

Abstract
Fibrogenesis is the active production of extracellular matrix in response to tissue injury. In many chronic diseases persistent fibrogenesis results in the accumulation of scar tissue, which can lead to organ failure and death. However no non-invasive technique exists to assess this key biological process. All tissue fibrogenesis results in the formation of allysine, which enables collagen cross-linking and leads to tissue stiffening and scar formation. We report here a novel allysine-binding gadolinium chelate (GdOA), that can non-invasively detect and quantify the extent of fibrogenesis using magnetic resonance imaging (MRI). We demonstrate that GdOA signal enhancement correlates with extent of disease and is sensitive to therapeutic response.
Keywords: fibrosis, aldehydes, Imaging agents, allysine, collagen
Graphical Abstract
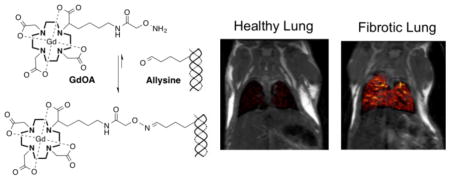
A key biochemical feature of fibrogenesis is the oxidition of lysine residues on collagen to give allysine, which is reponsible for collagen cross-linking, fibril formation, and scar tissue deposition in fibrotic diseases. The oxyamine functionalized molecular probe GdOA binds to allysine leading to MRI signal enhancement in a fibrotic lung disease model, providing a quantitative readout of fibrogenesis.
Almost half of deaths in the industrialized world can be attributed to diseases with a fibroproliferative component.[1] In the lung, pulmonary fibrosis constitutes a major cause of morbidity and mortality. Idiopathic pulmonary fibrosis (IPF), the most common and most lethal diffuse fibrosing lung disease, is responsible for an estimated 40,000 deaths per year in the US.[2] Pulmonary fibrosis is characterized by the accumulation of myofibroblasts and their production of an excess of extracellular matrix (ECM) proteins. Though this ECM accumulation and remodeling, termed fibrogenesis, is an innate part of the natural wound healing process, under persistent injury excess ECM results in a build up of scar tissue causing loss of tissue function and potential organ failure and mortality.[3]
High-resolution computed tomography is invaluable for the diagnosis of IPF,[4] but cannot distinguish regions of active fibrogenesis from stable scar. Molecular MRI, to quantify the extent of increased ECM components, by direct targeting of the collagen deposition that occurs during scar tissue formation, has previously been used to image fibrosis.[5] While this provides a means to quantify the changes in total fibrotic burden of a patient, collagen imaging does not provide a measure of the dynamic changes occurring at the molecular level during fibrogenesis. Imaging fibrogenesis would identify patients whose disease is progressing and would also enable monitoring of treatment with drugs that could stop or slow fibrotic progression.
A universal feature of fibrogenesis is the oxidation of ECM lysine residues, chiefly in collagen, to the aldehyde allysine by the lysyl-oxidase (LOX) family of enzymes. Allysine[6] undergoes a series of condensation reactions with other amino acids on neighboring collagen molecules (Fig. S1) to form irreversible cross-links that stabilize the ECM, and is a fundamental feature across all fibrotic diseases.[7] While lysine oxidation is catalyzed by LOX, the subsequent condensation reactions of allysine are slower. We reasoned that allysine could be a target for imaging fibrogenesis. In active fibrogenesis an increased pool of allysine would be generated, but in stable disease or with therapeutic intervention these allysine moieties would be converted to crosslinks.
To determine if allysine was sufficiently abundant for detection with MRI we quantified allysine in mouse lung tissue using HPLC. Normal and fibrotic lung tissue was digested at 110 °C in 6 M HCl for 24 h, and the allysine derivatized with 2-naphthol-7-sulfonate to yield a fluorescent molecule that could be detected and quantified by HPLC.[8] In normal mouse lung, there was 80±6 nmol allysine per gram of lung tissue, ~80 μM. In mice injured with bleomycin, the allysine concentration content was increased to 150±16 nmol/g. Such high micromolar concentrations are readily detectable by Gd-enhanced MRI.
In the design of a probe for MR imaging of fibrogenesis, certain criteria are essential. These include: i) high thermodynamic and kinetic chelate stability, ii) high water solubility, iii) rapid renal excretion, iv) low non-target background uptake, v) rapid penetration into the tissue interstitial space, and vi) target selectivity. To satisfy these criteria we developed GdOA, an oxyamine-functionalized derivative of GdDOTA for targeting allysine. The GdDOTA core provides a highly stable and inert Gd-chelate (Fig S2). The anionic and hydrophilic nature of GdOA results in high solubility, reduces nonspecific protein binding, and promotes rapid renal elimination. For target selectivity an oxyamine was selected, as the oxime formed from the reaction of an aldehyde with an oxyamine is known to be more stable to hydrolysis than their analogous hydrazone or imine,[9] and therefore expected to result in a strong MR signal enhancement on binding to allysine.
GdOA (Fig. 1a, SI Scheme 1) was prepared by coupling the NHS active-ester of N-Boc-aminooxyacetic acid[10] with an amine-functionalized derivative of DOTA (DOTA-NH2), followed by Boc-deprotection in 1 M HCl, with subsequent Gd chelation at pH 6.8. GdOA purity was assessed by HPLC-ICP-MS analysis, with a single Gd species identified and confirmed as GdOA by HPLC-MS analysis. A six-carbon chain was used as a linker to minimize interaction between the GdDOTA core and oxyamine. As a negative control, GdOX was synthesized which has the same pharmacokinetic properties of GdOA, but is incapable of undergoing a condensation reaction with allysine (Fig. 1a, SI Scheme 2).
Figure 1.

a) Structure of GdOA and GdOX, b) Relaxivity measuremeants for GdOA, and GdOX incubated in PBS (10 mM, pH 7.4, 37 °C) (white bar), BSA (3 mg protein, PBS, pH 7.4, 37 °C) (grey bar) and with BSA-Ald (3 mg protein, PBS, pH 7.4, 37 °C) (black bar), show a significant increase in relaxivity for GdOA with BSA-Ald indicating probe binding to aldehyde-rich BSA, c) binding curves of GdOA and GdOX incubated with BSA-Ald and BSA, show that GdOA binds to the aldehyde-rich BSA with a Kd of 164 μM (data from n=3). d) binding curves of GdOA and GdOX with aorta show that only GdOA binds to allysine-rich porcine aorta, with Kd of 360 μM (data from n=2).
The relaxivity (1.4 T, 37 °C) of GdOA was similar when measured in PBS solution (4.25 mM−1s−1) or in PBS with 3 mg bovine serum albumin (BSA, 150 μM) indicating very low nonspecific protein binding. However in the presence of 3 mg BSA that had been oxidized with FeCl3/aspartate to generate 16 nmol of aldehydes per mg of protein,[11] relaxivity increased by 90% to 8.10 mM−1s−1 (Fig. 1b); the protein-bound fraction of GdOA had a relaxivity of 16.87 mM−1s−1. GdOX showed negligible increase in relaxivity in the presence of BSA or oxidized BSA-Ald. Quantification of GdOA probe bound to BSA-Ald, following ultrafiltration and ICP-MS analysis, gave a binding constant of 164 μM (Fig. 1c). To assess inertness, GdOA was challenged with zinc and phosphate and showed no Gd release (Fig. S2).
Next the binding of GdOA to tissue was assessed. Aorta is rich in allysine as a result of high lysyl-oxidase activity and turnover of elastin and collagen. We measured 7.50 μmol of allysine per gram of porcine aorta using the HPLC assay. We then incubated GdOA or GdOX with segments of aorta (25 mg aorta, 37 °C, 24 h, pH 7). After repeat washing to remove non-specifically bound probe, the aorta associated Gd was quantified by ICP-MS analysis. GdOA gave a Kd of 360 μM, while GdOX showed no affinity (Fig. 1d).
The pharmacokinetics of GdOA and GdOX were assessed in naive mice using MR imaging to measure the blood MR signal wash-out from the left ventricle of the heart. Both probes displayed rapid and almost identical blood clearances with blood half-lives of 5.5 and 6.1 minutes for GdOA and GdOX respectively, indicating comparable probe pharmacokinetics (Fig. S3). Elimination was exclusively through the kidneys with minimal, transient liver enhancement observed. Biodistribution of Gd-OA at 1 hour after bolus intravenous injection in naïve C57Bl/6 mice showed that 95% of the injected dose had already been eliminated from the body (Table S1).
The ability of GdOA to detect and stage pulmonary fibrogenesis was then evaluated using a bleomycin lung injury mouse model. Bleomycin is a chemotherapeutic antibiotic,[12] but a major adverse effect of bleomycin is the overproduction of reactive oxygen species in the lung,[13] which can lead to fibrosis.[14] Mice injured with bleomycin rapidly and reliably develop pulmonary fibrosis.[15] We studied four groups of mice: Group 1) mice injured with a single intratracheal administration of bleomycin (Bleo); Group 2) age-matched healthy mice (Naive); Group 3) mice injured with bleomycin and then dosed daily with the pan-LOX inhibitor β-aminopropionitrile (BAPN, 100 mg/kg) (Bleo+BAPN);[16] and Group 4) mice injured with bleomycin and then dosed daily with PBS as a vehicle control (Bleo+PBS). After 14 days, mice were imaged before and after intravenous injection of 100 μmol/kg GdOA (all 4 cohorts) or GdOX (first 2 cohorts), the same dose used for most clinical GdDOTA enhanced MRI exams.[17]
Bleomycin-injured mice demonstrated increased pulmonary fibrosis as measured by the Ashcroft system of histology scoring (Fig. S4). Bleomycin-injured mice had 3.4-fold higher lung LOX activity (Fig. 2a, Fig. S5), 1.75-fold higher collagen content (Fig. S6), and 2.1-fold higher allysine content (Fig. 2b, Fig. S7) than in naive animals. Treatment with LOX inhibitor BAPN reduced lung LOX activity and allysine content to levels observed in naive mice, although BAPN had no effect on total lung collagen levels (Fig. 2a–b).
Figure 2.

Post imaging ex-vivo tissue analyses for GdOA and GdOX in Naïve, 14 day bleomycin (Bleo) mice, and 14 day bleomycin + daily BAPN (Bleo+BAPN) treated mice and coronal MR images with overlaid lung enhancement (shown in false color): a) total LOX activity levels increased in Bleo treated mice compared to naive animals. Daily BAPN dosing reduces LOX activity levels in Bleo+BAPN animals down to similar levels seen in naive mice, b) allysine lung concentration levels track with LOX activity, with a significant increase in Bleo mice compared to naive animals and a significant decrease in Bleo+BAPN animals, c) GdOA uptake in naive mouse, showing low MR signal enhancement in healthy lungs, d) GdOA uptake in bleomycin-treated mouse, showing strong lung enhancement in 14-day bleomycin-injured mice, e) Image quantification of Δ(lung:muscle) ratio, f) Gd concentration in lung tissue, g) GdOX uptake in bleomycin-challenged mouse, showing low lung enhancement in 14-day bleomycin-injured mice with negative control probe and h) GdOA in bleomycin-challenged mouse dosed daily for 14 days with BAPN, showing little lung enhancement, indicating an absence of allysine. (*:P < 0.05, **: P<0.01, ***: P< 0.001, n/s: not significant).
It is well known that T2* in the lung is very short (~1 ms) because of the magnetic susceptibility gradients caused by the air-tissue interface. In order to overcome this signal loss we used an ultrashort echo time (UTE) sequence. T1-weighted UTE MR images were taken before and after injection of GdOA or GdOX (Fig. 2c,d,g, Fig. S8), starting at 12 minutes when background blood signal was minimal and lung signal intensity was highest. The lung signal and adjacent skeletal muscle signal were measured before and after probe injection, and the change in lung-to-muscle signal ratio (ΔLMR) (Fig. 2e, Fig. S9) was calculated, where LMR = SIlung / SImuscle (SI = signal intensity), and ΔLMR = LMRpost-LMRpre. GdOA injection resulted in strong lung signal enhancement in bleomycin-injured lungs compared to naive mice. Signal enhancement correlated strongly with the extent of disease (Fig. S10). GdOX injection resulted in similar, weak lung enhancement in both bleomycin and naive mice, indicating that the oxyamine function was required for the higher signal observed in the bleomycin-injured mice. BAPN treatment did not inhibit collagen production (Fig. S6), but did prevent allysine production and crosslinking (Fig. 2b). MRI indicated that GdOA is sensitive to the allysine reduction caused by BAPN, further demonstrating the specificity of GdOA (Fig. 2h). We also compared the change in liver-to-muscle ratio between naive and bleomycin-groups after GdOA and saw no significant difference in signal, suggesting that the increased GdOA lung enhancement observed in bleomycin-mice is disease-dependent (Fig. S11). Ex vivo analysis of lung Gd confirmed the imaging results, with the micromolar Gd lung concentrations measured consistent with the image enhancement observed (Fig. 2f, Fig. S12).
In summary, we showed that allysine is a suitable target for molecular MR detection of fibrogenesis. The novel probe GdOA is stable with respect to Gd release, and is able to bind to oxidized collagen present during fibrogenesis. GdOA shows rapid uptake in a disease model of pulmonary fibrosis, and demonstrates specific allysine targeting resulting in enhanced MRI signal in fibrotic tissue. GdOA shows low nonspecific binding and rapid background clearance. GdOA imaging provides a quantitative non-invasive measure of the extent of active fibrogenesis in fibrotic diseases. Targeting allysine as a readout of the rate of fibrogenesis will allow for determination of fibrotic disease activity across all tissue types.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health grants: EB009062, DK104302, HL116315, HL131907, HL133153, OD010650 and RR023385 is gratefully acknowledged.
Footnotes
Electronic Supplementary Information (ESI) for this article is given via a link at the end of the document, containing detailed experimental procedures for compound syntheses, relaxivity measurements, binding and stability study measurements, animal model and MR protocols, full methods for analytical assays and full imaging and assay analysis.
References
- 1.a) Wynn T. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Friedman SL, Sheppard D, Duffield JS, Violette S. Science Transl Med. 2013;5:167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]; c) Rockey DC, Bell PD, Hill JA. New Engl J Med. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 2.a) Olson AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Am J Resp Crit Care. 2007;176:277–284. doi: 10.1164/rccm.200701-044OC. [DOI] [PubMed] [Google Scholar]; b) Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Am J Resp Crit Care. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 3.Rafii R, Juarez MM, Albertson TE, Chan AL. J Thorac Dis. 2013;5:48–73. doi: 10.3978/j.issn.2072-1439.2012.12.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noble PW, Barkauskas CE, Jiang D. J Clin Inv. 2012;122:2756–2762. doi: 10.1172/JCI60323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Caravan P, Das B, Deng Q, Dumas S, Jacques V, Koerner SK, Kolodziej A, Looby RJ, Sun WC, Zhang Z. Chemical Comm. 2009:430–432. doi: 10.1039/b819098d. [DOI] [PubMed] [Google Scholar]; b) Caravan P, Das B, Dumas S, Epstein FH, Helm PA, Jacques V, Koerner S, Kolodziej A, Shen L, Sun WC. Angew Chem Int Ed. 2007;46:8171–8173. doi: 10.1002/anie.200700700. [DOI] [PubMed] [Google Scholar]; c) Caravan P, Yang Y, Zachariah R, Schmitt A, Mino-Kenudson M, Chen HH, Sosnovik DE, Dai G, Fuchs BC, Lanuti M. Am J Resp Cell Mol. 2013;49:1120–1126. doi: 10.1165/rcmb.2013-0039OC. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Fuchs BC, Wang H, Yang Y, Wei L, Polasek M, Schühle DT, Lauwers GY, Parkar A, Sinskey AJ, Tanabe KK. J Hepatol. 2013;59:992–998. doi: 10.1016/j.jhep.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Polasek M, Fuchs BC, Uppal R, Schühle DT, Alford JK, Loving GS, Yamada S, Wei L, Lauwers GY, Guimaraes AR. J Hepatol. 2012;57:549–555. doi: 10.1016/j.jhep.2012.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Farrar CT, DePeralta DK, Day H, Rietz TA, Wei L, Lauwers GY, Keil B, Subramaniam A, Sinskey AJ, Tanabe KK. J Hepatol. 2015;63:689–696. doi: 10.1016/j.jhep.2015.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kagan HM, Li W. J Cell Biochem. 2003;88:660–672. doi: 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- 7.a) Pinnell SR, Martin GR. Proc Nat Acad Sci. 1968;61:708–716. doi: 10.1073/pnas.61.2.708. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanzer ML. Science. 1973;180:561–566. doi: 10.1126/science.180.4086.561. [DOI] [PubMed] [Google Scholar]; c) Siegel RC. Proc Natl Acad Sci. 1974;71:4826–4830. doi: 10.1073/pnas.71.12.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bornstein P, Kang AH, Piez KA. Proc Nat Acad Sc. 1966;55:417–424. doi: 10.1073/pnas.55.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umeda H, Kawamorita K, Suyama K. Amino acids. 2001;20:187–199. doi: 10.1007/s007260170059. [DOI] [PubMed] [Google Scholar]
- 9.Kalia J, Raines RT. Angew Chem. 2008;120:7633–7636. [Google Scholar]
- 10.Foillard S, Rasmussen MO, Razkin J, Boturyn D, Dumy P. J Org Chem. 2008;73:983–991. doi: 10.1021/jo701628k. [DOI] [PubMed] [Google Scholar]
- 11.a) Levine R. Method Enzymol. 1990:465–478. [Google Scholar]; b) Levine RL. J Biol Chem. 1983;258:11828–11833. [PubMed] [Google Scholar]; c) Chao CC, Ma YS, Stadtman ER. Proc Natl Acad Sci. 1997;94:2969–2974. doi: 10.1073/pnas.94.7.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Adamson I. Environmental Health Perspectives. 1976;16:119. doi: 10.1289/ehp.7616119. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Umezawa H, Ishizuka M, Maeda K, Takeuchi T. Cancer. 1967;20:891–895. doi: 10.1002/1097-0142(1967)20:5<891::aid-cncr2820200550>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 13.Claussen CA, Long EC. Chem Rev. 1999;99:2797–2816. doi: 10.1021/cr980449z. [DOI] [PubMed] [Google Scholar]
- 14.Chaudhary NI, Schnapp A, Park JE. Am J Resp Crit Care. 2006;173:769–776. doi: 10.1164/rccm.200505-717OC. [DOI] [PubMed] [Google Scholar]
- 15.Usuki J, Fukuda Y. Pathol Int. 1995;45:552–564. doi: 10.1111/j.1440-1827.1995.tb03503.x. [DOI] [PubMed] [Google Scholar]
- 16.Tang S-S, Trackman P, Kagan H. J Biol Chem. 1983;258:4331–4338. [PubMed] [Google Scholar]
- 17.Herborn CU, Honold E, Wolf M, Kemper J, Kinner S, Adam G, Barkhausen J. Invest Radiol. 2007;42:58–62. doi: 10.1097/01.rli.0000248893.01067.e5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.