

Abstract
This paper investigates how the properties of multi-block copolypeptides can be tuned by their block architecture, defined by the size and distribution of blocks along the polymer chain. These parameters were explored by the precise - genetically encoded - synthesis of recombinant elastin-like polypeptides (ELPs). A family of ELPs was synthesized in which the composition and length were conserved while the block length and distribution were varied, thus creating eleven ELPs with unique block architectures. To our knowledge, these polymers are unprecedented in their intricately and precisely varied architectures. ELPs exhibit lower critical solution temperature (LCST) behavior and micellar self-assembly, both of which impart easily measured physicochemical properties to the copolymers, providing insight into polymer hydrophobicity and self-assembly into higher order structures, as a function of solution temperature. Even subtle variation in block architecture changed the LCST phase behavior and morphology of these ELPs, measured by their-temperature-triggered phase transition and nanoscale self-assembly. Size and morphology of polypeptide micelles could be tuned solely by controlling the block architecture, thus demonstrating that when sequence can be precisely controlled, nanoscale self-assembly of polypeptides can be modulated by block architecture.
Keywords: Elastin-like polypeptides, lower critical solution temperature, copolymer, biopolymer, self-assembly
Graphical abstract
1. Introduction
Copolymers represent a family of macromolecules in which two or more chemically different monomeric units are covalently linked together. When the monomer units possess distinct hydrophobic and hydrophilic properties, copolymers can exhibit nanoscale self-assembly into a variety of morphologies, including lamellae, rods, micelles, and vesicles.1 In addition, copolymers serve a number of diverse applications,2 such as polymer blend compatibilizers,3 non-fouling surface coatings,4 ordered thin film templates,5 and nanoscale drug carriers.6 The sequence of monomeric building blocks in uncontrolled or random copolymers is reminiscent of proteins–the biological counterpart of synthetic polymers–in which amino acid monomers are spatially organized along a polypeptide chain and define the so-called primary structure. In spite of the short range order in the primary structure, proteins form higher order assemblies (i.e., secondary-, tertiary-, and quaternary-structures)7 that are mainly determined by hydrogen bonds and hydrophobic interactions8 and which ultimately dictate protein function. Elucidating the relationship between sequence distribution and polymer function in well-defined copolymer systems can thereby impact the broad use of such materials as well as potentially improve our ability to synthetically mimic biological structure-function relationships.
Thus far the main focus in the investigation of amphiphilic copolymers has been on di- and triblock copolymers. However, there have also been some experimental studies of multi-block copolymers. For instance, multi-block copolymers of poly(ethylene oxide) (PEO) and poly(methylphenylsilane) (PMPS) were observed to form vesicles and micellar rods.9 Similarly, for multi-block copolymers composed of five different monomer units, a pH dependent transition from spherical to rod-like micelles has been reported.10 The effect of the number of blocks was elucidated for multi-block copolymers composed of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and methyl methacrylate (MMA), which demonstrated that an increasing number of blocks led to increasingly large and complex micelles, as measured by light scattering and neutron scattering.11–12 In multi-block copolymers composed of poly(ethylene oxide) (PEO) and either poly(ε-caprolactone) (PCL) or poly(l-lactic acid) (PLLA), the crystalline melting temperatures of the PCL and PLLA were significantly lowered in the multi-block copolymer, due to their partial phase mixing with the PEO segments.13 Work has also been done to extend the classical, thermo-responsive Pluronic copolymers (PEO-PPO-PEO) to their multi-block analogs, which show interconnected structures and a corresponding increase in viscosity.14
In addition to these experimental studies, multi-block copolymers have also been explored from a theoretical perspective. Early theoretical work predicted that multi-block copolymers would form either flowerlike single intramolecular micelles or strings of molecular micelles.15 More recent work using Monte Carlo simulations of multi-block copolymers with regularly spaced hydrophobic and hydrophilic blocks demonstrated that, depending on the relative block lengths, a variety of different structures could be formed, including micelles, chains of micelles, tubes, and lamellar structures.16 Brownian dynamics simulations of multi-block copolymers showed that the extent of bridging of aggregates in solution was caused by an increasing flexibility of the polymer chains, and that a high content of the hydrophobic blocks favors formation of micelles with rich structural patterns.17
Despite this considerable amount of work, experimental studies of multi-block copolymers have thus far been hindered by the inherent limitations of chemical polymer synthesis. Regardless of the synthetic techniques used, it has not been possible to chemically synthesize perfectly sequence-defined and monodisperse multi-block copolymers. However, given the popularization of recombinant DNA techniques and the considerable research in the area of repeat polypeptides such as elastin-like polypeptides,18–19 resilin-like polypeptides,20–22 and silklike polypeptides23–24 it is now possible to synthesize perfectly sequence-defined and monodisperse multi-block copolypeptides using recombinant, rather than chemical, methods.
Herein, we report the synthesis and characterization of multi-block copolypeptides based on elastin-like polypeptides (ELPs), which are composed of the monomeric pentapeptide unit VPGXG, where the “guest residue” X can be any residue except proline. ELPs are of particular interest because they exhibit lower critical solution temperature (LCST) behavior25 and micellar self-assembly.26–27 Homopolymer and pseudorandom ELPs are soluble below their phase transition temperature (Tt) and coacervate into micron-scale insoluble aggregates above their Tt. ELP diblock copolymers, composed of ELP blocks with distinct Tts, can self assemble into spherical micelles above their critical micelle temperature (CMT) upon selective coacervation of their more hydrophobic block. The LCST behavior and self-assembly of multi-block ELPs have been used to develop a number of structures such as spherical and cylindrical micelles,26, 28–30 vesicles,31 and hydrogel networks,32–34 and have been exploited for a number of biomedical applications including drug delivery35–36 and tissue engineering.37–38 These existing material systems provide preliminary evidence that block architecture can manipulate the thermal properties and self-assembly of ELPs,31, 39 motivating us to further explore the impact of this design parameter using an ELP library with systematically varied block architecture.
We take a unique approach where the overall chemical composition and chain length of the ELP are held constant and the only parameter that is systematically varied is the size and distribution of the blocks along the polypeptide chain, which we term the block architecture. The LCST Tt and CMT provide sensitive outputs that are easily measured by optical scattering methods. We show that all multi-block ELPs exhibit LCST behavior that is modulated by their block architecture. The most interesting and unexpected observation from this new class of multi-block copolypeptides is that a subset exhibits temperature-triggered micellar self-assembly wherein the size and morphology of the micelles can be tuned solely by variations in block architecture. Overall, we show that controlling the block architecture of such ELPs allows direct control over both their temperature-responsive phase behavior and micellar self-assembly.
2. Experimental section
2.1 Materials
Oligonucleotides encoding ELP sequences were purchased from Integrated DNA Technologies (Coralville, IA). Competent E. coli EB5α cloning cells and BL21 expression cells were purchased from Edge BioSystems (Gaithersburg, MD). Terrific Broth (TB) Dry powder growth media was procured from MO BIO Laboratories (Carlsbad, CA). Kanamycin sulfate was purchased from EMD Millipore (Billerica, MA) and isopropyl-beta-D-thiogalactoside (IPTG) was purchased from Gold Biotechnology (St. Louis, MO). Calbiochem phosphate buffered saline (PBS) tablets (10 mM phosphate buffer, 140 mM NaCl, 3 mM KCl, pH 7.4 at 25°C) were procured from EMD Millipore (Billerica, MA). Low retention 0.02 µm and 0.1 µm Whatman Anotop 10 filters were purchased from GE Healthcare Life Sciences (Pittsburgh, PA).
2.2 Cloning and nomenclature
Oligonucleotides encoding 5 pentapeptide repeats of each monomer (VPGSG or VPGVG) or 4 pentapeptide repeats of alternating monomers (VPGSGVPGVG) were purchased with 2 base pair overhangs complementary to the BseRI cut site in a modified pET-24a(+) T7-lac cloning vector.40 These oligonucleotides were concatemerized in the cloning vector to achieve 1–4 repeats encoding 5–20 pentapeptides of each monomer or 1–5 repeats encoding 4–20 pentapeptides of the alternating monomers. Precise cloning of each ELP architecture was then achieved with recursive directional ligation by plasmid reconstruction (PRe-RDL) in EB5α E. coli.40 Complete DNA and polypeptide sequences of all ELP constructs are found in Supplementary Figures S1 and S20. Successful cloning was confirmed by diagnostic digest and plasmid DNA sequencing using T7 promoter and terminator primers.
Each ELP was designated by a name referring to its distinct architecture created by the genetically engineered sequence of hydrophilic VPGSG pentapeptide monomers (abbreviated ‘S’ for the hydrophilic serine guest residue) and hydrophobic VPGVG pentapeptide monomers (abbreviated ‘V’ for the hydrophobic valine guest residue). Several families of architecture were created by manipulating the placement of these monomers along the polypeptide chain.
ELPs were created in which blocks of each monomer were alternated along the polypeptide chain. These ELPs were designated ELP-SVBn where n represents the number of monomers in each block: 1, 5, 10, 20, or 30 pentapeptides. The triblock ELP family was created in which the three segments of the ELP were organized in each of two possible configurations: the hydrophilic ‘S’ block capped by two hydrophobic ‘V’ blocks, or the hydrophobic ‘V’ block capped by two hydrophilic ‘S’ blocks. These ELPs were designated ELP-SVTVSV and ELP-SVTSVS, respectively. The diblock ELP family was created in one configuration of the hydrophilic ‘S’ block followed by the hydrophobic ‘V’ block. This diblock ELP was designated ELP-SVDSV. The gradient ELP family was created such that the block sizes changed gradually over the length of the polypeptide chain. This gradual change in block size was varied over 50.0, 41.7, or 33.3% of the total ELP length. These represented low, intermediate, and high gradient changes in composition, such that these ELPs were designated ELP-SVGL, ELP-SVGI, and ELP-SVGH, respectively. Finally, a component ELP family was synthesized in which homopolymer ELPs, designated ELP-Scomp and ELP-Vcomp, were created for each individual monomer with a total length equivalent to the ELPs with various block architectures.
2.3 Expression and purification
BL21 E. coli were transformed with plasmids encoding the ELP genes. These cells were grown overnight in 50 mL TB media supplemented with 45 µg/mL kanamycin, and were then used to inoculate 1 L cultures, which were grown for an additional 24 hours. Induction with 0.2 mM IPTG was used to increase expression yield via the T7-lac promoter. E. coli were collected by centrifugation at 3,200 RPM and resuspended in PBS prior to lysis by sonication. 10% polyethylenimine (w/v) was added to condense genomic contaminants, then cellular debris was collected by centrifugation at 15,000 RPM at 4°C. The supernatant was heated to 60°C to denature protein contaminants, cooled on ice, and centrifuged at 15,000 RPM at 4°C to remove insoluble debris.
ELPs were then further purified using inverse transition cycling (ITC),18 in which centrifugation above and below the ELP’s Tt removed soluble and insoluble contaminants, respectively. The ELP solution was heated to 37°C (60°C for ELP-Scomp) and crystalline NaCl was added (<3 M) to induce the ELP transition. The coacervated ELP was collected by centrifugation at 15,000 RPM at room temperature. The supernatant was discarded and the ELP pellet was resuspended in cold PBS. The resuspended ELP solution was centrifuged at 15,000 RPM at 4°C. The insoluble contaminant pellet was discarded and the soluble ELP in the supernatant was retained. Four rounds of ITC yielded ELP products of sufficient purity, as confirmed by SDS-PAGE and CuCl2 staining Supplementary Figures S2 and S21).
2.4 Thermal characterization
The LCST behavior of each ELP was characterized by temperature-controlled turbidimetry. An Agilent Technologies Cary 300 UV-VIS spectrophotometer was used to measure the optical density (OD) at 350 nm of ELP solutions from 20–90°C at a heating rate of 1°C/min. Since the ELP transition is concentration dependent, such that the ELP’s Tt decreases with increasing concentration in a logarithmic relationship,41 ELP solutions were measured at concentrations of 5–100 µM in PBS. The Tt at each concentration was defined as the temperature that corresponded to the maximum of the Gaussian fit of the first derivative of OD, with respect to temperature (Supplementary Tables S4, S6, S11, and S13). For ELP-Scomp, which transitioned above 90°C, NaCl (1.0–2.5 M) was added to depress the Tt until it was observed below 90°C (Supplementary Tables S2 and S3). The Tts observed below 90°C with the addition of NaCl were used to extrapolate a hypothetical Tt above 90°C without additional NaCl. For those ELPs that displayed temperature-triggered micelle self-assembly the critical micelle temperature (CMT) was defined as the temperature corresponding to the positive deviation of OD from baseline (Supplementary Tables S5, S7, S12, and S14). Complete characterization of ELPs by temperature-regulated turbidimetry is found in Supplementary Figures S3–S15 and S22–24.
2.5 Light scattering
The self-assembly inferred from temperature-controlled turbidimetry was corroborated with dynamic light scattering (DLS). For those constructs that displayed self-assembly by UV-VIS measurement of OD, ELP solutions at a concentration of 100 µM were prepared in PBS and passed through 0.02 µm filters prior to analysis by DLS over a temperature range of 20–70°C using a Wyatt DynaPro temperature-controlled microsampler.
Static light scattering (SLS) measurements were then acquired simultaneously with DLS measurements on an ALV/CGS-3 goniometer (Supplementary Figures S16–S19 and S25–S27). Samples were prepared in PBS and passed through 0.1 µm filters prior to analysis. Refractive index increment (dn/dc) was determined from measurements made on an Abbemat 500 refractometer (Supplementary Tables S8 and S15). DLS and SLS analysis yielded radius of gyration (Rg), hydrodynamic radius (Rh), shape factor (ϱ), molecular weight, and aggregation number (Nagg) parameters (Supplementary Tables S9, S10, and S16).
2.6 Small angle neutron and X-ray scattering
Small angle neutron scattering (SANS) experiments were conducted at the NIST Center for Neutron Research (NCNR) on the NGB 30m SANS instrument. Samples were poured into quartz cuvettes and acquisitions were performed at 20°C. All samples were measured at 100 µM in PBS with D2O as a solvent in order to increase the contrast (difference in scattering length densities, ΔSLD) and decrease the incoherent background mainly caused by hydrogen. Three configurations were used with a fixed wavelength of 6 Å and sample-detector distances of 1.33 m, 4 m, and 13.17 m corresponding to q ranging from ~ 0.05 to 5 nm−1. Data were reduced using the NCNR SANS reduction macros in Igor Pro,42 which allows for the correction of intensities for the transmission, dead-time, detector background (with B4C as a neutron absorber at the sample position), and sample background (either the empty cuvette or the solvent). Absolute scale was obtained by measurement of the direct beam.
Small angle x-ray scattering (SAXS) experiments were conducted at Duke University’s Shared Materials and Instrumentation Facility using a SAXSLAB Ganesha instrument. The instrument has a Cu Kα X-ray source, a point-collimated pinhole system and 2D detector. Samples were measured in glass capillaries at 45°C and measurements were taken at sample-detector distances of 180, 480, and 1080 mm, corresponding to q ranging from 0.05 to 5 nm−1. Sample buffer was measured under identical conditions and subtracted from the sample scattering to account for incoherent background scattering. Data were reduced using SAXSLAB’s SAXSGUI interface.
3. Results and discussion
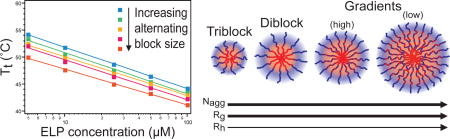
A library of eleven unique ELPs was synthesized, each with distinct architecture and a conserved composition, as visualized in Figure 1 and defined in Supplementary Table S1. The architectures included alternating, triblock, and diblock copolymers, as well as new architectures that we term gradient copolymers. Each ELP contained 60 repeats of the hydrophobic pentapeptide monomer VPGVG and contained 60 repeats of the hydrophilic pentapeptide monomer VPGSG. The length of each ELP was thus conserved at 120 total pentapeptide repeats, with each ELP having a molecular weight (MW) of 48.66 kDa. To understand the contribution of the hydrophobic and hydrophilic content to the block copolymer ELP behavior, single segment homopolymer ELPs were also synthesized, composed of 120 pentapeptides of VPGVG or VPGSG alone and were referred to as ELP-Vcomp and ELP-Scomp, respectively. Synthetic genes for each ELP were assembled by PRe-RDL in a T7-lac plasmid,40 recombinantly expressed in E. coli, and purified to homogeneity by exploiting their LCST behavior.18, 43
Figure 1.

Design of ELPs with various block architecture. Each ELP was composed of 60 pentapeptide repeats of the hydrophilic monomer VPGSG (blue) and 60 pentapeptide repeats of the hydrophobic monomer VPGVG (red). Varying the organization of pentapeptides along the polypeptide chain resulted in ELPs with precisely defined architectures, including ELPs with alternating blocks of increasing block size, as well as triblock, diblock, and gradient ELPs.
Preliminary characterization of the alternating copolymer ELPs yielded two interesting observations. The first surprising observation was that mixing hydrophilic and hydrophobic pentapeptide monomers within the copolymers in equal proportion in an alternating fashion did not yield a polymer whose thermal behaviors were an average of the behaviors of the two component ELPs, created as homopolymers of either monomer alone. Across all alternating copolymers, ranging from SVB1 (alternating blocks of one pentapeptide) to SVB30 (alternating blocks of 30 pentapeptides), the Tt was dominated by the hydrophobic block, as demonstrated by comparing the Tt of all copolymers as a function of concentration to their parent – hydrophobic (ELP-Vcomp) and hydrophilic (ELP-Scomp) – homopolymers. Even ELP-SVB1, the copolymer with the smallest block size of a single monomer unit, exhibited a Tt that was far more influenced by the hydrophobic block than the hydrophilic block, with a Tt that was close to that of the hydrophobic component alone (Figure 2A).
Figure 2.

Thermal characterization of component and alternating block copolymer ELPs. A) The hydrophilic (ELP-Scomp) and hydrophobic (ELP-Vcomp) component ELPs exhibited Tts that differed by 60–85°C. Homogeneous mixture of the components in ELP-SVB1 resulted in a Tt that was dominated by the hydrophobic component. B) Increasing the block size decreased the Tt across concentrations of 5–100 µM. Squares–experimental values; lines–logarithmic fit to experimental data.
The second observation from these data was that as the block size increased from 1 pentapeptide (ELP-SVB1), to 5 (ELP-SVB5), 10 (ELP-SVB10), 20 (ELP-SVB20), and 30 pentapeptides (ELP-SVB30) the Tt decreased. This trend was maintained over a range of concentrations from 5–100 µM (Figure 2B), such that increasing block size resulted in a Tt that was closer to the hydrophobic component at all concentrations. This clearly showed that the LCST behavior of copolymers ranging from alternating (ELP-SVB1) to tetrablock (ELP-SVB30) was dominated by the hydrophobic monomer, such that their Tt decreased with increasing length of their hydrophobic block. This trend was consistent with the known inverse dependence of Tt with respect to ELP chain length,41 wherein longer ELPs of a given sequence have a lower Tt than shorter ELPs.
In previous studies with diblock ELPs, increasing temperature resulted in preferential desolvation of the hydrophobic block, which triggered its self-assembly into micelles at a critical micellization temperature (CMT). In contrast, the ELP-SVB1 through ELP-SVB30 copolymers did not self-assemble into micelles at any temperature studied. Instead, these ELPs exhibited a single unimer-to-aggregate phase transition from soluble polypeptides into a polypeptide-rich phase and a solvent-rich phase, regardless of block size.
In order to investigate the size and conformation of the soluble polypeptides, small-angle neutron scattering (SANS) spectra were acquired at 20°C for ELP-Scomp, ELP-Vcomp, ELP-SVB1, ELP-SVB5, ELP-SVB10, and ELP-SVB20. The spectra were similar for all polypeptides (Figure 3A), indicating that despite differences in phase behavior, the conformation and size while soluble were similar. In the low-q regime, extrapolation to q = 0 allowed the radius of gyration (Rg) to be calculated using the Guinier approximation:
| (1) |
where I0, the forward scattering, is given by:
| (2) |
where N/V is the number concentration of polypeptide, ϱ is the scattering length density, and Vp is the molecular volume of the polypeptide. The RgS of the soluble polypeptides were ~ 8 nm (Table 1).
Figure 3.

SANS analysis of soluble component and alternating ELPs. (A) SANS spectra of soluble component and alternating ELPs. (B) Kratky-Porod plot of q2 * dΣ/dΩ vs q, showing q−2 scaling at high q (q > 0.5 nm−1).
Table 1.
Radii of gyration, high q scaling, and excluded volume parameter for soluble component and alternating ELPs, calculated from SANS spectra.
| ELP | Rg (nm) | n (for qn) | v (1/n) |
|---|---|---|---|
| Scomp | 7.72 | 2.14 | 0.47 |
| Vcomp | 8.52 | 2.20 | 0.45 |
| SVB1 | 8.21 | 2.00 | 0.50 |
| SVB5 | 7.22 | 2.23 | 0.45 |
| SVB10 | 7.87 | 2.25 | 0.44 |
| SVB20 | 7.52 | 2.18 | 0.46 |
In the higher q region, one no longer observes the overall size of the polypeptide chains, but instead this range contains information regarding the chain conformation. Here the scattering can be described by dΣ/dΩ ~ qn, where the exponent n is related to the excluded volume parameter ν by n = 1/v. For polymer chains, n = 2 (v = 1/2) characterizes an ideal Gaussian chain, n = 3 (v = 1/3) a collapsed polymer coil, and n = 4 (v = 1/4) a folded globular protein. The exponent n of the soluble polypeptides ranged from 2.00 to 2.25 (Table 1), indicating that the polypeptides adopted a conformation in between Gaussian chains (n = 2) and collapsed polymer coils (n = 3). Our general observation is consistent with a polypeptide that is largely unfolded but retains some backbone structure, and is similar to the value of n = 2.2 reported for resilin-mimetic protein polymers.21 The high-q scaling of the polypeptides could be observed visually with a Kratky- Porod plot (Figure 3B) of q2 * dΣ/dΩ vs q on a semi-logarithmic scale. The linear region of the Kratky-Porod plot indicates the region over which the scaling is approximately q−2, and the polypeptide has a random coil conformation.
Further increase in block size resulted in triblock ELPs in two configurations: One in which the hydrophilic block was flanked by two hydrophobic blocks (ELP-SVTVSV), and a second in which the hydrophobic block was flanked by two hydrophilic blocks (ELP-SVTSVS). Similar to the trend in Tt with respect to block size seen in Figure 2, ELP-SVTVSV displayed a unimer-to-aggregate transition at a Tt nearly identical to ELP-SVB30, whose block size matched the hydrophobic block size in this triblock configuration (Figure 4A).
Figure 4.

Thermal characterization of triblock ELPs. A) ELP-SVTVSV and ELP-SVTSVS displayed distinct thermal properties as determined by temperature-programmed turbidimetry at 100 µM (lines, left ordinate). Self-assembly of ELP-SVTSVS was confirmed by measurement of Rh by DLS (dots, right ordinate). Multiple Rh values for a single temperature indicate multiple relaxation times. B) The Tt of ELP-SVTVSV and the CMT of ELP-SVTSVS were similar. The Tt of ELP-SVTSVS, defining the micelle-to-aggregate phase transition, had decreased concentration dependence as compared to the Tt of the ELP-SVTVSV unimer-to-aggregate phase transition or CMT of the ELP-SVTSVS unimer-to-micelle transition. Squares–experimental values; lines–logarithmic fit to experimental data.
The alternative triblock configuration, ELP-SVTSVS, displayed a very different thermal behavior as indicated by turbidimetry, in which the gradual increase in OD with increasing temperature indicated submicron-scale self-assembly, prior to a rapid increase in OD that is a signature of phase separation of the ELP into micron-sized aggregates that with time coalesce into a macroscopic phase (Figure 4A). The self-assembly of ELP-SVTSVS was confirmed by DLS; with an increase in temperature, unimers (Rh ~ 6 nm) self-assembled into micelles (Rh ~ 15 nm) and upon further heating phase-transitioned into micron-sized aggregates. The CMT of ELP-SVTSVS, corresponding to the unimer-to-micelle transition, was nearly identical to the Tt of the alternative triblock configuration, ELP-SVTVSV, being only 1°C lower at all concentrations over a range of 10–100 µM (Figure 4B). The slope of ELP-SVTSVS’s CMT and ELP-SVTVSV’s Tt with respect to concentration (Figure 4B) confirmed that both unimer-to-micelle and unimer-to-aggregate transitions had a similar logarithmic dependence on concentration (Supplementary Tables S6 and S7). The decreased slope of Tt with respect to concentration for ELP-SVTSVS, however, demonstrates decreased concentration dependence for the micelle-to-aggregate transition. This is likely because once ELPs self-assemble into micelles, the local concentration of the corona ELP chains stays relatively constant, even as the global concentration of micelles changes.44
The maximum block size was attained in a diblock architecture in which the hydrophilic and hydrophobic monomers were completely segregated in blocks that each accounted for one half of the total polypeptide length. We investigated the diblock configuration of the hydrophilic block succeeded by the hydrophobic block (ELP-SVDSV). This architecture displayed temperature-triggered self-assembly as evident from turbidimetry and DLS measurements (Figure 5A). The diblock ELP exhibited a slightly lower CMT and a slightly higher Tt than the self-assembling triblock (ELP-SVTSVS) suggesting that the complete segregation of monomers in the diblock allowed the two blocks to behave more independently of one another. Similar to the selfassembling triblock, the Tt of the diblock’s micelle-to-aggregate transition displayed decreased concentration dependence, because the local concentration of the corona ELP chains stays constant, even as the global concentration varies (Figure 5B)
Figure 5.

Thermal characterization of diblock ELP. A) ELP-SVDSV displayed temperature-triggered self-assembly as determined by temperature-programmed turbidimetry at 100 µM (line, left ordinate). DLS measurement of Rh confirmed self-assembly into micelles prior to coacervation into micron-scale aggregates (dots, right ordinate). Multiple Rh values for a single temperature indicate multiple relaxation times. B) The ELP-SVDSV Tt exhibited decreased concentration dependence as compared to the CMT. Squares–experimental values; lines– logarithmic fit to experimental data.
The structure-function relationship of triblock and diblock ELPs suggested that temperature-triggered self-assembly in this material system occurred only when segregation of the copolymer’s hydrophobic components resulted in a block with greater than 30 contiguous hydrophobic pentapeptides. To further examine the role of block size and block distribution on ELP behavior, a family of gradient ELPs was investigated. Gradient ELPs were composed of blocks that changed gradually in length along the ELP chain. The percent of the ELP length over which the hydrophilic/hydrophobic character was mixed determined the gradient “steepness”, thus creating ELPs with low (ELP-SVGL), intermediate (ELP-SVGI), and high (ELP-SVGH) gradients (Figure 1 and Supplementary Table 1). The pure hydrophilic/hydrophobic terminal blocks of these gradient ELPs were 25.0, 29.2, or 33.3% of the total ELP length, respectively 30, 35, or 40 pentapeptides.
Interestingly, each gradient ELP exhibited unique thermal behavior. ELP-SVGL did not exhibit nanoscale self-assembly, and only exhibited a unimer-to-aggregate phase transition at a Tt very close to that measured for the alternating block copolymer ELPs (Figure 6A). ELP-SVGI and ELP-SVGH, however, exhibited two independent temperature triggered transitions, as observed by turbidimetry and confirmed by DLS: A unimer-to-micelle transition and a micelle-to-aggregate transition at higher temperatures. The unimer-to-micelle CMT decreased for both ELP-SVGI and ELP-SVGH, as compared to the Tt of ELP-SVGL, and the micelle-to-aggregate Tt increased from ELP-SVGI to ELP-SVGH (Figure 6B). These results supported the notion that greater spatial segregation of the hydrophilic and hydrophobic components enhanced their independent behavior, as evident in the expanded thermal range over which stable self-assembly was observed for ELP-SVGH, as compared to ELP-SVGI. Furthermore, characterization of these gradient ELPs clarified that the segregation of the copolymer’s hydrophobic components resulting in a block with at least 35 contiguous hydrophobic peptapeptides was necessary to drive self-assembly in this material system.
Figure 6.

Thermal characterization of gradient ELPs. A) Gradient ELPs were characterized by temperature-regulated turbidimetry (lines, left ordinate) and self-assembly of ELP-SVGI and ELP-SVGH was confirmed by measurement of Rh with DLS (dots, right ordinate). Multiple Rh values for a single temperature indicate multiple relaxation times. B) All micelle-to-aggregate Tts displayed decreased concentration dependence, as compared to unimer-to-aggregate Tts and unimer-to-micelle CMTs. ELP-SVGI achieved self-assembly only at concentrations above 25 µM, such that the slope of its Tt changed about this point. Squares–experimental values; lines– logarithmic fit to experimental data.
Comparing the Tts and CMTs of all ELPs at an equivalent concentration reinforced the trends in thermal behavior across ELPs of all architectures (Figure 7). For ELPs that did not exhibit self-assembly the Tts decreased monotonically with increasing block size, likely influenced by the inverse relationship between ELP length and ELP Tt.41 For ELPs that did exhibit selfassembly, the CMTs fell in line with this trend, where the transition of the hydrophobic ELP component acted independently from the transition of the hydrophilic ELP component. The Tts of self-assembled ELPs, corresponding to the micelle-to-aggregate transition, followed an opposite trend such that this Tt increased with increasing block size. This is counter-intuitive to the effect of block length on Tt, but rather speaks for the effect of increased spatial separation between blocks allowing hydrophilic and hydrophobic components to exhibit independent thermal behavior.
Figure 7.

Trends in Tt (squares) and CMT (triangles), dependent on ELP block copolymer architecture at concentrations of 100 µM. Increasing block size led to a decrease in unimer-to-aggregate phase transition Tt, a trend that also included the unimer-to-micelle CMT when selfassembly behavior emerged. In contrast, the micelle-to-aggregate Tt increased with increasing block size for self-assembled ELPs. The behavior of the mixed monomers in most block architectures were swayed toward the Tt of the hydrophobic ELP-Vcomp (red dashed line) rather than the hydrophilic ELP-Scomp (blue dashed line).
Self-assembled ELPs were further characterized with DLS and SLS to investigate the size and aggregation number of the micelles as a function of block architecture (Table 2). Each ELP was measured at the same concentration (100 µM) and temperature (43°C). Micelles formed by triblock ELP-SVTSVS were the smallest based on their Rg and Rh, likely a consequence of the “hairpin” configuration of chains in these micelles. Micelle size increased slightly from diblock ELP-SVDSV to gradient ELP-SVGH, while gradient ELP-SVGI formed the largest micelles. The aggregation number (Nagg=ELP chains/micelle) of each ELP micelle at 43°C was derived by SLS measurement of the MW of each micelle, divided by the theoretical MW of the individual ELP chains.
Table 2.
SLS and DLS characterization of self-assembled ELPs.1
| ELP | Rg (nm) | Rh (nm) | ρ (Rg/Rh) | Nagg | Nagg*2 |
|---|---|---|---|---|---|
| SVTSVS | 12.7 (±5.91%) | 16.2 | 0.78 | 23 | 16 |
| SVDSV | 13.8 (±4.21%) | 23.3 | 0.59 | 70 | 38 |
| SVGH | 17.0 (±2.78%) | 24.1 | 0.70 | 73 | 52 |
| SVGI | 26.1 (±2.00%) | 32.4 | 0.81 | 85 | 85 |
Measurements of 100 µM ELP were made at 43°C degrees.
Nagg* was measured for each ELP at 2°C above their respective CMT (ELP-SVTSVS at 42°C, ELP-SVDSV at 40°C, ELP-SVGH at 42°C, and ELP-SVGI at 43°C).
To more accurately compare Nagg between ELPs of differing CMTs,45 we obtained Nagg values for each ELP at equivalent concentrations, but at 2°C above each of their respective CMTs (denoted Nagg*). Normalization to the scattering at temperatures equidistant above the CMT was evident in the difference between Nagg and Nagg*, dependent on the temperature difference between 43°C and each ELP’s CMT. Nagg*, and to a lesser extent Nagg, increased with increasing micelle size, suggesting that larger micelles incorporated a greater number of ELP chains. All trends in Rg, Rh, and aggregation number held true regardless of measurement at 43°C, or at temperatures 2°C above each ELP’s CMT (Supplementary Tables S9 and S10).
The shape factor, ϱ, of the ELP micelles ranged from 0.59–0.81, reminiscent of a hard sphere (ϱsphere=0.774), suggesting the assembly of the ELPs into spherical micelles, regardless of block architecture. However, the variation in ϱ suggested slight differences in the compactness of the collapsed micelle core in relation to the hydrated micelle corona (Figure 8). Notably, ϱ increased between ELP-SVDSV, ELP-SVGH, and ELP-SVGI, indicating that Rg increased disproportionately to the increase in Rh between these assemblies. The lowest ϱ of ELP-SVDSV suggested that complete segregation of the hydrophobic and hydrophilic blocks created a micelle with a compact micelle core decorated by a swollen hydrophilic corona. The increase in ϱ accompanied the decrease in length of the terminal block consisting purely of hydrophilic or hydrophobic monomer, suggesting that decreasing terminal block size led to the least compact micelle core. The increased Rg evident with smaller terminal blocks was accompanied by an increase in Nagg, suggesting that a larger particle core with greater distribution of mass may permit increased recruitment of ELP chains to the assembly, as seen by the increased aggregation number of ELP-SVGI micelles.
Figure 8.

Trends in Rh, Rg, and Nagg, dependent on ELP block copolymer architecture. For those ELPs that exhibited temperature-triggered self-assembly, trends in Rh and Rg suggested changes in micelle size as well as changes in the distribution of mass within the micelle structure. Aggregation number, Nagg, increased with increasing micelle size.
In order to further characterize the ELP micelles, small-angle X-ray scattering (SAXS) spectra were acquired for the self-assembling polypeptides ELP-SVDSV and ELP-SVTSVS (Figure 9). The Guinier extrapolation was used to calculate Rg and Nagg (Table 3). Nagg was calculated by dividing the MW of the scatterer obtained from the Guinier extrapolation by the theoretical MW of a single polypeptide chain. The Rg and Nagg values calculated by SAXS (Table 3) were in good agreement with those calculated by SLS (Table 2).
Figure 9.

SAXS spectra and analytical model fits of self-assembled ELP-SVDSV and ELP-SVTSVS micelles. Relative to the spectrum for ELP-SVDSV, ELP-SVTSVS had a lower forwardscattering value, indicating a lower Nagg, and transitioned to q−2 at a higher q, indicating a smaller micelle size.
Table 3.
Characterization of self-assembled ELPs from Guinier fits of SAXS spectra.
| ELP | Nagg | Rg |
|---|---|---|
| SVTSVS | 34 | 10.3 |
| SVDSV | 127 | 16.7 |
Analytical shape models were then fit to the SAXS spectra in order to describe the morphology of the self-assembled micelles. The SAXS spectra were best fit by a block copolymer spherical micelle model (Figure 9, Table 4, model details in SI).46 The core radii and radii of gyration were broadly in agreement with both the Guinier approximation values (Table 3), indicating that the SAXS experiment was internally consistent, and with the static light scattering values (Table 2). A core water volume fraction of approximately 0.8 was fit for both micelles, indicating that the ELP micelle core was still highly hydrated, consistent with both small-angle scattering studies of other ELP micelles45 and micelles formed by classical PEO-PPO-PEO Pluronics.47
Table 4.
SAXS spectra analytical model fit parameters for self-assembled ELP-SVDSV and ELP-SVTSVS micelles.
Radius of the micelle core.
Rg of the corona chains.
Volume fraction water in the core.
To better understand the changes in Rg, Rh, and Nagg between ELP-SVDSV, ELP-SVGH, and ELP-SVGI we created a new family of self-assembling ELPs with terminal hydrophobic and hydrophilic blocks joined by a central blocky interface. Here the blocky interface was simply a short segment of alternating hydrophilic and hydrophobic blocks with sizes of 1, 5, or 10 pentapeptide monomer units, creating ELP-V-SVB1-S, ELP-V-SVB5-S, and ELP-V-SVB10-S, respectively (Figure 10A). These constructs mimicked the gradient ELP design, yet provided greater regularity in the architecture of the amphiphilic interface and provided a constant length of both the interface and the terminal blocks, thereby deconvoluting the effect of the interfacial block architecture on self-assembly. All blocky interface ELPs exhibited similar unimer-to-micelle CMTs, while their micelle-to-aggregate Tts increased between ELP-V-SVB10-S, ELP-V-SVB5-S, and ELP-V-SVB1-S (Figure 10B).
Figure 10.

Design and thermal characterization of blocky interface ELPs. A) Each ELP was composed of 40 terminal pentapeptides of the hydrophobic monomer VPGVG (red) and hydrophilic monomer VPGSG (blue), joined by a blocky region of alternating 1, 5, or 10 pentapeptide blocks. B) All blocky interface ELPs exhibited temperature-triggered self-assembly as determined by temperature-regulated turbidimetry at 100 µM (lines, left ordinate) and confirmed by DLS (dots, right ordinate). Multiple Rh values for a single temperature indicate multiple relaxation times.
These blocky interface ELPs displayed trends in Rg and Rh that depended on the interface architecture (Table 5). Micelles were smallest when the interface was composed of SVB1, the most highly mixed architecture of alternating amphiphilic monomers. The micelle size increased modestly as the block size in the interface increased to SVB5 and SVB10, suggesting that increased block size in the interface segment led to expansion of the self-assembled micelle. As seen in the previous family of self-assembled ELPs, Rg and Rh changed disproportionately between self-assembled structures such that ϱ increased with increasing block size in the interface, achieving a maximum value of 0.95 for ELP-V-SVB10-S. Interestingly, a trend in aggregation number was less evident for these ELPs, where only ELP-V-SVB10-S showed a slightly decreased Nagg relative to ELP-V-SVB1-S and ELP-V-SVB5-S, which had nearly equivalent Nagg. This suggested that size of the terminal blocks, a parameter that was held constant in this set of ELPs, might play an important role in determining aggregation number.
Table 5.
SLS and DLS characterization of blocky interface ELPs.
| ELP | Rg (nm) | Rh (nm) | ρ (Rg/Rh) | Nagg |
|---|---|---|---|---|
| V-SVB1-S | 19.9 (±2.86%) | 24.0 | 0.83 | 42 |
| V-SVB5-S | 22.1 (±1.29%) | 24.5 | 0.90 | 41 |
| V-SVB10-S | 23.7 (±1.83%) | 25.0 | 0.95 | 33 |
4. Conclusions
Creating polypeptides with precisely varied block architectures was made possible by using genetically designed and recombinantly synthesized ELPs, whose sequence and size are dictated with perfect precision. The LCST behavior and micellar self-assembly of ELPs provided easily observable measures to demonstrate the effect of block architecture on polymer characteristics. The sensitivity of the LCST behavior to even subtle changes in block architecture provided insight into the delicate relationship between block architecture and polypeptide behavior. Additionally, block architecture could control the size and morphology of self-assembled micelles, despite the invariant composition and length of the constituent polymers. Formation of micelles was observed with the presence of sufficiently long hydrophobic blocks, where micelle size and morphology could be controlled by building gradients between hydrophobic and hydrophilic blocks as well as by having a blocky interface with different block lengths. Exploring the behavior of this precisely defined model of block architecture serves to enrich the rational design of new multi-block copolymer materials with predictable behaviors tuned to their intended use in biomedical and industrial applications.
Supplementary Material
Acknowledgments
This work was supported by the NIH through grants to A.C. (R01GM061232, R01EB000188, and R01EB007205) and by the NSF through the Research Triangle MRSEC (DMR-11-21107). I.W. acknowledges support in the form of a National Science Foundation Graduate Research Fellowship (NSF-DGE-1106401). This work utilized facilities supported in part by the National Science Foundation under Agreement No. DMR-0944772. I.H. acknowledges support from the Bundesministerium für Bildung und Forschung (BMBF) via Project No. 05K13KT1. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. We acknowledge the Duke University Shared Materials and Instrumentation Facility for maintaining the SAXS instrument used in this work. We thank Michael Rubinstein (University of North Carolina, Chapel Hill) for useful discussions.
Footnotes
ELP sequences, SDS-PAGE of purified ELPs, turbidimetry plots, DLS and SLS analysis, and SAXS analytical model details. This material is available free of charge at http://pubs.acs.org.
References
- 1.Mai YY, Eisenberg A. Self-assembly of block copolymers. Chem Soc Rev. 2012;41(18):5969–5985. doi: 10.1039/c2cs35115c. [DOI] [PubMed] [Google Scholar]
- 2.Schacher FH, Rupar PA, Manners I. Functional Block Copolymers: Nanostructured Materials with Emerging Applications. Angew Chem Int Edit. 2012;51(32):7898–7921. doi: 10.1002/anie.201200310. [DOI] [PubMed] [Google Scholar]
- 3.Ruzette AV, Leibler L. Block copolymers in tomorrow's plastics. Nat Mater. 2005;4(1):19–31. doi: 10.1038/nmat1295. [DOI] [PubMed] [Google Scholar]
- 4.van Zoelen W, Buss HG, Ellebracht NC, Lynd NA, Fischer DA, Finlay J, Hill S, Callow ME, Callow JA, Kramer EJ, Zuckermann RN, Segalman RA. Sequence of Hydrophobic and Hydrophilic Residues in Amphiphilic Polymer Coatings Affects Surface Structure and Marine Antifouling/Fouling Release Properties. Acs Macro Lett. 2014;3(4):364–368. doi: 10.1021/mz500090n. [DOI] [PubMed] [Google Scholar]
- 5.Hamley IW. Ordering in thin films of block copolymers: Fundamentals to potential applications. Prog Polym Sci. 2009;34(11):1161–1210. [Google Scholar]
- 6.Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliver Rev. 2001;47(1):113–131. doi: 10.1016/s0169-409x(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 7.Dill KA. Dominant Forces in Protein Folding. Biochemistry. 1990;29(31):7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 8.Tanford C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. 2nd. Wiley-Interscience; New York: 1980. [Google Scholar]
- 9.Sommerdijk NA, Holder SJ, Hiorns RC, Jones RG, Nolte RJ. Self-assembled structures from an amphiphilic multiblock copolymer containing rigid semiconductor segments. Macromolecules. 2000;33(22):8289–8294. [Google Scholar]
- 10.Determan MD, Guo L, Thiyagarajan P, Mallapragada SK. Supramolecular selfassembly of multiblock copolymers in aqueous solution. Langmuir. 2006;22(4):1469–1473. doi: 10.1021/la0527691. [DOI] [PubMed] [Google Scholar]
- 11.Hadjiantoniou NA, Triftaridou AI, Kafouris D, Gradzielski M, Patrickios CS. Synthesis and characterization of amphiphilic multiblock copolymers: Effect of the number of blocks on micellization. Macromolecules. 2009;42(15):5492–5498. [Google Scholar]
- 12.Hadjiantoniou NA, Krasia-Christoforou T, Loizou E, Porcar L, Patrickios CS. Alternating amphiphilic multiblock copolymers: Controlled synthesis via RAFT polymerization and aqueous solution characterization. Macromolecules. 2010;43(6):2713–2720. [Google Scholar]
- 13.Bae YH, Huh KM, Kim Y, Park K-H. Biodegradable amphiphilic multiblock copolymers and their implications for biomedical applications. Journal of Controlled Release. 2000;64(1):3–13. doi: 10.1016/s0168-3659(99)00126-1. [DOI] [PubMed] [Google Scholar]
- 14.Sosnik A, Cohn D. Reverse thermo-responsive poly (ethylene oxide) and poly (propylene oxide) multiblock copolymers. Biomaterials. 2005;26(4):349–357. doi: 10.1016/j.biomaterials.2004.02.041. [DOI] [PubMed] [Google Scholar]
- 15.Halperin A. On the Collapse of Multiblock Copolymers. Macromolecules. 1991;24(6):1418–1419. [Google Scholar]
- 16.Hugouvieux V, Axelos MA, Kolb M. Amphiphilic multiblock copolymers: From intramolecular pearl necklace to layered structures. Macromolecules. 2008;42(1):392–400. [Google Scholar]
- 17.Zhang J, Lu Z-Y, Sun Z-Y. Self-assembly structures of amphiphilic multiblock copolymer in dilute solution. Soft Matter. 2013;9(6):1947–1954. [Google Scholar]
- 18.Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally- responsive polypeptides. Nature biotechnology. 1999;17(11):1112–5. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 19.MacEwan SR, Chilkoti A. Elastin-Like Polypeptides: Biomedical Applications of Tunable Biopolymers. Biopolymers. 2010;94(1):60–77. doi: 10.1002/bip.21327. [DOI] [PubMed] [Google Scholar]
- 20.Quiroz FG, Chilkoti A. Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat Mater. 2015;14(11):1164–1171. doi: 10.1038/nmat4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balu R, Knott R, Cowieson NP, Elvin CM, Hill AJ, Choudhury NR, Dutta NK. Structural ensembles reveal intrinsic disorder for the multi-stimuli responsive bio-mimetic protein Rec1-resilin. Scientific reports. 2015;5:10896. doi: 10.1038/srep10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elvin CM, Carr AG, Huson MG, Maxwell JM, Pearson RD, Vuocolo T, Liyou NE, Wong DC, Merritt DJ, Dixon NE. Synthesis and properties of crosslinked recombinant pro-resilin. Nature. 2005;437(7061):999–1002. doi: 10.1038/nature04085. [DOI] [PubMed] [Google Scholar]
- 23.Xia X-X, Wang M, Lin Y, Xu Q, Kaplan DL. Hydrophobic drug-triggered selfassembly of nanoparticles from silk-elastin-like protein polymers for drug delivery. Biomacromolecules. 2014;15(3):908–914. doi: 10.1021/bm4017594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beun LH, Storm IM, Werten MW, de Wolf FA, Cohen Stuart MA, de Vries R. From micelles to fibers: balancing self-assembling and random coiling domains in pH-responsive silk-collagen-like protein-based polymers. Biomacromolecules. 2014;15(9):3349–3357. doi: 10.1021/bm500826y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urry DW. Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J Phys Chem B. 1997;101(51):11007–11028. [Google Scholar]
- 26.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Temperature triggered self-assembly of polypeptides into multivalent spherical micelles. Journal of the American Chemical Society. 2008;130(2):687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hassouneh W, Zhulina EB, Chilkoti A, Rubinstein M. Elastin-like Polypeptide Diblock Copolymers Self-Assemble into Weak Micelles. Macromolecules. 2015;48(12):41834195. doi: 10.1021/acs.macromol.5b00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee TAT, Cooper A, Apkarian RP, Conticello VP. Thermo-reversible selfassembly of nanoparticles derived from elastin-mimetic polypeptides. Adv Mater. 2000;12(15) 1105-+ [Google Scholar]
- 29.Sallach RE, Wei M, Biswas N, Conticello VP, Lecommandoux S, Dluhy RA, Chaikof EL. Micelle density regulated by a reversible switch of protein secondary structure. Journal of the American Chemical Society. 2006;128(36):12014–12019. doi: 10.1021/ja0638509. [DOI] [PubMed] [Google Scholar]
- 30.McDaniel JR, Weitzhandler I, Prevost S, Vargo KB, Appavou MS, Hammer DA, Gradzielski M, Chilkoti A. Noncanonical Self-Assembly of Highly Asymmetric Genetically Encoded Polypeptide Amphiphiles into Cylindrical Micelles. Nano Lett. 2014;14(11):6590–6598. doi: 10.1021/nl503221p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin L, Castro E, Ribeiro A, Alonso M, Rodriguez-Cabello JC. Temperature- triggered self-assembly of elastin-like block co-recombinamers:the controlled formation of micelles and vesicles in an aqueous medium. Biomacromolecules. 2012;13(2):293–8. doi: 10.1021/bm201436y. [DOI] [PubMed] [Google Scholar]
- 32.Wright ER, McMillan RA, Cooper A, Apkarian RP, Conticello VP. Thermoplastic elastomer hydrogels via self-assembly of an elastin-mimetic triblock polypeptide. Adv Funct Mater. 2002;12(2):149–154. [Google Scholar]
- 33.Wu XY, Sallach R, Haller CA, Caves JA, Nagapudi K, Conticello VP, Levenston ME, Chaikof EL. Alterations in physical cross-linking modulate mechanical properties of two-phase protein polymer networks. Biomacromolecules. 2005;6(6):3037–3044. doi: 10.1021/bm0503468. [DOI] [PubMed] [Google Scholar]
- 34.Wu XY, Sallach RE, Caves JM, Conticello VP, Chaikof EL. Deformation responses of a physically cross-linked high molecular weight elastin-like protein polymer. Biomacromolecules. 2008;9(7):1787–1794. doi: 10.1021/bm800012x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi P, Aluri S, Lin YA, Shah M, Edman M, Dhandhukia J, Cui HG, MacKay JA. Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. Journal of Controlled Release. 2013;171(3):330–338. doi: 10.1016/j.jconrel.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacEwan SR, Chilkoti A. Controlled Apoptosis by a Thermally Toggled Nanoscale Amplifier of Cellular Uptake. Nano Lett. 2014;14(4):2058–2064. doi: 10.1021/nl5002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim DW, Nettles DL, Setton LA, Chilkoti A. In situ cross-linking of elastin-like polypeptide block copolymers for tissue repair. Biomacromolecules. 2008;9(1):222–30. doi: 10.1021/bm7007982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez-Osorio H, Juarez-Campo M, Diebold Y, Girotti A, Alonso M, Arias FJ, Rodriguez-Cabello JC, Garcia-Vazquez C, Calonge M. Genetically engineered elastin- like polymer as a substratum to culture cells from the ocular surface. Current eye research. 2009;34(1):48–56. doi: 10.1080/02713680802542053. [DOI] [PubMed] [Google Scholar]
- 39.Ribeiro A, Arias FJ, Reguera J, Alonso M, Rodriguez-Cabello JC. Influence of the Amino-Acid Sequence on the Inverse Temperature Transition of Elastin-Like Polymers. Biophys J. 2009;97(1):312–320. doi: 10.1016/j.bpj.2009.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDaniel JR, Mackay JA, Quiroz FG, Chilkoti A. Recursive directional ligation by plasmid reconstruction allows rapid and seamless cloning of oligomeric genes. Biomacromolecules. 2010;11(4):944–52. doi: 10.1021/bm901387t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyer DE, Chilkoti A. Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules. 2004;5(3):846–51. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 42.Kline S. Reduction and analysis of SANS and USANS data using IGOR Pro. Journal of Applied Crystallography. 2006;39(6):895–900. [Google Scholar]
- 43.MacEwan SR, Hassouneh W, Chilkoti A. Non-chromatographic purification of recombinant elastin-like polypeptides and their fusions with peptides and proteins from Escherichia coli. Journal of visualized experiments : JoVE. 2014;88 doi: 10.3791/51583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDaniel JR, Macewan SR, Dewhirst M, Chilkoti A. Doxorubicin-conjugated chimeric polypeptide nanoparticles that respond to mild hyperthermia. Journal of Controlled Release. 2012;159(3):362–7. doi: 10.1016/j.jconrel.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garanger E, MacEwan SR, Sandre O, Brûlet A, Bataille L, Chilkoti A, Lecommandoux S. Structural Evolution of a Stimulus-Responsive Diblock Polypeptide Micelle by Temperature Tunable Compaction of its Core. Macromolecules. 2015;48(18):6617–6627. [Google Scholar]
- 46.Pedersen JS, Gerstenberg MC. Scattering Form Factor of Block Copolymer Micelles. Macromolecules. 1996;29(4):1363–1365. [Google Scholar]
- 47.Svensson M, Alexandridis P, Linse P. Phase behavior and microstructure in binary block copolymer/selective solvent systems: Experiments and theory. Macromolecules. 1999;32:637–645. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.