMed] [Google Scholar]
- 8.Moser G, Lee SH, Hayes BJ, Goddard ME, Wray NR, Visscher PM. Simultaneous discovery, estimation and prediction analysis of complex traits using a Bayesian mixture model. PLoS Genet. 2015;11(4) doi: 10.1371/journal.pgen.1004969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacLeod IM, Hayes BJ, Goddard ME. The effects of demography and long-term selection on the accuracy of genomic prediction with sequence data. Genetics. 2014;198(4):1671–1684. doi: 10.1534/genetics.114.168344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolormaa S, Pryce JE, Kemper K, Savin K, Hayes BJ, Barendse W, Zhang Y, Reich CM, Mason BA, Bunch RJ, et al. Accuracy of prediction of genomic breeding values for residual feed intake and carcass and meat quality traits in Bos Taurus, Bos Indicus, and composite beef cattle1. J Anim Sci. 2013;91 [DOI] [PubMed]
- 11.VanRaden PM, Null DJ, Sargolzaei M, Wiggans GR, Tooker ME, Cole JB, Sonstegard TS, Connor EE, Winters M, van Kaam JBCHM, et al. Genomic imputation and evaluation using high-density Holstein genotypes. J Dairy Sci. 2013;96(1):668–678. doi: 10.3168/jds.2012-5702. [DOI] [PubMed] [Google Scholar]
- 12.VanRaden PM, O’Connell JR, Wiggans GR, Weigel KA. Genomic evaluations with many more genotypes. Genet Sel Evol. 2011;43(1):1–11. doi: 10.1186/1297-9686-43-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speed D, Balding DJ. MultiBLUP: improved SNP-based prediction for complex traits. Genome Res. 2014;24(9):1550–1557. doi: 10.1101/gr.169375.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loh P-R, Tucker G, Bulik-Sullivan BK, Vilhjalmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet. 2015;47(3):284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meuwissen THE, Solberg TR, Shepherd R, Woolliams JA. A fast algorithm for BayesB type of prediction of genome-wide estimates of genetic value. Genet Sel Evol. 2009;41:2. doi: 10.1186/1297-9686-41-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang T, Chen YP, Bowman PJ, Goddard ME, Hayes BJ. A hybrid expectation maximisation and MCMC sampling algorithm for Bayesian mixture model based genomic prediction and QTL mapping. BMC Genomics. 2016;17:744. doi: 10.1186/s12864-016-3082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84(2):210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Browning BL, Browning SR. A fast, powerful method for detecting identity by descent. Am J Hum Genet. 2011;88(2):173–182. doi: 10.1016/j.ajhg.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garrick D, Taylor J, Fernando R. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;41(1):55. doi: 10.1186/1297-9686-41-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen TTT, Bowman PJ, Haile-Mariam M, Pryce JE, Hayes BJ. Genomic selection for tolerance to heat stress in Australian dairy cattle. J Dairy Sci. 2016;99(4):2849–2862. doi: 10.3168/jds.2015-9685. [DOI] [PubMed] [Google Scholar]
- 22.Haile-Mariam M, Carrick MJ, Goddard ME. Genotype by environment interaction for fertility, survival, and milk production traits in Australian dairy cattle. J Dairy Sci. 2008;91(12):4840–4853. doi: 10.3168/jds.2008-1084. [DOI] [PubMed] [Google Scholar]
- 23.Hayes BJ, Carrick M, Bowman P, Goddard ME. Genotype × environment interaction for milk production of daughters of Australian dairy sires from test-day records. J Dairy Sci. 2003;86(11):3736–3744. doi: 10.3168/jds.S0022-0302(03)73980-0. [DOI] [PubMed] [Google Scholar]
- 24.Gilmour A, Cullis B, Welham S, Thompson R: ASReml Reference Manual 2nd edition. NSW Agriculture Biometrical Bulletin 3 2002.
- 25.Visscher PM, Hill WG, Wray NR. Heritability in the genomics era — concepts and misconceptions. Nat Rev Genet. 2008;9(4):255–66. [DOI] [PubMed]
- 26.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang T, Chen Y-PP, Goddard ME, Meuwissen THE, Kemper KE, Hayes BJ. A computationally efficient algorithm for genomic prediction using a Bayesian model. Genet Sel Evol. 2015;47:34. doi: 10.1186/s12711-014-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henderson C. Application of linear models in animal breeding. Canada: University of Guelph; 1984. [Google Scholar]
- 29.Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P, Cambisano N, Mni M, Reid S, Simon P, et al. positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 2002;12(2):222–231. doi: 10.1101/gr.224202. [DOI] [PubMed] [Google Scholar]
- 30.Schennink A, Heck JML, Bovenhuis H, Visker MHPW, van Valenberg HJF, van Arendonk JAM. Milk fatty acid unsaturation: genetic parameters and effects of Stearoyl-CoA desaturase (SCD1) and acyl CoA: diacylglycerol acyltransferase 1 (DGAT1) J Dairy Sci. 2008;91(5):2135–2143. doi: 10.3168/jds.2007-0825. [DOI] [PubMed] [Google Scholar]
- 31.Schennink A, Stoop WM, Visker MHPW, Heck JML, Bovenhuis H, Van Der Poel JJ, Van Valenberg HJF, Van Arendonk JAM. DGAT1 underlies large genetic variation in milk-fat composition of dairy cows. Anim Genet. 2007;38(5):467–473. doi: 10.1111/j.1365-2052.2007.01635.x. [DOI] [PubMed] [Google Scholar]
- 32.Roy R, Ordovas L, Zaragoza P, Romero A, Moreno C, Altarriba J, Rodellar C. Association of polymorphisms in the bovine FASN gene with milk-fat content. Anim Genet. 2006;37(3):215–218. doi: 10.1111/j.1365-2052.2006.01434.x. [DOI] [PubMed] [Google Scholar]
- 33.Mele M, Conte G, Castiglioni B, Chessa S, Macciotta NPP, Serra A, Buccioni A, Pagnacco G, Secchiari P. Stearoyl-coenzyme a desaturase gene polymorphism and milk fatty acid composition in Italian Holsteins. J Dairy Sci. 2007;90(9):4458–4465. doi: 10.3168/jds.2006-617. [DOI] [PubMed] [Google Scholar]
- 34.Ng-Kwai-Hang K: A Review of the Relationship between Milk Protein Polymorphism and Milk Composition/Milk Production. In: Proceedings of the International Dairy Federation Seminar: 25–27 Febuary, 1997 1997; Palmerston North, New Zealand; 1997: 22–37.
- 35.Wang X, Wurmser C, Pausch H, Jung S, Reinhardt F, Tetens J, Thaller G, Fries R. Identification and dissection of four major QTL affecting milk fat content in the German Holstein-Friesian population. PLoS One. 2012;7(7) doi: 10.1371/journal.pone.0040711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Littlejohn MD, Tiplady K, Lopdell T, Law TA, Scott A, Harland C, Sherlock R, Henty K, Obolonkin V, Lehnert K, et al. Expression variants of the Lipogenic AGPAT6 gene affect diverse milk composition phenotypes in Bos Taurus. PLoS One. 2014;9(1) doi: 10.1371/journal.pone.0085757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raven L-A, Cocks BG, Kemper KE, Chamberlain AJ, Jagt CJ, Goddard ME, Hayes BJ. Targeted imputation of sequence variants and gene expression profiling identifies twelve candidate genes associated with lactation volume, composition and calving interval in dairy cattle. Mamm Genome. 2015;27(1):81–97. doi: 10.1007/s00335-015-9613-8. [DOI] [PubMed] [Google Scholar]
- 38.Chamberlain AJ, Vander Jagt CJ, Hayes BJ, Khansefid M, Marett LC, Millen CA, Nguyen TTT, Goddard ME. Extensive variation between tissues in allele specific expression in an outbred mammal. BMC Genomics. 2015;16(1):1–20. doi: 10.1186/s12864-015-2174-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purfield DC, Bradley DG, Evans RD, Kearney FJ, Berry DP. Genome-wide association study for calving performance using high-density genotypes in dairy and beef cattle. Genet Sel Evol. 2015;47(1):1–13. doi: 10.1186/s12711-015-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao X, Kadri NK, Thomasen JR, De Koning DJ, Sahana G, Guldbrandtsen B: Fine mapping of a calving QTL on Bos taurus autosome 18 in Holstein cattle. J Anim Breed Genet 2015:n/a-n/a. [DOI] [PubMed]
- 41.Grinwald M, Ron EZ. The Escherichia Coli translation-associated heat shock protein YbeY is involved in rRNA transcription Antitermination. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0062297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rasouly A, Schonbrun M, Shenhar Y, Ron EZ. YbeY, a heat shock protein involved in translation in Escherichia Coli. J Bacteriol. 2009;191(8):2649–2655. doi: 10.1128/JB.01663-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dikmen S, Wang XZ, Ortega MS, Cole JB, Null DJ, Hansen PJ. single nucleotide polymorphisms associated with thermoregulation in lactating dairy cows exposed to heat stress. J Anim Breed Genet. 2015;132(6):409–419. doi: 10.1111/jbg.12176. [DOI] [PubMed] [Google Scholar]
- 44.Rabindran SK, Giorgi G, Clos J, Wu C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci U S A. 1991;88(16):6906–6910. doi: 10.1073/pnas.88.16.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li QL, Zhang ZF, Xia P, Wang YJ, Wu ZY, Jia YH, Chang SM, Chu MX. A SNP in the 3′-UTR of HSF1 in dairy cattle affects binding of target bta-miR-484. Genet Mol Res. 2015;14(4):12746–12755. doi: 10.4238/2015.October.19.18. [DOI] [PubMed] [Google Scholar]
- 46.Schmid AB, Lagleder S, Gräwert MA, Röhl A, Hagn F, Wandinger SK, Cox MB, Demmer O, Richter K, Groll M, et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/hop. EMBO J. 2012;31(6):1506–1517. doi: 10.1038/emboj.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mizrak SC, Bogerd J, Lopez-Casas PP, Párraga M, del Mazo J, de Rooij DG. Expression of stress inducible protein 1 (Stip1) in the mouse testis. Mol Reprod Dev. 2006;73(11):1361–1366. doi: 10.1002/mrd.20548. [DOI] [PubMed] [Google Scholar]
- 48.Hayes BJ, Bowman PJ, Chamberlain AJ, Savin K, van Tassell CP, Sonstegard TS, Goddard ME. A validated genome wide association study to breed cattle adapted to an environment altered by climate change. PLoS One. 2009;4(8) doi: 10.1371/journal.pone.0006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fritz S, Capitan A, Djari A, Rodriguez SC, Barbat A, Baur A, Grohs C, Weiss B, Boussaha M, EsquerrÈ D, et al. Detection of haplotypes associated with prenatal death in dairy cattle and identification of deleterious mutations in GART, SHBG and SLC37A2. PLoS One. 2013;8(6) doi: 10.1371/journal.pone.0065550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cochran SD, Cole JB, Null DJ, Hansen PJ. Discovery of single nucleotide polymorphisms in candidate genes associated with fertility and production traits in Holstein cattle. BMC Genet. 2013;14(1):1–23. doi: 10.1186/1471-2156-14-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmitt S, Küry S, Giraud M, Dréno B, Kharfi M, Bézieau S. An update on mutations of the SLC39A4 gene in acrodermatitis enteropathica. Hum Mutat. 2009;30(6):926–933. doi: 10.1002/humu.20988. [DOI] [PubMed] [Google Scholar]
- 52.Yuzbasiyan-Gurkan V, Bartlett E. Identification of a unique splice site variant in SLC39A4 in bovine hereditary zinc deficiency, lethal trait A46: an animal model of acrodermatitis enteropathica. Genomics. 2006;88(4):521–526. doi: 10.1016/j.ygeno.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 53.Blott S, Kim J-J, Moisio S, Schmidt-Küntzel A, Cornet A, Berzi P, Cambisano N, Ford C, Grisart B, Johnson D, et al. Molecular dissection of a quantitative trait locus: a phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics. 2003;163(1):253–266. doi: 10.1093/genetics/163.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyu J, Jho E-h, Lu W. Smek promotes histone deacetylation to suppress transcription of Wnt target gene brachyury in pluripotent embryonic stem cells. Cell Res. 2011;21(6):911–921. doi: 10.1038/cr.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wickramasinghe S, Hua S, Rincon G, Islas-Trejo A, German JB, Lebrilla CB, Medrano JF. Transcriptome profiling of bovine milk oligosaccharide metabolism genes using RNA-sequencing. PLoS One. 2011;6(4) doi: 10.1371/journal.pone.0018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.do Amaral BC, Connor EE, Tao S, Hayen MJ, Bubolz JW, Dahl GE. Heat stress abatement during the dry period influences metabolic gene expression and improves immune status in the transition period of dairy cows. J Dairy Sci. 2011;94(1):86–96. doi: 10.3168/jds.2009-3004. [DOI] [PubMed] [Google Scholar]
- 57.Collier RJ, Collier JL, Rhoads RP, Baumgard LH. Invited review: genes involved in the bovine heat stress response. J Dairy Sci. 2008;91(2):445–454. doi: 10.3168/jds.2007-0540. [DOI] [PubMed] [Google Scholar]
- 58.Woo SK, Lee SD, Na KY, Park WK, Kwon HM. TonEBP/NFAT5 stimulates transcription of HSP70 in response to hypertonicity. Mol Cell Biol. 2002;22(16):5753–5760. doi: 10.1128/MCB.22.16.5753-5760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raychaudhuri S, Loew C, Körner R, Pinkert S, Theis M, Hayer-Hartl M, Buchholz F, Hartl FU. Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell. 2014;156(5):975–985. doi: 10.1016/j.cell.2014.01.055. [DOI] [PubMed] [Google Scholar]
- 60.Neely GG, Hess A, Costigan M, Keene AC, Goulas S, Langeslag M, Griffin RS, Belfer I, Dai F, Smith SB, et al. A genome-wide drosophila screen for heat nociception identifies α2δ3 as an evolutionarily conserved pain Gene. Cell. 2010;143(4):628–638. doi: 10.1016/j.cell.2010.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paul E, Zhu ZI, Landsman D, Morse RH. Genome-wide association of mediator and RNA polymerase II in wild-type and mediator mutant yeast. Mol Cell Biol. 2015;35(1):331–342. doi: 10.1128/MCB.00991-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Djian-Caporalino C, Pijarowski L, Fazari A, Samson M, Gaveau L, O’Byrne C, Lefebvre V, Caranta C, Palloix A, Abad P. High-resolution genetic mapping of the pepper (Capsicum Annuum L.) resistance loci Me3 and Me4 conferring heat-stable resistance to root-knot nematodes (Meloidogyne spp.) Theor Appl Genet. 2001;103(4):592–600. doi: 10.1007/PL00002914. [DOI] [Google Scholar]
- 63.Petesch SJ, Lis JT. Activator-induced spread of poly(ADP-ribose) polymerase promotes nucleosome loss at Hsp70. Mol Cell. 2012;45(1):64–74. doi: 10.1016/j.molcel.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Di Giammartino DC, Shi Y, Manley James L. PARP1 represses PAP and inhibits polyadenylation during heat shock. Mol Cell. 2013;49(1):7–17. doi: 10.1016/j.molcel.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rao TVLN, Ramesha KP, Barani A, Chauhan SS, Basavaraju M. Association of GSTP1 gene polymorphisms with performance traits in Deoni cattle. Afr J Biotechnol. 2013;12(24):3768–3773. [Google Scholar]
- 66.Niskanen EA, Malinen M, Sutinen P, Toropainen S, Paakinaho V, Vihervaara A, Joutsen J, Kaikkonen MU, Sistonen L, Palvimo JJ. Global SUMOylation on active chromatin is an acute heat stress response restricting transcription. Genome Biol. 2015;16(1):1–19. doi: 10.1186/s13059-015-0717-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
For 600 K SNP chips, we can provide meta-analysis data related to our paper which can be easily used to conduct the analysis by other researchers.
For the sequence data set, as detailed by [5], the 1000 Bull Genomes Project (Run 2&3) has published the sequences of 136 Holstein and 27 Jersey bulls that were used as our reference for sequence imputation. The list of 2.875 million sequence variants used for the analysis is available on request.
The HyB_BR compiled program is available for request for non-commercial research.