

Abstract
Aqua ligands can undergo rapid internal rotation about the M-O bond. For magnetic resonance contrast agents, this rotation results in diminished relaxivity. Here we show that an intramolecular hydrogen bond to the aqua ligand can reduce this internal rotation and increase relaxivity. Molecular modeling was used to design a series of four Gd complexes capable of forming an intramolecular H-bond to the coordinated water ligand, and these complexes had anomalously high relaxivities compared to similar complexes lacking a H-bond acceptor. Molecular dynamics simulations supported the formation of a stable intramolecular H-bond while alternative hypotheses that could explain the higher relaxivity were systematically ruled out. Intramolecular H-bonding represents a useful strategy to limit internal water rotational motion and increase relaxivity of Gd complexes.
Keywords: gadolinium complexes, aquo-ion dynamics, relaxivity, molecular dynamics
Stop the spin
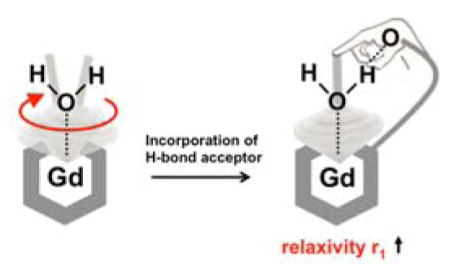
For Gd3+ complexes used in magnetic resonance imaging, rapid rotation of the aqua ligand about the Gd-O bond results in diminished relaxivity. Here, we demonstrate that this internal motion can be attenuated by an intramolecular hydrogen bond to the aqua ligand.
Introduction
Hydration is fundamental to the understanding of many chemical and biological processes.[1] Paramagnetic metal ions and complexes have been used to perturb the magnetic resonance properties of water molecules to interrogate questions of structure, kinetics, and dynamics.[2] Complexes of Gd3+ are widely used in medical imaging to shorten the relaxation times of solvent water to generate contrast in magnetic resonance imaging (MRI) exams.[3]
The magnitude of the relaxation effect depends strongly on the rotational correlation time of the molecule, and specifically on the reorientation of the Gd-H(water) vector. Over 40 years ago, Dwek pointed out that for water coordinated to a metal ion, fast rotation about the metal-oxygen bond will decrease the efficiency of the paramagnetic relaxation effect.[4] For MRI contrast agents, this fast intramolecular motion will reduce the relaxivity of the complex.
Direct experimental assessment of water rotation is challenging. Using the [Ln(DOTAM) (H2O) ]3+ system where the water exchange rate is very slow, Dunand et al. were able to measure the 17O and 1H relaxation times of the coordinated water for the Eu3+ complex and determine the effect of this fast rotation.[5] However water exchange in most lanthanide complexes is too fast for direct observation of the coordinated water ligand. Nonetheless, this fast intramolecular rotation is assumed to occur in many Gd3+ complexes, including those used as commercial MRI contrast agents.[6]
We hypothesized that this fast water rotation could be reduced by the formation of an intramolecular hydrogen bond between the coordinated water hydrogen and a suitable H-bond acceptor, and that the resultant complex would have a higher relaxivity than similar compounds lacking the H-bond acceptor. We previously reported the single amino acid Gd chelate Gd(DOTAla) as a rigid and modular synthon for contrast agent development.[7] Using in silico modeling we selected a panel of complexes with pendant groups that were geometrically capable of forming a H-bond interaction with the coordinated water ligand. We then synthesized four of these complexes and two closely related (“matched”) complexes incapable of forming the H-bond. We demonstrated that the complexes with an H-bond acceptor displayed an anomalously high relaxivity consistent with a reduction in the fast intramolecular water rotation rate. The mechanism was supported by molecular dynamics (MD) simulations, while alternative mechanisms for increased relaxivity were ruled out through a series of nuclear magnetic resonance (NMR) and electron-nuclear double resonance (ENDOR) studies as well as luminescence lifetime measurements on the analogous Tb3+ complexes.
Results and Discussion
Molecular Design and Synthesis
Molecular modeling using the Gd(DOTAla) complex indicated that the coordinated amide carbonyl orients substituents at the C-terminus of DOTAla such that they can hydrogen bond with the coordinated water ligand. Based on the modeling studies we selected a small library of complexes substituted at the DOTAla C-terminus with possible H-bond acceptors (see, Figure 1) . The H-bond acceptors were alcohol oxygen (4a) , carboxylate oxygen (4b, 4c) , and amine nitrogen (4e) . We also prepared 4d, the methyl ester of 4b as a control since esters are known to be poor H-bond acceptors.[8] Compound 4e can be converted from an H-bond acceptor amine to the H-bond donor ammonium by adjusting the pH.
Figure 1.

Structures of new compounds reported here with putative H-bond interactions.
We first built a model of Gd(DOTAla) from coordinates of the Cambridge Structural Database (CSD) entry for DOTA (code: JOPJIH01) . DOTAla was created from DOTA by building standard alanine internal coordinates and functionalizing DOTA appropriately in CHARMM.[9] The system was allowed to relax via a series of constrained minimizations with a harmonic potential using force constants of 10.0 kcal/(mol * Å2) and 5.0 kcal/(mol * Å2) . Structures 4a–4e were drawn using Avogadro[10] software and charges were fit via RESP.[11]
Rotamer libraries were generated for each derivative, sampling each dihedral angle every 15 degrees. Any rotamer that had a van der Waals clash with the scaffold was eliminated from consideration. Hydrogen bonding geometric parameters were then computed for remaining rotamers with all possible water orientations. The rotamer with the best hydrogen bonding geometry (H-bond distance, H-bond angle) was kept for each compound.
Compounds 4a, 4b, 4c and 4e were predicted to form a favorable hydrogen bond with the coordinated water ligand. A much weaker interaction was predicted for the methyl ester 4d. The predicted polar contact for 4d is mediated by the non-carbonyl oxygen of the ester, which is a weaker dipole compared to the carbonyl. Figure 2 shows the calculated best geometry for a H-bond interaction between the pendant carboxylate oxygen and the coordinated water hydrogen in 4b.
Figure 2.

The best hydrogen bond to Gd-bound water in 4b predicted by screening its rotamer library. This bond results from a strong H-bonding interaction with the pendant carboxylate oxygen.
We functionalized the N-terminus of each complex with ibuprofen. We previously reported a series of nine ibuprofen Gd(DOTAla) derivatives and that series showed a highly linear correlation between relaxivity and molecular weight (Figure 3, left panel, open circles, r2 = 0.99) as expected for a series of closely related compounds where the rotational correlation time should increase with increasing molecular weight.[7b] The previous data shown in Figure 3 serves as a calibration to predict what an expected relaxivity would be for a given molecular weight. The synthetic flexibility of DOTAla derivatives allowed us to produce the proposed molecules in 3 steps from common intermediate 1 (Supporting Information (SI) ) 1 was synthesized as previously reported and served as the common starting synthon for intermediates 2a–e.[7b]
Figure 3.


Left panel. Correlation between relaxivity and molecular weight for the Gd complexes discussed here. Open circles: Complexes reported in reference 4. The solid line represents a linear regression (r2 = 0.99) . Solid triangles: 4d, 4e (pH 6) . Open triangles: 4a, 4b, 4c, 4e (pH 9) . Right panel. pH – relaxivity titration of 4e between pH 4.8 and pH 9.4. Filled circles represent measured data points, the solid red line represents a fit by the Henderson-Hasselbach equation, with a calculated pKa of 7.98 ± 0.07.
Relaxivity
Measurement of the relaxivity of compounds 4a, 4b and 4c at 1.41 T, 310 K (37 °C) , in pH 7.4, 50 mM HEPES buffer showed similar values of 5.7 (4a) and 5.8 mM−1s−1 (4b, 4c) . These relaxivities (open triangles in Figure 3, left panel) were anomalously high based on the complexes’ molecular weight compared to the reference values shown in Figure 3 for related GdDOTAla complexes that lacked H-bond acceptors (open circles and straight line, values can be found in Table S3) . On the other hand, 4d displayed a relaxivity of 5.0 mM−1s−1, which is the expected value for a complex of its molecular weight. For compound 4e, we performed a pH titration to measure the relaxivity of the complex under conditions where the pendant group exists as either an amine or ammonium moiety, Figure 3b. As expected, the relaxivity of 4e was pH dependent with a pKa of 7.98 ± 0.07. In its ammonium form, the relaxivity of 4e was 4.9 mM−1s−1 and is exactly as predicted based on its molecular weight. However when measured in its amine, H-bond acceptor form, the relaxivity is anomalously higher at 5.5 mM−1s−1. The relaxivity results support our hypothesis that a Gd3+ complex with an appropriately positioned H-bond acceptor can form an intramolecular H-bond with the coordinated water ligand and restrict its rotation. Two matched pairs demonstrated the importance of the H-bond acceptor to the increased relaxivity. Weakening the carboxylate oxygen H-bond acceptor in 4b by forming its methyl ester 4d resulted in a reduction in relaxivity by 0.7 mM−1s−1. Protonation of the amine in 4e showed a similar drop in relaxivity.
Molecular Dynamics
Next, we performed MD simulations for 4b and its methyl ester 4d to understand the nature of the hydrogen bond between the coordinated water ligand and the pendant H-bond acceptor. For the dynamics trajectories, periodic boundary conditions were used with particle-mesh Ewald summations for long-range interactions.[12] For this, the complexes were solvated in a 30 Å octahedral waterbox. The system was heated from 0 K to 298 K in increments of 5 K after which it was equilibrated for 250 ps. A 10 ns NVT ensemble (constant number, volume, temperature) MD simulation was subsequently performed. The trajectories were appropriately oriented to a reference molecular frame, and subsequently, hydrogen bond properties were extracted.
We monitored the hydrogen bond between the coordinated water hydrogen and the H-bond acceptor for both 4b and 4d. 4b maintained a strong polar contact with the Gd-bound water. For 94% of the 10 ns simulation, 4b sustains a hydrogen-bond distance < 2.5 Å, indicative of a hydrogen bond (Figure 4) . Furthermore, the hydrogen bond is constantly maintained (100%) after the first 1.8 ns of simulation. In contrast, the ester oxygen in 4d does not interact with the Gd-bound water throughout the course of the trajectory, with a hydrogen-bond distance >5.0 Å for 83% of the simulation. These results, in combination with the computed optimal hydrogen-bond conformation, support the hypothesis that a strong H-bonding interaction with the Gd-bound water results in increased relaxivity of these compounds.
Figure 4.

Molecular dynamics showing the distance of the potential hydrogen bond to Gd-bound water over the course of 10 ns trajectory for 4b and 4d. 4b maintained a strong polar contact with the Gd-bound water with a relatively constant hydrogen-bond distance after the initial 200 ps. 4d does not interact with the Gd-bound water throughout the course of the trajectory, with a potential hydrogen-bond distance >5.0 Å for the majority of the simulation.
The structure-relaxivity correlations and MD simulations described above support the hypothesis that the increased relaxivity of compounds 4a–c and 4e (at pH 9) can result from hindering the rotation of the water ligand by an intramolecular H-bond. Before accepting this model, however, one has to consider and possibly rule out alternative mechanisms that could have a similar effect on relaxivity.
We studied hydration number (estimated using the corresponding Tb(III) complexes 5a–5e) . We predicted q that second-sphere effects would result in increased q as previously observed for phosphonate complexes.[13] Table S1 summarizes the calculated q for all complexes and shows that variation in q is small and indicates no significant, consistent second-sphere effect for compounds with enhanced relaxivity.
Next, we assessed if a shorter Gd-H distance could result in relaxivity increase as it would result in a stronger dipolar interaction between the electron spin of the Gd3+ and the nuclear spin of the water hydrogens. The anisotropic hyperfine interaction (hfi) constant T⊥ can be used as a direct measure of the Gd-H distance rGd-H. We have previously shown that T⊥ could be accurately determined from a 1H pulsed ENDOR spectrum collected at an operational microwave (mw) frequency (Uo) high enough to suppress unwanted distortions caused by zero field splitting.[14] A series of 1D and 2D 1H ENDOR studies were performed with 4b and 4d, but these indicated that the Gd-H distances for inner- and second-sphere water hydrogens were the same for these two complexes (Supporting Information, sections S4 and S5) .
We previously showed that the water exchange rate for the coordinated water ligand was very fast for the Gd(DOTAla) derivatives and that slow water exchange was not limiting relaxivity.[7a] We determined the water exchange kinetics of 4b, 4d and 4e (at pH 6 and pH 9) by performing variable temperature T2 measurements of water 17O at 9.4 T in the presence and absence of the Gd-complexes (Figure S2, Table S3) . At 37 °C, the water residency times TM were practically identical for the four studied complexes and ranged from 15 – 20 ns (Table S1, Supporting Information) . Thus differences in water exchange kinetics cannot explain the differences in relaxivity observed. The presence of the intramolecular H-bond did not significantly alter the water exchange rate. This is not surprising: water rotation is occurring on a short picosecond timescale while water exchange is 104 times slower.
There are several reports of gadolinium complexes having anomalously high relaxivity compared to related complexes of similar size, shape, and charge. For instance, work of Lowe et al.[15] describes Gd complexes with a pH-dependent, intramolecular ligation of a β-arylsulfonamide nitrogen which results in changes in the hydration state at the lanthanide center. The relaxivity observed for the q = 2 complexes was found to be anomalously high, and possibly indicative of interaction with the bound water by pendant, non-coordinating carboxylates.[15] Similarly, Dumas et al. described Gd complexes with unexpectedly enhanced relaxivity that possess pendant non-coordinating carboxylate donor arms.[13b, 16] Furthermore, numerous Gd complexes for Zn2+ sensing have been reported to display relaxivity values that exceed the expected values.[17] Previously, these cases of high relaxivity have usually been ascribed to a second-sphere effect wherein water molecules in the second hydration sphere have extended residency times because of interactions with H-bond acceptor(s) on the chelating ligand. However in many of those complexes it is likely that the H-bond acceptor could also be interacting with the inner-sphere coordinated water and hindering the water rotation.
Conclusions
Fast internal rotation of a coordinated water ligand is known to limit relaxivity in metal complexes. We show here that this fast rotation can be blocked/slowed by the incorporation of an appropriately positioned H-bond acceptor. Reduction of Gd-aqua ligand rotation results in increased relaxivity. Optimizing the inner-sphere water structure and dynamics of Gd complexes is a generalizable design strategy to enhance relaxivity. This work represents the first in depth structure-activity study to demonstrate the importance of intramolecular H-bond formation to controlling Gd-aqua ligand dynamics and augmenting relaxivity.
Supplementary Material
Scheme 1.

Restricting the water ligand mobility (specifically rotation about the Gd-O bond) , in Gd complexes through interaction with an intramolecular H-bond acceptor.
Acknowledgments
Financial support from the National Institutes of Health is gratefully acknowledged. P.C. acknowledges the National Institute for Biomedical Imaging and Bioengineering for funding for this project (NIBIB, Award No. R01EB009062) , and the National Center for Research Resources for instrumentation grants (S10OD010650, S10RR023385, P41RR14075) . E.B. acknowledges the NHLBI for support (K99HL125728) . H-K.K. acknowledges a fellowship from the National Research Foundation of Korea (NRF-2013R1A6A3A03064101) . Partially this material is based upon work supported by the US Department of Energy Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under Contract DE-AC02-06CH11357 at Argonne National Laboratory (J.N. and O.G.P) .
Footnotes
Electronic Supplementary Information (ESI) for this article is given via a link at the end of the document, containing detailed experimental procedures for compound syntheses, relaxivity measurements, ENDOR measurements, water exchange, complex hydration number and molecular dynamics calculations.
References
- 1.a) Nandi N, Bhattacharyya K, Bagchi B. Chem Rev. 2000;100:2013–2046. doi: 10.1021/cr980127v. [DOI] [PubMed] [Google Scholar]; b) Zhang L, Wang L, Kao YT, Qiu W, Yang Y, Okobiah O, Zhong D. Proc Natl Acad Sci. 2007;104:18461–18466. doi: 10.1073/pnas.0707647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Furie BC, Mann K, Furie B. J Biol Chem. 1976;251:3235–3241. [PubMed] [Google Scholar]; b) Naoto I, Kawakita M. J Biochem. 1984;95:661–669. doi: 10.1093/oxfordjournals.jbchem.a134655. [DOI] [PubMed] [Google Scholar]; c) Jona I, Martonosi A. Biochem Journ. 1986;234:363–371. doi: 10.1042/bj2340363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]; b) Merbach AE, Toth E. The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging. John Wiley & Sons Ltd; New York: 2001. [Google Scholar]; c) Hermann P, Kotek J, Kubíček V, Lukeš I. Dalton Trans. 2008:3027–3047. doi: 10.1039/b719704g. [DOI] [PubMed] [Google Scholar]
- 4.Dwek R. Nuclear magnetic resonance in biochemistry: applications to enzyme systems. Clarendon Press, Oxford University; 1973. p. 424. [Google Scholar]
- 5.a) Dunand FA, Aime S, Merbach AE. J Am Chem Soc. 2000;122:1506–1512. [Google Scholar]; b) Dunand FA, Borel A, Merbach AE. J Am Chem Soc. 2002;124:710–716. doi: 10.1021/ja016873q. [DOI] [PubMed] [Google Scholar]
- 6.a) Aime S, Castelli DD, Crich SG, Gianolio E, Terreno E. Acc Chem Res. 2009;42:822–831. doi: 10.1021/ar800192p. [DOI] [PubMed] [Google Scholar]; b) Caravan P, Farrar CT, Frullano L, Uppal R. Contrast Media Mol Imaging. 2009;4:89–100. doi: 10.1002/cmmi.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Boros E, Polasek M, Zhang Z, Caravan P. J Am Chem Soc. 2012;134:19858–19868. doi: 10.1021/ja309187m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Boros E, Caravan P. J Med Chem. 2013;56:1782–1786. doi: 10.1021/jm4000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lommerse JP, Price SL, Taylor R. J Comput Chem. 1997;18:757–774. [Google Scholar]
- 9.Brooks BR, Brooks CL, MacKerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. J Cheminform. 2012;4:1. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bayly CI, Cieplak P, Cornell W, Kollman PA. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 12.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 13.a) Vitha T, Kubıcek V, Kotek J, Hermann P, Elst LV, Muller RN, Lukes I, Peters JA. Dalton Trans. 2009:3204–3214. doi: 10.1039/b820705d. [DOI] [PubMed] [Google Scholar]; b) Dumas S, Jacques V, Sun W-C, Troughton JS, Welch JT, Chasse JM, Schmitt-Willich H, Caravan P. Invest Radiol. 2010;45:600–612. doi: 10.1097/RLI.0b013e3181ee5a9e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Boros E, Caravan P. Inorg Chem. 2015;54:2403–2410. doi: 10.1021/ic503035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Caravan P, Astashkin AV, Raitsimring AM. Inorg Chem. 2003;42:3972–3974. doi: 10.1021/ic034414f. [DOI] [PubMed] [Google Scholar]; b) Astashkin AV, Raitsimring AM, Caravan P. J Phys Chem A. 2004;108:1990–2001. [Google Scholar]
- 15.Lowe MP, Parker D, Reany O, Aime S, Botta M, Castellano G, Gianolio E, Pagliarin R. J Am Chem Soc. 2001;123:7601–7609. doi: 10.1021/ja0103647. [DOI] [PubMed] [Google Scholar]
- 16.Jacques V, Dumas S, Sun W-C, Troughton JS, Greenfield MT, Caravan P. Inves Radiol. 2010;45:613–624. doi: 10.1097/RLI.0b013e3181ee6a49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Regueiro-Figueroa M, Gündüz S, Patinec V, Logothetis NK, Esteban-Gómez D, Tripier R, Angelovski G, Platas-Iglesias C. Inorg Chem. 2015;54:10342–10350. doi: 10.1021/acs.inorgchem.5b01719. [DOI] [PubMed] [Google Scholar]; b) Matosziuk LM, Leibowitz JH, Heffern MC, MacRenaris KW, Ratner MA, Meade TJ. Inorg Chem. 2013;52:12250–12261. doi: 10.1021/ic400681j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.